

Abstract
A1 adenosine receptor (AR) agonists display antiischemic and antiepileptic neuroprotective activity, but peripheral cardiovascular side effects impeded their development. SAR study of N6-cycloalkylmethyl 4′-truncated (N)-methanocarba-adenosines identified 10 (MRS5474, N6-dicyclopropylmethyl, Ki 47.9 nM) as a moderately A1AR-selective full agonist. Two stereochemically defined N6-methynyl group substituents displayed narrow SAR; larger than cyclobutyl greatly reduced AR affinity, and larger or smaller than cyclopropyl reduced A1AR selectivity. Nucleoside docking to A1AR homology model characterized distinct hydrophobic cyclopropyl subpockets, the larger “A” forming contacts with Thr270 (7.35), Tyr271 (7.36), Ile274 (7.39) and carbon chains of glutamates (EL2), and smaller subpocket “B” between TM6 and TM7. 10 suppressed minimal clonic seizures (6 Hz mouse model) without typical rotarod impairment of A1AR agonists. Truncated nucleosides, an appealing preclinical approach, have more drug-like physicochemical properties than other A1AR agonists. Thus, we identified highly restricted regions for substitution around N6 suitable for an A1AR agonist with anticonvulsant activity.
Keywords: G protein-coupled receptor, purines, molecular modeling, seizures, in vivo
Extracellular adenosine acts through four subtypes of G protein-coupled adenosine receptors (ARs), i.e. at A1, A2A, A2B and A3AR subtypes.1 Endogenous adenosine begins to activate the A1AR at low concentrations (~10 nM) to induce cytoprotective and anti-ischemic functions. Full or partial agonists of the A1AR are being considered for treatment of various conditions, including seizures, stroke, diabetes, cardioprotection and cardiac arrhythmias.2–4 A1AR agonists are highly neuroprotective in ischemic5,6 and epileptic7–9 models. A1AR agonists are also being explored for antidepressant,10 antianxiety,11 and other neuropsychiatric effects. A1AR agonists are also useful for pain12 due to their presynaptic action to decrease the release of excitatory neurotransmitters in the brain.13 However, peripheral cardiovascular side effects have prevented the introduction of A1AR agonists for treating disorders of the central nervous system (CNS).14
Structural modification of adenosine to achieve selectivity in activating one or more AR subtypes has centered on the adenine C2 and N6 positions and on the ribose moiety.1,15 Modifications of the ribose moiety, such as 5′ amides and alternate carbocyclic ring systems, are especially useful for enhancing AR affinity. For example, replacement of the ribose tetrahydrofuryl ring with a methanocarba ([3.1.0]-bicyclohexane) ring system can enforce a North (N)-envelope conformation, depending on the fusion position of the cyclopropane ring. This replacement, which is highly preferred in AR binding over the isomer of the opposite South (S) conformation, maintains or enhances affinity at A1 and A3ARs with respect to the ribosides, but decreases affinity at A2AAR.16 Another widely explored modification, truncation at the 4′ carbon, i.e. removal of the 5′ group, is generally tolerated in A3AR binding for various chemical series, including 4′-thio (e.g. 1a, Chart 1) and (N)-methanocarba analogues (e.g. 2a).17,18 However, the observed effects of this truncation on the relative efficacy of nucleoside derivatives to activate ARs are variable for the different AR subtypes and chemotypes. For example, truncation in the 4′-thio adenosine series tends to produce nucleosides that antagonize the A3AR and in some cases may additionally agonize the A2AAR, such as 1b.19 Truncation in the (N)-methanocarba-adenosine series results in nucleosides that typically lose two orders of magnitude of affinity at the A1AR and A2AAR, depending on the N6 substituent, and display a wide range of efficacies at the A1AR and A3AR, e.g. A3AR antagonist 2a.18,20 Focusing on the neuroprotective A1AR, we scanned a wide range of N6 substituents known to enhance A1AR affinity21 and concluded that only certain N6-cycloalkylmethyl and dicycloalkylmethyl groups (e.g. 2b) maintain selectivity and agonist efficacy at this AR subtype.20
Chart 1.

Structures of representative 4′-truncated adenosine analogues and their receptor binding affinities in the series of 4′-thio-ribosides (1a, b) and ring-constrained (N)-methanocarba nucleosides (2a, b). Ki values in nM in binding to the hARs are indicated.17,18,20,44
A goal of this study was to characterize the structure activity relationship (SAR) of an expanded set of closely related and stereochemically defined N6-cycloalkylmethyl and dicycloalkylmethyl 4′-truncated (N)-methanocarba adenosine derivatives as human (h) A1AR agonists. Both binding and efficacy studies were performed on the novel derivatives, and selected nucleosides (including ribosides for comparison) were examined in in vivo anticonvulsant models.
Finally, receptor homology modeling and ligand docking were used to gain insight into the structural basis for AR recognition and activation in this series of closely related N6 derivatives. The modeling was based on an agonist-bound A2AAR X-ray structure recently reported by Xu et al.22 The interaction of ligands with the Gi-coupled A1AR was compared to the Gi-coupled A3AR, at which the affinity in this nucleoside series was also dramatically modulated. Pharmacological properties have been related to specific binding site interactions, especially in a small and sterically restricted region of the hydrophobic pocket where the N6 group has been proposed to reside. The loss of interactions in the ribose 5′ region was compensated by structural characteristics of a few N6 substituents in the 4′-truncated analogues. Thus, the present study had three objectives: to more clearly define the narrow structural limits for binding and full activation of the A1AR within the N6-cycloalkylmethyl series, to correlate these findings with molecular modeling based on an X-ray structure of the A2AAR and to characterize anticonvulsant activity known to be associated with the A1AR in the brain. Because the requirements for A1AR selectivity and full agonist efficacy in this series were found to be highly restricted, we identified a structural sweet spot within the SAR of 4′-truncated nucleosides.
Results
Chemical synthesis
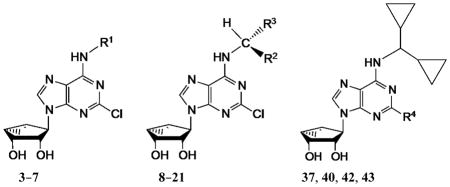
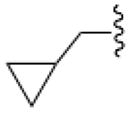
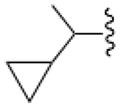
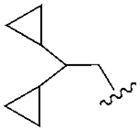
In an effort to increase A1AR affinity and selectivity, we explored a set of N6 substitutions of 4′-truncated (N)-methanocarba adenosines (Table 1), initially containing 2-chloro, that expanded upon our previous communication on an overlapping series of N6-modified truncated nucleosides.20 The present series included substitution of a N6-methynyl group that was either chiral or achiral, e.g. 7, 12–21, including a N6-dicyclopropylmethyl derivative 10 (associated previously with moderate A1AR selectivity) and a N6-dicyclopentylmethyl 11 derivative. In many cases, a N6-cyclopropylmethyl group was further substituted on the α-methynyl carbon with an acyclic (12–15) or cycloalkyl group (16–19), or a phenyl ring (20, 21). The stereochemistry at the methynyl carbon was clearly defined. A few previously reported N6-unsubstituted (3) and N6-alkyl/cycloalkyl derivatives (4–6, 8, and 9)20 were included for comparison in the biological assays. This fine tuning at the N6 position was followed by several modifications at the C2 position, incorporating 2-H, 2-iodo and 2-hydrazino substitutions (37, 40). Additionally, 2-pyrazolyl substitutions (42, 43) were patterned after a set of A1AR selective riboside agonists reported by Elzein et al.23
Table 1.
In vitro potency of a series of truncated (N)-methanocarba adenosine derivatives in binding to three subtypes of hARsand relative efficacy at hA 1AR.
| ||||||
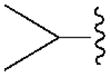
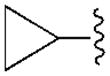
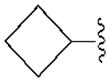
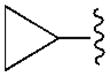
|---|---|---|---|---|---|---|
| Compound (configuration of Cα) | R1= | Affinity Ki, nM or (% inhibition)a | % Inhibition, cyclic AMPd | |||
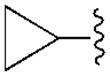
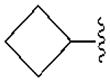
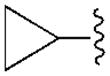
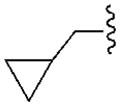
| A1 | A2A | A3 | A1 | |||
| 3b | H | 350±90 | 3140±450 | 160±42 | 68.1±4.4 | |
| 4b | CH2CH3 | 930±110 | (11%) | 6.6±1.6 | ND | |
| 5b |
|
68.4±8.9 | 4410±1090 | 8.9±1.9 | 81.0±21.1 | |
| 6b,c |
 (R,S) (R,S) |
86.8±23.7 | (41%) | 110±17 | 45.5±4.8 | |
| 7 |
|
780±100 | (45±3%) | 670±10 | −9.0±4.1 | |
| R2= | R3= | |||||
| 8b,e | CH3 | CH3 | 72.2±16.4 | (39%) | 12±1 | 50.5±6.4 |
| 9b,e | C2H5 | C2H5 | 78.8±15.6 | 3700±300 | 52±14 | 28.6±3.8 |
| 10b,e |
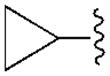
|
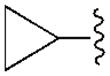
|
47.9±10.5 | 3950±410 | 470±15 | 94.3±5.3 |
| 11 |
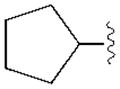
|
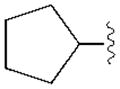
|
(34±3%) | (13±3%) | (48±2%) | ND |
| 12 (R) |
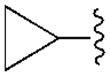
|
C2H5 | 68.1±5.0 | 3610±500 | 150±2 | 37.3±4.3 |
| 13 (S) | C2H5 |
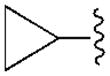
|
150±10 | 4910±430 | 780±70 | 57.9±1.1 |
| 14 (R) |
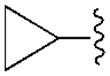
|
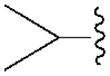
|
76.0±8.0 | 1570±180 | 780±80 | 33.8±2.3 |
| 15 (S) |
|
|
150±50 | 3650±370 | 1720±140 | 20.8±6.1 |
| 16 (R) |
|
|
270±60 | 5470±300 | 2930±480 | 33.4±5.2 |
| 17 (S) |
|
|
120±40 | 6450±720 | 2790±720 | 18.1±6.8 |
| 18 (R) |
|
|
490±90 | 4840±400 | 1760±210 | 18.4±2.1 |
| 19 (S) |
|
|
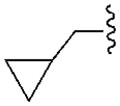
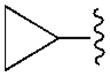
170±20 | 2550±170 | 550±50 | 24.0±5.2 |
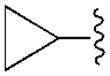
| 20 (R) | Ph |
|
3000±440 | (46±4%) | 790±100 | −7.4±6.0 |
| 21 (S) |
|
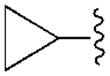
Ph | (50±5%) | (36±4%) | 2200±470 | −2.4±3.7 |
| R4= | ||||||
| 37a | H | 56.1±11.0 | (38±5%) | 17.0±2.0 | ND | |
| 37b | I | 488±95 | 2230±640 | 182±14 | ND | |
| 40 | NHNH2 | 210±80 | (43±7%) | 70.0±35.0 | ND | |
| 42 |
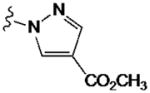
|
730±50 | 4110±750 | 490±150 | ND | |
| 43 |
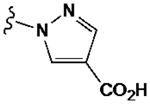
|
1080±230 | (17±5%) | 640±150 | ND | |
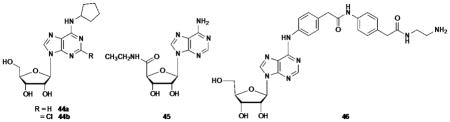
| 44a | j | 1.8±0.5f | 794h | 72±12e | 100±3 | |
| 44b | j | 0.83g | 2270h | 38±6e | ND | |
| 45 | j | 6.8±2.4f | 20h | 35±12e | 100±15 | |
| 46 | j | 10.4±3.8i | 370±100i | 12.4±4.1i | 94±26i | |
Using CHO or HEK293 (A 2Aonly) cells stably expressing a hAR (Supporting Information); affinity was expressed as Ki value (n = 3–5) or percent inhibition of radioligand binding at 10 μM. The radioligands used were: ([3H]N6-R-phenylisopropyladenosine 47, [3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine48, or [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide49, respectively), unless noted.
Compounds3 –6 and 8–10 were prepared previously.44
6 is a diastereomeric mixture.
Maximal efficacy (at 10 μM)in a n A1ARfunctional assay, unless noted determined by inhibition of forskolin-stimulated cyclic AMP production in AR-transfected CHO cells, expressed as percent inhibition (mean standard error, n = 3 –5) in comparison to effect(100%) of full agonist 44a at 10 μM.
Gao et al.59
Klotz et al.25
Müller and Jacobson60
Klutz et al.61 Functional assay in guanine nucleotide binding.
Structures as shown:
ND–not determined
The synthetic route to the truncated 2-chloro derivatives involved nucleophilic displacement by the appropriate amine of a 6-chloroadenine group in an 2′,3′-isopropylidene-protected precursor 22a (Scheme 1). A 2-chloro substitution of the adenine ring has been shown to increase affinity at either or both A1AR and A3AR, and in some cases, to alter AR efficacy.24 Finally, while maintaining constant the most effective N6 substituent dicyclopropylmethyl, substitution of the C2 position was varied by the synthetic routes shown in Scheme 1 (2-iodo, 37b) and Scheme 2 (2-H, 37a; 2-hydrazino, 40; 2-pyrazolo, 42, 43). The attempted synthesis of the 2-hydrazino derivative 40 by acid treatment to remove the isopropylidene group of the protected intermediate 39 resulted in decomposition. However, its preparation by hydrazine treatment of the unprotected nucleoside 10 was successful. Condensation of hydrazine compound 39 with methyl 2-formyl-3-oxo-propionate under reflux conditions23 gave the pyrazole derivative 41, which underwent an acid hydrolysis in the presence of Dowex50 to provide compound 42. Hydrolysis of the methyl ester of compound 42 in the presence of 1 N NaOH afforded acid derivative 43.
Scheme 1.

Synthesis of N6-substituted 4′-truncated derivatives in the ring-constrained (N)- methanocarba adenosine series. Intermediate 22a was prepared as described.44 Reagents and conditions: (a) RNH2, Et3N, MeOH, rt; (b) Dowex50, MeOH/H2O, rt. Compound 24 was used for the following step without isolation.
Scheme 2.

Substitution of the C2 position of truncated nucleosides containing N6- dicyclopropylmethyl substitution, i.e. the most effective for preservation of hA1AR binding affinity, selectivity, and efficacy. Reagents and conditions: (a) RNH2, DIPEA, i-PrOH, reflux; (b) Dowex50, MeOH/H2O, rt; (c) hydrazine, reflux; (d) methoxycarbonyl malondialdehyde, EtOH, reflux; (e) 1 N NaOH, MeOH, rt.
Pharmacological evaluation
Binding affinity at three hAR subtypes was measured in assays using standard agonist radioligands (47, [3H]R-PIA; 48, [3H]CGS21680; 49, [125I]I-AB-MECA) and membrane preparations from Chinese hamster ovary (CHO) cells (A1AR and A3AR) or human embryonic kidney (HEK) 293 cells (A2AAR) stably expressing a hAR subtype (Table 1).18 Because activity within the class of (N)-methanocarba nucleosides was previously noted to be very weak or absent at the hA2BAR,16,25 we did not include this receptor in the initial pharmacological screen. Known potent A1AR agonists, i.e. ribosides (44a, CPA; 44b, CCPA; 45, NECA; 46, ADAC), were included for comparison.20
In the series of 2-chloro derivatives, the A1AR affinity (Ki ≥50 nM) of 5′-truncated (N)-methanocarba-adenosine derivatives (7 –21) was often greater than the affinity at the A3AR (Ki typically ≥500 nM). A N6-dicyclopropylmethyl derivative 10 was the most potent in binding to the A1AR with a Ki value of 47.9 nM and 10-fold selectivity compared to the A3AR.20 Nevertheless, cycloalkyl derivative 7 was nonselective, and a cyclopropyl/ethyl derivative 12 displayed considerable affinity (Ki 150 nM) in A3AR binding. In comparing pairs of pure diastereoisomers differing only in the chirality of the α-methynyl carbon of the N6 group, compounds 12, 17, 19, and 20 were more potent in A1AR affinity than the opposite isomers, i.e. 13, 16, 18, and 21, respectively. N6-Cyclopropyl/isopropylmethyl diastereoisomers 14 and 15 differed by only 2-fold in A1AR binding affinity. A novel N6-(S)-((cyclopropyl-cyclobutyl) methyl) derivative 17 was 23-fold selective for human A1AR (Ki 120 nM) in comparison to A3AR, and 53-fold in comparison to A2AAR. Other potent analogues (Ki, in nM) in hA1AR binding were (R)-cyclopropyl/ethyl 12 (68) and (R)-cyclopropyl/isopropyl 14 (76) derivatives. Compounds 14 and 15, closely related in structure to 10 except for lacking a bond to close one of the cyclopropyl rings, were also moderately selective for the A1AR. Analogues containing a phenyl (20, 21) group on the N6-methynyl substituent bound weakly at the three ARs, and a dicyclopentyl analogue (11) inhibited less than 50% binding at 10 μM.
By introducing subtle structural changes, we attempted to indirectly characterize the environment of the receptor binding site surrounding each substituent on the chiral or prochiral N6-methynyl carbon atom. A graphical comparison of the most potent truncated adenosine derivatives arranged by rank order of hA1AR binding affinity, and illustrating hA1AR/hA3AR selectivity, is shown in Figure 1. We delineated separate binding preferences for the N6-methynyl substituents, i.e. R2 and R3 in Table 1, and the conformational effects of these groups. Compounds 10, 12, 14, 13, 17, and 15 displayed similar A1AR affinities in the range of 50 – 150 nM, but differed in degree of hA1AR/hA3AR selectivity. Dimethyl analogue 8 was an A3AR ligand (Ki 12 nM) with moderate selectivity in comparison to A1AR; diethyl analogue 9 showed comparable affinity at A1 and A3ARs. When R3 was enlarged from a 3-membered to a 4-membered ring (17), A1AR affinity was better preserved than when the same change occurs at R2 (16). Reducing the rigidity of R3 better preserves the A1AR selectivity than reducing the rigidity of R2 (cf. 12 – 15). Introduction of a methylene group that separates a cyclopropyl substituent from the N6-methynyl atom (18 and 19) did not maintain A1AR affinity. Thus, the optimal R3 substituent, i.e. cyclopropyl, could be reduced in size or rigidity to ethyl or isopropyl with only a minor reduction of A1AR affinity. Also, an enlargement of R3 to cyclobutyl reduced affinity by only 2.5-fold. However, when the R2 substituent was made smaller than the cyclopropyl ring, a more substantial loss of A1AR affinity occurred. Therefore, these two subpockets surrounding the N6 substituent had slightly different steric requirements. A greater than one bond deviation or C-atom alteration from the size of cyclopropyl either dramatically reduced the AR affinity or the A1AR selectivity.
Figure 1.

Graphical comparison of the most potent truncated adenosine derivatives arranged by rank order of hA1AR binding affinity (ranging from 48 nM to 270 nM). The lower plots represent the hA1AR selectivity in comparison to the hA3AR and maximal efficacy at the hA1AR (data in Table 1).
Replacement of 2-Cl on compound 10 with 2-H in 37a was tolerated at the A1AR, but affinity at the A3AR increased 28-fold. Substitution of 2-chloro of 10 with iodo greatly reduced affinity and selectivity in 37b. Efforts to combine the N6-dicyclopropylmethyl group with large C2 substituents based on the C2-pyrazolyl derivatives, known to be compatible with A1AR affinity, produced only weak nonselective ligands 42 and 43. The 2-hydrazino derivative 40 was also nonselective, and 10 remained the optimal A1AR-selective structure.
Functional data determined at a single concentration (10 μM) in an assay of adenylate cyclase (A1AR-induced inhibition of cyclic AMP production in CHO cells stably expressing the receptor26) are reported in Table 1. The potent and selective agonist 44a was used as the standard full agonist, and the nonselective AR agonist 5′-N-ethylcarboxamidoadenosine 45 was also a full agonist in this assay. Most of the N6-cycloakylmethyl analogues were partial agonists of the A1AR. However, a concentration-response analysis for 10 and 45 in A1AR-mediated inhibition of cyclic AMP indicated EC50 values were 40.7 ± 19.7 and 10.2 ± 3.3 nM, respectively, which was in close agreement with their A1AR binding affinities. Compound 13 was of intermediate maximal efficacy (57.9% of full agonist) at the A1AR based on results at a single saturating concentration. Other truncated analogues that proved to be of low efficacy in activation of the A1AR, but still within the range of 30–40%, were 12, 14, and 16 (for which affinity at the A1AR exceeded other AR subtypes). Curiously, a phenyl/cyclopropyl analogue 20 did not activate the A1AR at 10 μM, a concentration that exceeded its Ki value by 13-fold, suggesting possible antagonism. Compound 10 was tested for functional activity in stimulation of adenylate cyclase through the hA2BAR expressed in CHO cells and was found to be nearly inactive (13.7±4.4% of activity of full agonist 45 at 10 μM). Thus, the A1AR selectivity of 10 was maintained within the entire AR family.
Molecular modeling
The binding at ARs of the newly synthesized truncated (N)-methanocarba nucleosides was also evaluated through molecular modeling studies. Previously reported homology models of the hA1 and hA3 ARs,20 built using a recently reported agonist-bound A2AAR crystallographic structure (PDB ID: 3QAK) as a template,22 were used to perform docking simulations of each of the compounds 8 –21. In the A2AAR crystal structure, the agonist ligand contained a bulky N6 substituent, which opened EL3 to allow binding of other N6-substituted nucleosides. The thermostabilized agonist-bound A2AAR structure of Lebon et al.,27 in which EL3 is closer to EL2, and which contains mutations in the transmembrane (TM) region, including the ribose binding site, was not used as a template. In our previous study, the docking pose of compound 10 at the hA1AR was compared with that of 5′-N-ethylcarboxamidoadenosine 45.20 Here we give a more detailed description of the binding mode of this new series of truncated (N)-methanocarba nucleosides in terms of their affinity and selectivity profiles.
A docking pose of compound 10 inside the hA1AR binding site (Figure 2, panel A) featured most of the main receptor-ligand interactions observed in the agonist-bound hA2AAR crystal structures.22,27 These interactions, involving both the adenine core and the ribose ring, were noted in the docking poses of this new series of truncated (N)-methanocarba nucleosides at both the hA1 and hA3 ARs. In particular, the 3′- and 2′-hydroxyl groups formed H-bonds with residues (using a GPCR numbering convention28) at positions 7.42 (Thr277 in hA1AR and Ser271 in hA3AR) and 7.43 (His278 in hA1AR and His272 in hA3AR), respectively. The side chain of asparagine 6.55 (Asn254 in hA1AR and Asn250 in hA3AR) strongly interacted with these compounds through two H-bonds involving the 6-amino group and the N7 atom of the adenine ring. Moreover, the adenine core was anchored inside the binding site by a π–π stacking interaction with a phenylalanine in EL2 (Phe171 in hA1AR and Phe168 in hA3AR) and strong hydrophobic contacts with leucine 6.51 (Leu250 in hA1AR and Leu246 in hA3AR) and isoleucine 7.39 (Ile274 in hA1AR and Ile268 in hA3AR).
Figure 2.

(A) Side view and (B) top view of the docking pose of compound 10 (in magenta) inside the binding site of the hA1AR model. Side chains of some amino acids important for ligand recognition and H-bonding interactions are highlighted. Hydrogen atoms are not displayed. The Connolly surface of some amino acids surrounding the binding site is displayed. Surface color indicates hydrophobic regions (green), mildly polar regions (blue) and H-bonding regions (magenta). The boundaries of the two identified subpockets are highlighted (larger subpocket A in yellow and smaller subpocket B in orange). R2 (Table 1) is predicted to occupy subpocket A, and R3 is predicted to occupy subpocket B.
Two key interactions observed for the crystallographic poses of 45-like agonists (5′-N-ethylcarboxamido derivatives) at the hA2AAR were necessarily missing in these truncated derivatives. In fact, in the hA2AAR X-ray structures, Thr88 (3.36) and His250 (6.52) coordinated, through H-bonding interactions, the 5′-CO-NH-alkyl groups of the co-crystallized agonists 45 and 6-(2,2-diphenylethylamino)-9-((2R,3R,4S,5S)-5-(ethylcarbamoyl)-3,4-dihydroxy-tetrahydrofuran-2-yl)-N-(2-(3-(1-(pyridin-2-yl)piperidin-4-yl)ureido)ethyl)-9H-purine-2-carboxamide (50, UK-432097).22,27 The threonine at position 3.36 is conserved among all four AR subtypes, while the histidine at position 6.52 is conserved in hA2A, hA2B and hA1 ARs, but is substituted with a serine in the hA3AR. Therefore, these conserved residues were predicted to also be important in agonist binding at the hA1AR. The difference at position 6.52 could additionally be related to the different behavior of the truncated ring-constrained nucleosides at A1 and A3 ARs, as previously hypothesized.20
Moreover, the hydrophilic region of the receptor associated with ribose binding is key to the activation process, which likely involves essential residues of TM3, TM6, and TM7 throughout the AR family, as observed in the A2AAR.22,27 Thus, the loss of the 5′ substituent is expected to affect AR efficacy, which it clearly does at the A3AR, as observed for other previously reported truncated (N)-methanocarba nucleosides.31 However, the effect at the A1AR is highly variable with relative maximal efficacies of this series ranging from low to high (80–100% in compounds 5 and 10).
The structural basis for the full agonism of 10, in contrast to closely related compounds that have greatly reduced efficacy at this subtype, is difficult to identify but might be related to some interactions formed at the entrance of the binding site, i.e. the N6 binding region, that are crucial in orienting and stabilizing the compound inside the cavity.
A detailed analysis of ligand interactions with the upper region of the binding site has helped to clarify the affinity and selectivity profiles of this new series of truncated (N)-methanocarba derivatives. As shown in Figure 2 (panel B), the orientation of the N6-dicyclopropylmethyl substituent of compound 10 allowed us to identify two distinct upper subpockets in the hA1AR. The larger of the two subpocket, designated “A”, corresponds to the main entrance of the binding site and accommodated one cyclopropyl ring to form hydrophobic contacts with Thr270 (7.35), Tyr271 (7.36), Ile274 (7.39) and the carbon chains of Glu170 (EL2) and Glu172 (EL2). The other cyclopropyl ring of 10 perfectly fits a smaller side subpocket, designated “B”, which was located between TM6 and TM7 and delimited by Leu253 (6.54), Thr257 (6.58), Thr270 (7.35) and at the bottom by Leu250 (6.51). Subpocket A corresponds also to the region that accommodates the extended N6 and C2 groups of 50 in the A2AAR crystal structure.
With respect to this N6 region, docking results showed some differences between hA1 and hA3Rs in the binding of these new truncated (N)-methanocarba derivatives at ARs that could explain their different affinity profiles at these receptors. In fact, the orientation and the interactions of the N6 substituents were particular for each receptor subtype, mainly due to differences between the residues present in the upper region of the binding site. In particular, three residues delimiting subpocket B in the hA1AR are substituted in the hA3AR with amino acids bearing bulkier side chains, namely Ile249 (6.54), Ile253 (6.58) and Leu264 (7.35). While at the hA2AAR, these residues are substituted with Ile252 (6.54), Thr256 (6.58) and Met270 (7.35), respectively, giving rise to again a different scenario. Consequently, at the hA3AR there was no side pocket B between TM6 and TM7 able to accommodate a cyclopropyl ring, and this determined a different orientation of the N6 substituent inside the cavity (Figure 3). In fact, the upper region of the hA3AR binding site was overall more hydrophobic as compared to the hA1AR, but its shape was more suitable to accommodate unbranched hydrophobic substituents. Due to the more difficult fit of the branched N6 group in this region, the position of compound 10 was slightly shifted within the hA3AR binding site and so it established weaker interactions with the key residues deeper in the cavity. This finding can explain the low affinity for the hA3AR of the present set of (N)-methanocarba nucleosides bearing α-branched N6 substituents, with affinity generally decreasing with an increase in volume of the groups on the branches. This view is also consistent with the enhanced hA3AR affinity associated with reported truncated (N)- methanocarba analogs having unbranched hydrophobic substituents, such as benzyl, at the N6 position (also compare compounds 5 and 6).20
Figure 3.

Top view of the docking pose of compound 10 (in magenta) inside the binding site of the hA3AR model. Side chains of amino acids at the entrance of the binding site are highlighted and their Connolly surface is displayed. Surface color indicates hydrophobic regions (green), mildly polar regions (blue) and hydrogen bonding regions (magenta). Hydrogen atoms are not displayed.
Overall, increasing or decreasing the size of the groups on the α-carbon from cyclopropyl decreased hA1AR affinity. Thus, the steric hindrance of these larger groups disfavored the precise conformational arrangement necessary to occupy the two subpockets. Pocket A, although larger in its opening to the extracellular side of the receptor, did not tolerate deviation from R2 = cyclopropyl, while pocket B could accommodate R3 = cyclobutyl or ethyl with some cost in affinity.
Moreover, docking results and conformational analysis of the dihedral N6-Cα bond angles showed that the preferred binding conformation at the hA1AR for this series of truncated (N)-methanocarba nucleosides placed the hydrogen atom on the α-carbon pointing toward TM7, even though an alternative conformation, with the hydrogen oriented toward TM5, was also found (Figure S1, Supporting information). Therefore, in its preferred conformation, the R3 group on the Cα occupied the smaller subpocket B, while the R2 group was located in the larger subpocket A. Depending on the groups attached to the α-carbon and their steric complementarity with the two subpockets, two opposite diastereoisomers bound with different strength to the receptor binding site and so possessed different binding affinity. In fact, if a small group (e.g. ethyl) with low complementarity with the larger subpocket A, was present on the Cα, then the most potent diastereoisomer was the one that can more easily accommodate that group in subpocket B (compound 12, ethyl group in R3, as compared to compound 13, ethyl group in R2). On the other hand, if an extended group (e.g. cyclopropylmethyl) was present on the Cα, then the isomer able to locate this group in the larger subpocket A possesses higher affinity at the receptor (e.g. 19, cyclopropylmethyl group in R2, as compared to 18, cyclopropylmethyl group in R3).
Anticonvulsant testing
Three A1AR agonists were examined in models of electrically and chemically-induced seizures (Table 2).29 The anticonvulsant activity of 2-chloro-N6-cyclopentyladenosine (44b, CCPA) was studied previously,7,9 and 44b was included in the present study as a potent reference A1AR agonist that does not distinguish between central and peripheral action. N6-[4-[[[4-[[[(2-Aminoethyl)amino]carbonyl]methyl]anilino]carbonyl]methyl]phenyl]adenosine (46, ADAC) is a potent A1AR agonist that has been shown to be neuroprotective in various models.6,30 Full agonist 10 displayed efficacy in a seizure model in mice without the toxicity observed in the active dose range (tested up to 30 mg/kg, i.p.). Other A1AR agonists began to show toxicity, i.e. the side effects evident in the rotarod performance test (i.e. inability to remain on the rotarod) overlapping the active dose range.
Table 2.
Anticonvulsant activity in mice of A1AR agonists.
| Compounda | Behavioral toxicity TD50 (mg/kg) | 6 Hz model ED50 (mg/kg) | MES modelb (dose) | scMET modelb (dose) |
|---|---|---|---|---|
| 10 | > 30d | 2.74c | 1 out of 4 (3 mg/kg) | No protection (3 mg/kg) |
| 44b | 0.84c | 0.12c | 1 out of 4 at (1 mg/kg) | 1 out of 4 (1 mg/kg) |
| 46 | 0.14e | 0.03c | 4 out of 8 (2mg/kg) | 5 out of 8 (1 mg/kg) |
administered i.p.
qualitative results, expressed as number of animals protected from convulsions.
measured at 1 h (time of peak of effect) post injection, dose range for 10 was 0.75 – 10 mg/kg.
No rotarod toxicity at 30 mg/kg.
measured at 4 h (time of peak of effect) post injection.
The 6 Hz minimal clonic seizure model is an acute electroshock seizure test that produces a seizure with a limbic phenotype and displays a unique pharmacological profile to established antiepileptic drugs. In particular, the 6 Hz seizure is uniquely sensitive to the antiepileptic drug levetiracetam and partially resistant to the Na+ channel blockers.31 Three A1AR agonists, the two reference compounds 44b and 46 and the truncated derivative 10, displayed efficacy in the 6 Hz model. The activities of 10 and 44b were quantified at 1 h post administration of the compound, which was also equal to the time of peak effect (TPE). Complete dose-response curves using five different doses each of 10, 44b and 46 indicated ED50 values of 2.74, 0.12 and 0.03 mg/kg, respectively. Compound 10 protected in 4 out of 8 mice at 1.5 mg/kg. Compared to the activity in the 6 Hz model, these compounds had no or minimal effect in the traditional seizure models, the maximal electroshock (MES) model or the subcutaneous metrazol (pentylenetetrazol) model (scMET) at the same dosing range.
In the minimal behavioral toxicity test using the rotarod, the three compounds showed significant differences. The toxicity of 44b was observed roughly in the same dose range (0.84 vs. 0.12 mg/kg) as its protection in the 6 Hz model and at the same time point (1 h post injection). Animals became lethargic and were unable to stay on the rotarod following i.p. drug administration, as exhibited with other A1AR agonists.32 For compound 10, no toxicity (0 out of 8 mice) was observed at all doses tested up to 30 mg/kg, the highest dose tested, which was nearly completely protective (7 out of 8 animals) in the 6 Hz model. Therefore, the therapeutic window for 10 appeared to be superior to that of 44b.
Compound 10 was also tested in the corneal kindled mouse model to examine its effect on focal seizures. In a qualitative test, an EC50 (±SE) of 1.79±0.71 mg/kg was determined at 1 h post injection. This unique response profile of compound 10 makes it as an attractive candidate to treat drug-resistant epilepsy.
Discussion
Our previous closely related communication20 demonstrated that the affinity of truncated ring-constrained analogues, in comparison to ribosides, was less well preserved at the A1AR than at the A3AR. However, certain analogues, most notably a dicyclopropylmethyl analogue 10, were relatively well preserved in binding and activation of the A1AR. In this study, we built new analogues on the previous observation that 10 had 10-fold A1AR selectivity in comparison to the A3AR and 74-fold of selectivity vs. A2AAR. Moreover, this analogue was a full agonist at the hA1AR. The fine-tuning of the structure of 10 now indicates that even minor adjustments of the structure cause a loss of potency, selectivity, or efficacy at the hA1AR.
Our binding results at the hA1AR showed that only Cl was preferred at the adenine C2 position, and N6 substituents other than dicyclopropylmethyl displayed inferior pharmacological profiles. Separate substituents (R2 and R3) of the Cα methynyl group had distinct and very narrow SAR requirements. Groups that were much larger or smaller than cyclopropyl were not compatible with A1AR affinity, selectivity and efficacy. Therefore, we describe the compound with optimal pharmacological properties, compound 10, as a structural sweet spot for potent and selective activation of the A1AR.
Molecular docking studies of these truncated (N)-methanocarba nucleosides at the hA1AR highlighted how a precise complementarity in the N6 region was needed to determine a good affinity and selectivity profile and to compensate for the missing anchoring effect of the ribose 5′ region. In fact, a detailed modeling analysis of the upper part of the hA1AR binding site predicted two different subpockets, A and B, able to accommodate the N6 substituents of these derivatives, and it seemed that an optimal occupancy of both subpockets was required for enhanced affinity at this receptor subtype. In particular, the smaller subpocket B can readily accommodate up to a cyclobutyl group, while subpocket A can fit substituents that are extended in the direction of EL2. The steric restriction of the subpockets can be the reason for the null affinity at hA1AR of compound 11 bearing two cyclopentyl groups, as they are too bulky to fit in either pocket. On the other hand, smaller groups on the α-carbon of the N6 substituent, such as dimethyl or diethyl (compounds 8 and 9, respectively), even though they can occupy the pockets, possess lower complementarity as compared to the cyclopropyl group and consequently have no selectivity and lower affinity at the A1AR.
The identification of these two subpockets and the binding conformation proposed here are also in agreement with the binding data of others known A1AR agonists.21 For example, the moderately selective A1AR agonist R(−)-N6-(2-phenylisopropyl)-adenosine (47) is more potent than the corresponding opposite isomer (S-PIA). A hypothetical binding mode for these adenosine derivatives, similar to the one proposed for our new rigid, truncated nucleosides, would readily place the phenylmethyl group of the R isomer in the larger subpocket A and the methyl group in the smaller subpocket B. Thus, there is a close correspondence of our new findings to the previously explored preference for N6-Cα-branched R isomers in comparison to the corresponding S isomers.33 A summary of the characteristics of the two pockets surrounding substituents of the N6-methynyl carbon atom is provided in Table 3.
Table 3.
Summary of the characteristics of the two pockets in A1AR surrounding substituents of the N6-methynyl carbon atom, based on observed SAR and predictions from molecular modeling (see Table 1 for definition of R2 and R3).
| Characteristic | Pocket A | Pocket B |
|---|---|---|
| Size | Larger (extends upward, but has narrow dimensions near N6-methynyl atom) | Smaller |
| Location | TM7 – EL2 | Between TM6 and TM7 |
| Contact residuesa | Thr270 (7.35), Tyr271 (7.36), Ile274 (7.39), Glu170 (EL2), Glu172 (EL2) | Leu253 (6.54), Thr257 (6.58), Thr270 (7.35), Leu250 (6.51) |
| Correspondence to N6 group of 47b | site of PhCH2 binding | site of CH3 binding |
| Present in A3AR? | Yes | Noc |
| Correspondence to R (assuming most favorable docking with H toward TM7) | R2 | R3 |
| Preferred group | c-Pr | c-Pr |
| Accommodates group smaller than c-Pr? (Et) | No (exact steric complementarity is important) | Yes |
| Accommodates group less rigid than c-Pr? (i-Pr) | No | Yes |
| Accommodates group larger than c-Pr? (c-Bu) | Nod | Yes (only c-Bu) |
Using residue numbering convention of Ballesteros and Weinstein.28
47, (R)-N6-phenylisopropyladenosine.
Consistent with lower affinity of N6 α-branched analogues at the A3AR.
No clear explanation from modeling, except that the dimensions near the N6-methynyl atom might be sterically restrictive. A larger group is tolerated, with intermediate affinity, only if there is an α-CH2 spacer (19).
Complementary anchoring of the N6 substituent inside the two subpockets seems important in orienting and stabilizing the compound inside the cavity. It could also help in keeping the adenine-methanocarba moiety in an efficacious active conformation, able to form strong H-bonds and hydrophobic interactions with residues in TM6, TM7, and EL2, while compensating for the missing interactions due to the lack of a 5′ substituent.
In in vivo testing, compound 10 was the only A1AR agonist examined here that displayed a clear separation of anticonvulsant activity and toxicity. For example, a prototypical A1AR agonist 44b displayed toxicity in the rotarod assay (2 out of 8 animals) at a dose of 0.1 mg/kg. At a dose of 0.5 mg/kg 44b, 3 out of 8 animals displayed toxicity. Another known A1AR agonist 46 was more potent than 44b in both in vivo anticonvulsant activity and toxicity, but also failed to demonstrate a separation of the two activities.
The unique response profile of compound 10 (inactive in MES and scMET, active in 6 Hz and corneal kindling models) and its novel mechanism of action through the A1AR make it a potential candidate to treat drug-resistant epilepsy. Traditional anti-epileptic drugs (AEDs) carbamazepine, lamotrigine, phenytoin and topiramate are Na+ channel blockers, which have strong efficacy in the MES model.34 They are either inactive or only partially efficacious in the 6 Hz model. On the other hand, newer AEDs having different mechanisms of action, such as levetiracetam and retigabine, are potent and efficacious in the 6Hz model, which makes the 6 Hz model a model for identifying compounds that potentially target drug-resistant epilepsy.31 The kindling models are useful in searching for drugs to treat complex partial seizures, because kindled seizures not only provide an experimental model of focal seizures but also a means of testing drugs to stop seizure spread and generalization from a focus.35 The corneal kindled mouse model demonstrates a pharmacological profile consistent with the traditional hippocampal kindled rat model, while it requires no implantation surgery and less compound quantity for testing.36 The unique activities of compound 10 in the 6Hz and corneal kindled mouse model and its overcoming the limitations of other A1AR agonists (i.e. clear separation of anticonvulsant activity and toxicity) make it an attractive AED candidate for additional testing.
Many of the efforts to develop A1AR agonists for peripheral applications, such as treating cardiac arrhythmias, have tried to limit CNS penetration to avoid centrally mediated side effects. Conversely, other envisioned applications of A1 agonists, i.e. for neurodegenerative and neurological disorders, depend on brain entry. A1AR agonists have distinct neuroprotective and antinociceptive properties,5,6,12 and activation of the A1AR by endogenous adenosine mediates the protective effects of fractalkine/CX3CL1.37 However, the clinical development of previous generations of such agents has been limited by side effects, including cardiovascular effects. In previous studies of the activity of A1AR agonists in the CNS, only a small fraction of a peripherally administered agent crossed the blood-brain barrier (BBB).38 However, a similar attempt to alter the biodistribution by removing the 2′-hydroxyl group of 44a did not enhance brain uptake.35
The peripheral side effects of exogenously administered A1AR agonists that do not distinguish between the brain and periphery have impeded the development of such agents for the treatment of epileptic seizures. Recently, it was proposed that the anti-seizure effect of endogenous adenosine could be boosted indirectly by inhibiting formation of the neurabin-RGS4 complex for “fine-tuning adenosine receptor function in the nervous system”.39 Other approaches to solve this problem involved the design of ligands that favor the CNS over the periphery, such as N6-[R-(2-benzothiazolyl)thio-2-propyl]-2-chloroadenosine (NNC-21-0136, Kd = 1.16 nM at rat A1AR), that showed efficacy in in vivo stroke models and were reduced in their accompanying cardiovascular side effects.5 Prodrug approaches and localized adenosine delivery have also been explored.12,40
The physicochemical properties of nucleosides that act as AR agonists often lead to limited in vivo bioavailability and reduced passage across the BBB. The cLog P of compound 10 is 1.41 (more favorable than cLog P of 0.14 for A1AR-selective riboside and prototypical agonist 44a), with the optimal for small molecular pharmaceutical substances being typically 2 – 3.41 Also, the total polar surface area (tPSA) for 10 and 44a are calculated to be 92.8 and 122 Å2, respectively. Most drug-like small molecules have a PSA smaller than 120 Å2; thus, 10 is also preferred by this criterion. Compound 10 has fewer hydroxyl groups than 44a, which would favor bioavailability in brain, and the molecular weight of 10 (376 D) is comfortably within the most desirable range for pharmaceuticals.41 Therefore, by several criteria 10 is more drug-like than 44a.
We speculate on the basis for the apparent lack of peripheral side effects of compound 10 up to 10 mg/kg. Both 44b and 10 evidently enter the blood stream and pass the BBB into the brain. Although 44b was ~58-fold more potent than 10 in binding at the A1AR, 10 had better physicochemical properties (cLogP, tPSA, H-bond donors, molecular weight), so it might penetrate the BBB better than 44b. The bioavailability in the brain of peripherally administered 10, i.e. whether its altered physicochemical properties may facilitate its passage across the BBB, is undetermined. Normally a drug travels from the blood stream into brain, such that when the A1AR in brain is activated, the A1AR in heart should be activated within the same dose range as with 44b. For 10, the fact that no adverse effects were observed even at 30 mg/kg (ten times the ED50) suggests that the free concentration of 10 in plasma might be reduced relative to standard A1AR agonists, possibly due to plasma protein binding. If the free drug concentration in blood would be lower, its ability to cross the BBB may remain unaffected or even increased. This phenomenon has been observed for a number of CNS drugs.42 This hypothesis could be tested by pharmacokinetic and plasma protein binding assays.
A1AR agonists hold interest therapeutically for their cardio- and neuroprotective, antiarrhythmic, antiseizure, antilipolytic, antiglaucoma, and anxiolytic actions. Some of the novel derivatives were partial A1AR agonists, which are of interest for both cerebroprotective and cardiovascular application, depending on levels of endogenous adenosine and on receptor reserve.3,43 It is conceivable that the expanded range of physical properties in the present series of truncated derivatives would offer pharmacokinetic advantages. Therefore, this approach is appealing for preclinical development. This hypothesis will have to be evaluated in further in vivo studies.
Experimental procedures
Chemical synthesis
General Methods
All reagents and solvents (regular and anhydrous) were of analytical grade, obtained from commercial suppliers and used without further purification. All amines were purchased from Asiba Pharmatech (Edison, NJ), except 2,2-dicyclopropylethylamine, which was obtained from Ryan Scientific, Inc. (Mount Pleasant, SC), and dicyclopropylmethylamine, which was obtained from J&W PharmLab (Levittown, PA). Compounds 22a and 35 were synthesized as reported.44 Reactions were conducted under an atmosphere of nitrogen whenever anhydrous solvents were used. All reactions were monitored by thin-layer chromatography (TLC) using silica gel coated plates with a fluorescence indicator which were visualized: (a) under UV light, (b) by dipping in a mixture of anisaldehyde (2.5 mL)/conc. H2SO4 (5 mL)/methanol (425 mL) or (c) by dipping the plate in a solution of ninhydrin (0.3 g in 100 mL EtOH, containing AcOH, 1.3 mL) followed by heating. Silica gel column chromatography was performed with silica gel (SiO2, 200–400 mesh, 60 Å) using moderate air pressure. Evaporation of solvents was carried out under reduced pressure at a temperature below 50 °C. After column chromatography, appropriate fractions were pooled, evaporated, and dried at high vacuum for at least 12 h to give the desired products in high purity. 1H NMR spectra were recorded with a Bruker 400 MHz NMR spectrometer. Chemical shifts are reported in parts per million (ppm) relative to tetramethylsilane or using deuterated solvent as the internal standard (δH: CDCl3 7.26 ppm). ESI - High Resolution Mass Spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine. Observed mass accuracies are those expected on the basis of known performance of the instrument as well as the trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time-dependent drift in mass accuracy. TLC analysis was carried out on glass sheets precoated with silica gel F254 (0.2 mm) from Sigma-Aldrich (St. Louis, MO). The purity of final nucleoside derivatives was checked using a Hewlett–Packard 1100 HPLC equipped with a Zorbax SB-Aq 5 μm analytical column (50 × 4.6 mm; Agilent Technologies Inc., Palo Alto, CA). Mobile phase: linear gradient solvent system: 5 mM TBAP (tetrabutylammonium dihydrogenphosphate)-CH3CN from 80:20 to 0:100 in 13 min; the flow rate was 0.5 mL/min. Peaks were detected by UV absorption with a diode array detector at 230, 254, and 280 nm. All derivatives tested for biological activity showed >95% purity by HPLC analysis (detection at 254 nm).
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-(2,2-dicyclopropylethylamino)-9H-purin-9- yl)bicyclo[3.1.0]hexane-2,3-diol (7)
Dowex 50 resin (8 mg) was added to a solution of compound 23 (12 mg, 0.027 mmol) in methanol (0.5 mL) and water (0.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was filtered on a celite bed, the filtrate was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 35:1) to give the desired compound 7 (8.2 mg, 74%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 4.79 (s, 1H), 4.71 (t, J = 5.6 Hz, 1H), 4.90 (d, J = 6.8 Hz, 1H), 3.72 (d, J = 6.4 Hz, 2H), 2.01-1.96 (m, 1H), 1.71-1.64 (m, 1H), 1.34-1.30 (m 2H), 0.78-0.72 (m, 2H), 0.51-0.42 (m, 5H), 0.28-0.17 (m, 4H). HRMS calculated for C19H25ClN5O2 (M + H) +: 390.1697; found 390.1708.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-(dicyclopentylmethylamino)-9H-purin-9- yl)bicyclo[3.1.0]hexane-2,3-diol (11)
Dicyclopentylmethylamine (1.0 mg, 0.024 mmol) and triethylamine (0.1 mL, 0.16 mmol) were added to a solution of compound 22a (4.2 mg, 0.012 mmol) in methanol (0.8 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum and the residue was roughly purified on flash silica gel column chromatography. The resulting compound 24 was dissolved with methanol (0.6 mL) and water (0.3 mL). Dowex50 (4 mg) was added to the solution and stirring continued at room temperature overnight. After completion of starting material, the reaction mixture was filtered and the filtrate was evaporated under vacuum. The residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 30:1) to give the desired compound 11 (3.4 mg, 64%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.17 (s, 1H), 4.80 (s, 1H), 4.71 (t, J = 6.0 Hz, 1H), 4.40 (t, J = 6.8 Hz, 1H), 3.91 (d, J = 6.4 Hz, 1H), 2.20-1.18 (m, 1H), 1.98-1.96 (m, 1H), 1.81-1.74 (m, 4H), 1.69-1.53 (m, 10H), 1.42-1.29 (m 4H), 0.92-0.90 (m, 1H), 0.81-0.74 (m, 1H). HRMS calculated for C19H25ClN5O2 (M + H) +: 390.1697; found 390.1708.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((R)-1-cyclopropylpropylamino)-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol (12)
Dowex 50 resin (5 mg) was added to a solution of compound 25 (10 mg, 0.024 mmol) in methanol (0.5 mL) and water (0.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was filtered on a Celite bed, the filtrate was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 30:1) to give the desired compound 12 (6.1 mg, 69%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 4.79 (s, 1H), 4.71 (t, J = 5.6 Hz, 1H), 3.89 (d, J = 6.8 Hz, 1H), 3.66 (br s, 1H), 2.00-1.97 (m, 1H), 1.84-1.65 (m, 3H), 1.34-1.28 (m, 2H), 1.01 (t, J = 7.6 Hz, 3H), 0.80-0.74 (m, 1H), 0.60-0.58 (m, 1H), 0.45-0.42 (m, 2H), 0.36-0.32 (m, 1H). HRMS calculated for C17H23ClN5O2 (M + H) +: 364.1540; found 364.1538.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((S)-1-cyclopropylpropylamino)-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol (13)
Dowex 50 resin (3 mg) was added to a solution of compound 26 (7 mg, 0.017 mmol) in methanol (0.3 mL) and water (0.3 mL) and the mixture stirred at room temperature overnight. The reaction mixture was filtered on a Celite bed, the filtrate was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 30:1) to give the desired compound 13 (4.3 mg, 68%) as a syrup.1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 4.79 (s, 1H), 4.71 (t, J = 5.6 Hz, 1H), 3.89 (d, J = 6.8 Hz, 1H), 3.67 (br s, 1H), 2.02-1.98 (m, 1H), 1.86-1.59 (m, 3H), 1.32-1.24 (m, 2H), 1.01 (t, J = 7.6 Hz, 3H), 0.78-0.76 (m, 1H), 0.59-0.57 (m, 1H), 0.45-0.42 (m, 2H), 0.34-0.32 (m, 1H). HRMS calculated for C17H23ClN5O2 (M + H) +: 364.1540; found 364.1535.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((R)-1-cyclopropyl-2-methyl-propylamino)-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol (14)
Dowex 50 resin (7 mg) was added to a solution of compound 27 (10.71 mg, 0.025 mmol) in methanol (0.6 mL) and water (0.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was filtered on a Celite bed, the filtrate was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 35:1) to give the desired compound 14 (7.82 mg, 82%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.17 (s, 1H), 4.79 (s, 1H), 4.71 (t, J = 5.6 Hz, 1H), 3.89 (d, J = 6.4 Hz, 1H), 3.62 (t, J = 7.2 Hz, 1H), 2.07-1.97 (m, 2H), 1.88-1.64 (m, 1H), 1.36-1.31 (m, 1H), 1.08-1.05 (m, 8H), 0.80-0.74 (m, 1H), 0.66-0.61 (m, 1H), 0.45-0.36 (m, 2H). HRMS calculated for C18H25ClN5O2 (M + H) +: 378.1697; found 378.1691.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((S)-1-cyclopropyl-2-methyl-propylamino)-9H-purin-9- yl)bicyclo[3.1.0]hexane-2,3-diol (15)
Dowex 50 resin (5 mg) was added to a solution of compound 28 (6.8 mg, 0.016 mmol) in methanol (0.5 mL) and water (0.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was filtered on a Celite bed, the filtrate was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 35:1) to give the desired compound 15 (4.8 mg, 81%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.17 (s, 1H), 4.79 (s, 1H), 4.71 (t, J = 5.6 Hz, 1H), 3.89 (d, J = 6.4 Hz, 1H), 3.63 (t, J = 7.2 Hz, 1H), 2.09-1.96 (m, 2H), 1.86-1.64 (m, 1H), 1.34-1.30 (m, 1H), 1.08-1.05 (m, 8H), 0.80-0.74 (m, 1H), 0.65-0.61 (m, 1H), 0.45-0.36 (m, 2H). HRMS calculated for C18H25ClN5O2 (M + H) +: 378.1697; found 378.1694.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((R)-cyclopropylcyclobutylmethylamino)-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol (16)
Dowex 50 resin (4 mg) was added to a solution of compound 29 (5.9 mg, 0.013 mmol) in methanol (0.4 mL) and water (0.4 mL) and the mixture stirred at room temperature for 7 h. The reaction mixture was filtered on a Celite bed, the filtrate was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 30:1) to give the desired compound 16 (3.9 mg, 74%) as an oil. 1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 4.79 (s, 1H), 4.71 (t, J = 5.6 Hz, 1H), 3.89 (d, J = 6.4 Hz, 1H), 3.73 (br s, 1H), 2.69-2.64 (m, 1H), 2.10-2.07 (m, 1H), 2.01-1.91 (m, 4H), 1.89-1.80 (m, 1H), 1.69-1.65 (m, 1H), 1.34-1.30 (m, 3H), 0.95-0.91 (m, 1H), 0.78-0.74 (m, 1H), 0.56-0.53 (m, 1H), 0.40-0.36 (m, 2H). HRMS calculated for C19H25ClN5O2 (M + H) +: 390.1697; found 390.1711.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((S)-cyclopropylcyclobutylmethylamino)-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol (17)
Dowex 50 resin (6 mg) was added to a solution of compound 30 (7.95 mg, 0.018 mmol) in methanol (0.6 mL) and water (0.4 mL) and the mixture stirred at room temperature for 7 h. The reaction mixture was filtered on a Celite bed, the filtrate was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 30:1) to give the desired compound 17 (5.3 mg, 74%) as an oil.1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 4.79 (s, 1H), 4.71 (t, J = 5.6 Hz, 1H), 3.89 (d, J = 6.4 Hz, 1H), 3.73 (br s, 1H), 2.69-2.62 (m, 1H), 2.12-2.07 (m, 1H), 2.03-1.88 (m, 4H), 1.89-1.80 (m, 1H), 1.69-1.65 (m, 1H), 1.34-1.31 (m, 3H), 0.95-0.91 (m, 1H), 0.78-0.76 (m, 1H), 0.56-0.53 (m, 1H), 0.40-0.36 (m, 2H). HRMS calculated for C19H25ClN5O2 (M + H) +: 390.1697; found 390.1697.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((R)-1,2-dicyclopropylethylamino)-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol (18)
Dowex 50 resin (3 mg) was added to a solution of compound 31 (4.94 mg, 0.018 mmol) in methanol (0.3 mL) and water (0.3 mL) and the mixture stirred at room temperature for 7 h. The reaction mixture was filtered on a Celite bed, the filtrate was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 35:1) to give the desired compound 18 (3.4 mg, 78%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 4.79 (s, 1H), 4.71 (t, J = 5.6 Hz, 1H), 3.89 (d, J = 6.8 Hz, 1H), 3.80 (br s, 1H), 2.02-1.97 (m, 1H), 1.70-1.61 (m, 2H), 1.34-1.30 (m, 3H), 1.10-1.08 (m, 1H), 0.91-0.76 (m, 2H), 0.62-0.58 (m, 1H), 0.47-0.36 (m, 4H), 0.17-0.07 (m, 2H). HRMS calculated for C19H25ClN5O2 (M + H) +: 390.1697; found 390.1691.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((S)-1,2-dicyclopropylethylamino)-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol (19)
Dowex 50 resin (5 mg) was added to a solution of compound 32 (7.6 mg, 0.019 mmol) in methanol (0.5 mL) and water (0.5 mL) and the mixture stirred at room temperature for 7 h. The reaction mixture was filtered on a Celite bed, the filtrate was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 35:1) to give the desired compound 19 (5.2 mg, 76%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 4.79 (s, 1H), 4.71 (t, J = 5.6 Hz, 1H), 3.89 (d, J = 6.8 Hz, 1H), 3.80 (br s, 1H), 2.00-1.97 (m, 1H), 1.69-1.61 (m, 2H), 1.34-1.30 (m, 3H), 1.10-1.08 (m, 1H), 0.89-0.75 (m, 2H), 0.60-0.59 (m, 1H), 0.47-0.36 (m, 4H), 0.17-0.07 (m, 2H). HRMS calculated for C19H25ClN5O2 (M + H) +: 390.1697; found 390.1697.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((R)-cyclopropylphenylmethylamino)-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol (20)
Dowex 50 resin (5 mg) was added to a solution of compound 33 (6.6 mg, 0.014 mmol) in methanol (0.6 mL) and water (0.4 mL) and the mixture stirred at room temperature for 7 h. The reaction mixture was filtered on a Celite bed, the filtrate was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 30:1) to give the desired compound 20 (4.8 mg, 80%) as a syrup.1H NMR (CD3OD, 400 MHz) δ 8.16 (s, 1H), 7.48 (d, J = 7.6 Hz, 2H), 7.33 (t, J = 7.6 Hz, 2H), 7.26 (t, J = 7.6 Hz, 1H), 4.78 (s, 2H), 4.69 (t, J = 5.6 Hz, 1H), 3.88 (t, J = 7.2 Hz, 1H), 2.01-1.95 (m, 1H), 1.68-1.63 (m, 1H), 1.42-1.30 (m, 2H), 0.79-0.73 (m, 1H), 0.65-0.62 (m, 2H), 0.54-0.51 (m, 2H). HRMS calculated for C21H23ClN5O2 (M + H) +: 412.1540; found 412.1533.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((S)-cyclopropylphenylmethylamino)-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol (21)
Dowex 50 resin (7 mg) was added to a solution of compound 34 (8.6 mg, 0.019 mmol) in methanol (0.8 mL) and water (0.4 mL) and the mixture stirred at room temperature for 7 h. The reaction mixture was filtered on a celite bed, the filtrate was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 30:1) to give the desired compound 21 (6.2 mg, 79%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.17 (s, 1H), 7.48 (d, J = 7.6 Hz, 2H), 7.33 (t, J = 7.6 Hz, 2H), 7.24 (t, J = 7.2 Hz, 1H), 4.78 (s, 2H), 4.71 (t, J = 5.2 Hz, 1H), 3.88 (t, J = 7.6 Hz, 1H), 2.02-1.95 (m, 1H), 1.68-1.65 (m, 1H), 1.43-1.30 (m, 2H), 0.79-0.73 (m, 1H), 0.65-0.60 (m, 2H), 0.54-0.47 (m, 2H). HRMS calculated for C21H23ClN5O2 (M + H) +: 412.1540; found 412.1544.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-(2,2-dicyclopropylethylamino)-9H-purin-9-yl)-2,3-O- (isopropylidene)-bicyclo[3.1.0]hexane (23)
2,2-Dicyclopropylethylamine (38 mg, 0.30 mmol) and triethylamine (0.12 mL, 0.84 mmol) were added to a solution of compound 22a (20.91 mg, 0.061 mmol) in methanol (1.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum and the residue was purified on flash column chromatography (hexane:ethyl acetate = 1:1) to give the desired product 23 (22 mg, 84%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.14 (s, 1H), 5.36 (t, J = 6.0 Hz, 1H), 4.97 (s, 1H), 4.69 (d, J = 7.2 Hz, 1H), 3.73 (d, J = 6.4 Hz, 2H), 2.09-2.03 (m, 1H), 1.76-1.71 (m, 1H), 1.52 (s, 3H), 1.25 (s, 3H), 0.95-0.90 (m, 2H), 0.75-0.72 (m, 2H), 0.51-0.42 (m, 5H), 0.28-0.17 (m, 4H). HRMS calculated for C22H29ClN5O2 (M + H) +: 430.2010; found 430.2013.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((R)-1-cyclopropylpropylamino)-9H-purin-9-yl)-2,3-O- (isopropylidene)-bicyclo[3.1.0]hexane (25)
(R)-1-Cyclopropylpropylamine hydrochloride (16.16 mg, 0.11 mmol) and triethylamine (0.11 mL, 0.82 mmol) were added to a solution of compound 22a (20.33 mg, 0.05 mmol) in methanol (1.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum and the residue was purified on flash column chromatography (hexane:ethyl acetate = 1:1) to give the desired product 25 (18.7 mg, 78%) as a foamy syrup. 1H NMR (CD3OD, 400 MHz) δ 8.13 (s, 1H), 5.36 (t, J = 6.4 Hz, 1H), 4.96 (s, 1H), 4.69 (d, J = 7.2 Hz, 1H), 3.66 (br s, 1H), 2.07-2.04 (m, 1H), 1.86-1.79 (m, 1H), 1.77-1.70 (m, 2H), 1.52 (s, 3H), 1.25 (s, 3H), 1.03-0.99 (m, 4H), 0.95-0.90 (m, 2H), 0.62-0.56 (m, 1H), 0.45-0.41 (m, 2H), 0.36-0.33 (m, 1H). HRMS calculated for C20H27ClN5O2 (M + H) +: 404.1853; found 404.1855.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((S)-1-cyclopropylpropylamino)-9H-purin-9-yl)-2,3-O- (isopropylidene)-bicyclo[3.1.0]hexane (26)
(S)-1-Cyclopropylpropylamine hydrochloride (32.8 mg, 0.11 mmol) and triethylamine (0.11 mL, 0.82 mmol) were added to a solution of compound 22a (20.43 mg, 0.05 mmol) in methanol (1.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum and the residue was purified on flash column chromatography (hexane:ethyl acetate = 1:1) to give the desired product 26 (16.6 mg, 80%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.13 (s, 1H), 5.36 (t, J = 6.4 Hz, 1H), 4.96 (s, 1H), 4.69 (d, J = 7.2 Hz, 1H), 3.67 (br s, 1H), 2.09-2.03 (m, 1H), 1.88-1.79 (m, 1H), 1.77-1.72 (m, 2H), 1.52 (s, 3H), 1.25 (s, 3H), 1.03-0.99 (m, 4H), 0.96-0.89 (m, 2H), 0.61-0.56 (m, 1H), 0.47-0.41 (m, 2H), 0.36-0.30 (m, 1H). HRMS calculated for C20H27ClN5O2 (M + H) +: 404.1853; found 404.1854.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((R)-1-cyclopropyl-2-methyl-propylamino)-9H-purin-9-yl)- 2,3-O-(isopropylidene)-bicyclo[3.1.0]hexane (27)
(R)-1-Cyclopropyl-2-methyl-propylamine hydrochloride (18.22 mg, 0.12 mmol) and triethylamine (0.11 mL, 0.85 mmol) were added to a solution of compound 22a (20.78 mg, 0.06 mmol) in methanol (1.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum and the residue was purified on flash column chromatography (hexane:ethyl acetate = 1:1) to give the desired product 27 (19 mg, 75%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 5.36 (t, J = 6.0 Hz, 1H), 4.97 (s, 1H), 4.70 (d, J = 6.4 Hz, 1H), 3.62 (t, J = 7.2 Hz, 1H), 2.08-2.03 (m, 1H), 1.76-1.72 (m, 1H), 1.52 (s, 3H), 1.25 (m, 4H), 1.08-1.05 (m, 7H), 0.96-0.88 (m, 2H), 0.65-0.61 (m, 1H), 0.45-0.36 (m, 2H). HRMS calculated for C21H29ClN5O2 (M + H) +: 418.2010; found 418.2004.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((S)-1-cyclopropyl-2-methyl-propylamino)-9H-purin-9-yl)- 2,3-O-(isopropylidene)-bicyclo[3.1.0]hexane (28)
(S)-1-Cyclopropyl-2-methyl-propylamine hydrochloride (16.5 mg, 0.12 mmol) and triethylamine (0.12 mL, 0.84 mmol) were added to a solution of compound 22a (20.85 mg, 0.06 mmol) in methanol (1.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum and the residue was purified on flash column chromatography (hexane:ethyl acetate = 1:1) to give the desired product 28 (19.8 mg, 78%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 5.36 (t, J = 6.0 Hz, 1H), 4.97 (s, 1H), 4.71 (d, J = 6.4 Hz, 1H), 3.61 (t, J = 7.2 Hz, 1H), 2.08-2.04 (m, 1H), 1.76-1.71 (m, 1H), 1.51 (s, 3H), 1.25 (m, 4H), 1.08-1.05 (m, 7H), 0.96-0.88 (m, 2H), 0.65-0.61 (m, 1H), 0.45-0.36 (m, 2H). HRMS calculated for C21H29ClN5O2 (M + H) +: 418.2010; found 418.2017.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((R)-cyclopropylcyclobutylmethylamino)-9H-purin-9-yl)- 2,3-O-(isopropylidene)-bicyclo[3.1.0]hexane (29)
(R)-Cyclopropylcyclobutylmethylamine hydrochloride (28.3 mg, 0.17 mmol) and triethylamine (0.11 mL, 0.81 mmol) were added to a solution of compound 22a (19.92 mg, 0.05 mmol) in methanol (1.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum and the residue was purified on flash column chromatography (hexane:ethyl acetate = 1:1) to give the desired product 29 (20.8 mg, 83%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 5.36 (t, J = 6.0 Hz, 1H), 4.96 (s, 1H), 4.69 (d, J = 7.2 Hz, 1H), 3.85 (br s, 1H), 2.72-2.67 (m, 1H), 2.10-2.89 (m, 6H), 1.87-1.71 (m, 2H), 1.52 (s, 3H), 1.25 (s, 3H), 0.96-0.89 (m, 3H), 0.58-0.53 (m, 1H), 0.39-0.36 (m, 3H). HRMS calculated for C22H29ClN5O2 (M + H) +: 430.2010; found 430.2018.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((S)-cyclopropylcyclobutylmethylamino)-9H-purin-9-yl)- 2,3-O-(isopropylidene)-bicyclo[3.1.0]hexane (30)
(S)-Cyclopropylcyclobutylmethylamine hydrochloride (35.8 mg, 0.22 mmol) and triethylamine (0.14 mL, 1.03 mmol) were added to a solution of compound 22a (25.2 mg, 0.73 mmol) in methanol (1.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum and the residue was purified on flash column chromatography (hexane:ethyl acetate = 1:1) to give the desired product 30 (26 mg, 82%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.13 (s, 1H), 5.36 (t, J = 6.0 Hz, 1H), 4.96 (s, 1H), 4.69 (d, J = 7.2 Hz, 1H), 3.84 (br s, 1H), 2.71-2.67 (m, 1H), 2.09-2.89 (m, 6H), 1.87-1.71 (m, 2H), 1.51 (s, 3H), 1.25 (s, 3H), 0.95-0.88 (m, 3H), 0.58-0.54 (m, 1H), 0.37-0.36 (m, 3H). HRMS calculated for C22H29ClN5O2 (M + H) +: 430.2010; found 430.2008.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((R)-1,2-dicyclopropylethylamino)-9H-purin-9-yl)-2,3-O- (isopropylidene)-bicyclo[3.1.0]hexane (31)
(R)-1,2-Dicyclopropylethylamine hydrochloride (28.4 mg, 0.17 mmol) and triethylamine (0.11 mL, 0.82 mmol) were added to a solution of compound 22a (20.0 mg, 0.05 mmol) in methanol (1.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum and the residue was purified on flash column chromatography (hexane:ethyl acetate = 1:1) to give the desired product 31 (18.8 mg, 75%) as a foamy syrup. 1H NMR (CD3OD, 400 MHz) δ 8.13 (s, 1H), 5.36 (t, J = 6.4 Hz, 1H), 4.96 (s, 1H), 4.69 (d, J = 7.2 Hz, 1H), 3.80 (br s, 1H), 2.06-2.03 (m, 1H), 1.73-1.57 (m, 3H), 1.51 (s, 3H), 1.25 (s, 3H), 1.09-1.06 (m, 1H), 0.94-0.85 (m, 3H), 0.60-0.58 (m, 1H), 0.44-0.35 (m, 5H), 0.17-0.16 (m, 1H), 0.09-0.07 (m, 1H). HRMS calculated for C22H29ClN5O2 (M + H) +: 430.2010; found 430.2025.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((S)-1,2-dicyclopropylethylamino)-9H-purin-9-yl)-2,3-O- (isopropylidene)-bicyclo[3.1.0]hexane (32)
(S)-1,2-Dicyclopropylethylamine hydrochloride (31.5 mg, 0.19 mmol) and triethylamine (0.12 mL, 0.91 mmol) were added to a solution of compound 22a (22.21 mg, 0.05 mmol) in methanol (1.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum and the residue was purified on flash column chromatography (hexane:ethyl acetate = 1:1) to give the desired product 32 (21.5 mg, 77%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.13 (s, 1H), 5.36 (t, J = 6.4 Hz, 1H), 4.96 (s, 1H), 4.69 (d, J = 7.2 Hz, 1H), 3.80 (br s, 1H), 2.07-2.03 (m, 1H), 1.74-1.58 (m, 3H), 1.51 (s, 3H), 1.25 (s, 3H), 1.09-1.05 (m, 1H), 0.95-0.84 (m, 3H), 0.60-0.58 (m, 1H), 0.44-0.35 (m, 5H), 0.17-0.15 (m, 1H), 0.09-0.07 (m, 1H). HRMS calculated for C22H29ClN5O2 (M + H) +: 430.2010; found 430.2002.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((R)-N-cyclopropyl-N-phenylmethylamino)-9H-purin-9-yl)- 2,3-O-(isopropylidene)-bicyclo[3.1.0]hexane (33)
(R)-N-Cyclopropyl-N-phenylmethylamine hydrochloride (22 mg, 0.12 mmol) and triethylamine (0.11 mL, 0.84 mmol) were added to a solution of compound 22a (20.68 mg, 0.06 mmol) in methanol (1.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum and the residue was purified on flash column chromatography (hexane:ethyl acetate = 1:2) to give the desired product 33 (22.15 mg, 81%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 7.48 (d, J = 7.6 Hz, 2H), 7.33 (t, J = 7.6 Hz, 2H), 2.24 (t, J = 7.2 Hz, 1H), 5.35 (t, J = 6.0 Hz, 1H), 4.95 (s, 1H), 4.69 (d, J = 7.2 Hz, 1H), 3.60 (br s, 1H), 2.06-2.03 (m, 1H), 1.74-1.71 (m, 1H), 1.51 (s, 3H), 1.43-1.37 (m, 1H), 1.24 (s, 3H), 0.94-0.89 (m, 2H), 0.65-0.63 (m, 2H), 0.53-0.46 (m, 2H). HRMS calculated for C24H27ClN5O2 (M + H) +: 452.1853; found 452.1858.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((S)-N-cyclopropyl-N-phenylmethylamino)-9H-purin-9-yl)- 2,3-O-(isopropylidene)-bicyclo[3.1.0]hexane (34)
(S)-N-Cyclopropyl-N-phenylmethylamine hydrochloride (32.8 mg, 0.17 mmol) and triethylamine (0.11 mL, 0.84 mmol) were added to a solution of compound 22a (20.43 mg, 0.06 mmol) in methanol (1.5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum and the residue was purified on flash column chromatography (hexane:ethyl acetate = 1:2) to give the desired product 34 (22.1 mg, 82%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 7.48 (d, J = 7.2 Hz, 2H), 7.33 (t, J = 7.6 Hz, 2H), 2.24 (t, J = 7.2 Hz, 1H), 5.35 (t, J = 6.8 Hz, 1H), 4.95 (s, 1H), 4.69 (d, J = 7.2 Hz, 1H), 3.60 (br s, 1H), 2.06-2.03 (m, 1H), 1.75-1.71 (m, 1H), 1.51 (s, 3H), 1.42-1.37 (m, 1H), 1.24 (s, 3H), 0.96-0.88 (m, 2H), 0.66-0.62 (m, 2H), 0.54-0.43 (m, 2H). HRMS calculated for C24H27ClN5O2 (M + H) +: 452.1853; found 452.1856.
Molecular modeling
hA1AR and hA3AR homology models
Previously reported molecular models of the hA1AR and hA3AR, built using the alignment and the homology modeling tools implemented in the program Molecular Operating Environment (MOE),45 were used in this study. Both models were built using as template the crystal structure of the human A2AAR co-crystallized with the agonist 50 (PDB ID: 3QAK),22 as described by Tosh et al.20 In particular, hA1AR and hA3AR sequences were retrieved form the UniProt database46 and aligned against the sequence of the A2AAR template, taking into account the highly conserved residues in each TM domain. Then, homology models were built using the automated Homology Modeling protocol implemented in the MOE suite.
Molecular docking of truncated methanocarba derivatives at the hA1AR and hA3AR models
Compounds structures were built using the builder tool implemented in the MOE suite48 and subjected to energy minimization using the MMFF94x force field, until a RMS gradient of 0.05. The minimized conformations of each compound were used as starting point for the docking study. The flexible docking of the ligands at the hA1AR and hA3AR models was performed by means of the Glide47 package part of the Schrödinger suite.48
The docking site was defined with key residues in the binding pocket of the hA1AR and hA3AR models, namely Phe(EL2), Asn(6.55), Trp(6.48) and His(7.43), and a 20 Å × 20 Å × 20 Å box was centered on those residues. Docking of the ligands was performed in the rigid binding site of the models with Glide using the XP (extra precision) procedure.
The top scoring docking conformations were retained and subjected to the receptor sampling by means of the Refinement module in Prime.49 The Prime side-chain sampling was performed on all the residues within a 6Å of the ligand. The refined model for each ligand was chosen as final binding conformation.
In vivo testing
Animals and test substances used for seizure testing
Adult male CF No 1 albino mice (26–30 g, 6 Hz; 18–25 g all other tests), were obtained from Charles River, Portage, Michigan. Animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care, International (AAALAC)-accredited temperature and humidity controlled facility and maintained on a standard 12h: 12h light-dark cycle (lights on at 0600) with free access to standard laboratory chow Prolab RMH 3000 and water ad libitum. All animal experiments were performed in accordance with the guidelines set by National Institutes of Health and the University of Utah Institutional Animal Care and Use Committee (IACUC) committee. All animals were allowed free access to both food (and water except when they were removed from their cages for the experimental procedure. Except for the kindling studies, animals were used once. All animals were euthanized in accordance with the Institute of Laboratory Resources policies on the humane care of laboratory animals.
Anticonvulsant tests
In vivo anticonvulsant activity was established by both electrical and chemoconvulsant seizure tests which have been described previously.50–52 The electrical tests used were the maximal electroshock (MES) seizure test, the 6 Hz minimal clonic seizure test, and the corneal kindled mouse test. The chemical test was the s.c. metrazol seizure tests. TPE is deduced from data generated in initial qualitative test procedures. Five groups of 4 animals each are administered the test compound, and each group is tested at one of five different time intervals: 1/4, 1/2, 1, 2 and 4 h. The time point at which the compound produces the most activity/toxicity was chosen as TPE.
MES test and 6 Hz test
For the MES and 6 Hz tests, a drop of anesthetic/electrolyte solution (0.5% tetracaine hydrochloride in 0.9% saline) was applied to the eyes of each animal prior to placement of the corneal electrodes. The electrical stimulus in the MES test was 50 mA, 60 Hz, for mice. Abolition of the hind leg tonic extensor component of the seizure was used as the endpoint.
The ability of the test substance to prevent seizures in mice induced by 6 Hz corneal stimulation (32 mA, 3 sec duration) was evaluated at a convulsive current that evokes a seizure in 97% of the population tested (CC97). Six Hz seizures are characterized by a minimal clonic phase that is followed by stereotyped, automatistic behaviors described originally as being similar to the aura of human patients with partial seizures.53,54 Animals not displaying this behavior were considered protected.
Corneal-kindled mouse model of partial seizures
Mice were kindled according to the methods described by Matagne and Klitgaard.55 Briefly, mice were stimulated twice daily with a corneal stimulation of 3 mA for 3 seconds for an average of 12 days. Prior to each stimulation, a drop of 0.9% saline containing 0.5% tetracaine hydrochloride (Sigma-Aldrich, St. Louis, MO) was applied to the cornea to ensure local anesthesia and good electrical conductivity. Stimulations were at least four hours apart. Animals were considered kindled when they displayed five consecutive stage five seizures according to the Racine scale.56 At the completion of the kindling acquisition, mice were permitted at least a 3-day stimulation-free period prior to any drug testing. Mice were stimulated once the day before drug testing to ensure they had achieved and maintained a kindled state. On the day of the drug study, corneal kindled mice (n=4, or 8) received a single i.p. dose of test compound. Mice were challenged with the corneal kindling stimulus of 3 mA for 3 seconds at TPE after test compound administration. Mice were scored as protected (seizure score of ≤ 3) or not protected, (seizure score ≥ 4) based on the Racine scoring.56
Minimal behavioral toxicity tests
Minimal behavioral toxicity was identified in mice by the rotarod performance test.57 When a mouse is placed on a 1-inch knurled rod that rotates at a speed of 6 r.p.m., the animal can maintain its equilibrium for long periods of time. The animal was considered toxic if it fell off this rotating rod three times during a 1-min period.
Determination of median effective (ED50) or behavioral toxic dose (TD50)
All quantitative in vivo anticonvulsant/toxicity studies were conducted at the previously determined TPE. Groups of at least eight mice were tested with various doses of the candidate drug until at least two points were established between the limits of 100% protection or minimal toxicity and 0% protection or minimal toxicity. The dose of drug required to produce the desired endpoint in 50% of animals (ED50 or TD50) in each test, the 95% confidence interval, the slope of the regression line, and the S.E.M. of the slope were then calculated by a computer program based on the method described by Finney.58
Compound administration
The test compound was administered at a concentration that permitted optimal accuracy of dosing without the volume contributing excessively to total body fluid. Thus, test compounds are administered to mice in a volume of 0.01 ml/g of body weight. Compound 10 or other AR agonist was initially dissolved in DMSO (50 mg/ml) as a stock solution. To prepare the formulation for testing, an appropriate amount of stock solution was first diluted in DMSO to achieve 10% DMSO (v/v) in the final volume. Then, to the aqueous DMSO solution, 30% PEG400 (J.T. Baker) was gradually was added to make the final formulation.
Supplementary Material
Acknowledgments
We thank Prof. Ray Stevens and Dr. Vsevolod Katrich (Scripps Res. Inst., La Jolla, CA) for helpful discussions, Dr. Noel Whittaker (NIDDK) for mass spectral determinations, Steven Moss for binding assays, and Intramural Research Program of the NIH, NIDDK for support. The in vivo anti-seizure studies conducted at the University of Utah were supported by NINDS, NIH Contract No. HHSN271201100029C.
Abbreviations
- ADAC
N6-[4-[[[4-[[[(2-aminoethyl)amino]carbonyl]methyl]anilino]carbonyl]-methyl]phenyl]adenosine
- AR
adenosine receptor
- cyclic AMP
adenosine 3′,5′-cyclic phosphate
- BBB
blood-brain barrier
- CCPA
2-chloro-N6-cyclopentyladenosine
- CHO
Chinese hamster ovary
- GPCR
G protein-coupled receptor
- HEK
human embryonic kidney
- i.p
intraperitoneal
- MES
maximal electroshock
- MRS5127
(1′S,2′R,3′S,4′R,5′S)-4′-[2-chloro- 6-(3-iodobenzylamino)-purine]-2′,3′-O-dihydroxybicyclo-[3.1.0]hexane
- NECA
5′-N-ethylcarboxamidoadenosine
- PIA
N6-phenylisopropyladenosine
- scMET
subcutaneous metrazol model
- TM
transmembrane domain
- TPE
time of peak effect
- tPSA
total polar surface area
Footnotes
Supporting information available
Synthetic procedures for compounds 37 and 39 –43, spectroscopic characterization, in vitro bioassay procedures, anticonvulsant data, and modeling figure. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller C. Nomenclature and classification of adenosine receptors – An update. Pharmacol Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giorgi I, Nieri P. Therapeutic potential of A1 adenosine receptor ligands: a survey of recent patent literature. Expert Opin Therap Patents. 2008;18:677–691. [Google Scholar]
- 3.Albrecht-Küpper BE, Leineweber K, Nell PG. Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic Signal. 2012;8(Suppl 1):91–99. doi: 10.1007/s11302-011-9274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Zablocki JA, Wu L, Shryock J, Belardinelli L. Partial A1 adenosine receptor agonists from a molecular perspective and their potential use as chronic ventricular rate control agents during atrial fibrillation (AF) Curr Top Med Chem. 2004;4:839–854. doi: 10.2174/1568026043450998. [DOI] [PubMed] [Google Scholar]; b) Elzein E, Zablocki J. A1 adenosine receptor agonists and their potential therapeutic applications. Expert Opin Investig Drugs. 2008;17:1901–1910. doi: 10.1517/13543780802497284. [DOI] [PubMed] [Google Scholar]
- 5.Knutsen LJ, Lau J, Petersen H, Thomsen C, Weis JU, Shalmi M, Judge ME, Hansen AJ, Sheardown MJ. N-substituted adenosines as novel neuroprotective A1 agonists with diminished hypotensive effects. J Med Chem. 1999;42:3463–3477. doi: 10.1021/jm960682u. [DOI] [PubMed] [Google Scholar]
- 6.von Lubitz DKJE, Lin RCS, Paul IA, Beenhakker M, Boyd M, Bischofberger N, Jacobson KA. Postischemic administration of adenosine amine congener (ADAC): analysis of recovery in gerbils. Eur J Pharmacol. 1996;316:171–179. doi: 10.1016/s0014-2999(96)00667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Concas A, Santoro G, Mascia MP, Maciocco E, Dazzi L, Ongini E, Biggio G. Anticonvulsant doses of 2-chloro-N6-cyclopentyladenosine, an adenosine A1 receptor agonist, reduce GABAergic transmission in different areas of the mouse brain. J Pharmacol Exp Therap. 1993;267:844–851. [PubMed] [Google Scholar]
- 8.Gouder N, Fritschy JM, Boison D. Seizure suppression by adenosine A1 receptor activation in a mouse model of pharmacoresistant epilepsy. Epilepsia. 2003;44:877–885. doi: 10.1046/j.1528-1157.2003.03603.x. [DOI] [PubMed] [Google Scholar]
- 9.Fedele DE, Li T, Lan JQ, Fredholm BB, Boison D. Adenosine A1 receptors are crucial in keeping an epileptic focus localized. Exp Neurol. 2006;200:184–190. doi: 10.1016/j.expneurol.2006.02.133. [DOI] [PubMed] [Google Scholar]
- 10.Marek GJ. Activation of adenosine1 (A1) receptors induces antidepressant-like, anti-impulsive effects on differential-reinforcement-of-low rate 72-s behavior in rats. J Pharmacol Exp Therap. 2012;341:564–570. doi: 10.1124/jpet.112.191718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prediger RDS, da Silva GE, Batista LC, Bittencourt AL, Takahashi RN. Activation of adenosine A1 receptors reduces anxiety-like behavior during acute ethanol withdrawal (Hangover) in mice. Neuropsychopharmacology. 2006;31:2210–2220. doi: 10.1038/sj.npp.1301001. [DOI] [PubMed] [Google Scholar]
- 12.Korboukh I, Hull-Ryde EA, Rittner J, Randhawa AS, Coleman J, Fitzpatrick BJ, Setola V, Janzen WP, Frye SV, Zylka MJ, Jin J. Orally active adenosine A1 receptor agonists with antinociceptive effects in mice. J Med Chem. 2012;55:6467–6477. doi: 10.1021/jm3004834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunwiddie TV, Fredholm BB. Adenosine A1 receptors inhibit adenylate cyclase activity and neurotransmitter release and hyperpolarize pyramidal neurons in rat hippocampus. J Pharm Exp Therap. 1989;249:31–37. [PubMed] [Google Scholar]
- 14.a) Paul S, Elsinga PH, Ishiwata K, Dierckx RAJO, van Waarde A. Adenosine A1 receptors in the central nervous system: Their functions in health and disease, and possible elucidation by PET imaging. Current Med Chem. 2011;18:4820–4835. doi: 10.2174/092986711797535335. [DOI] [PubMed] [Google Scholar]; b) Nam HW, McIver SR, Hinton DJ, Thakkar MM, Sari Y, Parkinson FE, Haydon PG, Choi DS. Adenosine and Glutamate Signaling in Neuron-Glial Interactions: Implications in Alcoholism and Sleep Disorders. Alcoholism: Clin Exp Res. 2012;36:1117–1125. doi: 10.1111/j.1530-0277.2011.01722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan L, Burbiel JC, Maass A, Müller CE. Adenosine receptor agonists: from basic medicinal chemistry to clinical development. Expert Opin Emerg Drugs. 2003;8:537–576. doi: 10.1517/14728214.8.2.537. [DOI] [PubMed] [Google Scholar]
- 16.Jacobson KA, Ji XD, Li AH, Melman N, Siddiqui MA, Shin KJ, Marquez VE, Ravi RG. Methanocarba analogues of purine nucleosides as potent and selective adenosine receptor agonists. J Med Chem. 2000;43:2196–2203. doi: 10.1021/jm9905965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeong LS, Pal S, Choe SA, Choi WJ, Jacobson KA, Gao ZG, Klutz AM, Hou X, Kim HO, Lee HW, Lee SK, Tosh DK, Moon HR. Structure activity relationships of truncated D- and L- 4′-thioadenosine derivatives as species-independent A3 adenosine receptor antagonists. J Med Chem. 2008;51:6609–6613. doi: 10.1021/jm8008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melman A, Wang B, Joshi BV, Gao ZG, de Castro S, Heller CL, Kim SK, Jeong LS, Jacobson KA. Selective A3 adenosine receptor antagonists derived from nucleosides containing a bicyclo[3.1.0]hexane ring system. Bioorg Med Chem. 2008;16:8546–8556. doi: 10.1016/j.bmc.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hou X, Kim HO, Alexander V, Kim K, Choi S, Park SG, Lee JH, Yoo LS, Gao ZG, Jacobson KA, Jeong LS. Discovery of a new human A2A adenosine receptor agonist, truncated 2-hexynyl 4′-thioadenosine. ACS Med Chem Lett. 2010;1:516–520. doi: 10.1021/ml1001823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tosh DK, Phan K, Deflorian F, Wei Q, Gao ZG, Jacobson KA. Truncated (N)- methanocarba nucleosides as A1 adenosine receptor agonists and partial agonists: Overcoming lack of a recognition element. ACS Med Chem Lett. 2011;2:626–631. doi: 10.1021/ml200114q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao ZG, Blaustein J, Gross AS, Melman N, Jacobson KA. N6-Substituted adenosine derivatives: Selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem Pharmacol. 2003;65:1675–1684. doi: 10.1016/s0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elzein E, Kalla R, Li X, Perry T, Marquart T, Micklatcher M, Li Y, Wu Y, Zeng D, Zablocki J. N6-Cycloalkyl-2-substituted adenosine derivatives as selective, high affinity A1 adenosine receptor agonists. Bioorg Med Chem Lett. 2007;17:162–166. doi: 10.1016/j.bmcl.2006.09.065. [DOI] [PubMed] [Google Scholar]
- 24.Gao ZG, Kim SK, Biadatti T, Chen W, Lee K, Barak D, Kim SG, Johnson CR, Jacobson KA. Structural determinants of A3 adenosine receptor activation: Nucleoside ligands at the agonist/antagonist boundary. J Med Chem. 2002;45:4471–4484. doi: 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tosh DK, Paoletta S, Phan K, Gao ZG, Jacobson KA. Truncated nucleosides as A3 adenosine receptor ligands: Combined 2-arylethynyl and bicyclohexane substitutions. ACS Med Chem Lett. 2012;3:596–601. doi: 10.1021/ml300107e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ. Comparative pharmacology of human adenosine receptor subtypes - characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg’s Arch Pharmacol. 1998;357:1–9. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- 27.Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AGW, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 29.Swinyard EA, Woodhead JH, White HS, Franklin MR. General principles: experimental selection, quantification, and evaluation of anticonvulsants. In: Levy RH, Mattson RH, Meldrum B, Penry JK, Dreifuss FE, editors. Antiepileptic Drugs. Raven Press; New York: 1989. pp. 85–102. [Google Scholar]
- 30.Vlajkovic SM, Lee KH, Wong ACY, Guo CX, Gupta R, Housley GD, Thorne PR. Adenosine amine congener mitigates noise-induced cochlear injury. Purinergic Signal. 2010;6:273–281. doi: 10.1007/s11302-010-9188-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barton ME, Klein BD, Wolf HH, White HS. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001;47:217–227. doi: 10.1016/s0920-1211(01)00302-3. [DOI] [PubMed] [Google Scholar]
- 32.Nikodijevic O, Sarges R, Daly JW, Jacobson KA. Behavioral effects of A1-and A2- selective adenosine agonists and antagonists: Evidence for synergism and antagonism. J Pharm Exp Therap. 1991;259:286–294. [PMC free article] [PubMed] [Google Scholar]
- 33.Daly JW, Padgett W, Thompson RD, Kusachi S, Bugni WJ, Olsson RA. Structure-activity relationships for N6-substituted adenosines at a brain A1-adenosine receptor with a comparison to an A2-adenosine receptor regulating coronary blood flow. Biochem Pharmacol. 1986;35:2467–2481. doi: 10.1016/0006-2952(86)90042-0. [DOI] [PubMed] [Google Scholar]
- 34.White HS, Woodhead JH, Wilcox KS, Stables JP, Kupferberg HJ, Wolf HH. Discovery and Preclinical Development of Antiepileptic Drugs. In: Levy RH, Mattson RH, Meldrum B, Perucca E, editors. Antiepileptic Drugs. Lippincott Williams & Wilkins; New York: 2002. pp. 36–48. [Google Scholar]
- 35.Lothman EW, Salerno RA, Perlin JB, Kaiser DL. Screening and characterization of antiepileptic drugs with rapidly recurring hippocampal seizures in rats. Epilepsy Res. 1988;2:366–379. doi: 10.1016/0920-1211(88)90048-4. [DOI] [PubMed] [Google Scholar]
- 36.Rowley NM, White HS. Comparative anticonvulsant efficacy in the corneal kindled mouse model of partial epilepsy: Correlation with other seizure and epilepsy models. Epilepsy Res. 2010;92:163–169. doi: 10.1016/j.eplepsyres.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 37.Lauro C, Cipriani R, Catalano M, Trettel F, Chece G, Brusadin V, Antonilli L, van Rooijen N, Eusebi F, Fredholm BB, Limatola C. Adenosine A1 Receptors and Microglial Cells Mediate CX3CL1-Induced Protection of Hippocampal Neurons Against Glu-Induced Death. Neuropsychopharmacol. 2010;35:1550–1559. doi: 10.1038/npp.2010.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bueters TJ, IJzerman AP, van Helden HP, Danhof M. Characterization of the pharmacokinetics, brain distribution, and therapeutic efficacy of the adenosine A1 receptor partial agonist 2′-deoxy-N6-cyclopentyladenosine in sarin-poisoned rats. Toxicol Appl Pharmacol. 2003;192:86–94. doi: 10.1016/s0041-008x(03)00252-7. [DOI] [PubMed] [Google Scholar]
- 39.Chen Y, Liu Y, Cottingham C, McMahon L, Jiao K, Greengard P, Wang Q. Neurabin scaffolding of adenosine receptor and RGS4 regulates anti-seizure effect of endogenous adenosine. J Neurosci. 2012;32:2683–2695. doi: 10.1523/JNEUROSCI.4125-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maillard MC, Nikodijevic O, LaNoue KF, Berkich DA, Ji XD, Bartus R, Jacobson KA. Adenosine receptor prodrugs: Synthesis and biological activity of derivatives of potent, A1-selective agonists. J Pharmaceutical Sci. 1994;83:46–53. doi: 10.1002/jps.2600830112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.a) Hann MM, Keserü GM. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nature Rev Drug Discov. 2012;11:355–365. doi: 10.1038/nrd3701. [DOI] [PubMed] [Google Scholar]; b) Riley RJ, Martin IJ, Cooper AE. The influence of DMPK as an integrated partner in modern drug discovery. Curr Drug Metab. 2002;3:527–550. doi: 10.2174/1389200023337135. [DOI] [PubMed] [Google Scholar]
- 42.Dubey RK, McAllister CB, Inoue M, Wilkinson GR. Plasma binding and transport of diazepam across the blood-brain barrier. No evidence for in vivo enhanced dissociation. J Clin Invest. 1989;84:1155–1159. doi: 10.1172/JCI114279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harrison PK, Bueters TJH, IJzerman AP, van Helden HPM, Tattersall JEH. Partial adenosine A1 receptor agonists inhibit sarin-induced epileptiform activity in the hippocampal slice. Eur J Pharmacol. 2003;471:97–104. doi: 10.1016/s0014-2999(03)01783-7. [DOI] [PubMed] [Google Scholar]
- 44.a) Tosh DK, Chinn M, Ivanov AA, Klutz AM, Gao ZG, Jacobson KA. Functionalized congeners of A3 adenosine receptor-selective nucleosides containing a bicyclo[3.1.0]hexane ring system. J Med Chem. 2009;52:7580–7592. doi: 10.1021/jm900426g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Moon HR, Choi WJ, Kim HO, Jeong LS. Improved and alternative synthesis of D- and L-cyclopentenone derivatives, the versatile intermediates for the synthesis of carbocyclic nucleosides. Tetrahedron: Asymm. 2002;13:1189–1193. [Google Scholar]
- 45.Molecular Operating Environment (MOE), version 2011.10. Chemical Computing Group Inc; 1255 University St., Suite 1600; Montreal, QC, H3B 3X3 (Canada): [Google Scholar]
- 46.The UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shaw DE, Shelley M, Perry JK, Francis P, Shenkin PS, Glide A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 48.Schrödinger, LLC, Portland, OR.
- 49.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the Role of Crystal Packing Forces in Determining Protein Side-Chain Conformations. J Mol Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 50.White HS, Johnson M, Wolf HH, Kupferberg HJ. (1995) The early identification of anticonvulsant activity: role of the maximal electroshock and subcutaneous pentylenetetrazol seizure models. Ital J Neurol Sci. 1995;16:73–77. doi: 10.1007/BF02229077. [DOI] [PubMed] [Google Scholar]
- 51.White HS, Woodhead JH, Franklin MR. General principles: experimental selection, quantification, and evaluation of antiepileptic drugs. In: Levy RH, Mattson RH, Meldrum B, editors. Antiepileptic Drugs. Raven Press; New York: 1995. pp. 99–110. [Google Scholar]
- 52.Smith M, Wilcox KS, White HS. Discovery of antiepileptic drugs. Neurotherapeutics. 2007;4:12–17. doi: 10.1016/j.nurt.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toman JE, Everett GM, Richards RK. The search for new drugs against epilepsy. Tex Rep Biol Med. 1952;10:96–104. [PubMed] [Google Scholar]
- 54.Barton ME, Klein BD, Wolf HH, White HS. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001;47:217–227. doi: 10.1016/s0920-1211(01)00302-3. [DOI] [PubMed] [Google Scholar]
- 55.Matagne A, Klitgaard H. Validation of corneally kindled mice: a sensitive screening model for partial epilepsy in man. Epilepsy Res. 1998;31:59–71. doi: 10.1016/s0920-1211(98)00016-3. [DOI] [PubMed] [Google Scholar]
- 56.Racine R. Modification of seizure activity by electrical stimulation. 2. Motor Seizures. Electroencephalogr Clin Neurophysiol. 1972:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 57.Dunham MS, Miya TA. A note on a simple apparatus for detecting neurological deficit in rats and mice. J Amer Pharm Assoc Sci Ed. 1957;46:208–209. doi: 10.1002/jps.3030460322. [DOI] [PubMed] [Google Scholar]
- 58.Finney DJ. Statistical logic in the monitoring of reactions to therapeutic drugs. Methods Inf Med. 1971;10:237–245. [PubMed] [Google Scholar]
- 59.Gao ZG, Mamedova LK, Chen P, Jacobson KA. 2-Substituted adenosine derivatives: Affinity and efficacy at four subtypes of human adenosine receptors. Biochem Pharmacol. 2004;68:1985–1993. doi: 10.1016/j.bcp.2004.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Müller C, Jacobson KA. Recent developments in adenosine receptor ligands and their potential as novel drugs. BBA Biomembr. 2011;1808:1290–1308. doi: 10.1016/j.bbamem.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Klutz AM, Gao ZG, Lloyd J, Shainberg A, Jacobson KA. Enhanced A3 adenosine receptor selectivity of multivalent nucleoside-dendrimer conjugates. J Nanobiotechnol. 2008;6:12. doi: 10.1186/1477-3155-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.