

Abstract
In our effort to investigate further texaphyrin conjugation as a means of increasing delivery and accumulation of known anticancer platinum agents in cancer cells, we have continued our studies on the mode of action of a texaphyrin-platinum conjugate, particularly in cisplatin-resistant tumor cells that are characterized by several mechanisms of resistance, including reduced drug accumulation. Our results provide support for the proposal that intracellular platinum and Pt-DNA adduct levels were significantly increased using our conjugate relative to corresponding Pt controls. Moreover, no differences were found in cellular accumulation and Pt-DNA adduct formation between Pt sensitive and Pt resistant ovarian cells. As a result, resistance to the conjugate was lower than cisplatin in resistant cells. Based on these results we conclude that texaphyrin conjugation provides a promising strategy for overcoming biochemical pharmacologic mechanisms of resistance.
Keywords: Drug delivery, Cisplatin, texaphyrin, Cell studies
Cytotoxic platinum anticancer complexes, including the FDA-approved Pt(II)-based drugs cisplatin, carboplatin, and oxaliplatin, are thought to mediate their action through inducing persistent DNA cross-link adducts.1-3 This mode of action has made them mainstays in cancer chemotherapy, including the treatment of ovarian cancer. However, reduction in drug uptake and the resultant lowered levels of adduct formation accounts, in part, for the resistance to Pt drugs seen after initial treatments.1 This biochemical resistance mode limits the clinical utility of the platinum drugs. In the case of ovarian cancer, the average 5-year survival rate is 46%; however, most cases (67%) are detected at a late stage and the 5-year survival rate for this population drops to 31%.4 The resistance to Pt drugs seen in the clinic is also ascribed in part to the fact that cell death machinery becomes tolerant to DNA damage, and this can occur through several molecular mechanisms, including mutations in p53, a transcriptional factor with tumor suppressor function when present in its wild-type state. However, the situation is clearly complicated in that resistance is also seen in ovarian tumor cells due to a failure of cisplatin to activate wild-type p53.1 Pt drug resistance is also correlated with upregulation of multidrug resistance (MDR) transporters and increased production of glutathione (which mitigates the effects of oxidative stress) and other platinophiles.5,6
Texaphyrins (e.g., motexafin gadolinium; MGd; Figure 1) have a clinically validated ability to localize well to many cancers.7 They have been studied in recent years as potential anticancer agents and appear to operate via a novel mechanism of action that involves perturbations of redox balance and metal homeostasis.8,9 Texaphyrins do not induce MDR and are known to enhance the effects of oxidative stress.8 The fact that they also localize well to tumors means that they could be used to deliver Pt-type drugs to cancerous sites, thereby potentially increasing the local concentration and overcoming at least some of the pathways responsible for Pt resistance.
Figure 1.
Various Pt(II) complexes and MGd.
Many strategies have been put forth in an effort to increase the tumor specificity and accumulation of platinum drugs, including conjugation to potentially site-directing molecules, such as folate, poly(ethyleneglycol) (PEG), porphyrins, and peptides among others.10-14 While increased delivery of the Pt drug can augment tumor cell killing effects, we believe that the Pt drug delivered to resistant tumor cells should also have intrinsic properties to overcome multifactorial mechanisms of resistance. To this effect we have recently developed Texaphyrin-Pt conjugate 1 as a potential means of increased tumor uptake versus conventional methods of lone platinum complexes.15 Described herein, we compared its activity in the A2780 human ovarian cancer cell line and its ostensibly isogenic cisplatin-resistant cell line (2780CP). As detailed below, the associated analysis has allowed us to assess the potential utility of this complex against resistant cells and elucidate which mechanisms of resistance may be circumvented by this generalized conjugation strategy.
With such considerations in mind, tests were carried out using the MTT cytotoxic assay following exposure of cells to the Pt agents, and the effect of conjugate 1 on cell proliferation was compared to that of methylmalonatoplatinum (MMPt), carboplatin, and cisplatin (Table 1).15 Conjugate 1 (IC50 = 1.4 ± 0.3 μM) provided a dose potency in the A2780 cell line similar to that of carboplatin (IC50 = 1.6 ± 0.3 μM).16 This similarity in activity is encouraging considering the many known platinum conjugates with reduced activity.17,18 MMPt,19 a Pt complex similar to that present in conjugate 1, provided similar antiproliferative activity relative to both 1 and carboplatin. Cisplatin displayed the greatest cytotoxicity towards this cell line with an IC50 of 0.31 ± 0.06 μM.20
Table 1.
IC50 Values of platinum complexes with cisplatin sensitive A2780 ovarian and its isogenic cisplatin resistant cell line (2780CP). Data are shown as Mean ± SD.
| Complex | IC50 (μM) A2780 | IC50 (μM) 2780CP | Resistance Factor |
|---|---|---|---|
| 1 (MGd-Pt) | 1.4 ± 0.3 | 14.4 ± 1.7 | 10.3 ± 1.3 |
| MMPt | 1.4 ± 0.2 | 21.3 ± 2.2 | 15.2 ± 3.7a |
| Carboplatin | 1.6 ± 0.3 | 26.3 ± 4.1 | 16.4 ± 5.2a |
| Cisplatin | 0.31 ± 0.06 | 7.1 ± 0.9 | 22.9 ± 5.3a |
p<0.05 by Student’s t-test vs. resistance factor for conjugate 1.
Cell proliferation studies were also performed with platinum resistant ovarian cancer cells (2780CP). In this instance, cells incubated in the presence of conjugate 1, MMPt, carboplatin, and cisplatin were inhibited, providing IC50 values of 14.4 ± 1.7, 21.3 ± 2.2, 26.3 ± 4.1, and 7.1 ± 0.9 μM, respectively. Notably, the activities of carboplatin and MMPt proved lower (i.e., higher IC50) than that of conjugate 1, and the differences were significant (p<0.05).
To gain a better understanding of relative activity of complexes against resistant cells, the resistance factor was calculated (Table 1). Cisplatin, while potent against sensitive cells (i.e. A2780) demonstrated a significantly reduced activity (by almost 23-fold) against resistant cells (i.e. 2780CP). The resulting resistance factor (ca. 23) is attributed to both biochemical pharmacologic and molecular mechanisms of resistance. Both carboplatin and MMPt provided similar resistance factors, which were about 25% less than cisplatin, but this was not statistically significant. Conjugate 1 provided the lowest resistance factor and about 32-55% lower resistance relative to other platinum complexes involved in the study. This improvement was significant (p<0.05; Table 1) and strongly indicative of partial circumvention of cisplatin resistance.
Relative potencies of Pt complexes in the A2780 cell line, as shown in Table 1, may in part be due to differences in activation of the agents and/or cellular levels of Pt and DNA adducts. To determine whether relative activation rates could be involved, samples of fetal bovine serum (FBS) were incubated with various Pt based agents and the amount of free (unbound) Pt was determined over time, with the half-life (t1/2) in question being derived from linear regression analyses (Figure 3).
Figure 3.
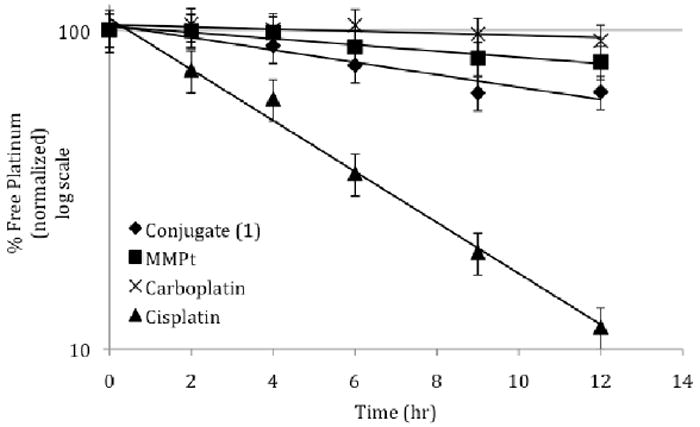
Platination of FBS proteins with various platinum agents. Platinum concentrations were measured to 5 ug/mL upon incubated at 37 °C. Aliquots of the samples subject to incubation were diluted with 4x the original volume of cold methanol so as to precipitate proteins, which were removed by centrifugation. The supernatant was then subjected to flameless atomic absorption spectrometry (FAAS), which allowed for the determination of free platinum.
As expected, the activation of cisplatin by aquation proved rapid, and correlated with fast protein platination and a corresponding accelerated decrease in free Pt (t1/2 = 4 hours). Conjugate 1 was expected to provide a much more stable platinum species. However, it still gave rise to a reasonably rapid activation and protein platination (t1/2 = 16 hours). The longer half-life (t1/2 ≥ 36 hours) of the platinum control MMPt and carboplatin is taken as evidence of their relatively greater stability under these experimental conditions. The difference in activation rate between 1, MMPt, and carboplatin is interpreted in terms of either a) the texaphyrin core or b) the linkage between the texaphyrin and the Pt moiety resulting in a greater reactivity with protein. In clinical practice, the low reactivity of carboplatin is compensated for by use of higher drug concentrations (relative to cisplatin).21 An increase in reactivity of 1 relative to carboplatin could account for its greater potency. To the extent this is true, it may prove possible to use lower concentrations of 1 in vivo than would be expected based on simple Pt(II) dosage considerations. Thus, conjugate 1 would have advantages from both the presence of a tumor-localizing carrier (i.e., a texaphyrin core) and reduced risks of Pt-based toxicity. In due course, this could translate to improved clinical outcomes.
The origins of cisplatin resistance are multifactorial and reflects a variety of biochemical pharmacologic mechanisms, including reductions in cellular uptake and retention, decreased DNA adduct formation and a greater tolerance for DNA damage.22-25 The improved resistance factor obtained for conjugate 1 presumably reflects an ability to overcome some, perhaps, many of these forms of resistance. To gain insights into which, if any, of these putative determinants was most important, the intracellular uptake of 1 was analyzed, along with that of the control complexes MMPt, carboplatin, and cisplatin (Figure 4).26 These latter agents demonstrated a 50% or greater reduction in intracellular uptake in resistant 2780CP cells as compared to sensitive A2780 cells. Such findings are consistent with the reported 2-fold reduction in uptake seen in resistant cells exposed to platinum anticancer agents.22 In striking contrast to this, no difference in uptake was observed for conjugate 1 between A2780 and 2780CP. This is a highly encouraging result in that resistance due to reduced platinum uptake has been overcome in resistant cells. Conjugate 1 provided an 8- and 12-fold increase in uptake over the most similar control (MMPt) for A2780 and 2780CP, respectively. Additionally, it should be noted that conjugate 1 provided greater uptake than cisplatin for 2780CP over the allotted 4-hour incubation period.
Figure 4.
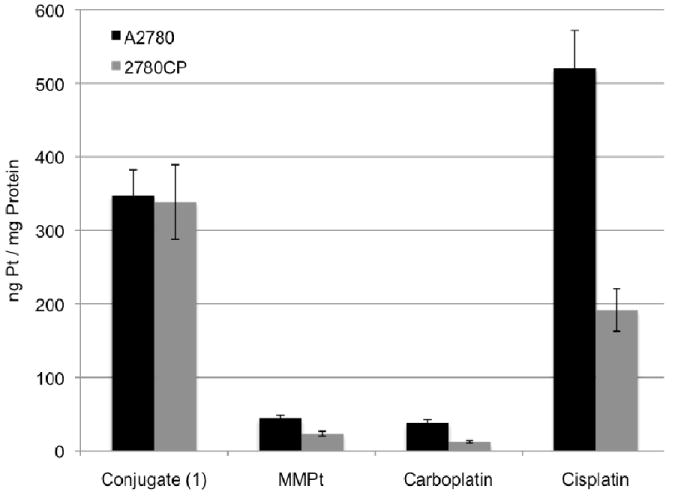
Intracellular platinum concentration of sensitive (A2780) and resistant (2780CP) ovarian cancer cells after a 4-hour incubation with 200 μM of each complex.
One tell-tale sign of platinum resistance is a significant decrease in Pt adducts formed with DNA as a result of reduced drug accumulation. Pt-DNA adducts formed by conjugate 1, as well as the Pt control complexes MMPt, carboplatin, and cisplatin, were quantified (Figure 5).27 Here, both the A2780 and 2780CP cell lines were incubated with 200 μM of the respective drug for a 4-hour period. The DNA was then isolated and subjected to FAAS for quantification of the Pt adducts. Cisplatin provided a control against which efforts to overcome resistance due to reduced adducts could be calibrated. Indeed, using cisplatin, a 50% reduction in Pt-DNA adducts was seen in 2780CP cells. Reduction in adducts were also observed for MMPt and carboplatin, although data for MMPt and carboplatin adducts with DNA in 2780CP cells fell below detection limits. The generally reduced Pt-DNA adducts of Pt complexes containing bidentate ligands in both cell lines relative to cisplatin is thought to reflect a decrease in the rate of adduct formation resulting from a reduction in reactivity.21 In the case of conjugate 1 such a reduction was not seen; indeed, in contrast to what is normally seen, the number of Pt-DNA adducts formed by conjugate 1 proved to be high in both the cisplatin sensitive and resistant cell line. This result, which is consistent with the conjugation strategy providing a benefit, is ascribed to the greater reactivity of the conjugate (Table 1), as well as to enhanced cellular uptake (Fig. 4). Also noteworthy is the fact that there was at least a 4-fold increase in Pt adducts induced by 1 as compared to MMPt; this was true in both the sensitive and resistant cell lines. These are key findings that we interpret as full circumvention of platinum resistance at the level of adduct formation.
Figure 5.
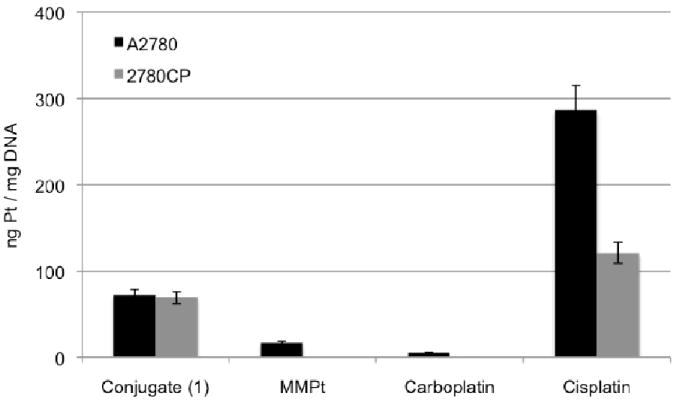
Pt-DNA adducts formed in vitro upon a 4-hour incubation period with the representative drug. Platinum concentration was determined by FAAS.
DNA-platinum adduct repair has been observed within many resistant cancers and is a biochemical pharmacologic form of resistance that reduces adduct persistence.23, 24 We hypothesized that adducts formed by conjugate 1 are similar to those formed by cisplatin and carboplatin. If this is the case, it is expected that the Pt-DNA lesions formed as the result of treating with conjugate 1 will be subject to the same lesion repair mechanisms as operative in the case of cisplatin and carboplatin. To test this hypothesis, A2780 and 2780CP cells were exposed to conjugate 1 for a 4-hour period followed by re-incubation by drug-free media for an additional 8 hours. The Pt-DNA levels were quantified by FAAS before and after the 8-hour repair period, and the results are shown in Figure 6.28 As mentioned previously, no difference in adduct levels between A2780 and 2780CP were observed after exposure to the drug, indicating no resistance at the level of Pt adduct formation. After the 8-hour repair period, similar Pt-DNA adduct levels were observed in sensitive A2780 cells; however, reduced levels were observed in platinum resistant 2780CP cells. This result, which we interpret in terms of repair of the Pt-DNA adducts in platinum resistant cell lines during the 8 hour repair period, is in accordance with the Pt-DNA adduct repair known to occur after exposure to cisplatin.29 On this basis, we conclude that the increased cytotoxicity of conjugate 1 relative to carboplatin and MMPt in 2780CP is not due to reduced repair of the resulting Pt-DNA adducts, rather it is ascribed to increases in uptake and/or reactivity, as noted above.
Figure 6.
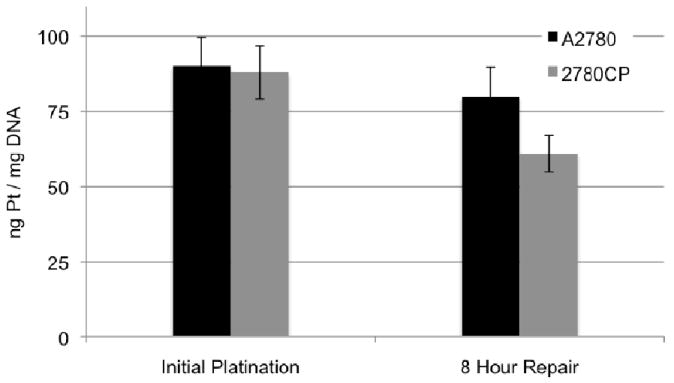
Levels of Pt-DNA adducts formed in each ovarian cell line due to exposure of 200 μM conjugate 1 for 4 hours followed by removal of 1 and 8-hour repair.
As indicated earlier, the origin of cisplatin resistance is multifactorial. However, increased tolerance to cisplatin-induced DNA adducts appears to suggest that cumulative resistance from molecular mechanisms predominates.25 The ability of a cell to tolerate DNA damage caused by conjugate 1 and cisplatin is provided in Figure 7. The bars in the graph represent adduct levels normalized to the number of adducts needed to kill 50% of cells (i.e., the data is adjusted to reflect the relative IC50 values). A 10-fold increase in adducts, formed by cisplatin, between A2780 and 2780CP is indicative that molecular mechanisms of resistance (such as failure to activate p53)29 are not overcome. The data for conjugate 1 in this specific regard is similar to that of cisplatin. We thus appreciate that molecular-based mechanisms of Pt resistance represent a challenge that is not yet met by conjugate 1.
Figure 7.
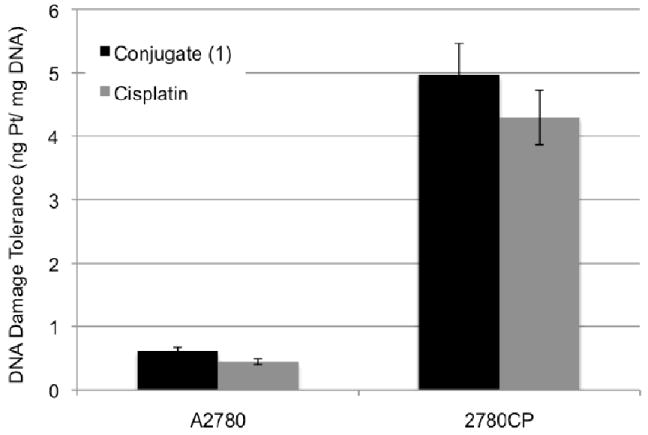
DNA damage tolerance seen in A2780 and 2780CP cells upon exposure to conjugate 1 and cisplatin.
In summary, with a human ovarian cancer cell model, conjugate 1 was shown to provide an increase in intracellular platinum as well as a greater number of Pt-DNA adducts relative to similar controls (i.e. MMPt, carboplatin). No difference in the biochemical pharmacology of conjugate 1 was observed between the A2780 and 2780CP cell lines, which is consistent with the underlying design hypothesis, namely that certain pharmacologic mechanisms of resistance, including specifically uptake, can be overcome successfully through conjugation of a platinum(II) center to a texaphyrin core. As expected, the DNA damage tolerance of conjugate 1 proved similar to that of cisplatin, leading us to suggest that other molecular mechanisms of resistance persist and are yet to be overcome. Current work in our lab involves addressing this limitation.
Figure 2.
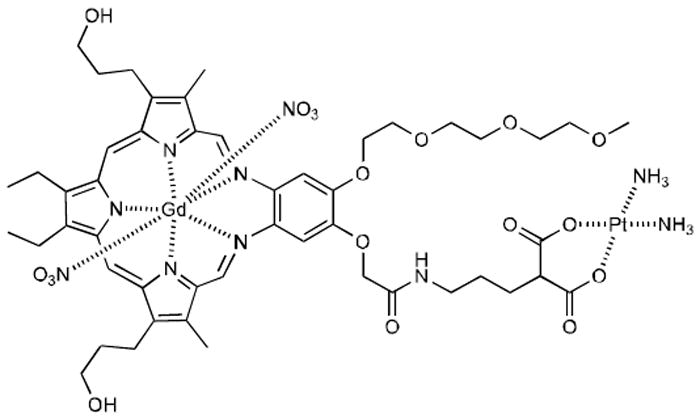
Texaphyrin-Platinum Conjugate 1.
Acknowledgments
Supported in part by grants from the NIH (Grant CA 68682 to J.L.S; CA127263 to Z.H.S.), the Texas Institute for Drug and Diagnostics Development, the Center for Targeted Therapeutics and the Robert A. Welch Foundation (F-1018 and H-F-0032 to J.L.S. and Z.H.S., respectively.)
The authors gratefully acknowledge the expert guidance of Dr. Guangan He and Dr. Ismail Meraz in experimental procedural discussions.
References and notes
- 1.Siddik ZH. Oncogene. 2003;22:7265. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 2.Kelland L. Nature Rev Cancer. 2007;7:573. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 3.Wong E, Giandomenico CM. Chem Rev. 1999;99:2451. doi: 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]
- 4.American Cancer Society. Cancer Facts and Figures 2009. ACS; Atlanta, GA: 2009. [Google Scholar]
- 5.Kasherman Y, Sturup S, Gibson D. J Med Chem. 2009;52:4319. doi: 10.1021/jm900138u. [DOI] [PubMed] [Google Scholar]
- 6.Gibson D. Dalton Trans. 2009:10681. doi: 10.1039/b918871c. [DOI] [PubMed] [Google Scholar]
- 7.Miller RA, Woodburn K, Fan Q, Renschler MF, Sessler JL, Koutcher JA. Int J Biol Radiat Oncol. 1999;45:981. doi: 10.1016/s0360-3016(99)00274-6. [DOI] [PubMed] [Google Scholar]
- 8.Magda D, et al. Gadolium(III) Texaphyrin (Xcytrin®): A New Class of Redox Active Drug Leads. In: Sessler JL, Doctrow S, McMurry T, Lippard SJ, editors. Medicinal Inorganic Chemistry. American Chemical Society Symposium Series 903, Oxford University Press; 2005. [Google Scholar]
- 9.Magda D, Lecane P, Miller RA, Lepp C, Miles D, Mesfin M, Biaglow JE, Ho VV, Chawannakul D, Nagpal S, Karaman MW, Hacia JG. Cancer Res. 2005;65:93837. doi: 10.1158/0008-5472.CAN-04-4099. [DOI] [PubMed] [Google Scholar]
- 10.Aronov O, Horowitz AT, Gabizon A, Fuertes MA, Perez JM, Gibson D. Bioconjugate Chem. 2003;14:563. doi: 10.1021/bc025642l. [DOI] [PubMed] [Google Scholar]
- 11.Lottner C, Knuechel R, Bernhardt G, Brunner H. Cancer Lett. 2004;215:167. doi: 10.1016/j.canlet.2004.06.035. [DOI] [PubMed] [Google Scholar]
- 12.Mukhopadhyay S, Barnes CM, Haskel A, Short SM, Barnes K, Lippard SJ. Bioconjugate Chem. 2008;19:39. doi: 10.1021/bc070031k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galanski M, Keppler BK. Anti-Cancer Agents in Med Chem. 2007;7:55. doi: 10.2174/187152007779314017. [DOI] [PubMed] [Google Scholar]
- 14.van Zutphen S, Reedijk J. Coord Chem Rev. 2005;249:2845. [Google Scholar]
- 15.Arambula JF, Sessler JL, Fountain ME, Wei W, Magda D, Siddik ZH. Dalton Trans. 2009;48:10834. doi: 10.1039/b912089k. IC50 values presented were determined as previously described and are a mean of 3 separate experiments. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelland LR, Abel G, McKeage MJ, Jones M, Goddard PM, Valenti M, Murrer BA, Harrap KR. Cancer Res. 1993;53:2581. [PubMed] [Google Scholar]
- 17.Aronov O, Horowitz AT, Gabizon A, Fuertes MA, Perez JM, Gibson D. Bioconjugate Chem. 2004;15:814. doi: 10.1021/bc0499331. [DOI] [PubMed] [Google Scholar]
- 18.Brunner H, Arndt MR, Treittinger B. Inorg Chim Acta. 2004;357:1649. [Google Scholar]
- 19.Cleare MJ, Hoeschele JD, Rosenberg B, Van Camp L. U S Patent 4657927. 1987. Synthesis of MMPt was adapted from a procedure previously reported: Rochon FD, Gruia LM. Inorg Chim Acta. 2000;306:193.. Briefly, an aqueous solution of Ba(OH)2 (0.75 mmol) and 2-methylpropanedioic acid (0.75 mmol) was added dropwise to a stirring solution of (NH2)2Pt(H2O)2•SO4 (0.75 mmol) and allowed to stir for 1 h in the dark. The resulting precipitate was then filtered and the filtrate was condensed in vacuo to produce a white fluffy solid. The solid obtained in this way was sequentially washed with 10 mL of MeOH and 10 mL of diethylether and dried in vacuo to produce 180 mg (0.52 mmol, 69%) of the product as a white fluffy solid.
- 20.Mujoo K, Watanabe M, Nakamura J, Khokhar AR, Siddik ZH. J Cancer Res Clin Oncol. 2003;129:709. doi: 10.1007/s00432-003-0480-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knox RJ, Friedlos F, Lydall DA, Roberts JJ. Cancer Res. 1986;46:1972. [PubMed] [Google Scholar]
- 22.Kelland LR. Crit Rev Oncol Hematol. 1993;15:191. doi: 10.1016/1040-8428(93)90042-3. [DOI] [PubMed] [Google Scholar]
- 23.Heiger-Bernays WJ, Essigmann JM, Lippard SJ. Biochemistry. 1990;29:8461. doi: 10.1021/bi00488a037. [DOI] [PubMed] [Google Scholar]
- 24.Vaisman A, Varchenko M, Umar A, Kunkel TA, Risinger JI, Barrett JC, Hamilton TC, Chaney SG. Cancer Res. 1998;58:3579. [PubMed] [Google Scholar]
- 25.Johnson SW, Laub PB, Beesley JS, Ozols RF, Hamilton TC. Cancer Res. 1997;57:850. [PubMed] [Google Scholar]
- 26.In brief, 5 × 106 cells were incubated with 200 μM of the Pt complex in question for 4 h. The cells were then washed with cold PBS, trypsinized, and pelleted by centrifugation. Cells were resuspended in 200 μL PBS and 200 μL of ATL buffer (Qiagen). The suspension was then heated in a water bath (56 °C) for 1 h. The lysis suspension was then diluted with H2O and subjected to flameless atomic absorption spectrphotometry (FAAS) for Pt detection. Total protein concentration was determined through the Bradford assay using a protein assay kit (Bio-Rad).
- 27.For this study, 5 × 106 cells were incubated with 200 μM of the Pt complex in question for 4 h. The cells were then washed with cold PBS, trypsinized, and pelleted by centrifugation. Cells were resuspended in 200 μL PBS and subjected to DNA purification using a Qiagen DNeasy Blood and Tissue Kit. The isolated platinated DNA was quantified by UV-Vis spectroscopy and Pt-adducts were quantified through FAAS.
- 28.Cells were treated in a manner similar to that described in Reference 27. However, the cells subjected to an 8-hour repair period were washed once with cold PBS to ensure complete removal of the agent subject to study (initially at 200 μM) from the media before drug free media was added prior to commencing the 8-hour repair period.
- 29.Siddik ZH, Mims B, Lozano G, Thai G. Cancer Res. 1998;58:698. [PubMed] [Google Scholar]