

Abstract
Increased inducible nitric oxide synthase (NOS2) expression in breast tumors is associated with decreased survival of estrogen receptor-negative (ER−) breast cancer patients. We recently communicated the preliminary observation that nitric oxide (NO) signaling results in EGFR tyrosine phosphorylation. To further define the role of NO in the pathogenesis of ER− breast cancer, we examined the mechanism of NO-induced EGFR activation in human ER− breast cancer. NO was found to activate EGFR and Src by a mechanism that includes S-nitrosylation. NO, at physiologically relevant concentrations, induced an EGFR/Src-mediated activation of oncogenic signal transduction pathways (including c-Myc, Akt and β-catenin) and the loss of PP2A tumor suppressor activity. Additionally, NO signaling increased cellular EMT, expression and activity of COX-2 and chemoresistance to adriamycin and paclitaxel. When connected into a network, these concerted events link NO to the development of a stem cell-like phenotype, resulting in the upregulation of CD44 and STAT3 phosphorylation. Our observations are also consistent with the finding that NOS2 is associated with a basal-like transcription pattern in human breast tumors. These results indicate that the inhibition of NOS2 activity or NO signaling networks may have beneficial effects in treating basal-like breast cancer patients.
Keywords: S-Nitrosylation, nitric oxide, inducible nitric oxide synthase, basal-like, breast cancer
Introduction
Breast tumors exhibit distinct molecular and therapeutic differences and the subtypes of breast cancer are defined by their unique gene expression profiles (1). The basal-like subtype is a particularly aggressive form of breast cancer that is associated with poor patient survival, increased rates of recurrence and more prevalently affects premenopausal and African-American women (1–5). Furthermore, basal-like tumors do not respond to conventional therapies and commonly present with the triple-negative phenotype (negative expression of estrogen receptor-α, progesterone receptor and Her2/neu) (2, 6). Basal-like breast tumors are characterized by high expression of cytokeratin 5/6, but also epidermal growth factor receptor (EGFR) (7) which contributes to the aggressive nature of basal-like tumors (8). Another common feature of the basal-like phenotype is activated β-catenin signaling as nuclear β-catenin is associated with the expression of basal-like markers and decreased patient survival (9, 10). β-catenin signaling is critical for cellular epithelial-to-mesenchymal transition (EMT), an essential process for tumor invasion and metastasis (11, 12), which is commonly observed in the basal-like subtype (3). Additionally, the basal-like disease is associated with CD44+/CD24− cells, indicative of a breast cancer stem cell population (13). Thus, enhanced EGFR and β-catenin signaling, increased EMT and cancer stem cell populations and resistance to therapies are hallmarks of basal-like breast cancer and likely contribute to its aggressiveness. However the factors that promote and maintain a basal-like phenotype in breast cancer etiology are poorly understood.
Inducible nitric oxide synthase (NOS2) is pro-inflammatory enzyme which is expressed in many types of human cancers (e.g. colon, glioma, melanoma) (14–16). We recently reported that high NOS2 expression is a candidate predictor of poor survival in estrogen receptor (ER)-negative breast cancer patients and also among those patients who present with a basal-like disease (17). High tumor NOS2 expression is associated with a basal-like transcription pattern, EGFR phosphorylation and increased CD44 expression (17). Furthermore, NO signaling increases ER− cell migration (17). These observations suggest that NOS2/NO signaling is involved in basal-like breast cancer development and disease aggressiveness.
Because 1) high NOS2 expression is associated with a basal-like phenotype and poor disease outcome and 2) high NOS2 expression significantly correlates with high EGFR tyrosine 1173 phosphorylation in human breast tumors (17), we examined the effects of NO signaling on EGFR activation and whether additional pathways may intersect with NO-induced EGFR signaling to promote a basal-like/stem cell-like phenotype in breast cancer. Here, we report that NO activates EGFR and Src kinase activity via S-nitrosyl (SNO) post-translational modification in breast cancer cells. NO-induced EGFR and Src activation subsequently activated Akt, c-Myc, STAT3 and β-catenin pathways. These effects of NO were sensitive to either gefitinib, a clinically available EGFR inhibitor, or PP2, an inhibitor of both Src and EGFR kinases, as well as chemical inhibitors of S-nitrosylation. We also observed that nitrosative levels of NO signaling result in EMT and increased chemoresistance. We conclude that the NO/EGFR signaling axis described here promotes an aggressive cancer phenotype with basal-like/stem cell-like characteristics and that NOS2 inhibition may have clinically beneficial effect.
Materials and Methods
Cell culture
Human breast adenocarcinoma cell lines MDA-MB-231 and MDA-MB-468 and murine RAW 264.7 macrophages were obtained from American Type Culture Collection (Manassas, VA, USA). The murine basal carcinoma M6 cell line was provided by Dr. Jeffrey Green (NCI). All cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (Atlanta Biologics, Norcross, GA, USA) and 100 IU penicillin and 100 µg/ml streptomycin (Invitrogen). Cells were cultured at 37°C in 5% CO2 and passaged two to three times per week. Cell lines were authenticated by ATCC using short tandem repeat profiling within the past six months.
Macrophage co-culture
RAW264.7 cells were serum-starved overnight before stimulation with IFN-γ/LPS for 6 hours. Cells were collected on ice by gently lifting the cells off of the plate. Cells were then pelleted in serum-free RPMI supplemented with either 5 mM AG or 1 mM L-Arginine. Cells were counted and seeded into plates previously seeded with 50,000 MDA-MB-468 cells and the co-cultures were then incubated for 24 hours. Cells were then rinsed with cold PBS and the MDA-MB-468 cells were isolated by trypsinization and resuspension in RPMI + 10% FBS. Cells were then pelleted by centrifugation and lysed as described for western blotting. TCF-luciferase co-culture experiments used MDA-MB-468 cells transiently transfected with TopFlash reporter plasmid as described in Supplementary Data.
Nitrate and nitrite analysis
Cell culture media was analyzed for total nitrate and nitrite (NOx−) concentrations by using a modified Griess assay as previously described (47). Briefly, conditioned media were added to acidified N-(1-Naphthyl)ethylenediamine dihydrochloride, sulfanilamide and vanadium (III) chloride solutions and incubated at 37°C for 30–45 minutes. Product formation was measured at 540 nm and concentrations were determined from a sodium nitrite standard curve. Values shown are means from at least three independent experiments ± sd.
Steady state NO analysis
DETANO stock solutions were diluted into DMEM and incubated at 37°C under humidified air supplemented with 5% CO2. Aliquots of media were removed at indicated times using a gas-tight syringe and NO concentrations were measured using a Seivers NO chemiluminescence analyzer as previously described (47). Concentrations were determined from a standard curve of serial dilutions of a saturated NO solution. Values shown are means of three independent measurements ± sd.
Detection of nitrosation
Nitrosative capacity of DETANO was measured by the 2,3-diaminonaphthalene (DAN) assay as previously described (19). Briefly, DETANO was diluted into DMEM containing DAN and incubated at 37°C for 24 hours. DAN-triazole product formation was measured by fluorescence spectroscopy and values shown are mean relative fluorescent units (RFU) from three independent reactions.
Protein S-nitrosothiol (SNO) detection
Cells were exposed to DETANO for 24 hours and harvested under low light conditions using a commercially available protein-SNO detection kit (Cayman Chemical). This kit uses the biotin-switch approach as described by Jaffrey, et al (22). Individual protein-SNO detection was done by immunoprecipitation of target proteins after the biotin-switch assay is performed. Protein-SNO is compared to total protein content as determined by comassie stained gels.
Statistical analyses
Data analysis was performed using GraphPad Prism software (La Jolla, CA, USA). Statistical significance from control samples was calculated for c-Myc activity, PP2A activity, cellular proliferation, PGE2 formation and select densitometric analyses by one-way ANOVA with Dunett’s post-test. The degree of significance was determined by P < 0.05 or P < 0.01 as mentioned in the figure legends. DAN nitrosation data was analyzed by linear regression calculations. Linear correlation was calculated to be r2 = 0.991 (± 95% CI). Drug resistance data was fit to a sigmoidal dose response curve and IC50 concentrations were calculated.
Additional Material and Methods are located in Supplementary Data.
Results
NOS2 and nitric oxide signaling activate EGFR
Recently we reported that high NOS2 expression correlates with EGFR Tyr1173 phosphorylation in human breast tumors, indicative of kinase activation by NO (17). Therefore we examined the effect of NOS2 activity on EGFR activation in MDA-MB-468 human ER− breast cancer cell lines. Because human breast cancer cell lines, like most other human cell lines, do not constitutively express this enzyme in culture despite overexpression in human breast tumors, we employed a murine macrophage co-culture strategy (18). NOS2 expression was absent in serum starved RAW 264.7 cells but was induced upon interferon-gamma (IFN-γ) and lipopolysaccharide (LPS) stimulation prior to co-culture (Figure 1A). To examine the effects of NOS2 activity on EGFR activation, IFN-γ/LPS-stimulated RAW 264.7 murine macrophages were co-cultured with MDA-MB-468 cells in increasing ratios. In addition to NO, activated macrophages secrete various cytokines and prostaglandins; therefore the role of macrophage-expressed NOS2 was also examined using the NOS2-selective inhibitor, aminoguanidine (AG). After exposure to macrophages, MDA-MB-468 cells were isolated and EGFR Tyr 1173 phosphorylation was examined by western blot. EGFR phosphorylation was markedly increased in cells cultured at a 1:4 and 1:8 ratio, similar to monocultured, EGF-stimulated MDA-MB-468 cells (Figure 1B). The effect of activated macrophages was eliminated in the presence of AG (Figure 1B), indicating that a product of NOS2 catalysis was responsible for EGFR activation. Additionally, EGFR phosphorylation was not observed in similar experiments using unstimulated macrophages (data not shown). Nitrate and nitrite (NOx−) accumulation in the culture media was measured as an aggregate indicator of total NOS2 activity. Consistent with EGFR activation, NOx− concentrations in media from unstimulated and AG treated RAW cells were near control levels indicating that NOS2 activity was negligible under these conditions (Figure 1C). IFN-γ/LPS stimulated macrophages produced NOx− in a nearly linear manner with respect to macrophage number, consistent with our previous findings (19).
Figure 1. NOS2 activity and NO-donor increases EGFR tyrosine phosphorylation.
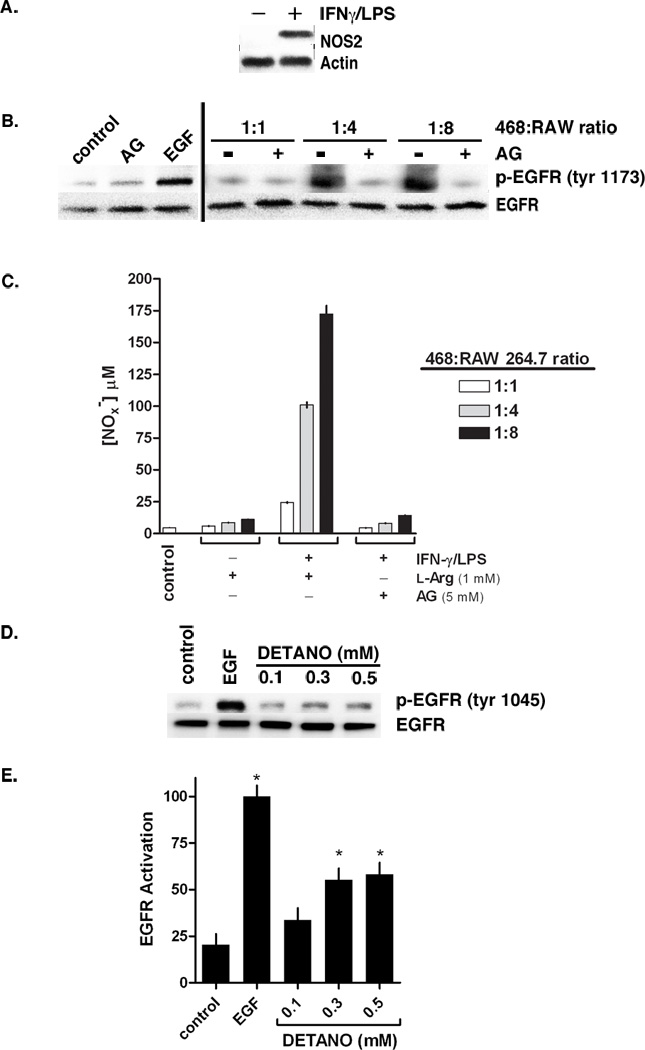
(A) Relative NOS2 expression in RAW 264.7 murine macrophages pretreated with or without IFN-γ/LPS. (B) Western blot showing EGFR tyrosine 1173 phosphorylation in MDA-MB-468 cells co-cultured with IFN-γ/LPS activated RAW cells in increasing ratios and in media containing or lacking the NOS2 inhibitor aminoguanidine (AG). Co-cultured samples are compared to monocultured MDA-MB-468 cells treated with AG or EGF. (C) Total nitrate and nitrite (NOx−) concentrations in conditioned media from co-culture experiments as an aggregate indicator of NOS2 activity. (D) Western blot of EGFR tyrosine 1045 phosphorylation in serum-starved MDA-MB-468 cells treated with either EGF (10 ng/ml) or DETANO for 24 hours. (E) Densitometric analyses of EGFR tyrosine 1173 phosphorylation indicating that EGF and 0.3 and 0.5 mM DETANO resulted in significant increases in EGFR activation compared to untreated controls (*P < 0.05).
MDA-MB-468 cells treated with 0.5 mM of the chemical NO-donor, (Z)-1-[N-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate (DETANO, alternatively named NOC-18), also results in EGFR Tyr1173 and Tyr1045 phosphorylation (17). Here we examined EGFR Tyr1045 phosphorylation in MDA-MB-468 cells exposed to a physiologically-relevant range of DETANO concentrations by western blot (Figure 1C). DETANO resulted in EGFR Tyr1045 phosphorylation in a concentration-threshold manner as 0.1 mM DETANO resulted in similar phosphorylation levels as serum-starved, untreated control cells while EGFR phosphorylation markedly increased with 0.3 and 0.5 mM donor as determined by densitometric analyses (Figure 1D). Similar results were observed in the HER2+/ER− SKBR3 cell line (data not shown).
NO activates EGFR via S-nitrosylation
The NO concentration-threshold effect on EGFR activation is indicative of a indirect NO mechanism, as the rate of NO autoxidation is proportional to the square of NO concentration. Because the biochemistry of NO is largely determined by its concentration (20, 21), the threshold concentration range for NO to activate EGFR was examined. Since 0.3 mM but not 0.1 mM DETANO was able to activate EGFR, we assumed that a threshold NO concentration exists between the two donor concentrations. To establish this threshold NO concentration, DETANO was decomposed in media and steady state NO concentration were measured over time by chemiluminescence. DETANO at 0.1 mM produced a maximum NO concentration of approximately 200 nM with a steady state NO concentration between 100–150 nM over most of the incubation time (Figure 2A). DETANO at 0.3 mM produced a maximum of 400 nM with a steady state NO concentration around 300 nM. This places the threshold NO concentration to be between 200–300 nM and indicates that the autoxidation of NO may be responsible for the activation of EGFR. NO autoxidation produces nitrosating species such as N2O3. To measure the ability of DETANO to form nitrosating species, the NO donor was decomposed in the presence of 2,3-diaminonaphthalene (DAN) (19). DETANO decomposition resulted in DAN nitrosation in a linear concentration-dependent manner (Figure 2B). Because DETANO activation of EGFR is correlated to nitrosative capacity, we examined the effect of protein S-nitrosyl (SNO) post-translational modification in response to DETANO concentration. MDA-MB-231 cells were exposed to DETANO and total protein-SNO formation was determined by the biotin-switch assay (22). Consistent with DAN nitrosation and EGFR activation, only modest protein-SNO occurred with 0.1 mM DETANO, however robust protein-SNO was detected in cells exposed to 0.3 and 0.5 mM DETANO (Figure 2C). Therefore the threshold levels of NO required to activate EGFR are consistent with protein-SNO formation. To examine if EGFR is S-nitrosylated, EGFR was immunoprecipitated from cells treated with DETANO and SNO modification was measured using the biotin-switch assay. EGFR-SNO was detected at 0.3 and 0.5 mM, but not at 0.1 mM DETANO (Figure 2D). To further address the nitrosative mechanism of EGFR activation, cells were exposed to 0.5 mM DETANO alone or in the presence of chemical inhibitors of nitrosation, glutathione (GSH), sodium azide (N3−) and sodium thiocyanate (SCN−). Similarly, cells were exposed to 0.5 mM DETANO in the presence of the thiol-blocking agent iodoacetacetate (IAA) to examine the role of protein cysteine residues in this mechanism. Both chemical inhibitors of nitrosation (GSH, N3− and SCN−) and the thiol-blocking agent (IAA) eliminated the ability of 0.5 mM DETANO to result in EGFR Tyr1173 phosphorylation and EGFR-SNO modification (Figure 2E).
Figure 2. NO activates EGFR via S-nitrosation post-translational modification.

(A) Steady-state NO concentration in DETANO-supplemented media as determined by chemiluminescence. Mean [NO] (± sem) values are shown. (B) Nitrosative ability of DETANO as determined by DAN fluorescence assay. Mean DAN-triazole fluorescence (RFU) values (± sd) are shown and linear regression analysis (± 95% CI) indicates a linear response between DETANO concentration and nitrosative capacity. (C) Total cellular protein S-nitrosation from MDA-MB-468 cells treated with DETANO for 24 hours compared to total cellular protein content. (D) Total protein, immunoprecipitated EGFR and EGFR-SNO from MDA-MB-468 cells treated with DETANO for 24 hours. (E) Western blot of relative EGFR tyrosine 1173 phosphorylation and EGFR-SNO modification from MDA-MB-468 cells treated with EGF (10 ng/mL) or DETANO and chemical inhibitors of S-nitrosation.
NO activates EGFR and Src kinase signaling
Because NOS2 and NO signaling activated EGFR, we examined the effect of NO signaling on other receptor tyrosine kinases (RTK) using a spot-ELISA technique. Serum-starved MDA-MB-468 cells were treated with EGF or DETANO for 24 hours and whole cell lysates were analyzed using a RTK Pathscan kit. EGFR was the only RTK examined that responded to NO signaling, suggesting that NO functionally replaces EGF as a growth factor (data not shown). Additionally, the spot-ELISA experiments confirm that NO increases EGFR tyrosine phosphorylation compared to control, however NO appears to be less efficacious than EGF, similar to the results shown in Figure 1. In addition to RTK activation, the spot-ELISA experiments show that NO signaling increases Src and Akt kinase phosphorylation (Figure 3A).
Figure 3. NO activates EGFR and Src kinase signaling pathways in ER− breast cancer cells.

(A) Densitometric analyses from receptor tyrosine kinase (RTK) Pathscan spot-ELISA experiment in MDA-MB-468 cells treated with EGF or DETANO compared to serum-starved controls. EGFR, Src and Akt phosphorylation was increased by EGF and 0.5 mM DETANO. (B) Total input protein and Src, immunoprecipitated and S-nitrosated (SNO) Src from MDA-MB-468 cells treated with DETANO. (C) Relative Akt, Src and STAT3 phosphorylation from MDA-MB-468 cells treated with either EGF or DETANO compared to untreated serum-starved controls. (D) Akt, Src, STAT3 phosphorylation and CD44 expression in the presence of chemical inhibitors of nitrosation. (E) Western blot showing the stabilization of c-Myc in MDA-MB-468 cells in response to DETANO concentration. (F) Relative c-Myc-DNA binding from MDA-MB-468 cells treated with 0.5 mM DETANO and/or PP2 (100 nM). Normalized mean c-Myc activity (± sd) is shown. Statistical significance was determined by *P < 0.05. (G) Representative western blots from Her2+ SKBR3 cells exposed to EGF or DETANO showing NO-induced increases in EGFR and STAT3 tyrosine phosphorylation and increased CD44 expression.
Src kinase is activated upon S-nitrosyl formation (23). We show here that DETANO results in Src-SNO at 0.3 and 0.5 mM, but not at 0.1 mM, consistent with EGFR-SNO formation (Figure 3B). Similarly, Src Tyr419 phosphorylation is observed by western blot at the same DETANO concentrations that form Src-SNO (Figure 3C). In addition to Src activation, DETANO also increased Akt Ser473 and STAT3 Tyr705 phosphorylation with the same concentration-threshold, suggesting that nitrosative NO signaling is responsible for the observed effects. Furthermore NO-mediated Akt, Src and STAT3 phosphorylation and CD44 expression was reduced in the presence of inhibitors of nitrosation (GSH, N3−, and SCN−) and the thiol-blocking agent iodoacetic acid (IAA), consistent with a nitrosative mechanism (Figure 3D).
NO signaling (0.5 mM DETANO) stabilizes c-Myc protein and increases c-Myc nuclear activity (17). Here we show the concentration effect of DETANO on c-Myc protein stabilization. DETANO resulted in a concentration-threshold effect on c-Myc stability as 0.3 and 05 mM DETANO strongly stabilized c-Myc relative to actin controls (Figure 3E). c-Myc activation is downstream from EGFR and Src signaling; thus we examined c-Myc nuclear activation in response to DETANO and EGFR/Src inhibition. MDA-MB-468 cells were exposed to 0.5 mM DETANO and/or PP2 (100 nM) for 24 hours and nuclear extracts were analyzed by an ELISA-based assay. DETANO significantly increased c-Myc-DNA binding compared to control, while PP2 alone reduced c-Myc activation (Figure 3F). The combination of DETANO and PP2 resulted in serum-starved control levels of c-Myc actvation, indicating that EGFR and/or Src activity is required for DETANO to increase c-Myc signaling (Figure 3F).
EGFR and STAT3 activation and CD44 overexpression are hallmarks of the basal-like phenotype (13, 24) and are characteristic of a breast cancer stem cell-like phenotype (25, 26). To examine the effect of NO signaling on EGFR and STAT3 phosphorylation and CD44 expression in a non-basal-like (HER2+) breast cancer cell line, serum-starved SKBR3 cells were treated with DETANO or EGF for 24 hours. DETANO caused a concentration-threshold increase in both EGFR Tyr1173 and STAT3 Tyr705 phosphorylation, similar to EGF controls (Figure 3G). DETANO also resulted in increased CD44 expression, similar to EGF controls (Figure 3G). These data suggest that nitrosative NO signaling increases the basal-like and stem cell-like phenotypes.
β-catenin contributes to the expression of basal-like signature genes by NOS2
NOS2 expression is a candidate predictor of poor outcome in both ER− breast cancer patients and those with basal-like breast cancer (17). NOS2 expression was also associated with a distinct set of 44 genes overexpressed in human tumors, many of which are established basal-like signature genes (17). Because β-catenin activation is commonly observed in basal-like tumors (9, 10), we examined the relationship between NOS2 expression and β-catenin activation by analyzing the promoter region of all 44 genes overexpressed in high NOS2 tumors for Lef consensus sequences. Thirty four of the 44 genes (77%) where found to have Lef binding sites and 15 of these candidate β-catenin-regulated genes are basal-like signature genes (Supplemental Figure 1). Thus, the majority of basal-like genes upregulated in high NOS2 tumors appear to be regulated by β-catenin (15/21; 71%), which suggests that β-catenin may be a major contributor to the NOS2-induced basal-like signature gene. These observations led us to examine NOS2 and NO signaling effects on β-catenin transcriptional activity in basal-like breast cancer cells.
NOS2/NO activate β-catenin signaling
To examine the effect of NOS2 activity on breast cancer cell β-catenin signaling, we employed the murine macrophage co-culture experimental design with MDA-MB-468 cells transiently transfected with a TCF-luciferase plasmid. Unstimulated RAW 264.7 cells did not activate β-catenin, as measured by luciferase activity, at all MDA-MB-468:macrophage ratios, similar to untreated monocultured negative control cells (Figure 4A). However, IFN-γ/LPS stimulated RAW 264.7 cells expressing NOS2 increased breast cancer cell luciferase activity in a biphasic manner. At a 1:1 ratio there was no increase in luciferase activity compared to control. However at 1:4 ratio there was very high luciferase activity that was comparable to LiCl-treated positive control cells. The luciferase activity dropped at a 1:8 ratio to approximately half of the 1:4 effect, but still remained markedly higher than the negative control cells. The effect of IFN-γ/LPS stimulated RAW 264.7 macrophages on β-catenin activity was abolished in the presence of the NOS2 inhibitor aminoguanidine (AG), indicating that NO or another NOS2 product is responsible for activating β-catenin.
Figure 4. NO activates the β-catenin signaling pathway in basal-like breast cancer cells.

(A) TCF-luciferase reporter activity of MDA-MB-468 cells alone or co-cultured with RAW 264.7 cells under the conditions shown. Data are normalized to untreated controls and represent the fold-increase of mean relative luminescence units (RLU) (± sd). (B) TCF-luciferase reporter activity from breast cancer cell lines (MDA-MB-468, MDA-MB-231 and M6) exposed to LiCl or DETANO for 20 hours. Data represent mean fold-increase RLU (± sd) compared to untreated controls. (C) Representative immunofluorescent micrographs of MDA-MB-231 β-catenin cellular localization in response to LiCl or DETANO exposure. The yellow pixels in merged images represent nuclear β-catenin localization and the scale bars represent 10 µm. (D) Representative western blot of MDA-MB-468 nuclear fractions showing an increase in nuclear β-catenin in response to DETANO compared to tata-binding protein (TBP) nuclear content.
To examine the effect of NO on β-catenin activation, basal-like breast cancer cells were exposed to DETANO. Human breast cancer cell lines (MDA-MB-231 and MDA-MB-468) and a murine basal mammary cell line (M6), were transiently transfected with a β-catenin/TCF luciferase reporter construct as described above. As a positive control, cells were treated with LiCl to measure the extent of β-catenin activation. β-catenin activation by LiCl was strongly induced in both MDA-MB-468 and M6 cell lines but was only modestly activated in MDA-MB-231 cells compared to untreated controls (Figure 4B). DETANO exposure resulted in a biphasic response in the human cell lines as 0.1 mM donor did not induce β-catenin activity compared to untreated control cells. However at 0.3 mM DETANO, β-catenin was activated close to LiCl controls. At a higher 0.5 mM DETANO concentration, β-catenin activity was reduced in both human cell lines. In contrast, DETANO induced β-catenin pathway activation in a concentration dependent manner in the M6 cell line which did not follow the biphasic NO effect of the other cell lines.
To verify the nuclear accumulation of β-catenin in response to NO, serum-starved MDA-MB-231 cells were exposed to either LiCl or DETANO for 24 hours. Immunofluorescent micrographs of cellular β-catenin localization relative to nuclei are shown in Figure 4C. LiCl and 0.3 mM DETANO cause marked nuclear accumulation of β-catenin that is absent in untreated and 0.1 mM DETANO treated cells. This is further confirmed in western blot analysis of nuclear fractions of DETANO treated MDA-MB-468 cells (Figure 4D). Consistent with the luciferase assay results, untreated and 0.1 mM DEATNO treated cells had basal amounts of β-catenin in the nuclei compared to TATA-binding protein (TBP) expression, however 0.3 mM and to a lesser extent 0.5 mM DETANO caused increased nuclear β-catenin protein content.
Protein-SNO is required for NO to activate β-catenin
The activation of β-catenin by NO was examined in the presence of chemical inhibitors of nitrosation. Using the luciferase assay described above, MDA-MB-468 cells were incubated with 0.3 mM DETANO alone or with azide, thiocyanate, glutathione or urate. Similar to above, 0.3 mM donor activated β-catenin, but this effect was abolished by inhibitors of nitrosation (Figure 5A). Pretreating the cells with NEM prior to DETANO blocked the ability of NO to activate β-catenin signaling (Figure 5B). These data indicate that β-catenin activation requires a nitrosative species formed from NO autoxidation and that this species reacts with target protein thiols to form a S-nitrosothiol post-translational modification.
Figure 5. S-nitrosation is required for NO to activate β-catenin signaling.

(A) TCF-luciferase activity from MDA-MB-468 cells exposed to LiCl or 0.3 mM DETANO and inhibitors of nitrosation. (B) TCF-luciferase activity from MDA-MB-468 cells pretreated with NEM (black) or untreated (grey) followed by DETANO incubation. (C) TCF-luciferase activity from MDA-MB-468 cells exposed to 0.3 mM DETANO and kinase (Gefitinib, PP2, wortmannin and triciribine) and soluble guanylate cyclase (ODQ) inhibitors. Data represent fold-RLU relative to control (± sd). (D) Representative immunofluorescent images of β-catenin localization in MDA-MB-231 cells treated with 0.3 mM DETANO ± 100 nM PP2.
To examine the biochemical mechanism of NO activation of β-catenin, cells were exposed to 0.3 mM DETANO alone or with inhibitors of EGFR (Gefitinib), EGFR/Src (PP2), PI3K (wortmannin), Akt (triciribine) and sGC (ODQ). Gefitinib, PP2, wortmannin and triciribine inhibited the ability of NO to activate β-catenin, however ODQ had no effect (Figure 5C). Immunofluorescent photographs show β-catenin localization in MDA-MB-231 cells exposed to DETANO alone and with 100 nM PP2. DETANO caused β-catenin to translocate to the nuclei, however this effect is diminished in the presence of PP2 (Figure 5D). These data indicate that NO exerts its effects on β-catenin via EGFR and/or Src kinase activation.
NO inhibits PP2A-c tumor suppressor activity
Akt, c-Myc and β-catenin signaling pathways share a common endogenous negative regulator, protein phosphatase 2A (PP2A) (27). PP2A is a highly regulated family of proteins with tumor suppressor functions and cancer cells must limit PP2A activity to allow for dysregulated signal transduction and cellular proliferation (28, 29). The catalytic subunit of PP2A (PP2A-c) Tyr307 is phosphorylated by multiple kinasaes including EGFR and Src, which causes marked reduction in phosphatase activity (30). Because NO signaling activates both EGFR and Src kinases and activates Akt, c-Myc and β-catenin signaling, we examined the effects of NO signaling on PP2A-c Tyr307 phosphorylation and phosphatase activity. NO signaling increased PP2A-c Tyr307 phosphorylation in a concentration-threshold manner in MDA-MB-468 cells (Supplemental Figure 2). At 0.1 mM DETANO, PP2A-c phosphorylation was similar to serum-starved controls while PP2A-c phosphorylation was similar to EGF controls at 0.5 mM DETANO. Consistent with phosphorylation, PP2A-c phosphatase activity was significantly decreased in cells treated with 0.3 and 0.5 mM DETANO, similar to EGF treatment (Supplemental Figure 2). PP2A-c Tyr307 phosphoarylation was inhibited in the presence of PP2, indicating that EGFR and/or Src kinases are required for PP2A-c phosphorylation (Supplemental Figure 2). Furthermore in the presence of PP2, PP2A-c activity was not significantly altered from control cells (Supplemental Figure 2). Similar results (PP2A-c phosphorylation and activity) were observed in SKBR3 cells. These results indicate that NO signaling via EGFR and/or Src decrease PP2A tumor suppressor function to allow NO-mediated Akt, c-Myc and β-catenin signaling.
NO induces epithelial-to-mesenchymal transition
Epithelial-to-mesenchymal transition (EMT) is associated with the basal-like phenotype and tumor metastasis (3, 31, 32). Because NO results in oncogenic pathway activation and increased cellular migration, we examined the effect of NO on EMT in MDA-MB-468 cells. Serum-starved cells were treated with or without 0.5 mM DETANO for 24 hours and cellular morphology was examined. Bright field microphotographs show that untreated cells retain cell-to-cell adhesion while DETANO treatment reduced adhesion (Figure 6A). DETANO treatment also resulted in decreased expression of E-cadherin and increased expression of vimentin relative to control and this effect was eliminated in the presence of GSH, N3− or PP2 (Figure 6B). DETANO also resulted in significantly increased proliferation in both MDA-MB-468 and SKBR3 cell lines as compared to serum-starved controls (Figure 6C). These results indicate that NO signaling results in ER− breast cancer cell EMT.
Figure 6. NO increases cellular EMT.

(A) Representative microphotographs of MDA-MB-468 cells compared to cells exposed to 0.5 mM DETANO showing altered cellular morphology and cell-to-cell adhesion. (B) Representative western blot of E-cadherin and vimentin expression in MDA-MB-468 cells exposed to 0.5 mM DETANO alone or with GSH, N3− or PP2. (C) Cellular proliferation in serum-starved MDA-MB-468 or SKBR3 cells exposed to EGF or 0.5 mM DETANO. Significance from control was determined by one-way ANOVA with Dunnett’s post-test analysis (*P < 0.01). (D) Western blot of relative COX-2 expression in MDA-MB-468 cells treated with DETANO for 24 hours as compared to actin. (E) COX-2 activity as determined by PGE2 measurements in DETANO-treated MDA-MB-468 conditioned media. Data represent mean PGE2 concentrations (± sd). Significance from control was determined by one-way ANOVA with Dunnett’s post-test analysis (*P < 0.05).
EMT is associated with overexpression of the pro-inflammatory enzyme cyclooxygenase-2 (COX-2) in many tumor types (33–35). Furthermore, the basal-like disease is associated with COX-2 overexpression (4). To examine the effects of NO signaling on COX-2 expression, MDA-MB-468 cells were treated with DETANO. NO signaling induced COX-2 overexpression in a concentration-dependent manner (Figure 6D) and DETANO-treated cells produced significantly more PGE2 compared to control (Figure 6E), indicating that NO signaling results in a pro-inflammatory condition associated with cancer cell EMT.
NO Increases Cellular Chemoresistance
Patients with triple-negative or basal-like breast cancer respond better to anhtracycline/taxane based chemotherapy (36), however overall prognosis remains poor for patients with basal-like tumors. Basal-like tumors have increased expression of P-glycoprotein, a protein related to multidrug resistance, compared to non-basal-like tumors (37). To examine the effect of NO on ER− breast cancer cell chemoresistance, serum-starved MDA-MB-231 cells were treated with DETANO or EGF for 24 hours and P-glycoprotein expression was measured by western blot. DETANO resulted in a concentration-dependent increase in P-glycoprotein expression (Figure 7A) and densitometric analyses show that 0.5 mM DETANO significant increased protein expression compared to both serum-starved and EGF controls (Figure 7B). The effect of adriamycin and paclitaxel resistance was measured by in DETANO-pretreated MDA-MB-231 cells by BrDU incorporation. Cells were treated with or without 0.5 mM DETANO for 24 hours, rinsed with serum-free media and exposed to drug for 18 hours in the presence of BrDU. Cells pretreated with DETANO were less sensitive to both adriamycin and paclitaxel (Figure 7C). The IC50 of adriamycin increased from 3.7 nM in control cells to 240 nM in DETANO pre-treated cells representing a 65-fold increase in sensitivity. The IC50 of paclitaxel increased from 0.31 nM in control cells to 2.5 nM in DETANO pre-treated cells representing an 8-fold increase in sensitivity. These results indicate that this oncogenic level of NO signaling (200–500 nM) contributes to cellular chemoresistance.
Figure 7. NO increases chemoresistance in basal-like breast cancer cells.

(A) Representative western blot of P-glycoprotein expression in MDA-MB-231 cells exposed to wither EGF or DETANO. (B) Densitometric analyses of P-glycoprotein expression in response to EGF or DETANO. Significance from control was determined by one-way ANOVA with Dunnett’s post-test analysis (*P < 0.05). (C) Survival of MDA-MB-231 cells pre-treated with or without 0.5 mM DETANO in response to adriamycin and paclitaxel exposure. Data is shown as percent proliferation compared to cells not treated with chemotherapeutic.
Discussion
The strong correlation between NOS2 expression and ER− breast cancer patient survival is a compelling observation that suggests NO signaling is a major promoter of aggressive breast cancer. In this report, we made the novel observation that NO activates an oncogenic signaling network in human breast cancer cells involving EGFR and Src activation as the initiating events. Our results provide a previously unrecognized chemical and biochemical mechanism in support of the clinically significant observation that tumor NOS2 expression is associated with the basal-like phenotype in breast cancer patients and predicts poor patient survival (17). NO signaling activated EGFR and Src kinases via a SNO-modification resulted in characteristic signal transduction pathways and phenotypes associated with basal-like breast cancer. The activation of EGFR and Src resulted in oncogenic c-Myc, Akt, STAT3 and β-catenin signaling, while inhibiting the tumor suppressor PP2A, demonstrating an important role of NO in simultaneously activating oncogenes and limiting tumor suppressors. As previously shown, the level of NO required to activate EGFR and Src are the same NO levels that promote an aggressive cellular phenotype (17). Our mechanistic and cell culture-based results are consistent with our previously reported patient data and suggest that NOS2 inhibition may be a novel strategy to treating basal-like breast cancer patients.
NO has been shown to possess both pro- and anti-tumor effects. This dichotomy of NO signaling is resolved by the effects of different NO concentrations and their specific cellular signaling mechanisms. Levels of NO that lead to nitrosative stress and apoptosis are observed at NO concentrations > 1 µM, while physiological regulation of the cardiovascular system of NO is observed < 50 nM (27, 38). The effects observed here correlate to NO concentrations between 200–500 nM and represent a guanylate cyclase-independent signaling mechanism. Since NO and reactive nitrogen species concentrate in the lipid bilayer (39, 40), membrane associated proteins such as EGFR and Src are ideal targets of NO-mediated modification.
NO activation of EGFR appears to be via a protein-SNO mechanism. Protein-SNO modification has been proposed to have biochemical signaling effects (41, 42); here we provide evidence of protein-SNO promoting an oncogeneic signaling network and the basal-like breast cancer phenotype. The stability and structural effects of protein-SNO formation is expected to be context dependent (43, 44). However the effect of SNO formation has been described for Src kinase, as Rahman et al. show that nitrosation of cysteine 498 results in increased kinase activity (23). EGFR is predicted to have SNO sites at the ligand interaction site and is thought to limit EGFR activity (43) and other reports suggest that EGFR-SNO results in kinase inhibition (45). In contrast we reported that DETANO increases EGFR tyrosine phosphorylation consistent with activation (17) and show here that EGFR-SNO formation parallels tyrosine phosphorylation and downstream EGFR signaling.
NO activation of EGFR and Src resulted in increased β-catenin transcriptional activity. Recent patient data indicate that two molecules associated with cancer progression, NOS2 and β-catenin, are both associated with the basal-like phenotype and are individually strong predictors of outcome for ER− breast cancer including the basal-like subtype (9, 17). β-catenin signaling is a feature of basal-like tumors and nuclear β-catenin localization is predictive of poor survival (9, 10). The results presented here indicate that two prognostic markers of basal-like breast cancer, NOS2 and β-catenin, are mechanistically linked in the progression of basal-like breast cancer, as we show that NOS2 activity leads to β-catenin signaling and the promotion of the basal-like phenotype.
Clinically relevant phenotypes of basal-like breast cancer include EMT, cancer stem-cell markers and drug resistance. The data shown here indicate that NO signaling recapitulate these clinical observations in ER− cell lines. The basal-like subtype is enriched for the CD44+/CD24− cancer stem cell population (13), which require STAT3 signaling for proliferation (26). Here we show that NO signaling via S-nitrosylation results in CD44 expression and STAT3 phosphorylation. EMT is related to the basal-like phenotype (3, 4) and here we show that NO signaling results in EMT. Basal-like patients respond to preoperative taxane and adriamycin chemotherapies (36, 46). The NO-induced increase in P-glycoprotein expression and increased resistance to both paclitaxel and adriamycin indicate that NOS2/NO signaling may have a clinically relevant role in patient outcome. These observations suggest that “nitrosative signaling” from NOS2 promote an aggressive phenotype that is clinically challenging.
In conclusion, NOS2 activity and NO signaling, via SNO modification, activates EGFR and Src signaling to induce a basal-like breast cancer phenotype in human cell lines by activating oncogenic signaling pathways, inhibiting PP2A tumor suppressor activity, inducing COX-2 expression and activity, inducing EMT and increasing cellular chemoresistance. The reaction of NO with EGFR and Src form a nexus for driving multiple molecular pathways that promote basal-like breast cancer phenotypes. Because NOS2 expression is highly associated with poor patient survival and physiologically relevant NO concentrations result in pro-tumor signaling profiles, NOS2 inhibition may be a novel strategy in treating basal-like/triple-negative breast cancer.
Supplementary Material
Acknowledgments
We would like to thank Dr. Larry Keefer (National Cancer Institute, Frederick, MD, USA) for generously providing DETANO. This research was supported by the intramural program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (1ZIASC007281-18).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol. 2008;26(15):2568–2581. doi: 10.1200/JCO.2007.13.1748. [DOI] [PubMed] [Google Scholar]
- 3.Sarrio D, Rodriguez-Pinilla SMa, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-Mesenchymal Transition in Breast Cancer Relates to the Basal-like Phenotype. Cancer Research. 2008;68(4):989–997. doi: 10.1158/0008-5472.CAN-07-2017. [DOI] [PubMed] [Google Scholar]
- 4.Marchini C, Montani M, Konstantinidou G, et al. Mesenchymal/Stromal Gene Expression Signature Relates to Basal-Like Breast Cancers, Identifies Bone Metastasis and Predicts Resistance to Therapies. PLoS One. 5(11):e14131. doi: 10.1371/journal.pone.0014131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carey LA, Perou CM, Livasy CA, et al. Race, Breast Cancer Subtypes, and Survival in the Carolina Breast Cancer Study. JAMA. 2006;295(21):2492–2502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 6.Reis-Filho JS, Tutt ANJ. Triple negative tumours: a critical review. Histopathology. 2008;52(1):108–118. doi: 10.1111/j.1365-2559.2007.02889.x. [DOI] [PubMed] [Google Scholar]
- 7.Cheang MCU, Voduc D, Bajdik C, et al. Basal-Like Breast Cancer Defined by Five Biomarkers Has Superior Prognostic Value than Triple-Negative Phenotype. Clinical Cancer Research. 2008;14(5):1368–1376. doi: 10.1158/1078-0432.CCR-07-1658. [DOI] [PubMed] [Google Scholar]
- 8.Hoadley KA, Weigman VJ, Fan C, et al. EGFR associated expression profiles vary with breast tumor subtype. BMC Genomics. 2007;8:258. doi: 10.1186/1471-2164-8-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH. Wnt/{beta}-Catenin Pathway Activation Is Enriched in Basal-Like Breast Cancers and Predicts Poor Outcome. Am J Pathol. 2010;176(6):2911–2920. doi: 10.2353/ajpath.2010.091125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geyer FC, Lacroix-Triki M, Savage K, et al. [beta]-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod Pathol. 2011;24(2):209–231. doi: 10.1038/modpathol.2010.205. [DOI] [PubMed] [Google Scholar]
- 11.Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer and Metastasis Reviews. 2009;28(1):151–166. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 12.Polette M, Mestdagt M, Bindels S, et al. Beta-catenin and ZO-1: shuttle molecules involved in tumor invasion-associated epithelial-mesenchymal transition processes. Cells Tissues Organs. 2007;185(1–3):61–65. doi: 10.1159/000101304. [DOI] [PubMed] [Google Scholar]
- 13.Honeth G, Bendahl P-O, Ringner M, et al. The CD44+/CD24− phenotype is enriched in basal-like breast tumors. Breast Cancer Research. 2008;10(3):R53. doi: 10.1186/bcr2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eyler CE, Wu Q, Yan K, et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell. 2011;146(1):53–66. doi: 10.1016/j.cell.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ekmekcioglu S, Ellerhorst J, Smid CM, et al. Inducible nitric oxide synthase and nitrotyrosine in human metastatic melanoma tumors correlate with poor survival. Clin Cancer Res. 2000;6(12):4768–4775. [PubMed] [Google Scholar]
- 16.Wink DA, Ridnour LA, Hussain SP, Harris CC. The reemergence of nitric oxide and cancer. Nitric Oxide. 2008;19(2):65–67. doi: 10.1016/j.niox.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glynn SA, Boersma BJ, Dorsey TH, et al. Increased NOS2 predicts poor survival in estrogen receptor-negative breast cancer patients. J Clin Invest. 2010;120(11):3843–3854. doi: 10.1172/JCI42059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas DD, Ridnour LA, Espey MG, et al. Superoxide fluxes limit nitric oxide-induced signaling. J Biol Chem. 2006;281(36):25984–25993. doi: 10.1074/jbc.M602242200. [DOI] [PubMed] [Google Scholar]
- 19.Espey MG, Miranda KM, Pluta RM, Wink DA. Nitrosative Capacity of Macrophages Is Dependent on Nitric-oxide Synthase Induction Signals. Journal of Biological Chemistry. 2000;275(15):11341–11347. doi: 10.1074/jbc.275.15.11341. [DOI] [PubMed] [Google Scholar]
- 20.Ridnour LA, Thomas DD, Switzer C, et al. Molecular mechanisms for discrete nitric oxide levels in cancer. Nitric Oxide. 2008;19(2):73–76. doi: 10.1016/j.niox.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ridnour LA, Thomas DD, Donzelli S, et al. The biphasic nature of nitric oxide responses in tumor biology. Antioxid Redox Signal. 2006;8(7–8):1329–1337. doi: 10.1089/ars.2006.8.1329. [DOI] [PubMed] [Google Scholar]
- 22.Jaffrey SR, Snyder SH. The Biotin Switch Method for the Detection of S-Nitrosylated Proteins. Sci STKE. 2001;2001(86) doi: 10.1126/stke.2001.86.pl1. pl1-. [DOI] [PubMed] [Google Scholar]
- 23.Rahman MA, Senga T, Ito S, Hyodo T, Hasegawa H, Hamaguchi M. S-nitrosylation at cysteine 498 of c-Src tyrosine kinase regulates nitric oxide-mediated cell invasion. J Biol Chem. 2010;285(6):3806–3814. doi: 10.1074/jbc.M109.059782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ingthorsson S, Halldorsson T, Sigurdsson V, et al. Selection for EGFR gene amplification in a breast epithelial cell line with basal-like phenotype and hereditary background. In Vitro Cell Dev Biol Anim. 2011;47(2):139–148. doi: 10.1007/s11626-010-9371-6. [DOI] [PubMed] [Google Scholar]
- 25.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marotta LL, Almendro V, Marusyk A, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44CD24 stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121(7):2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Switzer CH, Glynn SA, Ridnour LA, et al. Nitric oxide and protein phosphatase 2A provide novel therapeutic opportunities in ER-negative breast cancer. Trends in Pharmacological Sciences. 2011;32(11):644–651. doi: 10.1016/j.tips.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med. 2008;14(4):152–160. doi: 10.1016/j.molmed.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Eichhorn PJA, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2009;1795(1):1–15. doi: 10.1016/j.bbcan.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 30.Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science. 1992;257(5074):1261–1264. doi: 10.1126/science.1325671. [DOI] [PubMed] [Google Scholar]
- 31.DiMeo TA, Anderson K, Phadke P, et al. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009;69(13):5364–5373. doi: 10.1158/0008-5472.CAN-08-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foroni C, Broggini M, Generali D, Damia G. Epithelial-mesenchymal transition and breast cancer: Role, molecular mechanisms and clinical impact. Cancer Treat Rev. doi: 10.1016/j.ctrv.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 33.Ogunwobi OO, Liu C. Hepatocyte growth factor upregulation promotes carcinogenesis and epithelial-mesenchymal transition in hepatocellular carcinoma via Akt and COX-2 pathways. Clin Exp Metastasis. 2011;28(8):721–731. doi: 10.1007/s10585-011-9404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.St John MA, Dohadwala M, Luo J, et al. Proinflammatory mediators upregulate snail in head and neck squamous cell carcinoma. Clin Cancer Res. 2009;15(19):6018–6027. doi: 10.1158/1078-0432.CCR-09-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neil JR, Johnson KM, Nemenoff RA, Schiemann WP. Cox-2 inactivates Smad signaling and enhances EMT stimulated by TGF-beta through a PGE2-dependent mechanisms. Carcinogenesis. 2008;29(11):2227–2235. doi: 10.1093/carcin/bgn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rouzier R, Perou CM, Symmans WF, et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin Cancer Res. 2005;11(16):5678–5685. doi: 10.1158/1078-0432.CCR-04-2421. [DOI] [PubMed] [Google Scholar]
- 37.Kuroda H, Ishida F, Nakai M, Ohnisi K, Itoyama S. Basal cytokeratin expression in relation to biological factors in breast cancer. Hum Pathol. 2008;39(12):1744–1750. doi: 10.1016/j.humpath.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 38.Cheng R, Ridnour LA, Glynn SA, et al. In: Nitric Oxide and Cancer: An Overview. Bonavida B, editor. Springer New York: Nitric Oxide (NO) and Cancer; 2010. pp. 3–20. [Google Scholar]
- 39.Moller MN, Li Q, Vitturi DA, Robinson JM, Lancaster JR, Jr, Denicola A. Membrane "lens" effect: focusing the formation of reactive nitrogen oxides from the *NO/O2 reaction. Chem Res Toxicol. 2007;20(4):709–714. doi: 10.1021/tx700010h. [DOI] [PubMed] [Google Scholar]
- 40.Liu X, Miller MJ, Joshi MS, Thomas DD, Lancaster JR., Jr Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc Natl Acad Sci U S A. 1998;95(5):2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foster MW, Hess DT, Stamler JS. Protein, S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009;15(9):391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marozkina NV, Gaston B. S-Nitrosylation signaling regulates cellular protein interactions. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbagen.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lam YW, Yuan Y, Isaac J, Babu CVS, Meller J, Ho S-M. Comprehensive Identification and Modified-Site Mapping of <italic>S-</italic>Nitrosylated Targets in Prostate Epithelial Cells. PLoS One. 2010;5(2):e9075. doi: 10.1371/journal.pone.0009075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee TY, Chen YJ, Lu TC, Huang HD. SNOSite: exploiting maximal dependence decomposition to identify cysteine S-nitrosylation with substrate site specificity. PLoS One. 2011;6(7):e21849. doi: 10.1371/journal.pone.0021849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Estrada C, Gomez C, Martin-Nieto J, De Frutos T, Jimenez A, Villalobo A. Nitric oxide reversibly inhibits the epidermal growth factor receptor tyrosine kinase. Biochem J. 1997;326(Pt 2):369–376. doi: 10.1042/bj3260369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maegawa RO, Tang SC. Triple-negative breast cancer: unique biology and its management. Cancer Invest. 2010;28(8):878–883. doi: 10.3109/07357907.2010.483507. [DOI] [PubMed] [Google Scholar]
- 47.Miranda KM, Espey MG, Wink DA. A Rapid, Simple Spectrophotometric Method for Simultaneous Detection of Nitrate and Nitrite. Nitric Oxide. 2001;5(1):62–71. doi: 10.1006/niox.2000.0319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.