

Abstract
Bacterial resistance to β-lactam antibiotics caused by class B metallo-β-lactamases (MBL), especially for certain hospital-acquired, Gram-negative pathogens, poses a significant threat to public health. We report several 2-substituted 4,5-dihydrothiazole-4-carboxylic acids to be novel MBL inhibitors. Structure activity relationship (SAR) and molecular modeling studies were performed and implications for further inhibitor design are discussed.
Keywords: Metallo-β-lactamase, Inhibitor, Rational design, Molecular modeling
The development of β-lactam antibiotics over the past 60 years has led to the availability of drugs to treat a wide range of Gram-positive and Gram-negative bacterial infections. However, during the past few decades, populations of drug resistant bacteria have increased significantly.1 One important mechanism bacteria use to resist β-lactam antibiotics is the production of β-lactamases, which can hydrolyze the 4-membered β-lactam ring and render the drugs inactive. There are two mechanistically distinct types of β-lactamases: serine β-lactamases and metallo-β-lactamases (MBLs).2 The class A, C and D β-lactamases belong to the first type and use a serine −OH group as a nucleophile to attack and hydrolyze the amide bond in the β-lactam ring. In contrast, the class B β-lactamases are Zn2+ dependent metalloenzymes, with one or two Zn ions at the active site.3,4 MBLs are able to hydrolyze essentially all β-lactams, including imipenem, which have the widest antibacterial spectrum and are one of the very few effective drugs to treat some Gram-negative bacterial infections, such as Pseudomonas aeruginosa. The clinical significance of these enzymes has only recently been recognized, when plasmid-encoded, transferable MBLs were found to spread quickly among many species of Gram-negative bacteria around the world and confer resistance to imipenem and extended-spectrum cephalosporins.5–7
There has been much interest in discovering and developing MBL inhibitors during the past decade.8 As representatively shown in Figure 1, a number of structurally diverse inhibitors have been identified, such as thiol-containing compounds,9,10 trifluoromethyl ketones,11 tetrazoles,12 and succinates.13 However, none of these compounds have been used in clinical trials to sensitize β-lactam antibiotics due to chemical instability, a narrow spectrum of activity or side effects. For example, captopril (Fig. 1) was discovered to be very active against IMP-1 from P. aeruginosa, one of the most prevalent forms of transferable MBLs.5–7,14 It is also used in the clinic to treat hypertension as a potent inhibitor of angiotensin-converting enzyme. There is, therefore, a pressing need to find potent, drug-like inhibitors that have a new scaffold for further development. Here, we report the discovery and structure activity relationships (SAR) of 2-substituted 4,5-dihydrothiazole-4-carboxylic acids as a new class of MBL inhibitors.
Figure 1.

Representative MBL inhibitors.
High-throughput screening (HTS) has been widely used to find inhibitors of enzymes, including MBLs, in the pharmaceutical industry. However, due to the high costs, HTS is often not available in many academic institutes, where alternative methods have to be used. For metalloenzymes, a coordination chemistry based approach has been successfully applied, by us15 and other groups,16,17 to discover new inhibitors. In order to find novel MBL inhibitors, we synthesized or purchased the following compounds:

Each of these compounds features a different potential Zn2+-binding group. In addition, they also contain a hydrophobic group, such as a phenyl or benzyl, that could potentially extend to the lipophilic pocket of MBLs to enhance the binding. These compounds were tested for their inhibitory activities against recombinant IMP-1,18,19 together with captopril as a positive control, which was found to be a good inhibitor with an IC50 value of 5.0 µM (Table 1), or a Ki value of 2.4 µM. Another control is ethylenediaminetetraacetic acid (EDTA), a non-competitive, broad inhibitor of MBLs, which acts as a metal chelator depleting Zn2+ from the active site of the metalloenzyme. EDTA was found to inhibit the activity of IMP-1 with an IC50 value of 27.9 µM. Compound 1, 2-benzylthiazole-4-carboxlic acid was identified to be a novel IMP-1 inhibitor having an IC50 of 34.7 µM.
Table 1.
MBL inhibitory activities (IC50 in µM) of 1 – 7.
| Compound | IC50 for IMP-1 | IC50 for Bla2 |
|---|---|---|
| Captopril | 5.0 | 25.8 |
| EDTAa | 27.9 | 1060 |
| 1 | 34.7 | >200 |
| 2 | >200 | >200 |
| 3 | >200 | >200 |
| 4 | 5.5 | >200 |
| 5 | >200 | >200 |
| 6 | >200 | >200 |
| 7 | >200 | >200 |
with a 12-hour incubation.
Since compound 1 potentially represents a new class of MBL inhibitors, we performed a medicinal chemistry study in an effort to find compounds with improved activity as well as structure activity relationships (SAR). First, compounds 2 – 7 (Figure 2) were synthesized to examine the effects of related core structures as well as those of phenyl versus benzyl at the 2-position. The inhibitory activities of these compounds against IMP-1 are listed in Table 1. For the fully aromatic thiazole ring, 1 with a 2-benzyl is more active than 2 with a phenyl (IC50 > 200 µM). However, for the partially saturated 4,5-dihydrothiazole ring, compound 3 with a 2-benzyl group is inactive, while compound 4, (R)-2-phenyl-4,5-dihydrothiazole-4-carboxylic acid, was found to be a good IMP-1 inhibitor with an IC50 value of 5.5 µM (or a Ki of 3.3 µM), ~6× more active than 1. However, its enantiomer 5, (S)-2-phenyl-4,5-dihydrothiazole-4-carboxylic acid, was found to be inactive. In addition, none of compounds 6 and 7 having a thiazolidine ring possess inhibitory activity against IMP-1.
Figure 2.

Structures of compounds 1 – 7.
We next synthesized compounds 8 – 13 shown in Table 2 to investigate the SAR for a substituent on the 2-phenyl ring of compound 4. Compounds 8 and 9 with a 2-(4-chlorophenyl) and 2-(4-bromophenyl), respectively, were found to completely lose the inhibory activity. Compound 10 with a 3-NH2 substituent was observed to have a weak inhibitory activity with an IC50 of 77.0 µM. Its derivatives 11 and 12 containing an additional acetyl and tert-butoxylcarbonyl (BOC) group, respectively, are inactive. Compound 13 with a 2-OH substituent on the phenyl ring has also no activity. Compounds 14 – 17 (Table 2) were synthesized to find the effects of replacing 2-phenyl of 4 with an lipophilic amide or carbamate group. These groups were designed to mimic the sidechain of penicillin, one of the substrates of IMP-1. However, as can be seen in Table 2, none of these compounds show activity against IMP-1.
Table 2.
Structures and activities of compounds 8 – 17.
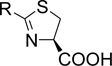 | |||
|---|---|---|---|
| R- | IC50 for IMP-1 | IC50 for Bla2 | |
| 8 | 4-Cl-phenyl | >200 | >200 |
| 9 | 4-Br-phenyl | >200 | >200 |
| 10 | 3-NH2-phenyl | 77.0 | 4.9 |
| 11 | 3-acetamido-phenyl | >200 | >200 |
| 12 | 3-BOCNH-phenyl | >200 | 74.1 |
| 13 | 2-OH-phenyl | >200 | >200 |
| 14 | 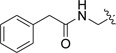 |
>200 | >200 |
| 15 | 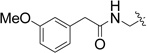 |
>200 | >200 |
| 16 | 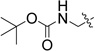 |
>200 | >200 |
| 17 | 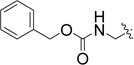 |
>200 | >200 |
Molecular modeling (docking) was used to rationalize the above observed SARs. The crystal structure10 (PDB code: 1DD6) of IMP-1 in complex with the thiol inhibitor MCI (Figure 1) was chosen to be the docking template, which was prepared, according to our previous method,15,20,21 by removing only the inhibitor using the program Glide22 in Schrödinger (version 2010).23 The two Zn2+ ions were treated as an integrated part of the protein. Compound 4 was built, energy-minimized using OPLS_2005 force field in Schrödinger, and docked into the prepared IMP-1 structure using Glide. Figure 3a shows the 20 docking structures of 4 with the lowest energies, which can be basically classified into two binding conformations A and B. Conformation A represents 7 tightly clustered docking structures with a lower average energy, and there are 13 docking poses in conformation B, as representatively shown in Figures 3b and 3c. The common feature of conformations A and B is that one O atom of the carboxylate group binds to both Zn2+ ions, behaving similarly as the S atom of MCI. The other O atom of 4 in conformation A also interacts with the tightly bound Zn1 (coordinated by three imidazoles of His77, 79 and 139), while that of B binds to the Zn2 (which is bound to IMP-1 via coordination by Asp81, Cys158 and His197 with less affinity). None of the N atoms of 4 in conformations A and B are predicted to bind to Zn2+, with the distances being >3 Å. It is of interest that the binding of the 2-phenyl group of 4 in conformations A and B is different. The phenyl of A is located almost in the same position as the phenyl of MCI, and the mainly hydrophobic pocket is tightly surrounded by Val25, Val31, Glu23 (the hydrophobic carbon skeleton), Ser21, Pro32, Phe51 and His197 (Figure 3b). The phenyl group of conformation B is situated in a much larger and longer channel that holds the two aromatic rings of MCI (Figure 3c). Our SAR results suggest the conformation A better mimics the real binding structure of compound 4 to IMP-1, because the pocket shown in Figure 3b could only accommodate a phenyl group favorably. Any additional substituent, even as small as a −Cl or −OH, could result in an unfavored steric and/or hydrophobic interaction. In addition, our SAR and docking studies could provide useful implications for future inhibitor design. For example, derivatives of compound 4 that possess an additional group that extends to the other binding site of IMP-1 could be more active.
Figure 3.

(A) Stereoview of the 20 lowest-energy docking structures of compound 4 in the active site of the crystal structure of IMP-1:MCI complex, with the protein shown as an electrostatic potential surface and Zn2+ as a light blue sphere; (B) The lowest-energy docking structure of conformation A of 4 (the ball and stick model in green) in the active site, superimposed with the crystal structure of MCI (in orange); (C) The lowest-energy docking structure of conformation B of 4 superimposed with the structure of MCI.
Finally, we tested the inhibitory activities of newly synthesized compounds against another MBL, Bacillus anthracis Bla2,24 in order to see whether our inhibitors have a broad spectrum of activity. B. anthracis is the causative agent of anthrax and expression of Bla2 can result in resistance to β-lactam antibiotics. Therefore, inhibitors of this enzyme have potential therapeutic value. The results are shown in Tables 1 and 2. Captopril was also found to be a weak inhibitor of Bla2 with an IC50 value of 25.8 µM, or a Ki value of 17.9 µM, while EDTA is almost inactive against this enzyme (IC50 > 1 mM). Although compound 4 is a good IMP-1 inhibitor, it was observed to have essentially no activity against Bla2 with an IC50 of >200 µM. However, analogous compound 10 with a 3-NH2 substituent showed improved inhibitory activity against Bla2 as compared to captopril, with an IC50 value of 4.9 µM (Table 2) or a Ki value of 5.1 µM. All other compounds, except for compound 12 being a weak inhibitor (IC50 = 74.1 µM), are also inactive against Bla2. Nevertheless, the activity of compound 10 shows that it is also possible to develop 4,5-dihydrothiazole-4-carboxylic acids to find a compound that have a broad activity against other MBLs.
General methods for synthesizing compounds 3 – 17 are illustrated in Scheme 1.25 For 4,5-dihydrothiazole compounds 3 – 5 and 8 – 13, an aromatic nitrile, 1.2 equivalent amount of L-cysteine (or D-cysteine) hydrochloride and NaHCO3 was refluxed in methanol and phosphate buffer (pH = 6) for 72h,26 to give these compounds in 40 – 70% yield. However, this method does not work for making analogous compounds 14 – 17. Rather, the methyl esters of these compounds were synthesized using a similar procedure,27 which were carefully undergone a mild hydrolysis using 0.9 equivalent of LiOH to afford compounds 14 – 17 in 70 – 80% yield. Excess amount of LiOH caused decomposition of the products. Thiazolidine compounds 6 and 7 were prepared in ~80% yield by reacting benzaldehyde or phenylacetaldehyde with L-cysteine at room temperature in a mixture of ethanol and water.28
Scheme 1.

General synthesis for compounds 3 – 17.a
aReagents and conditions: (i) NaHCO3, phosphate buffer (pH = 6), MeOH, 66 °C, 72h; (ii) RCOCl or (BOC)2O, Et3N, CH2Cl2; (iii) L-Cys-OMe, Et3N, MeOH; (iv) 0.9 equiv. 1 M LiOH, 0 °C – room temperature, 1h; (v) EtOH, H2O, room temperature, 18h.
In summary, this work is of interest for several reasons. First, using rational compound screening followed by medicinal chemistry, 2-phenyl-4,5-dihydrothiazole-4-carboxylic acid (4) was identified to be a novel inhibitor of P. aeruginosa MBL IMP-1 with an IC50 value of 5.5 µM. Second, docking studies were explored to rationalize the structure activity relationships of this class of compounds and provide implications for future inhibitor design. Third, 2-(3-aminophenyl)-4,5-dihydrothiazole-4-carboxylic acid (10) was identified to be an inhibitor of B. anthracis MBL Bla2 (IC50 = 4.9 µM), showing the promise of further developing this class of compounds in the context of overcoming β-lactam resistance.
Supplementary Material
Acknowledgements
This work was supported by a grant (R21AI090190) from National Institute of Allergy and Infectious Disease (NIAID/NIH) to Y.S. and a grant (R01AI32956) from NIAID/NIH to T.P. L.B.H. was supported by a training grant (T90DK070109) from NIH/NIDDK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Leeb M. Nature. 2004;431:892. doi: 10.1038/431892a. [DOI] [PubMed] [Google Scholar]
- 2.Fisher JF, Meroueh SO, Mobashery S. Chem. Rev. 2005;105:395. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 3.Crowder MW, Spencer J, Vila A. J. Acc. Chem. Res. 2006;39:721. doi: 10.1021/ar0400241. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z, Fast W, Valentine AM, Benkovic S. J. Curr. Opin. Chem. Biol. 1999;3:614. doi: 10.1016/s1367-5931(99)00017-4. [DOI] [PubMed] [Google Scholar]
- 5.Walsh TR, Toleman MA, Poirel L, Nordmann P. Clin. Microbiol. Rev. 2005;18:306. doi: 10.1128/CMR.18.2.306-325.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bebrone C. Biochem. Pharmacol. 2007;74:1686. doi: 10.1016/j.bcp.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 7.Walsh TR. Clin. Microbiol. Infect. 2005;11(Suppl 6):2. doi: 10.1111/j.1469-0691.2005.01264.x. [DOI] [PubMed] [Google Scholar]
- 8.Toney JH, Moloughney JG. Curr. Opin. Investig. Drugs. 2004;5:823. [PubMed] [Google Scholar]
- 9.Garcia-Saez I, Hopkins J, Papamicael C, Franceschini N, Amicosante G, Rossolini GM, Galleni M, Frere JM, Dideberg O. J. Biol. Chem. 2003;278:23868. doi: 10.1074/jbc.M301062200. [DOI] [PubMed] [Google Scholar]
- 10.Concha NO, Janson CA, Rowling P, Pearson S, Cheever CA, Clarke BP, Lewis C, Galleni M, Frere JM, Payne DJ, Bateson JH, Abdel-Meguid SS. Biochemistry. 2000;39:4288. doi: 10.1021/bi992569m. [DOI] [PubMed] [Google Scholar]
- 11.Walter MW, Felici A, Galleni M, Soto RP, Adlington RM, Baldwin JE, Frère J-M, Gololobov M, Schofield C. J. Bioorg. Med. Chem. Lett. 1996;6:2455. [Google Scholar]
- 12.Toney JH, Fitzgerald PM, Grover-Sharma N, Olson SH, May WJ, Sundelof JG, Vanderwall DE, Cleary KA, Grant SK, Wu JK, Kozarich JW, Pompliano DL, Hammond GG. Chem. Biol. 1998;5:185. doi: 10.1016/s1074-5521(98)90632-9. [DOI] [PubMed] [Google Scholar]
- 13.Toney JH, Hammond GG, Fitzgerald PM, Sharma N, Balkovec JM, Rouen GP, Olson SH, Hammond ML, Greenlee ML, Gao YD. J. Biol. Chem. 2001;276:31913. doi: 10.1074/jbc.M104742200. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe M, Iyobe S, Inoue M, Mitsuhashi S. Antimicrob. Agents Chemother. 1991;35:147. doi: 10.1128/aac.35.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng L, Sundriyal S, Rubio V, Shi Z, Song Y. J. Med. Chem. 2009;52:6539. doi: 10.1021/jm9012592. [DOI] [PubMed] [Google Scholar]
- 16.Jacobsen FE, Lewis JA, Cohen SM. J. Am. Chem. Soc. 2006;128:3156. doi: 10.1021/ja057957s. [DOI] [PubMed] [Google Scholar]
- 17.Jacobsen FE, Lewis JA, Cohen SM. ChemMedChem. 2007;2:152. doi: 10.1002/cmdc.200600204. [DOI] [PubMed] [Google Scholar]
- 18.Brown NG, Horton LB, Huang W, Vongpunsawad S, Palzkill T. Antimicrob. Agents Chemother. 2011;55:5696. doi: 10.1128/AAC.00340-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The enzyme assay was performed using 2 nM IMP-1 (or 10 nM Bla2) and 25 µM nitrocefin in 50 mM HEPES buffer (pH = 7.0) containing 20 µg/mL BSA and 0.01% triton in a final volume of 300 µL. For inhibition assay, compounds were pre-incubated with the enzyme for 20 min at room temperature, before initiation of the reaction by adding nitrocefin. The increasing absorbance at 482 nm was monitored using a Beckman DU640 UV spectrometer. The initial velocities for different concentrations of an inhibitor were imported into Prism (version 5.0, GraphPad, La Jolla, CA). The IC50 as well as Ki values were calculated by using a standard dose response curve fitting in the software.
- 20.Deng L, Endo K, Kato M, Cheng G, Yajima S, Song Y. ACS Med. Chem. Lett. 2011;2:165. doi: 10.1021/ml100243r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng L, Diao J, Chen P, Pujari V, Yao Y, Cheng G, Crick DC, Prasad BVV, Song Y. J. Med. Chem. 2011;54:4721. doi: 10.1021/jm200363d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glide, version 5.5. New York, NY: Schrödinger, LLC; 2010. [Google Scholar]
- 23.Schrödinger Suite, version 2010. New York, NY: Schrödinger, LLC; 2010. [Google Scholar]
- 24.Materon IC, Queenan AM, Koehler TM, Bush K, Palzkill T. Antimicrob. Agents Chemother. 2003;47:2040. doi: 10.1128/AAC.47.6.2040-2042.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Experimental Section can be found in Supplemental Data on-line.
- 26.Lu Y, Li C, Wang Z, Chen J, Mohler ML, Li W, Dalton JT, Miller DD. J. Med. Chem. 2011;54:4678. doi: 10.1021/jm2003427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merino P, Tejero T, Unzurrunzaga FJ, Franco S, Chiacchio U, Saita MG, Iannazzo D, Piperno A, Romeo G. Tetrahedron Asymmetry. 2005;16:3865. [Google Scholar]
- 28.Lu Y, Li C, Wang Z, Ross CR, Chen J, Dalton JT, Li W, Miller DD. J. Med. Chem. 2009;52:1701. doi: 10.1021/jm801449a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.