

Abstract
Summary: Genetic correlations are the genome-wide aggregate effects of causal variants affecting multiple traits. Traditionally, genetic correlations between complex traits are estimated from pedigree studies, but such estimates can be confounded by shared environmental factors. Moreover, for diseases, low prevalence rates imply that even if the true genetic correlation between disorders was high, co-aggregation of disorders in families might not occur or could not be distinguished from chance. We have developed and implemented statistical methods based on linear mixed models to obtain unbiased estimates of the genetic correlation between pairs of quantitative traits or pairs of binary traits of complex diseases using population-based case–control studies with genome-wide single-nucleotide polymorphism data. The method is validated in a simulation study and applied to estimate genetic correlation between various diseases from Wellcome Trust Case Control Consortium data in a series of bivariate analyses. We estimate a significant positive genetic correlation between risk of Type 2 diabetes and hypertension of ~0.31 (SE 0.14, P = 0.024).
Availability: Our methods, appropriate for both quantitative and binary traits, are implemented in the freely available software GCTA (http://www.complextraitgenomics.com/software/gcta/reml_bivar.html).
Contact: hong.lee@uq.edu.au
Supplementary Information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Recently, we have developed new methods to estimate the proportion of variation in quantitative traits (Yang et al., 2010, 2011) or in liability to disease that is associated with single-nucleotide polymorphisms (SNPs) (Lee et al., 2012, 2011). The methods use very distant relationships between individuals so that estimates are unlikely to be confounded with shared family environment effects. The methodology can be extended to estimation of the genetic covariance and hence genetic correlation between different disorders that is tagged by SNPs to provide estimates of genome-wide pleiotropy. Evidence for a genetic correlation between disorders estimated directly by interrogation of the genome could have an important impact on the design of future genetic and functional studies for medical nosology and may provide new insights for novel treatments across disorders.
The aim of this study is to estimate genome-wide pleiotropy using genome-wide association studies (GWAS) case–control data for different diseases or disorders. For binary disease traits, we derive valid statistical approaches to obtain unbiased estimates of comorbidity interpretable on the scale of liability to disease. We develop computationally efficient algorithms for estimation. The method is applied to estimate the genetic correlation between hypertension (HT) and type 2 diabetes (T2D), bipolar disorder (BD) and rheumatoid arthritis (RA), BD and T2D or HT and RA from Wellcome Trust Case Control Consortium (WTCCC) GWAS data.
2 METHODS
2.1. Bivariate linear mixed model and efficient AIREML
We used a standard bivariate linear mixed model (Thompson, 1973). The models can be written as
where y is a vector of observations for trait, b1 and b2 are vectors of fixed effects, g1 and g2 are vectors of random polygenic effects for each individual in both trait 1 and 2 and e1 and e2 are residuals for trait 1 and 2, respectively. X and Z are incidence matrices for the effects b and g, respectively. The variance covariance matrix (V) is defined as
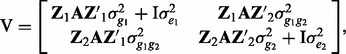 |
where A is the genomic similarity relationship matrix based on SNP information (Yang et al., 2010) and I is an identity matrix, 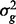 ,
, 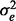 and
and 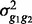 , which are genetic variance, residual variance and covariance between g1 and g2. Lee and Van der Werf (2006) showed that the method of average information (AI) matrices derived directly from the V is much more efficient computationally than the original AI algorithms (Gilmour et al., 1995; Johnson and Thompson, 1995). Following equation (8) in Lee and Van der Werf (2006) , the AI matrix for the bivariate model can be derived as
, which are genetic variance, residual variance and covariance between g1 and g2. Lee and Van der Werf (2006) showed that the method of average information (AI) matrices derived directly from the V is much more efficient computationally than the original AI algorithms (Gilmour et al., 1995; Johnson and Thompson, 1995). Following equation (8) in Lee and Van der Werf (2006) , the AI matrix for the bivariate model can be derived as
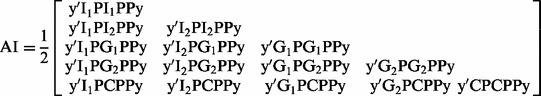 |
where 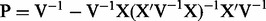 ,
, 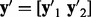 ,
, 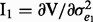 ,
, 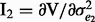 ,
, 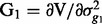 ,
, 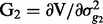 and
and 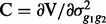 .
.
2.2. Correlation on the scale of liability is approximately the same as that on the observed risk scale
For disease traits when the y phenotype vectors contain only 1 for cases and 0 for controls, a liability threshold model can be written to link unobserved continuous liability to the observed discrete scale of disease (Falconer, 1965)
| (1) |
where l is a vector of liability phenotypes which are distributed as N(0, 1) in the population, g* is a vector of random additive genetic effects on the liability scale which are distributed N (0, 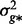 ) and e* is a vector of random residuals on the liability scale distributed with N(0,
) and e* is a vector of random residuals on the liability scale distributed with N(0, 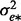 ). The probit link function links liability to the probability of y = 1, and g* on the scale of liability can be approximated by a linear function of g on the observed 0–1 scale (Dempster and Lerner, 1950). Using this linear approximation, the correlation between two diseases is the same on both the observed and liability scale (Gianola, 1982; Höschele et al., 1987). When samples are ascertained (typical in case–control studies), the genetic value on the observed scale can be defined with an ascertainment correcting factor as (Lee et al., 2011)
). The probit link function links liability to the probability of y = 1, and g* on the scale of liability can be approximated by a linear function of g on the observed 0–1 scale (Dempster and Lerner, 1950). Using this linear approximation, the correlation between two diseases is the same on both the observed and liability scale (Gianola, 1982; Höschele et al., 1987). When samples are ascertained (typical in case–control studies), the genetic value on the observed scale can be defined with an ascertainment correcting factor as (Lee et al., 2011)
| (2) |
where gcc is genetic values on the observed scale in a case–control study, c is a constant, K is the disease prevalence in the population, P is the proportion of the sample that are cases and z is the height of the standard normal probability density function that truncates the proportion K. From equation (2), the covariance between genetic values on the observed scale with ascertained samples can be written as
| (3) |
From equation (3) it is clear that that even when samples are ascertained, the correlation is the same on both observed and liability scales, because an approximate linear relationship exists between the genetic values on the different scales.
2.3. Simulation study
In order to confirm the derivation that the genetic correlation is approximately the same on both observed and liability scales when samples are ascertained, we performed a simulation study. The simulation procedure was similar to that in Lee et al. (2011) except that two traits were simulated with a genetic correlation between them (described in the Supplementary material).
2.4. Application to genome-wide genotype data
We applied our method to estimate genetic correlation between HT and T2D, BD and RA, BD and T2D, or HT and RA using WTCCC GWAS data (WTCCC, 2007) , following stringent quality control (QC) as described in the supplementary material. Since there are two control groups in the WTCCC data, i.e. 1958 cohort controls and NBS controls, we used 1958 cohort controls for the first trait, and NBS controls for the second trait. In a confirmation study, we swapped the control groups i.e. NBS for the first trait and 1958 cohort for the second trait.
We estimated a test statistic by dividing the square of the estimated genetic correlation coefficient by its approximate sampling variance and calculated a p-value from this test statistic assuming that it is distributed as a chi-square with 1 degree of freedom.
3 RESULTS
In simulations the estimated genetic correlation on the observed scale was close to the true values when using various combinations of true heritability and population prevalence (Supplementary Table S1). This confirms that the estimated genetic correlation is approximately the same on both observed and liability scales [Equation (3)]. Previously we have shown that if misdiagnosis occurs between the two disorders, then the expectation of the estimate of the genetic correlation coefficient can be non-zero even when the true genetic correlation is zero (Wray et al., 2012).
The estimated genetic correlation between HT and T2D was 0.31 (SE = 0.14 and p-value = 0.023) (Supplementary Table S2), indicating that genetic factors for HT and T2D are positively correlated. However, estimates for genetic correlation between BD and RA, BD and T2D, or HT and RA were not significantly different from zero (Supplementary Table S2). In a confirmation study switching control groups between the first and second trait (Supplementary Table S2), the genetic correlation between HT and T2D was 0.32 (SE = 0.14 and P = 0.024). Again, none of other analyses had significant genetic correlations (Supplementary Table S2). None of the parameter estimates differed significantly between our original and confirmation analyses. We previously demonstrated that the application of our stringent QC process resulted in estimated genetic variance not significantly different from zero if we conduct a dummy case-control analysis using these two control sets but treating one set as a cases (Lee et al., 2011) (h2 = 0.06, SE 0.11).
Supplementary Material
ACKNOWLEDGEMENTS
We thank QBI IT team. S.H.L. acknowledges the use of the Genetic Cluster Computer for carrying out a part of simulations. The cluster is financially supported by the Netherlands Scientific Organization (NOW 480-05-003). This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the WTCCC data is available from www.wtccc.org.uk. Funding for the WTCCC project was provided by the Wellcome Trust under award 076113.
Funding: The Australian National Health and Medical Research Council (613672, 613601, 613608 and 1011506), the Australian Research Council (DP1093502 and FT0991360) and the US National Institute of Health (GM075091).
Conflict of Interest: none declared.
REFERENCES
- Dempster ER, Lerner IM. Heritability of threshold characters. Genetics. 1950;35:212–236. doi: 10.1093/genetics/35.2.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer DS. The inheritance of liability to certain diseases, estimated from the incidence among relatives. Ann. Hum. Genet. 1965;29:51–71. [Google Scholar]
- Gianola D. Theory and analysis of threshold characters. J. Anim. Sci. 1982;54:1079–1096. [Google Scholar]
- Gilmour AR, et al. Average information REML: an efficient algorithm for variance parameters estimation in linear mixed models. Biometrics. 1995;51:1440–1450. [Google Scholar]
- Höschele I, et al. Estimation of variance components with quasi-continuous data using Bayesian methods. J. Anim. Breed. Genet. 1987;104:334–349. [Google Scholar]
- Johnson DL, Thompson R. Restricted maximum likelihood estimation of variance components for univariate animal models using sparse matrix techniques and average information. J. Dairy Sci. 1995;78:449–456. [Google Scholar]
- Lee SH, et al. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat. Genet. 2012;44:247–250. doi: 10.1038/ng.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Van der Werf JHJ. An efficient variance component approach implementing an average information REML suitable for combined LD and linkage mapping with a general complex pedigree. Genet. Sel. Evol. 2006;38:25–43. doi: 10.1186/1297-9686-38-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, et al. Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet. 2011;88:294–305. doi: 10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R. The estimation of variance and covariance components with an application when records are subject to culling. Biometrics. 1973;29:527–550. [Google Scholar]
- Wray NR, et al. Impact of diagnostic misclassification on estimation of genetic correlations using genome-wide genotypes. Eur. J. Hum. Genet. 2012;20:668–674. doi: 10.1038/ejhg.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WTCCC Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, et al. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.