

Abstract
Motivation: We previously reported the development of a highly accurate statistical algorithm for identifying β-barrel outer membrane proteins or transmembrane β-barrels (TMBBs), from genomic sequence data of Gram-negative bacteria (Freeman,T.C. and Wimley,W.C. (2010) Bioinformatics, 26, 1965–1974). We have now applied this identification algorithm to all available Gram-negative bacterial genomes (over 600 chromosomes) and have constructed a publicly available, searchable, up-to-date, database of all proteins in these genomes.
Results: For each protein in the database, there is information on (i) β-barrel membrane protein probability for identification of β-barrels, (ii) β-strand and β-hairpin propensity for structure and topology prediction, (iii) signal sequence score because most TMBBs are secreted through the inner membrane translocon and, thus, have a signal sequence, and (iv) transmembrane α-helix predictions, for reducing false positive predictions. This information is sufficient for the accurate identification of most β-barrel membrane proteins in these genomes. In the database there are nearly 50 000 predicted TMBBs (out of 1.9 million total putative proteins). Of those, more than 15 000 are ‘hypothetical’ or ‘putative’ proteins, not previously identified as TMBBs. This wealth of genomic information is not available anywhere else.
Availability: The TMBB genomic database is available at http://beta-barrel.tulane.edu/.
Contact: wwimley@tulane.edu
1 INTRODUCTION
The transmembrane β-barrel (TMBB) is the dominant architecture of the membrane-spanning proteins found in the outer membranes of Gram-negative bacteria (Schulz, 2000; Wimley, 2003). Although it has been estimated that approximately 3% of the proteins in Gram-negative organisms encode TMBBs (Freeman, Jr and Wimley, 2010; Wimley, 2002, 2003), fewer than 100 unique structures have been determined experimentally (Jayasinghe et al., 2001). The relatively slow progress in TMBB structural characterization is partially a consequence of the hydrophobic nature of membrane proteins, which makes standard techniques for purification and crystallization, or structure determination by nuclear magnetic resonance (NMR), more difficult. While structural information is scarce, genome sequencing is advancing (and accelerating) rapidly. Thus, computational approaches that can utilize the available structural data to predict and identify TMBBs, even in the absence of efficient structure determination, are needed.
The complete genomic sequences of thousands of organisms have become available in recent years, and a variety of computational tools have been developed to sift through the abundance of genomic data toward the goal of identifying the structures and functions of the genes that are expressed as proteins. Since this process relies strongly on homology to known structures, the resultant genomic database annotations for TMBBs are noticeably sparse. In the last decade, many computational prediction tools have proven at least partially successful in using the information from the limited examples of TMBBs to distinguish between TMBBs and non-TMBBs or to predict the structure and topology of TMBBs (Bagos et al., 2004a, b; Bagos et al., 2005; Bigelow and Rost, 2006; Bigelow et al., 2004; Freeman, Jr. and Wimley, 2010; Garrow et al., 2005a, b; Gromiha and Suwa, 2006a, b; Gromiha et al., 2005; Hayat and Elofsson, 2012; Hayat et al., 2011a, b; Imai et al., 2011; Jacoboni et al., 2001; Martelli et al., 2002; Mirus and Schleiff, 2005; Ou et al., 2008, 2010; Park et al., 2005; Randall et al., 2008; Remmert et al., 2009; Savojardo et al., 2011; Schleiff et al., 2003; Singh et al., 2011; Tsirigos et al., 2011; Waldispuhl et al., 2006; Wimley, 2002). Among the various approaches are statistical models, neural networks, hidden Markov models, k-nearest neighbor, and support vector machines. We recently published a prediction method (Freeman, Jr and Wimley, 2010) based on the statistical prevalence of the amino acids in the transmembrane segments of known structures, which was shown to accurately discriminate TMBBs from non-TMBBs.
Here, we describe a comprehensive bioinformatics database (TMBB-DB: the transmembrane beta barrel database: http://beta-barrel.tulane.edu) generated by using the Freeman–Wimley prediction method (Freeman, Jr and Wimley, 2010) to analyze the protein-coding sequences of all bacterial chromosomes belonging to Gram-negative bacteria (currently over 600). For each of the 1.9 million proteins in the database, we provide an overall β-barrel score that can be used to predict if a sequence is likely to encode a TMBB. We also provide the Freeman–Wimley β-strand and β-hairpin score profiles, which are useful for structure and topology prediction. Furthermore, the sequences were analyzed for the presence of an N-terminal signal sequence because most known TMBB precursors encode export signals at the N-terminus that allow translocon-dependent transport across the inner membrane (Petersen et al., 2011). Finally, we have also included a prediction for transmembrane α-helices. This helps to eliminate false positives because TMBBs generally do not also have TM helices. The information in the database is sufficient for accurate identification of most β-barrel membrane proteins in known Gram-negative genomes. There are more than 15 000 ‘hypothetical’ or ‘putative’ proteins in the database which are almost certainly β-barrel membrane proteins. This wealth of genomic information is not available anywhere else.
2 METHODS
2.1 Proteomic dataset
In order to create a database with the most complete set of TMBB predictions, we downloaded complete chromosomal data from NCBI Entrez. The pre-translated, FASTA-formatted protein-coding sequences from 610 chromosomes were downloaded as text from NCBI. This set mostly includes sequences from Gram-negative bacteria, but sequences from some acid-fast bacteria (mycobacteria) and related Gram-positive bacteria are also included. This dataset represents the proteomes of 540 organisms, as there are several bacterial species, which have more than one complete chromosome. There are currently 1 881 712 protein sequences for which predictions were made, and thus number will increase with annual updates to the database.
2.2 Predictions
All sequences in the database were analyzed using the Freeman–Wimley algorithm and given a β-barrel score, which was shown to be one of the most accurate predictors of TMBBs available (Freeman, Jr and Wimley, 2010; Wimley, 2002). Briefly, the Freeman–Wimley algorithm uses the amino acid abundances found in the transmembrane strands of TMBBs of known structure to identify patterns in a test sequence consistent with TMBB architecture, namely a β-hairpin with two 10-residue dyad repeat motifs separated by a turn sequence of about five residues. Most TMBBs also encode N-terminal signal peptides in the precursor sequence because they are exported into the periplasmic space through the translocon machinery in the inner membrane. Thus, we used the SignalP 4 server to predict whether or not a sequence was likely exported through the inner membrane (Claros et al., 1997; Petersen et al., 2011). While users of the database can choose whatever identification criteria they deem appropriate, we have shown that for a sequence to be positively predicted as a TMBB it should have a β-barrel score >45 [the TMBB score is on an arbitrary scale as discussed elsewhere (Freeman, Jr and Wimley, 2010)]. It must also have a signal peptide predicted by the SignalP 4 algorithm, which uses different algorithms for helical transmembrane (TM) proteins and non-TM proteins (Petersen et al., 2011).
2.3 Score conversion
The predictions made in this database use an updated version of the Freeman–Wimley analysis algorithm. Previously, the β-barrel score was a positive integer value that ranged from zero to an indeterminate maximum value of ~500. In the TMBB-DB, the scores have been converted to a probability such that the value ranges from 0 to 1 and are more representative of the probability function, indicating a positive prediction. The β-barrel score modification is based on the positive predictive value (PPV) of the β-barrel score observed in genomic sequences from Escherichia coli. To calculate the PPV function, we analyzed all proteins in E. coli that have definitive annotations (not ‘putative’, ‘predicted’, ‘unknown’ or ‘hypothetical’). This well-annotated dataset contains 2418 proteins, including 40 TMBBs. Importantly, all 40 positives are identified as TMBBs in the both the National Center for Biotechnology Information (NCBI) (RefSeq) and the UniProt sequence databases. The area under the receiver operator characteristic (ROC) curve (true positive rate versus false-positive rate) for our prediction algorithm, using this annotated E. coli dataset is 0.998, showing again that the Freeman–Wimley β-barrel score is a powerful TMBB identification tool:
| (1) |
The PPV is the proportion of true positives (TP) predicted to all positive predictions, including false positives (FP). This measurement can be evaluated at a prediction threshold to estimate the probability that a positive prediction is correct at that threshold. The PPV of the E. coli dataset was fit with a sigmoidal model [equation (2)] where f(β) is the probability that the β-barrel score corresponds to a correct positive prediction, k is a growth constant, xc is the center of the curve through the portion with maximum slope, and β is the β-barrel score of a sequence:
| (2) |
By fitting the known proteins of E. coli we obtain k = 0.04596 and Xc = 66, where, Xc represents the midpoint of the sigmoidal PPV curve.
2.4 Database design
All prediction results have been deposited into a publicly available database (http://beta-barrel.tulane.edu/). The website is constructed in two major layers where the data layer is a MySQL database, and the user interface layer is driven by Apache/PHP. Users may navigate to a sequence by browsing the list of chromosomes and then browsing the prediction data for that chromosome. Alternately, the user can search by β-barrel score range and/or SignalP 4 score cutoff. Proteins outside of the β-barrel score range or below the SignalP cutoff can either be hidden or shown at the users’ discretion. An advanced search feature allows the user to combine search terms with Boolean functions. In addition to β-barrel score and SignalP score, valid search terms include as follows: GI (NCBI) accession number, UniProt accession number, RefSeq accession number (protein or genome), protein name and organism. Full data and sequence files are downloadable at any stage. We have also made the entire database available for download as a flat text file.
When the user selects a sequence of interest, they are redirected to a page that has graphical representations of the Freeman–Wimley analysis profile and the Wimley–White hydrophobicity profile (Wimley and White, 1996) of the sequence. The raw data are also available. For each protein, the user may also follow links to the UniProt database entry or the NCBI entry for that sequence. The user may also conduct a BLAST search where the accession number for the sequence is provided as the search query. If a sequence of interest to a user is not included in the database, user-friendly web version of the Freeman–Wimley analysis software is available as is a downloadable, standalone version for single sequences or collections. Updates to the database will be done annually using a script-based, semi-automatic updater that we have developed.
3 RESULTS AND DISCUSSION
3.1 TMBB-DB: the transmembrane β-barrel database
We have employed a novel approach to increase the number of correctly identified TMBBs in all available Gram-negative chromosomes using an orthogonal prediction strategy (Fig. 1). Our structurally based, statistical prediction method (Freeman, Jr and Wimley, 2010) was used to score the protein sequences with the probability that they encode TMBB domains. There are a number of available TMBB prediction algorithms (see above), and although they cannot always be directly compared, it appears that the Freeman–Wimley algorithm (Freeman, Jr and Wimley, 2010) and the BetaWare algorithm (Savojardo et al., 2011) are the most accurate available. Here we use the Freeman–Wimley algorithm because it is very easy to adapt it to analyze millions of sequences rapidly. The software for single sequence or whole genome analysis is freely available on the TMBB-DB page (http://beta-barrel.tulane.edu) and on our main β-barrel page (http://www.tulane.edu/~biochem/WW/Barrel.html).
Fig. 1.
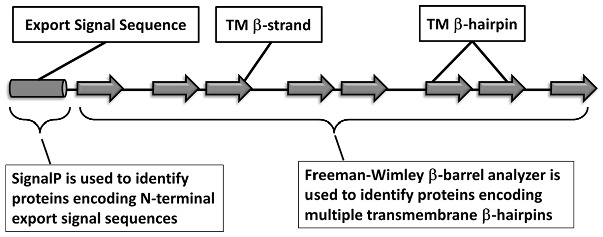
Prediction of TMBBs using signal peptide prediction and TMBB structure prediction. Schematic of a TMBB-encoding protein shows signal peptide predicted using SignalP (Petersen et al., 2011) and TMBB domain using Freeman–Wimley β-barrel analysis (Freeman, Jr and Wimley, 2010). The Freeman–Wimley algorithm is as follows: (i) amino acid abundances are assigned to each residue within a 10-residue sliding window. The three terminal residues at either end are assigned as interfacial residues and the remainder as bilayer core residues. (ii) The β-strand score is the sum of scores within the window, where peaks indicate the middle of predicted β-strands. (iii) The β-hairpin score is a sum of β-strand scores, where two β-strand peaks are separated by a five-residue gap (representing the hairpin turn). (iv) The topology prediction shown in the β-hairpin score is simplified to a single value called the β-barrel score
A revised scoring convention has been adopted for TMBBs to simplify the interpretation of the Freeman–Wimley β-barrel score. The β-barrel score is a cardinal value that ranges from zero to an indefinite maximum of around 500. The known sequences from the proteome of E. coli were used as a test case to evaluate the relationship between the β-barrel score and the probability of making a correct positive prediction. This dataset included 40 known TMBBs and 2378 known non-TMBBs. Other proteins were ignored for this calculation. The PPV or probability that a positive prediction is correct increases as the β-barrel score threshold increases (Fig. 2). The data fit a sigmoidal model [equation (2)] with an R2 of 0.996. The parameter values given by the fit are k = 0.04596, and Xc = 66. This model is used to convert a β-barrel score into a probability between 0 and 1, which simplifies the interpretation of a β-barrel score. Using this equation, the threshold β-barrel score that we use in our work (45) corresponds to a converted probability score of 0.28. However, we note that the TMBB-DB database contains all data for all proteins so that users can identify TMBBs using any criteria they choose.
Fig. 2.
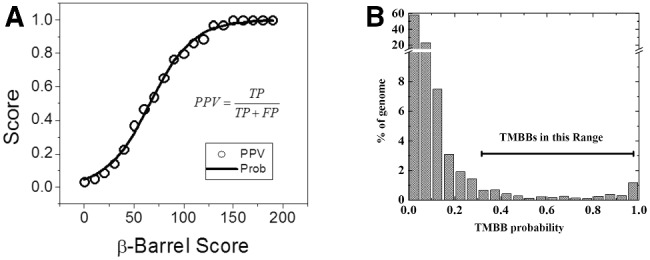
From β-barrel score to probability. (A) The probability that a particular β-barrel score is a positive prediction can be estimated from an assessment of the PPV and a function of the arbitrary β-barrel score for a given dataset. The dataset used to assess the PPV of the β-barrel score included the annotated genes from an E. coli chromosome. There were 40 TMBBs and 2378 non-TMBBs identified out of 5253 total sequences (see the text). Proteins annotated as hypothetical, putative, or predicted were excluded. The PPV was plotted as a function of β-barrel score and was fit with a sigmoidal function. (B) Histogram of β-barrel probability for the E. coli O157 genome. Based on our previous work, a protein with probability value above 0.28 (β-barrel score above 45) is a strong candidate TMBB
The majority of TMBBs also encode an N-terminal export signal peptide in the translated precursor, which signals secretion across the inner membrane via the Sec translocon machinery (White and von Heijne, 2004). The SignalP 4 server is the most accurate signal peptide prediction tool available (Petersen et al., 2011). It follows that an ideal positive prediction for a TMBB is a sequence that has a high β-barrel score and high SignalP score. Based on our previous work (Freeman, Jr and Wimley, 2010), we use a threshold β-barrel score of 45 (probability score of 0.28, see above) for putatively identifying TMBBs. While the user can select any desired signalP cutoff, we wanted to use an inclusive SignalP score cutoff as default in the database to match the inclusive β-barrel probability cutoff (PPV) of 0.28. To find an appropriate value, we examined the scores for known TMBBs and non-TMBBs. A SignalP cutoff of 0.3 identifies 93% of all known, classical, certain TMBBs (annotated as ‘porin’, ‘outer membrane protein’ or TonB-dependent receptor/transporter and having a very high β-barrel probability between 0.99 and 1.00) as having a signal sequence, while identifying only 13% of non-TMBBs as having one, a value that is similar to the percentage in the entire genome database.
A test case was performed to illustrate how the combined Freeman–Wimley and SignalP analyses can be used to predict unidentified TMBB-encoding sequences. The analysis data of a known and predicted TMBB are shown in Figure 3. The known TMBB, the 8-stranded colicin S4 receptor in Escherichia coli (OmpW), received strongly positive scores in SignalP and has a β-barrel score ranked in the 64th percentile among positively predicted sequences, with a β-barrel probability score of 0.97. ECS5270 (gi 38 704 255) a hypothetical protein in E. coli of similar length to OmpW received similarly high scores in SignalP and has a β-barrel score ranked in the 74th percentile among positively predicted sequences with a β-barrel probability score of 0.99. A BLAST search suggested that the hypothetical sequence is a member of the KdgM superfamily which is a family of porins associated with acidic sugar transport (Blot et al., 2002) and biofilm formation (Blot et al., 2002; Freeman, Jr et al., 2011). This test demonstrates how potential biologically relevant TMBB sequences can be identified using the outlined prediction strategy despite the uninformative annotations commonly found in genomic databases.
Fig. 3.
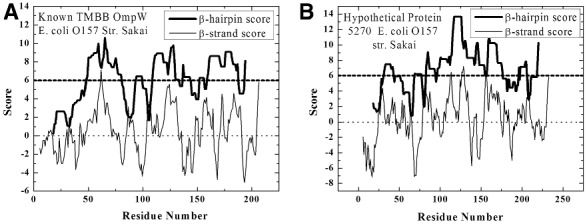
TMBB prediction analysis. Sample protein sequences were analyzed for propensity to fold into TMBBs. The Freeman–Wimley prediction plots show the β-strand and β-hairpin prediction scores over the sequences of OmpW and ECS5270 (gi 38704255), a predicted TMBB from E. coli O157 (strain Sakai). Threshold values are indicated for each. The β-hairpin threshold is an empirical value. Most TMBBs have a significant portion of their sequence above the threshold. The structure of OmpW has been solved (Hong et al., 2006). It has eight transmembrane β-strands arranged in four hairpins. The topology prediction of the hypothetical protein looks very similar to OmpW, which suggests it has a similar structure. The signal peptide scores for both sequences indicate that a signal peptide is present. Although it has not been studied experimentally, ECS5270 is predicted with high confidence to be a TMBB using this orthogonal strategy of TMBB and signal peptide prediction
The β-barrel score profile of an entire genome can provide useful insights into the biology of an organism. The plots shown in Figure 4 exemplify the scoring profiles of entire genomes. The legend in the upper right shows the quadrants based on the TMBB score and signal peptide score cutoff values that we use. The three organisms chosen were Francisella tularensis, which causes the sometimes fatal disease, tularemia, an enteropathogenic strain of E. coli, and Flavobacterium johnsoniae. Approximately 2.3% of the proteins in E. coli and in F. tularensis have β-barrel probabilities greater than 0.28 and signal sequence scores greater than 0.3 and thus are predicted to encode TMBBs. The genome of F. tularensis has only about 1/3 as many proteins as E. coli, but it also has about 1/3 as many TMBBs. In contrast, while having a slightly smaller proteome than E. coli, 10.6% of the proteins in F. johnsoniae are predicted to encode TMBBs, one of the highest proportions observed in any genome. The surprisingly large proportion of outer membrane proteins in F. johnsoniae may correlate with two of its highly unusual capabilities, gliding cell motility, and cell surface-localized chitin digestion (McBride, 2004). An indication of the discriminatory power of the combined prediction analysis is shown by the fact that sequences that concurrently have predicted signal sequences and high β-barrel scores typically include mostly known TMBBs and unknown or hypothetical proteins. Obvious false positives are rare in this quadrant.
Fig. 4.
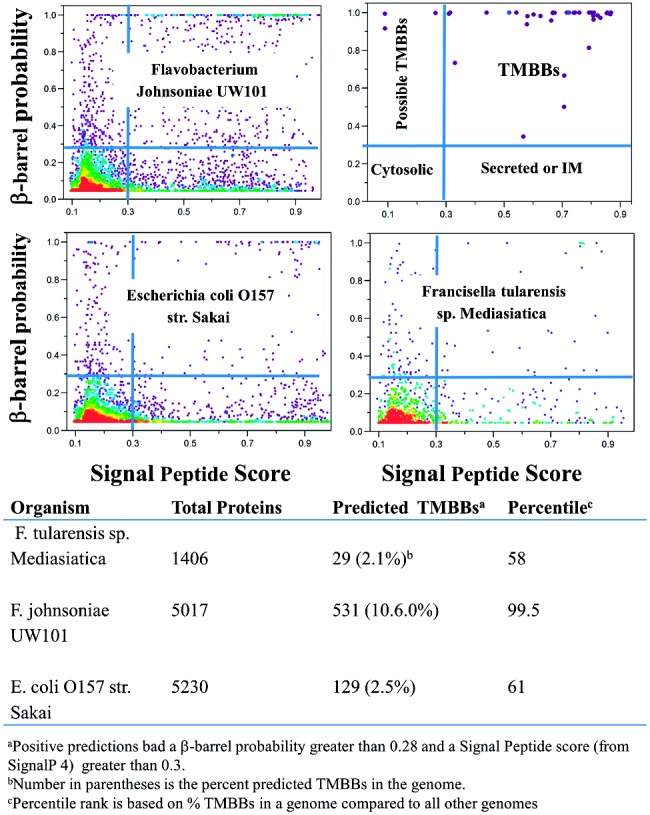
Analysis of sample genomes. Three sample genomes of Gram-negative organisms were analyzed using the dual strategy of TMBB prediction and signal peptide prediction. The results for each protein in each genome are plotted in the two-dimensional scatter plot. The coloring of the plots indicates the density of points in an area, with red being the most dense, and purple being the least dense. The plot in the upper right shows a legend identifying where certain classes of proteins will populate the scatterplots. In this panel, we also show values for the 40 known TMBBs of E. coli. These genomic data show that most proteins score near zero using both prediction methods (Signal peptide and β-barrel). TMBBs, i.e. sequences with high β-barrel scores and high signal peptide prediction probability, range in these examples from 2.1 to 10.6% of the genomes
However, a small proportion of known TMBBs do not have recognizable signal sequences, and these will fall in the upper left quadrant. To assess the rate of false negative predictions that could result from this, we used the TMBB-DB database. We searched for all proteins annotated as ‘outer membrane protein,’ which are mostly defined by homology to known TMBBs and are thus true positives. We found that 4508 of 5348 (84%) of these proteins have a signal sequence. If the search is restricted to the very high-scoring sequences (β-barrel probability >0.9), the proportion is even higher: 2141/2361 or 91% of the sequences have a signal sequence. Proteins annotated as ‘porin’ have signal sequences at a rate of 94% (2585/2757) and proteins annotated as ‘TonB-dependent transporters’ or ‘TonB-dependent receptors’ have signal sequences at a rate of 3890/4293 or 91%. We conclude that >90% of the classical, well-described classes of constitutive TMBBs have signal sequences that are recognized by SignalP, compared to about 16% of the proteins in the genomes, overall.
We then examined ‘autotransporter’ proteins in the database because members of that class of transmembrane β-barrel protein are thought to have signal sequences less frequently (Kim et al., 2006). Autotransporters have an N-terminal secreted protein domain and a C-terminal β-barrel domain, which specifically transports the secreted portion of the chain across the outer membrane (Kim et al., 2006). In the TMBB-DB, 96% (647/672) of proteins annotated as ‘autotransporter’ have β-barrel probability score over the threshold of 0.28, but only 73% of those have signal sequence scores greater than 0.3. Even in the autotransporters with the highest β-barrels core (β-barrel probability >0.9), which are very likely to be true positives, only 71% (377/552) contain recognizable signal sequences. While the proportion of autotransporters with signal sequences is lower than for classical TMBBs, it is still much higher than the background abundance of about 16%. We conclude that the combination of β-barrel probability and signal sequence allows for the identification of most TMBBs in these genomes, including autotransporters.
3.2 Comparison to other databases
There are many algorithms available online for the identification of TMBBs or for structure/topology prediction (Bagos et al., 2004a, b; Bagos et al., 2005; Bigelow et al., 2004; Bigelow and Rost, 2006; Freeman, Jr and Wimley, 2010; Garrow et al., 2005a, b; Gromiha and Suwa, 2006a, b; Gromiha et al., 2005; Hayat et al., 2011a, b; Hayat and Elofsson, 2012; Imai et al., 2011; Jacoboni et al., 2001; Martelli et al., 2002; Mirus and Schleiff, 2005; Ou et al., 2008, 2010; Park et al., 2005; Randall et al., 2008; Remmert et al., 2009; Savojardo et al., 2011; Schleiff et al., 2003; Singh et al., 2011; Tsirigos et al., 2011; Waldispuhl et al., 2006; Wimley, 2002). These include the Freeman–Wimley statistical algorithm we used here (Freeman, Jr and Wimley, 2010; Wimley, 2002), which is one of the most accurate and easiest to use for the analysis of whole genomes. There are other databases containing predicted TMBBs in genomes (Remmert et al., 2009; Tsirigos et al., 2011). The information available in these published resources is also useful. However, what we have provided by constructing the TMBB-DB database is unique, and thus complements and extends existing databases (Figure 5). We have used a highly accurate algorithm to score all of the proteins in all available Gram-negative genomes and have added signal peptide and transmembrane helix predictions for added stringency. Our annotated, and up-to-date, database of all proteins in Gram-negative genomes enables the most accurate and comprehensive identification of transmembrane β-barrel membrane proteins available. This information has utility in fields ranging from bioinformatics (e.g. genome annotation) to medicine (e.g. vaccine design).
Fig. 5.
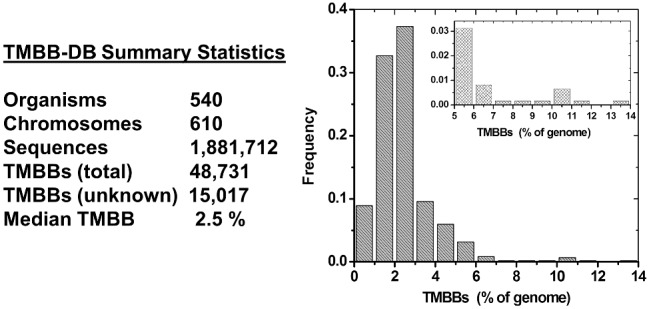
Overall database statistics. ‘Left’: Current database coverage. A positively predicted TMBBs has a β-barrel probability >0.28 and a SignalP score >0.3. Unknown TMBBs are positively predicted unknown or hypothetical proteins. ‘Right’: Histogram of TMBB in % of genome. ‘Inset’: The region above 5%, highlighting the few genomes with high-TMBB content. Most genomes have between 1 and 5% TMBBs and the median value is 2.5%
Funding: NIH (GM060000).
Conflict of Interest: none declared.
REFERENCES
- Bagos PG, et al. Evaluation of methods for predicting the topology of beta-barrel outer membrane proteins and a consensus prediction method. BMC Bioinformatics. 2005;6:7. doi: 10.1186/1471-2105-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagos PG, et al. A Hidden Markov Model method, capable of predicting and discriminating beta-barrel outer membrane proteins. BMC Bioinformatics. 2004a;5:29. doi: 10.1186/1471-2105-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagos PG, et al. PRED-TMBB: a web server for predicting the topology of beta-barrel outer membrane proteins. Nucleic Acids Res. 2004b;32:W400–W404. doi: 10.1093/nar/gkh417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigelow H, Rost B. PROFtmb: a web server for predicting bacterial transmembrane beta barrel proteins. Nucleic Acids Res. 2006;34:W186–W188. doi: 10.1093/nar/gkl262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigelow HR, et al. Predicting transmembrane beta-barrels in proteomes. Nucleic Acids Res. 2004;32:2566–2577. doi: 10.1093/nar/gkh580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blot N, et al. The oligogalacturonate-specific porin KdgM of Erwinia chrysanthemi belongs to a new porin family. J. Biol. Chem. 2002;277:7936–7944. doi: 10.1074/jbc.M109193200. [DOI] [PubMed] [Google Scholar]
- Claros MG, et al. Prediction of N-terminal protein sorting signals. Curr. Opin. Struct. Biol. 1997;7:394–398. doi: 10.1016/s0959-440x(97)80057-7. [DOI] [PubMed] [Google Scholar]
- Freeman TC, Jr, et al. The prediction and characterization of YshA, an unknown outer-membrane protein from Salmonella typhimurium. Biochim. Biophys. Acta. 2011;1808:287–297. doi: 10.1016/j.bbamem.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman TC, Jr, Wimley WC. A highly accurate statistical approach for the prediction of transmembrane beta-barrels. Bioinformatics. 2010;26:1965–1974. doi: 10.1093/bioinformatics/btq308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrow AG, et al. TMB-Hunt: a web server to screen sequence sets for transmembrane beta-barrel proteins. Nucleic Acids Res. 2005a;33:W188–W192. doi: 10.1093/nar/gki384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrow AG, et al. TMB-Hunt: an amino acid composition based method to screen proteomes for beta-barrel transmembrane proteins. BMC Bioinformatics. 2005b;6:56. doi: 10.1186/1471-2105-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromiha MM, et al. TMBETA-NET: discrimination and prediction of membrane spanning beta-strands in outer membrane proteins. Nucleic Acids Res. 2005;33:W164–W167. doi: 10.1093/nar/gki367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromiha MM, Suwa M. Discrimination of outer membrane proteins using machine learning algorithms. Proteins. 2006a;63:1031–1037. doi: 10.1002/prot.20929. [DOI] [PubMed] [Google Scholar]
- Gromiha MM, Suwa M. Influence of amino acid properties for discriminating outer membrane proteins at better accuracy. Biochim. Biophys. Acta. 2006b;1764:1493–1497. doi: 10.1016/j.bbapap.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Hayat S, Elofsson A. BOCTOPUS: improved topology prediction of transmembrane beta barrel proteins. Bioinformatics. 2012;28:516–522. doi: 10.1093/bioinformatics/btr710. [DOI] [PubMed] [Google Scholar]
- Hayat S, et al. Statistical analysis and exposure status classification of transmembrane beta barrel residues. Comput. Biol. Chem. 2011a;35:96–107. doi: 10.1016/j.compbiolchem.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Hayat S, et al. Prediction of the exposure status of transmembrane beta barrel residues from protein sequence. J. Bioinform. Comput. Biol. 2011b;9:43–65. doi: 10.1142/s0219720011005240. [DOI] [PubMed] [Google Scholar]
- Hong H, et al. The outer membrane protein OmpW forms an eight-stranded beta-barrel with a hydrophobic channel. J. Biol. Chem. 2006;281:7568–7577. doi: 10.1074/jbc.M512365200. [DOI] [PubMed] [Google Scholar]
- Imai K, et al. Eukaryote-wide sequence analysis of mitochondrial beta-barrel outer membrane proteins. BMC Genomics. 2011;12:79. doi: 10.1186/1471-2164-12-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoboni I, et al. Prediction of the transmembrane regions of beta-barrel membrane proteins with a neural network-based predictor. Protein Sci. 2001;10:779–787. doi: 10.1110/ps.37201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasinghe S, et al. MPtopo: a database of membrane protein topology. Protein Sci. 2001;10:455–458. doi: 10.1110/ps.43501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, et al. Protein-translocating trimeric autotransporters of gram-negative bacteria. J. Bacteriol. 2006;188:5655–5667. doi: 10.1128/JB.01596-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli PL, et al. A sequence-profile-based HMM for predicting and discriminating beta barrel membrane proteins. Bioinformatics. 2002;18 (Suppl. 1):S46–S53. doi: 10.1093/bioinformatics/18.suppl_1.s46. [DOI] [PubMed] [Google Scholar]
- McBride MJ. Cytophaga-flavobacterium gliding motility. J. Mol. Microbiol. Biotechnol. 2004;7:63–71. doi: 10.1159/000077870. [DOI] [PubMed] [Google Scholar]
- Mirus O, Schleiff E. Prediction of beta-barrel membrane proteins by searching for restricted domains. BMC Bioinformatics. 2005;6:254. doi: 10.1186/1471-2105-6-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou YY, et al. TMBETADISC-RBF: discrimination of beta-barrel membrane proteins using RBF networks and PSSM profiles. Comput. Biol. Chem. 2008;32:227–231. doi: 10.1016/j.compbiolchem.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Ou YY, et al. Prediction of membrane spanning segments and topology in beta-barrel membrane proteins at better accuracy. J. Comput. Chem. 2010;31:217–223. doi: 10.1002/jcc.21281. [DOI] [PubMed] [Google Scholar]
- Park KJ, et al. Discrimination of outer membrane proteins using support vector machines. Bioinformatics. 2005;21:4223–4229. doi: 10.1093/bioinformatics/bti697. [DOI] [PubMed] [Google Scholar]
- Petersen TN, et al. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Randall A, et al. TMBpro: secondary structure, beta-contact and tertiary structure prediction of transmembrane beta-barrel proteins. Bioinformatics. 2008;24:513–520. doi: 10.1093/bioinformatics/btm548. [DOI] [PubMed] [Google Scholar]
- Remmert M, et al. HHomp—prediction and classification of outer membrane proteins. Nucleic Acids Res. 2009;37:W446–W451. doi: 10.1093/nar/gkp325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savojardo C, et al. Improving the detection of transmembrane beta-barrel chains with N-to-1 extreme learning machines. Bioinformatics. 2011;27:3123–3128. doi: 10.1093/bioinformatics/btr549. [DOI] [PubMed] [Google Scholar]
- Schleiff E, et al. Prediction of the plant beta-barrel proteome: a case study of the chloroplast outer envelope. Protein Sci. 2003;12:748–759. doi: 10.1110/ps.0237503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz GE. b-Barrel membrane proteins. Curr. Opin. Struct. Biol. 2000;10:443–447. doi: 10.1016/s0959-440x(00)00120-2. [DOI] [PubMed] [Google Scholar]
- Singh NK, et al. TMBHMM: a frequency profile based HMM for predicting the topology of transmembrane beta barrel proteins and the exposure status of transmembrane residues. Biochim. Biophys. Acta. 2011;1814:664–670. doi: 10.1016/j.bbapap.2011.03.004. [DOI] [PubMed] [Google Scholar]
- Tsirigos KD, et al. OMPdb: a database of {beta}-barrel outer membrane proteins from Gram-negative bacteria. Nucleic Acids Res. 2011;39:D324–D331. doi: 10.1093/nar/gkq863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldispuhl J, et al. Predicting transmembrane beta-barrels and interstrand residue interactions from sequence. Proteins. 2006;65:61–74. doi: 10.1002/prot.21046. [DOI] [PubMed] [Google Scholar]
- White SH, von Heijne G. The machinery of membrane protein assembly. Curr. Opin. Struct. Biol. 2004;14:397–404. doi: 10.1016/j.sbi.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Wimley WC. Toward genomic identification of beta-barrel membrane proteins: composition and architecture of known structures. Protein Sci. 2002;11:301–312. doi: 10.1110/ps.29402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley WC. The versatile beta-barrel membrane protein. Curr. Opin. Struct. Biol. 2003;13:404–411. doi: 10.1016/s0959-440x(03)00099-x. [DOI] [PubMed] [Google Scholar]
- Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996;3:842–884. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]