

Non-ribosomal peptides are a large class of microbial metabolites that incorporate both proteinogenic and non-proteinogenic amino acids. The elegant assembly-line enzymology of NRPS systems allows diverse monomeric units to be assembled into a broad array of mature metabolites.[1] Molecules derived from NRPS systems and related hybrid systems have both proven and postulated utility as therapeutics for infectious disease, cancer, and a variety of other human ailments. Efforts to discover and engineer new variants constitute a vigorous field of research.
The kutzneride family of antifungal antibiotics exemplifies the type of chemical diversity that can be found in NRPS metabolites: five residues in these hexadepsipeptides are non-proteinogenic amino acids, and the sixth is a hydroxy acid (Scheme 1A and Figure S1). [2] Among the unusual building blocks is piperazate (Piz), a 6-member cyclic amino acid containing a rare N–N bond. Piperazate is found in a number of additional NRPS derived metabolites including the immunosuppressant sanglifehrin,[3] the antibiotic monamycin,[4] and the anticancer agents polyoxypeptin,[5] and himastatin.[6] It is typically linked to neighboring residues via amide bond formation at N2, though bond formation at N1 is reported for the sanglifehrin family. [3] The Piz core skeleton is observed to be further tailored by desaturation, hydroxylation, and chlorination; four variants are seen in the kutzneride family alone (Figure 1B). [7]
Scheme 1.
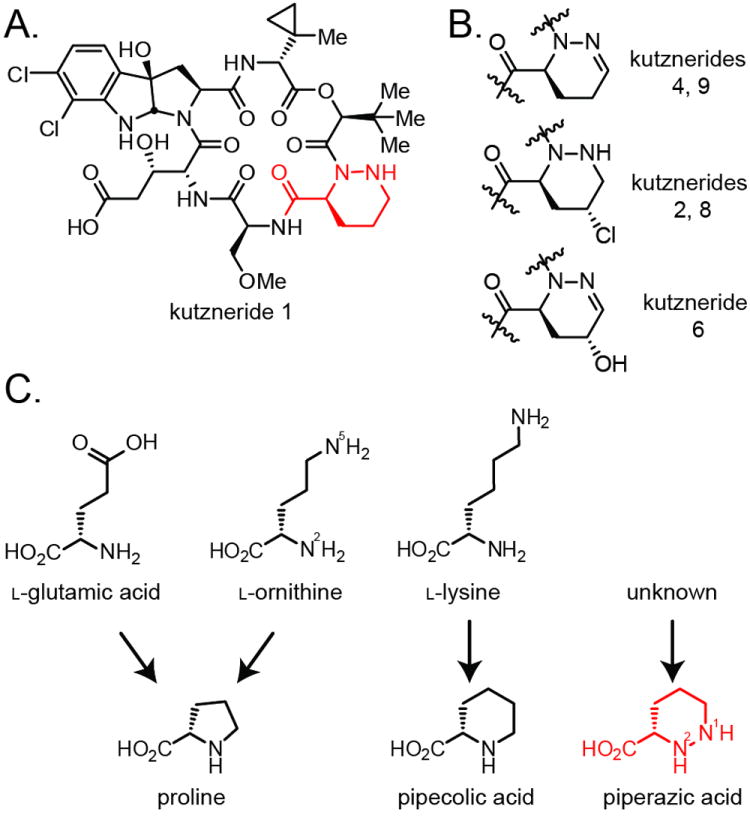
A) Kutzneride 1 displays a diverse set of monomeric building blocks. The piperazate residue (red) is also present in kutznerides 3, 5, and 7. B) Three additional Piz variants are found as indicated in the kutzneride family of compounds. C) Biosynthetic precursors to cyclic amino acids proline and pipecolic acid. The origin of piperazic acid has remained poorly defined.
Figure 1.
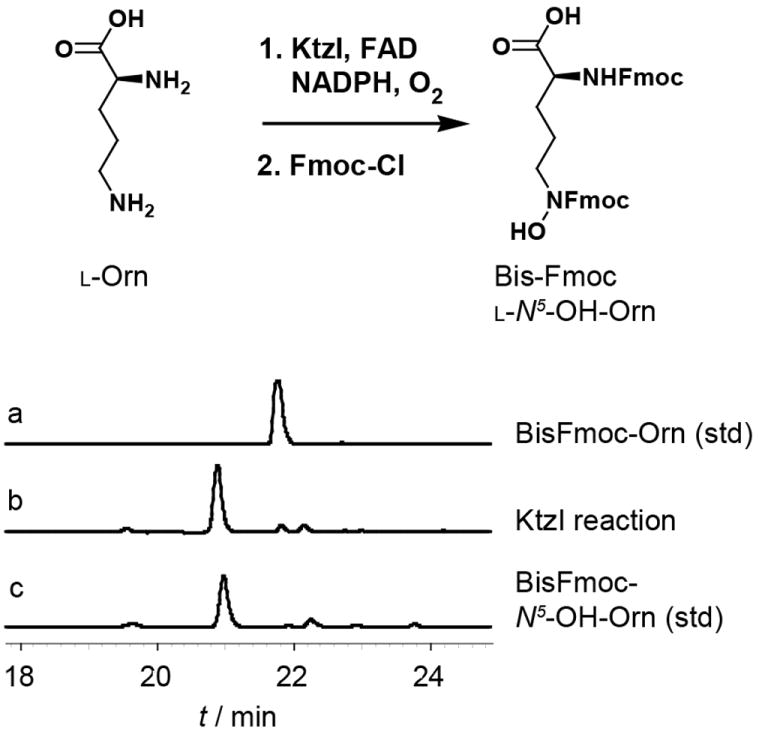
Purified KtzI catalyses the conversion of Orn to N5-OH-Orn using FAD as cofactor and NAD(P)H and O2 as cosubstrates. HPLC-UV (280 nm) traces indicate product formation relative to standards as the bisFmoc-derivatives.
Cyclic amino acids can have profound effects on the structure of mature peptide metabolites by adopting conformations that support turns in peptide chains. Proline has a well-defined role promoting turns in protein structure. Experimental and computational studies of model dipeptides indicate that piperazate residues, and especially the 1,6-dehydro-Piz derivative, show a marked preference for the turn-inducing conformation relative to pipecolic acid and even proline. [8] Cyclic amino acids are therefore recognized as unique elements of the NRPS family and their biosynthesis has been studied in varying contexts. As a primary metabolite, proline is derived from either reduction of glutamate to the γ-semialdehyde or oxidation of ornithine to the same (Scheme 1C). [9] Spontaneous cyclization and further reduction complete the transformation. Alternatively, ornithine can be converted to proline in a single step by the action of cyclodeaminases. [10] Pipecolate, a one-carbon homologation of proline, is observed as a component of secondary metabolites including the immunosuppressive rapamycin where it serves as a mimic of proline when bound to the target prolyl isomerases of the FKBP family. In Streptomyces hygroscopicus, pipecolate is derived directly from lysine via a cyclodeaminase transformation catalyzed by RapL.[11]
While piperazoyl moieties have been targets of synthetic chemistry efforts,[12] relatively little is known about the biosynthesis of piperazate itself. As the aza-analogue of pipecolic acid, piperazate could plausibly derive from ornithine via net oxidation of N2 and N5 (ornithine numbering system) to generate the N1–N2 bond (piperazic acid numbering system). The biosynthetic origin of piperazic acid was investigated previously in the context of the monamycin and polyoxypeptin metabolites. [13] In each report isotopically-labeled ornithine was tested as a precursor to piperazate but the lack of label incorporation led to the conclusion that ornithine was not converted to piperazate. Instead, both studies noted incorporation of glutamine and glutamic acid and proposed these as possible branch points from primary metabolism into the piperazate biosynthetic pathway. Separate studies of valanimycin biosynthesis indicated that an N–O bond is a likely precursor to the N–N bond observed in this antibiotic. [14] These various insights led to an early proposal for the biosynthesis of C5-substituted piperazates wherein glutamine is first reduced to an ornithine-like enamine intermediate prior to N-hydroxylation, C5-modification and cyclization to Piz (Scheme S1). [15]
The kutzneride gene cluster was the first Piz-producing cluster to be cloned and sequenced allowing a unique opportunity to revisit Piz biosynthesis. [16] Analysis of the kutzneride gene cluster led to the identification of a gene ktzI whose protein product is homologous to lysine and ornithine N-hydroxylases such as IucD, CchB, VbsO, and PvdA. These four homologues are found in the biosynthetic pathways of siderophores from E. coli,[17] S. coelicolor,[18] R. etli,[19] and P. aeruginosa,[20] respectively, where they are involved in the formation of hydroxamic acid moieties. Because the kutznerides do not contain any N–O bonds in the mature metabolites, we initially proposed that KtzI could function as an N-hydroxylase participating in Piz biosynthesis. [16]
The current studies were initiated with biochemical characterization of the putative N-hydroxylase KtzI. A His-tag fusion of the enzyme was efficiently overproduced in E. coli and purified to homogeneity in a two-step procedure. Enzymatic activity was reconstituted in vitro by aerobic incubation in the presence of FAD, NADPH, and l-Orn (Figure 1). Product formation was assayed by derivatization with Fmoc-chloroformate followed by HPLC analysis and comparison to synthetic standards. Formation of the N5-OH-Orn product was dependent on FAD as the flavin cofactor (FMN was inactive) though the enzyme could accept either NADPH or NADH as the source of reducing equivalents (Figure S2). l-Lys and d-Orn were observed to be non-substrate effectors of the enzyme, modestly increasing the background rate of NADPH oxidation (Figure S3) but not leading to the formation of N-hydroxy products. d-Lys, l/d-Glu, l/d-Gln were not observed to act as substrates or effectors of KtzI. The previously proposed enamine substrate was not prepared for testing as a substrate due to its presumed instability in aqueous solution. [21] Pseudo-first order kinetic analysis of the enzyme with l-Orn as substrate revealed an apparent kcat of 6.96 ± 0.13 min-1 and KM of 0.091 ± 0.008 mM to provide an overall efficiency of 76.48 min-1mM-1 (Figure S2). This value is within the range reported for similar Lys/Orn N-hydroxylases[17-20] and suggests that Orn is a physiological substrate for KtzI.
Based on the results of our biochemical study, we have revisited the role of Orn and the KtzI product N5-OH-Orn in Piz biosynthesis. In the absence of a genetic system to assess the requirement for ktzI in kutzneride production by gene deletion tests, we developed a stable-isotope feeding strategy to assess incorporation of either ornithine or N5-OH-Orn into the piperazate moiety. Kutzneria cultures (10 mL) grown in MMN media for 5 days were extracted with organic solvent and analyzed by LCMS. Through targeted formula searching we positively identified peaks corresponding to the masses of all reported kutznerides (Figure S4). Selection of the dominant [M+H-H2O]+ ion for each formula and fragmentation via collision-induced dissociation yielded a small number of product ions (Figure S5). Among these ions, one varied between kutzneride family members based on the tailoring modifications predicted at the piperazate and glycolate residues. Correlation of these modifications to the observed masses allowed assignment of the fragmentations shown in Figure 3. Using nomenclature guidelines previously proposed for MSMS of cyclic peptides, we annotate this as the b02G’M’ ion, where G’ (glycolate) and M’ (methylcyclopropyl-glycine) denote the initial site of ring fragmentation (Figure S5).[22] The LC-MS/MS analysis provides two demonstrable benefits for this system. First, it is a technically simple methodology that allowed analysis of multiple kutzneride variants with minimal sample processing and no purification. Second, it is highly sensitive and therefore allowed us to miniaturize our culture conditions, maximize the concentrations of the isotopically-labeled precursors in rich media, yet still analyse metabolites produced at levels near 1 μg/mL.
Figure 3.
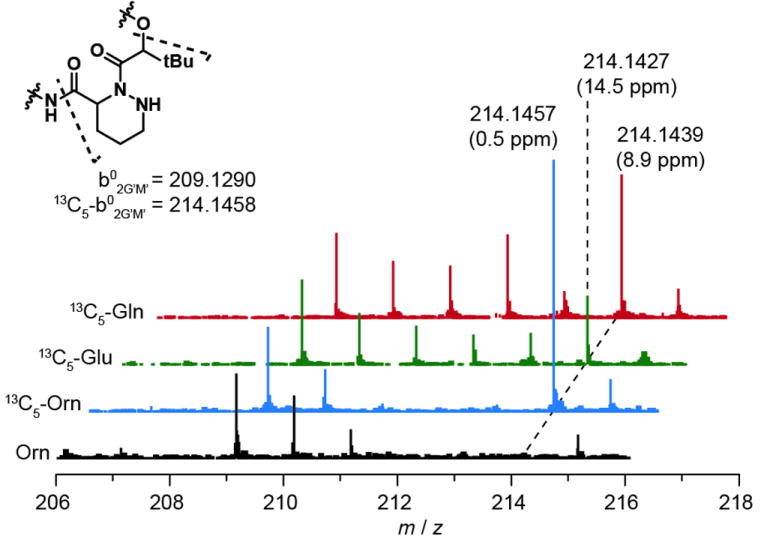
Feedings studies with 13C5-Gln and 13C5-Glu show evidence of intact incorporation of these precursors into the Piz residue. Each spectrum is normalized to signal at 209, the 37Cl213C1 isotopomer of unlabelled compound. Data were acquired on an Agilent 6520 Q-TOF mass spectrometer. Observed masses and errors are indicated for select peaks.
To test the hypothesis that ornithine and N5-OH-Orn are each biosynthetic precursors to piperazate, we purchased 13C5-Orn and prepared 13C5-N5-OH-Orn via a synthetic procedure (Scheme S2). Despite multiple steps to initially protect the N2 of 13C5-Orn with Boc, we were able to obtain high overall yields (16%) of 13C5-N5-OH-Orn for feeding studies. The use of 13C5-labelled compounds was necessary to shift the isotopic envelope of labelled kutznerides beyond the ~4 dalton envelope typically observed for the dichlorinated metabolites. Labelled precursors were added to 10 mL production cultures 18–48 hours post inoculation. Compounds extracted at day 5 were analysed by LC/MS and LC/MSMS. Relative to a control feeding of unlabelled amino acids, the MS spectra of kutznerides produced in the presence of 13C5-Orn and 13C5-N5-OH-Orn showed the appearance of new isotope envelopes shifted 5 mass units (Figure S6). Under optimal conditions labelling reached an estimated 35% of total compound; labelling levels ranged from 2-35% across experimental replicates, but even low level incorporation could be clearly analysed by our method and the overall conclusions were consistent across experiments.
Localizing the sites of label incorporation in kutznerides 1/3 (diasteroemers) was achieved by MSMS analysis of the predicted envelope of the labelled compounds (i.e., starting 5 units above the unlabelled parent mass). In controls with unlabelled amino acids, the predominant parent ion selected by this method is the natural 37Cl213C1 isotopomer and a small amount of b02G’M’ ion is observed at m/z = 209 (Figure 2A). When produced in cultures supplemented with either 13C5-Orn or 13C5-N5-OH-Orn, kutznerides 1/3 fragment to generate strong peaks at 214.1429 (10.7 ppm error) and 214.1452 (0.00 ppm error) corresponding to 13C5-b02G’M’ ion; peaks at 209/210 are evident when label incorporation is low due to the concomitant selection of parent 37Cl213C1 ions from unlabelled kutznerides though their relative intensity is significantly diminished. The b3W’E’ ion (Figure S5), whose structure includes the glycolate residue but not the piperazate residue, does not show label incorporation after feeding experiments (Figure S7). Based on this observation, we conclude that the labels are specifically incorporated into the Piz residue and not the t-Bu-glycolate residue of the b02G’M’ ion.
Figure 2.
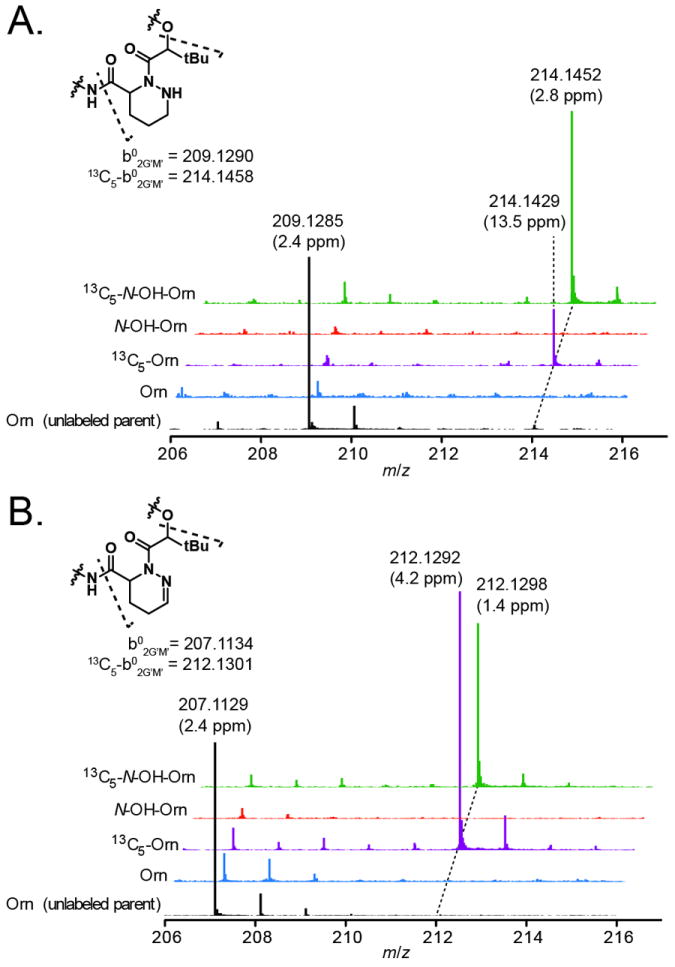
MS/MS analysis of kutznerides 1/3 (panel A) and kutznerides 4/6 (panel B) following indicated feedings of amino acid. Black spectra indicate reference b02G’M’ ion formation (inset structure) from unlabelled parent compound, and are shown on a reduced scale for clarity; colored spectra indicate ion formation from the expected mass envelope of 13C5-labeled compounds. Data were acquired on an Agilent 6520 Q-TOF mass spectrometer. Observed masses and errors are indicated for select peaks.
Analysis of MSMS spectra for kutznerides 4/9 (diastereomers) shows a similar pattern (Figure 2B). The b02G’M’ ion at 207.1129 (2.4 ppm error) containing dehydro-Piz is shifted 5 mass units to 212.1292 (4.2 ppm error) and 212.1298 (1.4 ppm error) when the media is supplemented with 13C5-Orn or 13C5-N5-OH-Orn. Generation and interpretation of the MSMS spectra for kutznerides 2/8 is complicated by the overlapping Cl3 isotope envelopes for labelled and unlabelled compounds and by the low ion yield for the chlorinated b02G’M’ ion. With the highest label incorporation levels, production of labelled fragment ions was evident at 248 and 250 (Figure S8). The data support the conclusion that these three variations of the Piz residues are derived from a common acyclic precursor. Analysis of b02G’M’ ion from kutzneride 6 containing 5-hydroxy-dehydro-Piz was not possible due to low production of kutzneride 6 coupled with low yield of the b02G’M’ ion during fragmentation. However it might be reasonably assumed that this variant is produced via the same pathway.
For comparison to previous studies on monamycin and polyoxypeptin, we also supplemented production cultures of Kutzneria with either 13C5-Glu or 13C5-Gln to assess their incorporation. MS spectra of metabolites from these cultures showed label incorporation spread out over a broad isotope window (Figure S9). This expanded envelope is likely due to incorporation of these labels into both the hydroxyglutamate and piperazate residues, as well from scrambling of the labels by primary metabolism. MSMS analysis shows increased intensity at +1 intervals from the base 209 (kutznerides 1/3, Figure 3) and 207 (kutznerides 4/9, Figure S10) b02G’M’ ions indicating incorporation of label after metabolic cleavage of the C5 backbone; that substantially stronger signal is seen at 214 (kutznerides 1/3) and 212 (kutznerides 4/9) is suggestive of some intact incorporation of Glu and Gln into the b02G’M’ ion.
The apparent incorporation of Glu and Gln into Piz raises questions about the ordering of chemical intermediates in the pathway; Orn could, in theory, be converted to Glu via proline metabolism independent of N-hydroxylation. To determine if N5-OH-Orn is downstream of Orn in the Piz pathway, we performed a label suppression experiment. [23] Cultures were fed with 13C5-Orn and increasing amounts of unlabelled N5-OH-Orn. At a 9:1 ratio of 13C5-Orn to N5-OH-Orn (30 mg : 3.3 mg), label incorporation into the b02G’M’ ion of kutznerides 1/3 is reduced to ~23% intensity. At a 3:1 ratio (30 mg : 10 mg), label incorporation is barely detectable (Figure 4). A more dramatic effect is seen in the analysis of kutznerides 4/9 where label incorporation into the b02G’M’ ion is barely detectable at the 9:1 ratio (Figure S11). These data are consistent with N5-OH-Orn being a downstream metabolite from Orn.
Figure 4.
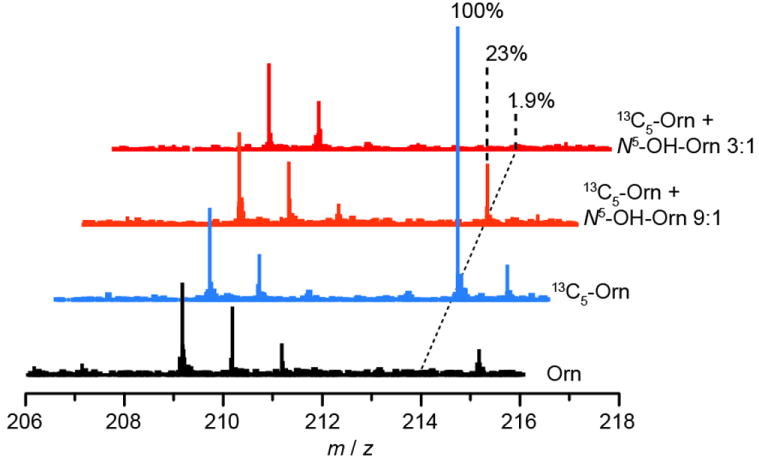
MSMS analysis of label suppression experiment. Addition of unlabelled N5-OH-Orn to cultures fed concurrently with 13C5-Orn suppresses label incorporation into the b02G’M’ ion. The magnitude of the effect, with substoichiometric addition of N5-OH-Orn, is consistent with N5-OH-Orn being a downstream metabolite in Piz biosynthesis. Data were acquired on an Agilent 6520 Q-TOF mass spectrometer. Each spectrum is normalized to signal at 209, the 37Cl213C1 isotopomer of unlabelled compound. Percentages indicate signal intensity at ~212.1296 relative to 13C5-Orn feeding.
Our results in this work argue against the initial model of Piz biosynthesis which posited an enamine intermediate derived from the direct reduction of glutamine (Scheme S1).[15] Instead, the current data support a model that includes Orn as the branch point from primary to secondary metabolism and N5-OH-Orn as a vital intermediate on the pathway (Scheme 2A). Our key finding is that Orn and N5-OH-Orn are incorporated intact into the Piz residues of the kutznerides. While Orn could plausibly be incorporated by initial conversion to Gln via proline metabolism, two observations suggest this is not the case. First, our biochemical experiments show that KtzI is an Orn N-hydroxylase with physiologically-relevant kinetic parameters. Second, the label suppression experiments suggest that N5-OH-Orn is downstream of Orn in the pathway. Taken together these data point to conversion of Orn to N5-OH-Orn as an early and essential step in Piz biosynthesis. Incorporation of Glu and Gln is plausibly explained by the known conversion of these compounds to Orn in primary metabolism.
Scheme 2.
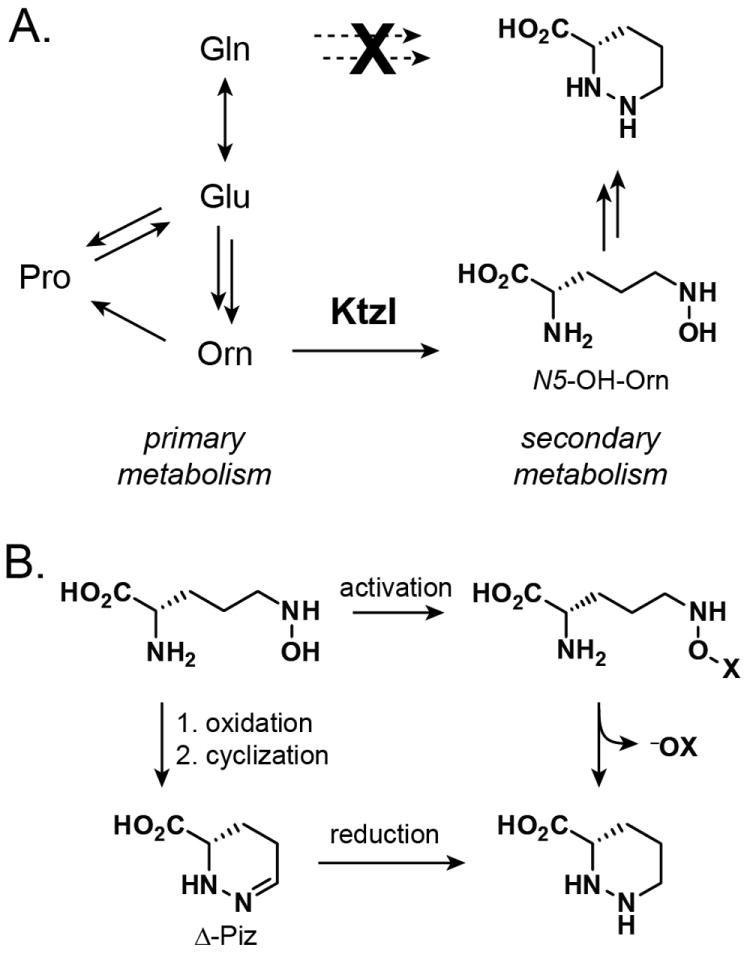
A) A model for Piz biosynthesis in Kutzneria spp. 744. Ornithine—not glutamine—serves as a branch point from primary metabolism, with formation of N5-OH-Orn by KtzI serving as the initial step in the pathway. B) Conversion of N5-OH-Orn to Piz could possibly be achieved by activation of the N-OH group and displacement by N2 (top) or by further oxidation to a nitroso species, cyclization, and reduction (bottom).
The subsequent chemical steps that may connect N5-OH-Orn to piperazate remain unclear. Nucleophilic substitution of the N5-OH by N2 seems unlikely based on leaving group considerations. Experimentally, spontaneous or KtzI-mediated cyclization of N-OH-Orn to Piz is not observed in aqueous solution. Activation of the hydroxyl group (e.g., by phosphorylation or sulfation) might facilitate such a transformation, though no candidate enzymes for such a transformation are encoded in the ktz cluster (Scheme 2B). An alternative hypothesis involves a second two-electron oxidation of N5-OH-Orn to the corresponding nitroso species which might cyclize, spontaneously or enzymatically, to give the dehydropiperazate residue. This molecule could then undergo reduction to give the core piperazate scaffold. KtzI was not observed to catalyze 4-electron oxidation of Orn, so any potential nitroso-producing activity would likely be encoded elsewhere in the genome.
It is unclear why the previous studies did not see incorporation of ornithine during their feeding experiments.[12] One possible explanation is that the strains which produce monamycin and polyoxypeptin do not take up exogenous ornithine readily leading to label exclusion. Alternatively, we cannot rule out the possibility that these systems biosynthesize piperazate using a distinct pathway that does not utilize ornithine as a starting point. However, two recent studies suggest that this newly proposed route for piperazate biosynthesis is not specific to Kutzneria. The genome of Streptomyces hygroscopicus contains a homologue of KtzI within a gene cluster encoding the production of the dimeric NRPS metabolite himastatin. [24] Deletion of the ktzI homolog was observed to abrogate production of himistatin, though a direct link to Piz biosynthesis has not yet been established experimentally. In another instance, a recently described gene cluster for the structurally distinct sanglifehrin family of hybrid NRPS/PKS metabolites also shows a predicted Orn N-hydroxylase, SfaB, which could be involved in production of the piperazate residue. [25]
The cyclic structure of piperazate and its defining N–N bond make this molecule a unique building block in NRPS-derived metabolites. Though its biosynthesis has been a topic of inquiry in the past in the context of monamycin and polyoxypeptin, the ability to analyse the genetic basis of kutzneride production has allowed us to revisit and expand upon previous hypotheses that lacked experimental support. From our biochemical analysis of KtzI and isotopic-labelling experiments, we can now define ornithine as a starting point for the pathway, and identify N5-OH-Orn as an early intermediate in the conversion of Orn to Piz. This insight can now serve to guide continuing efforts in this system and others to fully describe the chemical logic that underpins the N–N bond formation in this unique amino acid.
Supplementary Material
Acknowledgments
We thank William Wuest, Albert Bowers, Steven Malcolmson, Paul Thomas, and Neil Kelleher for helpful advice during the course of this work. Emma Doud is gratefully acknowledged for technical assistance with instrumentation. Funding for the work was provided by NIH grants GM020011 (C.T.W.) and GM082137 (R.K.). Fellowship support was provided by the NIH for J.R.H. (F32GM083464) and E.A.G. (F32GM087941).
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 2.a Broberg A, Menkis A, Vasiliauskas R. J Nat Prod. 2006;69:97–102. doi: 10.1021/np050378g. [DOI] [PubMed] [Google Scholar]; b Pohanka A, Menkis A, Levenfors J, Broberg A. J Nat Prod. 2006;69:1776–1781. doi: 10.1021/np0604331. [DOI] [PubMed] [Google Scholar]
- 3.Fehr T, Kallen J, Oberer L, Sanglier JJ, Schilling W. J Antibiot (Tokyo) 1999;52:474–479. doi: 10.7164/antibiotics.52.474. [DOI] [PubMed] [Google Scholar]
- 4.Bevan, Davies JS, Hassall CH, Morton RB, Phillips DAS. J Chem Soc C. 1971:514–522. [Google Scholar]
- 5.Umezawa K, Nakazawa K, Ikeda Y, Naganawa H, Kondo S. J Org Chem. 1999;64:3034–3038. doi: 10.1021/jo981512n. [DOI] [PubMed] [Google Scholar]
- 6.Leet JE, Schroeder DR, Golik J, Matson JA, Doyle TW, Lam KS, Hill SE, Lee MS, Whitney JL, Krishnan BS. J Antibiot (Tokyo) 1996;49:299–311. doi: 10.7164/antibiotics.49.299. [DOI] [PubMed] [Google Scholar]
- 7.The stereochemistry of 5-Cl-Piz is shown as assigned by Broberg, et al (ref 2a). Recent work on the enzymology of halogenation suggests the stereochemistry may differ: Jiang W, Heemstra JR, Jr, Forseth RR, Neumann CS, Manaviazar S, Schroeder FC, Hale KJ, Walsh CT. Biochemistry. 2011;50:6063–6072. doi: 10.1021/bi200656k.
- 8.Xi N, Alemany LB, Ciufolini MA. J Am Chem Soc. 1998;120:80–86. [Google Scholar]
- 9.Adams E, Frank L. Annu Rev Biochem. 1980;49:1005–1061. doi: 10.1146/annurev.bi.49.070180.005041. [DOI] [PubMed] [Google Scholar]
- 10.Goodman JL, Wang S, Alam S, Ruzicka FJ, Frey PA, Wedekind JE. Biochemistry. 2004;43:13883–13891. doi: 10.1021/bi048207i. [DOI] [PubMed] [Google Scholar]
- 11.Gatto GJ, Boyne MT, Kelleher NL, Walsh CT. J Am Chem Soc. 2006;128:3838–3847. doi: 10.1021/ja0587603. [DOI] [PubMed] [Google Scholar]
- 12.Oelke AJ, France DJ, Hofmann T, Wuitschik G, Ley SV. Nat Prod Rep. 2011;28:1445–1471. doi: 10.1039/c1np00041a. [DOI] [PubMed] [Google Scholar]
- 13.a Arroyo V, Hall MJ, Hassall CH, Yamasaki K. J Chem Soc Chem Commun. 1976:845–846. [Google Scholar]; b Umezawa K, Ikeda Y, Kawase O, Naganawa H, Kondo S. J Chem Soc Perkin Trans. 12001:1550–1553. [Google Scholar]
- 14.a Parry RJ, Li Y, Lii FL. J Am Chem Soc. 1992;114:10062–10064. [Google Scholar]; b Parry RJ, Li W, Cooper HN. J Bacteriol. 1997;179:409–416. doi: 10.1128/jb.179.2.409-416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Parry RJ, Li W. J Chem Soc Chem Commun. 1994:995–996. [Google Scholar]
- 15.Miller ED, Kauffman CA, Jensen PR, Fenical W. J Org Chem. 2007;72:323–330. doi: 10.1021/jo061064g. [DOI] [PubMed] [Google Scholar]
- 16.Fujimori DG, Hrvatin S, Neumann CS, Strieker M, Marahiel MA, Walsh CT. Proc Natl Acad Sci USA. 2007;104:16498–16503. doi: 10.1073/pnas.0708242104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thariath A, Socha D, Valvano MA, Viswanatha T. J Bacteriol. 1993;175:589–596. doi: 10.1128/jb.175.3.589-596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pohlmann V, Marahiel MA. Org Biomol Chem. 2008;6:1843–1848. doi: 10.1039/b801016a. [DOI] [PubMed] [Google Scholar]
- 19.Heemstra JR, Walsh CT, Sattely ES. J Am Chem Soc. 2009;131:15317–15329. doi: 10.1021/ja9056008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a Ge L, Seah SYK. J Bacteriol. 2006;188:7205–7210. doi: 10.1128/JB.00949-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Meneely KM, Barr EW, Bollinger JM, Lamb AL. Biochemistry. 2009;48:4371–4376. doi: 10.1021/bi900442z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Meneely KM, Lamb AL. Biochemistry. 2007;46:11930–11937. doi: 10.1021/bi700932q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Studies of imine hydrolysis in buffered aqueous conditions indicate a half-life of ~0.5 sec for a model compound that is a reasonable approximation for an ornithine-like enamine: Hine J, Craig JC, Underwood JG, Via FA. J Am Chem Soc. 1970;92:5194–5199.
- 22.Briefly, the b02G’M’ ion is formed by initial fragmentation of the cyclic molecule between the alkyl glycolate (G’) and methylcyclopropyl-glycine (M’) residues to give the acylium ion (b series), followed by neutral loss of four C-terminal residues and water to give a two-residue ion consisting of glycolate and piperazate. For nomenclature guidelines, see: Ngoka LCM, Gross ML. J Amer Soc Mass Spectrom. 1999;10:360–363. doi: 10.1016/S1044-0305(99)00006-9..; and Liu W-T, Ngo J, Meluzzi D, Bandeira N, Gutierrez M, Simmons TL, Schultz AW, Linington RG, Moore BS, Gerwick WH, Pevzner PA, Dorrestein PC. Anal Chem. 2009;81:4200–4209. doi: 10.1021/ac900114t.
- 23.For a previous application of this technique, see: Zeidler J, Sayer BG, Spenser ID. J Am Chem Soc. 2003;125:13094–13105. doi: 10.1021/ja030261j.
- 24.Ma J, Wang Z, Huang H, Luo M, Zuo D, Wang B, Sun A, Cheng YQ, Zhang C, Ju J. Angew Chem. 2011;50:7797–7802. doi: 10.1002/anie.201102305. [DOI] [PubMed] [Google Scholar]
- 25.Qu X, Jiang N, Xu F, Shao L, Tang G, Wilkinson B, Liu W. Mol Biosyst. 2011;7:852–861. doi: 10.1039/c0mb00234h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.