

Abstract
Styrene is one of the most important industrial intermediates consumed in the world and is mainly used as a monomer for reinforced plastics and rubber. Styrene has been found to be hepatotoxic and pneumotoxic in humans and experimental animals. The toxicity of styrene is suggested to be metabolism-dependent. Styrene-7,8-oxide has been considered as the major metabolite responsible for styrene-induced cytotoxicity. The objective of the study was to investigate the correlation between cytotoxicity of styrene and chemical and biochemical properties of the vinyl group of styrene by development of structure activity relationships (SAR). 4-Fluorostyrene, 4-chlorostyrene and 4-bromostyrene were selected for the SAR study. Cytotoxicity of styrene and the halogenated styrene derivatives with an order of 4-bromostyrene > 4-chlorostyrene > 4-fluorostyrene ≈ styrene was observed in CYP2E1 transgenic cells. Similar orders in the efficiency of the metabolism of styrene and the halogenated styrene analogues to their oxides and in the electrophilicity of the corresponding oxides were observed. Additionally, the order of the potency of cellular glutathione depletion and the degree of protein adduction induced by styrene and the halogenated styrenes were consistent with that of their cytotoxicities. The wild-type cells were less susceptible to the toxicity of the corresponding model compounds than CYP2E1 cells. The present study provided insight into the roles of the biochemical and chemical properties of styrene in its cytotoxicity.
Keywords: Styrene, Styrene Oxide, Bioactivation, Protein Adduct
1. Introduction
Styrene is a high production volume chemical with around 20-30 million tons produced each year to make products such as rubber, plastic, fiberglass, pipes, automobile parts and food containers. Occupational exposures to styrene occur mainly in the reinforced plastic industry (IARC, 1994; IARC, 2002; Leibman, 1995; Scott and Preston, 1994). Human exposure to styrene may also result from cigarette smoke, engine exhausts, heating systems, newly installed carpets, and painting (Hodgson et al., 1993; Teixeira et al., 2010; Huff et al., 2011). Styrene has also been detected in the foods and drinking water stored in polystyrene containers (Fleming-Jones and Smith, 2003; Tang et al., 2000). Animal studies showed that intraperitoneal administration of styrene in mice caused elevated activities of γ-glutamyltranspeptidase (GGT) and lactate dehydrogenase (LDH) in bronchoalveolar lavage fluid (BALF) (Carlson, 1997; Gadberry et al., 1996). Multifocal necrosis and cell loss in bronchiolar epithelium were observed in CD-1 mice after exposure to 40 or 160 ppm inhaled styrene (Green et al., 2001). Upon repeated exposures, there was decreased cytoplasmic staining of Clara cells and cell crowding in the terminal bronchioles (Cruzan et al., 1997). Studies in CD-1 and B6C3F1 mice consistently showed cell crowding, decreased staining, and increased cell replication in the Clara cells of the mouse bronchiolar epithelium (Cruzan et al., 1997; Green et al., 2001). Green at el. reported increased proliferation in Clara cells after mice were administrated orally with styrene (Green et al., 2001). Recently, Harvilchuck et al. found elevated reactive oxygen species in Clara cells after exposure to styrene (Harvilchuck et al., 2009).
Styrene is primarily metabolized by P450 isozymes such as CYP2F2 and CYP2E1 (Guengerich et al., 1991; Nakajima et al., 1994; Carlson, 1997; Green et al., 2001; Cruzan et al., 2012) to form a chemically active metabolite, styrene-7,8-oxide (5, Scheme 1), which has been suggested to be responsible for styrene toxicity in mice (Bond, 1989). Styrene oxide as an electrophilic species can react with the nucleophilic sites of biomolecules to form adducts. Various DNA adducts have been identified in humans exposed to styrene (Maki-Paakkanen et al., 1991). Styrene was found to induce glutathione depletion in both the liver and lung (Carlson et al., 2006). Styrene oxide derived albumin and hemoglobin modifications were detected in workers exposed to styrene occupationally (Christakopoulos et al., 1993; Fustinoni et al., 1998; Johanson et al., 2000; Osterman-Golkar et al., 1995; Vodicka et al., 1999; Vodicka et al., 2001; Vodicka et al., 2003), and the protein modifications correlated with the levels of styrene exposure (Vodicka et al., 1999). Lanosa et al. found a close correlation between metabolic activation of styrene and the sensory irritation response to styrene (Lanosa et al., 2010). Recently, our group reported cellular protein modification by styrene oxide in cultured micro-dissected mouse airways and cells after exposure to styrene (Yuan et al., 2010). Mörbt et al. also identified modified thioredoxin reductase by styrene oxide in cultured A297 cells incubated with styrene (Mörbt et al., 2009).
Scheme 1.
The objectives of the present study were to develop structure toxicity relationships of selected halogenated styrenes and to investigate the role of the vinyl group of styrene in cytotoxicity induced by styrene. This allowed us to better understand the biochemical mechanisms of pulmonary toxicity induced by styrene.
2. Materials and methods
2.1 Materials
Styrene (1, 99%), 4-fluorostyrene (2, 99%), 4-chlorostyrene (3, 97%), 4-bromostyrene (4, 98%), styrene oxide (5, 98%, racemic), 4-fluorostyrene oxide (6, 99%), 4-chlorostyrene oxide (7, 98%), and 4-bromostyrene oxide (8, 98%), trifluoroacetic acid, CDCl3, dimethylformamide (DMF), N,O-bis(trimethylsilyl)trifluoroacetamide, cysteamine, β-nicotinamide adenine dinucleotide phosphate (NADPH), and glutathione (GSH) were purchased from Sigma Chemical Co. (St. Louis, MO). Ethyl acetate, anhydrous Na2SO4, HPLC-grade acetonitrile and secureseal were obtained from Fisher Scientific Co. (Pittsburgh, PA). DNase kit and RNase I were from Qiagen (Valencia, CA). 14C-Styrene (β-14C), 14C-4-fluorostyrene (β-14C), and 14C-4-bromostyrene (β-14C) were custom synthesized by American Radiolabeled Chemical, Inc., (St. Louis, MO) with chemical purity of 99+% and radioactive purity of 99+%. CellTiter 96 Aqueous One solution kit was from Promega (Sunnyvale, CA). Trypan Blue stain was purchased from Bio Whittaker (Walkersville, MD). Transgenic cell line expressing CYP2E1 (h2E1) and the wild-type cell line (cHo1, human B-lymphoblastoid) were obtained from BD-Gentest (Palo Alto, CA). The cell lines were used in our earlier styrene toxicity studies (Chung et al., 2006; Yuan et al., 2010). Recombinant human CYP2E1 was purchased from BD-Gentest (Palo Alto, CA).
2.2 Synthesis of styrene, 4-fluorostyrene, 4-chlorostyrene, and 4-bromostyrene oxide-derived glycols (9-12)
The individual oxides (100 mg each) were mixed with 20 mL of 1.0 N HCl containing 40% acetonitrile, followed by stirring at room temperature overnight. The organic solvent was evaporated by a rotary evaporator, and the remaining aqueous was extracted with acetyl acetate (15 mL × 3). The organic layers were combined, dried over anhydrous Na2SO4, and evaporated to dryness in vacuum. The residues were chromatographed in a silica gel column to give the desired compounds. Styrene glycol (9): 1H-NMR (300 MHz, CDCl3) δ 3.48-2.82 (br, 2H, OH), 3.65 (dd, J = 8.4 and 11.5 Hz, 1H, CH2), 3.74 (dd, J = 3.4 and 11.5 Hz, 1H, CH2), 4.81 (dd, J = 3.4 and 8.4 Hz, 1H, CH), 7.52-7.18 (m, 5H, Ar). 4-Fluorostyrene glycol (10): 1H-NMR (300 MHz, CDCl3) δ 3.36-2.68 (br, 2H, OH), 3.65 (dd, J = 8.3 and 11.4 Hz, 1H, CH2), 3.75 (dd, J = 3.4 and 11.4 Hz, 1H, CH2), 4.82 (dd, J = 3.4 and 8.3 Hz, 1H, CH), 7.07(dt, J = 2 and 8.5 Hz, 2H, Ar), 7.35 (dd, J = 5.4 and 8.5 Hz, 2H, Ar). 4-Chlorostyrene glycol (11): 1H-NMR (300 MHz, CDCl3) δ 3.22-2.62 (br, 2H, OH), 3.64 (dd, J = 8.2 and 11.2 Hz, 1H, CH2), 3.76 (dd, J = 3.2 and 11.2 Hz, 1H, CH2), 4.82 (dd, J = 3.2 and 8.2 Hz, 1H, CH), 7.32(d, J = 8.4 Hz, 2H, Ar), 7.36 (d, J = 8.4 Hz, 2H, Ar). 4-Bromostyrene glycol (12): 1H-NMR (300 MHz, CDCl3) δ 3.32-2.74 (br, 2H, OH), 3.63 (dd, J = 8.2 and 11.4 Hz, 1H, CH2), 3.77 (dd, J = 3.4 and 11.4 Hz, 1H, CH2), 4.81 (dd, J = 3.4 and 8.2 Hz, 1H, CH), 7.27(d, J = 8.4 Hz, 2H, Ar), 7.51 (d, J = 8.4 Hz, 2H, Ar).
2.3 Synthesis of styrene, 4-fluorostyrene, 4-chlorostyrene, and 4-bromostyrene oxide-derived cysteamine adducts (13-20)
General procedure: the oxides (5 mmol) were individually mixed with cysteamine (7.5 mmol) in 10 mL of acetonitrile-water solution (6 : 1) containing triethylamine (10 mmol). The mixtures were stirred at room temperature under an atmosphere of nitrogen for 36 h. The resulting reaction mixtures were diluted with ethyl acetate (30 mL) and then washed with water (10 mL × 3). The organic layers were combined, dried over anhydrous Na2SO4, and evaporated under vacuum. The residues were subjected to column chromatography on silica gel to afford the following cysteamine adducts.
The reaction of styrene oxide with cysteamine gave a mixture of 2-(2-aminoethylthio)-2-phenylethanol and 2-(2-aminoethylthio)-1-phenylethanol (13 and 17, approximate 1:1) as light yellow oil (197 mg, 20% in yield). 1H-NMR (300 MHz, CDCl3): δ 2.48-3.04 (m, 6H + 8H), 3.80-3.90 (m, 2H), 3.96 (dd, J = 5.7, 7.9Hz, 1H), 4.78 (dd, J = 3.6, 9Hz, 1H), 5.29 (brs, 2H), 7.20-7.45 (m, 5H × 2); m/z = 198.2 [M+H]+.
The reaction of 4-fluorostyrene oxide with cysteamine gave a mixture of 2-(2-aminoethylthio)-2-(4-fluorophenyl)ethanol and 2-(2-aminoethylthio)-1-(4-fluorophenyl)ethanol (14 and 18, approximate 1:1) as light yellow oil (194 mg, 18%). 1H-NMR (300 MHz, CDCl3): δ 2.45-3.05 (m, 6H + 8H), 3.78-3.90 (m, 2H), 3.95 (dd, J = 6, 7.2Hz, 1H), 4.78 (dd, J = 3.48, 8.9Hz, 1H), 5.30 (brs, 2H), 6.94-7.14 (m, 2H × 2), 7.24-7.42 (m, 2H × 2); m/z = 216.3 [M+H]+.
The reaction of 4-chlorostyrene oxide with cysteamine gave a mixture of 2-(2-aminoethylthio)-2-(4-chloroophenyl)ethanol and 2-(2-aminoethylthio)-1-(4-chlorophenyl)ethanol (15 and 19, approximate 1:1) as light yellow oil (150 mg, 13% in yield). 1H-NMR (300 MHz, CDCl3): δ 2.50-3.16 (m, 6H + 8H), 3.72-4.06 (m, 2H), 4.12 (dd, J = 7.1, 14Hz, 1H), 4.78 (dd, J = 3.4, 8.8Hz, 1H), 5.30 (brs, 2H), 7.18-7.42 (m, 4H × 2); m/z = 232.1 [M+H]+.
The reaction of 4-bromostyrene oxide with cysteamine gave a mixture of 2-(2-aminoethylthio)-2-(4-bromophenyl)ethanol and 2-(2-aminoethylthio)-1-(4-bromophenyl)ethanol (16 and 20, approximate 1:1) as light yellow oil (206 mg, 16% in yield). 1H-NMR (300 MHz, CDCl3): δ 2.50-3.00 (m, 6H + 8H), 3.79-3.87 (m, 2H), 3.92 (dd, J = 5.6, 7.6Hz, 1H), 4.75 (dd, J = 3.5, 9.0Hz, 1H), 5.30 (brs, 2H), 7.14-7.36 (m, 2H × 2), 7.40-7.56 (m, 2H × 2); m/z = 276.0 [M+H]+.
2.4 Instrumentation
The HPLC system consisted of an Agilent 1100 LC binary pump system and an Agilent 1100 Autosampler (Palo Alto, CA). The HPLC system was interfaced with a PE Sciex API 2000 LC/MS/MS System including an electrospray ionization source and a triple quadrupole mass analyzer (Applied Biosystems, Foster City, CA). Data were gathered by software Analyst 1.2. Microplate readers VERSAMax and GeminiXS (Molecular Devices, Sunnyvale, CA) were utilized for cell viability studies and protein assays.
2.5 Cell culture
Parental and h2E1 cells were cultured in RPMI 1640 supplemented with L-glutamine and 10% fetal bovine serum (Hyclone Laboratory, Logan, UT) under 95% air/5% CO2 at 37 °C, according to the protocol our lab published earlier (Chung et al., 2006; Yuan et al., 2010).
2.6 Cytotoxicity study in vitro
Before treatment, cells were counted with Trypan Blue staining by use of a hemocytometer to reach a density of 2 × 105 cells/mL. Styrene or halogenated styrenes (100 μM) dissolved in DMF were added to 24-well microplates containing CYP2E1 or wild-type cells. The final concentration of the organic solvent was < 0.5%. The resulting plates were sealed with a secureseal to prevent possible evaporation of the chemicals tested. After 24 h incubation, cell viability was determined by CellTiter 96 Aqueous One solution assay.
2.7 Cellular protein covalent binding
Parental and CYP2E1 cells were cultured in 10-cm Petri dishes at a density of 5 × 105 cells/mL (10 mL per dish). The cells were incubated with 14C-labeled styrene, 4-fluorostyrene, or 4-bromostyrene (chemical concentration: 100 μM; specific radioactivity: 6.25 Ci/mol). After 24 h incubation, the cells were washed three times with PBS (pH 7.4), lysed with 500 μL of lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton) at room temperature for 15 min, and centrifuged at 19,000 g for 20 min. The supernatant was collected and incubated with one unit of DNase and RNase I at room temperature for 20 min, followed by extensive dialysis (MWCO: 3.5KDa, Fisher brand) against PBS buffer with 0.1% SDS at room temperature until the radioactivity in the dialysis solution reached the background. An aliquot of 200 μL of the dialyzed sample was mixed with 5 ml of scintillation cocktail solution and the mixture was subjected to radioactivity assessment using a scintillation counter. Protein assay was performed using a bicinchoninic acid protein assay kit (Pierce, Rockford, IL). Bovine serum albumin was used as a protein standard.
2.8 SDS-PAGE and radioactivity imaging
Protein concentrations of the dialyzed protein samples were measured using bicinchoninic acid protein kit (Pierce Chemical, Rockford, IL), and equal amounts (20 μg) of protein were loaded and resolved by SDS-polyacrylamide gel electrophoresis using pre-cast 10% Tris-glycine gels (Invitrogen, Carlsbad, CA). The gel was dried in a gel dryer (Bio-Rad, Hercules, CA) at 80 °C for 1 h. And the dried gel was exposed to a tritium storage phosphor screen (GE Healthcare, Piscataway, NJ) for 7 days. The exposed screen was scanned by a Typhoon Trio imaginer (GE Healthcare, Piscataway, NJ).
2.9 Bioactivation by recombinant human CYP2E1
Enzyme kinetic studies were performed following the protocol established in our lab (Shen et al., 2010). Briefly, a 0.5 mL solution containing recombinant human CYP2E1 (50 pmol), 5 mM MgCl2, and 2 mM NADPH in 50 mM phosphate buffer (pH 7.4) was preincubated in a 37 °C water bath shaker for 3 min. The reaction was initiated by addition of 5.0 μL of various concentrations of styrene, 4-fluorostyrene, 4-chlorostyrene or 4-bromostyrene (final concentration: 0.1–100 μM). The incubation continued for 20 min and was quenched by addition of the same volume of ethyl acetate, followed by spiking with deuterated styrene glycol (styrene glycol-d8, prepared in our lab) as internal standard (Shen et al., 2010). The resulting mixture was vortexed and the organic phase was collected. The collected extract was dried by blowing with N2, and the remaining was silylated by N,O-bis(trimethylsilyl)trifluoroacetamide and subjected to GC/MS analysis. The production of the resulting glycols was quantified by comparing their peak areas vs. that of the internal standard. Vmax and Km values were calculated by non-linear least-squares regression analysis using SigmaPlot software.
2.10 Reactivity of styrene oxide and halogenated styrene oxides to cysteamine
Styrene oxide, 4-fluorostyrene oxide, 4-chlorostyrene oxide and 4-bromostyrene oxide (1.0 μM) were individually incubated with cysteamine (100 μM) in phosphate buffer in a 37 °C water bath. A 100 μL aliquot of the reaction mixture was withdrawn at 1, 3, 7, 15, 30 and 60 min and was immediately mixed with 100 μL of 1 N HCl, followed by LC/MS analysis. An LC/MS scan operated in a positive and selective ion monitoring (SIM) mode was acquired. The ions with m/z 198, 216, 232, and 276 were monitored, and these ions were responsible for the respective [M+H]+ of the corresponding cysteamine adducts. The LC conditions and mass spectrometer parameters were applied as follows: twenty microliter samples were injected into a C18 column (3 μ, 2.1 × 100 mm, Alltech, Deerfield, IL); the samples were eluted from the column with a gradient of solvent A (0.1% aqueous trifluoroacetic acid) and solvent B (0.1% trifluoroacetic acid in acetonitrile) at a flow rate of 200 μL/min, and the flow was directed to the mass spectrometer. Mass spectrometer operating conditions: positive ionization mode, ionization voltage at 5kV, orifice potential at 30 volts, and ion source temperature at 250 °C.
2.11 Cellular GSH contents
CYP2E1 cells were cultured in 6-well plates at a density of 1 × 106 cells/mL. The cells were incubated with styrene, 4-fluorostyrene, or 4-bromostyrene (100 μM) and harvested at various time points. Cellular GSH were assessed using an ApoGSH™ glutathione detection kit (BioVision, CA). The assay was performed based on the protocol provided by the manufacturer. In brief, the harvested cells were resuspended in 100 μL cell lysis buffer. After 15 min incubation at room temperature, the supernatants were collected and transferred to a test tube containing 2 μL of 25 mM monochlorobimane and 2 μL of the 50 U/mL glutathione S-transferase solution. After incubating for 30 min at 37 °C, fluorescence at Ex/Em = 380/460 nm was measured using a GeminiXS fluorescence plate reader.
2.12 Statistics
All values were presented as mean ± SE. Analysis of Variance (ANOVA) with post hoc pairwise comparison of the means using a Tukey test for multiple comparisons (p < 0.01) was performed using PRISM (GraphPad Software, Inc., La Jolla, CA).
3. Results
3.1 Cytotoxicity of styrene and halogenated styrenes
Styrene, 4-fluorostyrene, 4-chlorostyrene, and 4-bromostyrene were selected for the structure-toxicity relationship study, and their toxicities were assessed in CYP2E1 transgenic cells following the procedure we applied early (Chung et al., 2006). Cells were incubated with 100 μM styrene or the halogenated styrenes. Cell viability was determined after 24 h incubation by CellTiter 96 Aqueous One solution assay. Relative to the group treated with vehicle, 4-chlorostyrene and 4-bromostyrene induced 57.7 % and 39.6 % decreased in cell viability, respectively. Comparing with styrene-treated group, 4-fluorostyrene showed little difference in toxicity to CYP2E1 cells.
Susceptibility of the wild-type cells to styrene and the halogenated styrenes was also evaluated at concentration of 100 μM. As expected, the wild-type cells were found to be less susceptible to styrene and the corresponding halogenated styrenes than CYP2E1 transgenic cells (Figure 1).
Figure 1.
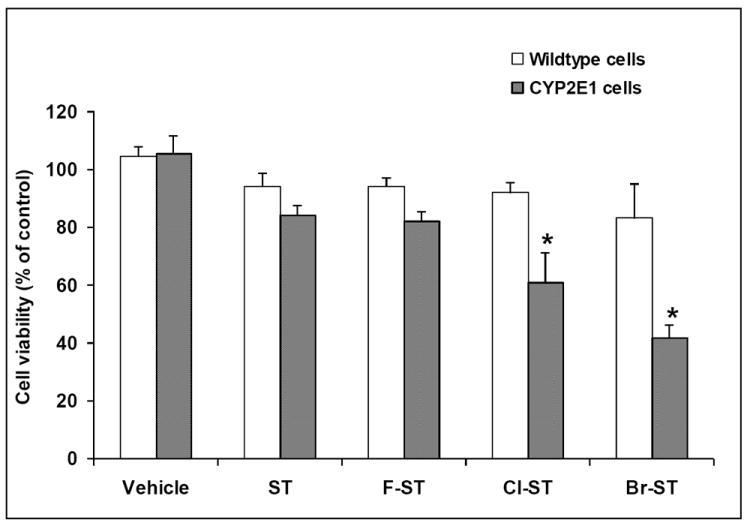
CYP2E1 and wild-type cells were exposed to vehicle, styrene (ST), 4-fluorostyrene (F-ST), 4-chlorostyrene (Cl-ST) or 4-bromostyrene (Br-ST) at concentration of 100 μM at 37 °C for 24 h. Cell viability was assessed by use of CellTiter 96 Aqueous One solution assay. Control: cells were treated with vehicle. (* indicates significant difference from the styrene group, p < 0.01, means ± SE, n = 4)
3.2 Protein covalent binding
Cellular protein covalent binding levels were measured by assessment of radioactivity bound to cellular proteins. To minimize the cost of protein covalent binding study, we selected 4-fluorostyrene and 4-bromostyrene to represent halogenated styrenes, and we had 14C-styrene, 14C-4-fluorostyrene, and 14C-4-bromostyrene custom synthesized. CYP2E1 cells were exposed to 100 μM the individual 14C-labeled compounds for 24 h. Figure 2 shows the radioactivity bound to proteins obtained from cells after exposure to the individual 14C-labeled styrenes. The binding load was 2.02, 2.27 and 6.17 nmol/mg protein for styrene, 4-fluorostyrene and 4-bromostyrene, respectively (Table 1). In parallel, the wild-type cells were incubated with individual 14C-labeled styrene, 4-fluorostyrene and 4-bromostyrene at 100 μM for 24 h. Less radioactivity bound to cellular proteins was observed in the wild-type cells relative to that in CYP2E1 cells after exposure to the individual radioactive styrenes (Figure 2). Particularly, 5-fold lower radioactivity was bound to proteins in the wild-type cells than CYP2E1 cells treated with 14C-labeled 4-bromostyrene.
Figure 2.

CYP2E1 and wild-type cells were incubated with 14C-labeled styrene (ST), 4-fluorostyrene (F-ST) or 4-bromostyrene (Br-ST) at concentration of 100 μM at 37 °C for 24 h. Cellular proteins were harvested, extensively dialyzed against PBS buffer with 0.1% SDS, and subjected to radioactivity assessment and protein assay (* indicates significant difference from the styrene group, p < 0.01, means ± SE, n = 4)
Table 1.
Summary of cytotoxicity in human CYP2E1 cells and biochemical and chemical properties of styrene (ST), 4-fluorostyrene (F-ST), 4-chlorostyrene (Cl-ST) and 4-bromostyrene (Br-ST) in a recombinant human CYP2E1 system, and of the reactivity of the corresponding oxides to cysteamine.
| Cell viability (% of control after 24 h exposure) | Vmax/Km (mL/min/mg) | Reactivity of oxides to cysteamine (k, min-1) | Bioactivation index (k × Vmax/Km) | GSH depletion (% of control after 3 h exposure) | |
|---|---|---|---|---|---|
| ST | 80.0 ± 3.0a | 0.62 ± 0.03a | 13.4 ± 1.88a | 8.30 ± 0.57a | 83.7 ± 2.1a |
| F-ST | 78.8 ± 3.2a | 0.66 ± 0.05a | 15.7 ± 1.57a | 10.4 ± 0.64a | 82.4 ± 2.2a |
| Cl-ST | 57.7 ± 9.9b | 0.98 ± 0.04b | 16.1 ± 2.08a | 15.8 ± 2.03b | 71.2 ± 5.8b |
| Br-ST | 39.6 ± 4.1c | 1.09 ± 0.05b | 29.3 ± 3.19b | 31.9 ± 3.48c | 61.0 ± 6.5b |
Control: vehicle-treated.
Values are expressed as mean ± SD, n = 4.
values with different superscripts are significantly different from one another (p<0.05).
In addition, the radioactive protein samples were resolved by SDS-PAGE, and radioactivity of the resolved proteins was imaged. As expected, much more radioactivity was bound to proteins obtained from cells treated with 4-bromostyrene than that of cells after treatment with styrene or 4-fluorostyrene (Figure 3). In addition, similar patterns in radioactive bands were observed in the gel loaded with protein samples obtained from cells after exposure to radioactive styrene, 4-fluorostyrene or 4-bromostyrene.
Figure 3.
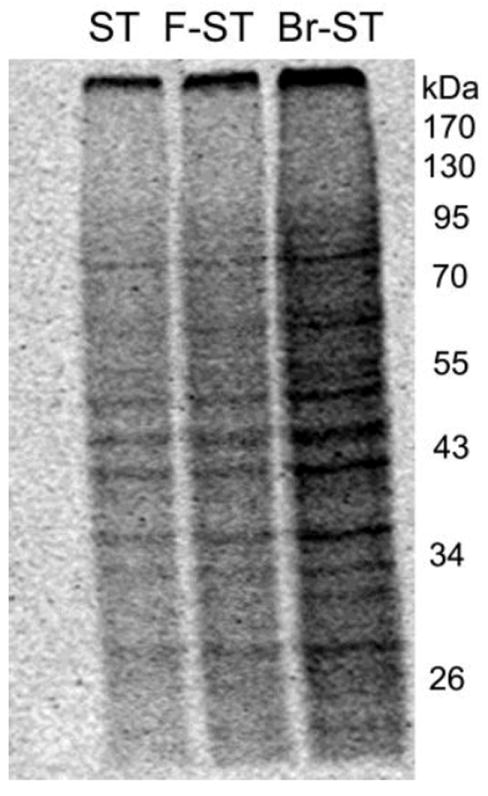
CYP2E1 cells were incubated with 14C-labeled styrene (ST), 4-fluorostyrene (F-ST) or 4-bromostyrene (Br-ST) at concentration of 100 μM at 37 °C for 24 h. Cellular proteins were harvested, extensively dialyzed, and subjected to SDS-polyacrylamide gel electrophoresis. Radioactive protein bands were detected by exposure of gel to a phosphor screen.
3.3 Bioactivation by recombinant CYP2E1
Bioactivation of styrene and the halogenated styrenes to the corresponding oxides was evaluated in a recombinant human CYP2E1 system. The halogenated styrenes were exclusively metabolized to oxides 6-8, and no phenols were detected. The resulting kinetic parameters were calculated and summarized in Table 2. 4-Bromostyrene and 4-chlorostyrene showed the lowest Km value, followed by 4-fluorostyrene and styrene. The same observation was found in the order of Vmax values for 4-bromostyrene, 4-chlorostyrene, 4-fluorostyrene and styrene. Values of Vmax/Km were calculated and found to follow the order of 4-bromostyrene > 4-chlorostyrene > 4-fluorostyrene ≈ styrene (Table 1).
Table 2.
Parameters Km and Vmax of metabolism of styrene (ST), 4-fluorostyrene (F-ST), 4-chlorostyrene (Cl-ST) and 4-bromostyrene (Br-ST) to the corresponding oxides in a recombinant human CYP2E1 system.
| ST | F-ST | Cl-ST | Br-ST | |
|---|---|---|---|---|
| Km (μM) | 56.1 ± 4.26a | 54.2 ± 5.06a | 39.4 ± 1.44b | 39.0 ± 3.78b |
| Vmax (pmol/min/mg) | 34.7 ± 1.77a | 36.3 ± 1.94a | 38.5 ± 0.97a | 42.6 ± 3.18b |
Values are expressed as mean + SD.
values with different superscripts are significantly different from one another (p < 0.05), n = 4.
3.4 Reactivity to cysteamine
Thio groups (-SH) have been suggested to be mainly responsible for the nucleophilic attacks by styrene oxide in protein covalent modification (Rappaport et al., 1993). To evaluate the potentials of protein adduction by the oxides derived from styrene and the halogenated styrenes, we measured the reactivities of the oxides toward cysteamine. The formation of the cysteamine adducts was monitored by LC/MS at various time points. Authentic cysteamine adducts derived from the oxides were synthesized and characterized. The reaction of cysteamine with styrene oxide and the halogenated styrene oxides produced two regioisomers of the corresponding adducts at a ratio of approximate 1:1 estimated by their 1H-NMR spectra (Scheme 1). The two regioisomers were eluted out as a single peak in chromatography of LC/MS and could not be separated from each other. The formation of the two regioisomers was assessed by integration of the peak areas responsible for the two isomers of the cysteamine adducts. A time-dependent formation of the corresponding adducts was observed, and the production of the adducts reached a plateau in 15 min. The kinetic data were processed and reaction rate constants were calculated, using XLfit of an Exponential Association Model. The rate constants for production of the cysteamine adducts derived from 4-bromostyrene oxide, 4-chlorostyrene oxide, 4-fluorostyrene oxide and styrene oxide were 29.3 ± 3.19, 16.1 ± 2.08, 15.7 ± 1.57 and 13.4 ± 1.88 (min-1), respectively (Table 1).
3.5 Glutathione depletion
Depletion of cellular GSH has often been considered as an important biomarker of early stage cell injury by many electrophilic toxicants. Styrene has been reported to induce GSH depletion in vitro and in vivo (Carlson et al., 2006; Gamer et al., 2004; Harvilchuck and Carlson, 2008; Turner et al., 2005). We compared the potencies of GSH depletion induced by styrene and the halogenated styrenes. Cellular GSH contents of CYP2E1 cells were monitored at 0, 3, 6, 12, and 24 h after exposure to 100 μM styrene or the halogenated styrenes. As expected, the three halogenated styrenes along with styrene induced GSH depletion (Figure 4). The GSH contents reached the corresponding lowest levels at 3 h, then bounced back, and recovered at 12 h. The group treated with 4-bromostyrene showed the greatest degree in GSH depletion, followed by 4-chlorostyrene and 4-fluorostyrene. Styrene showed similar potency of GSH depletion as 4-fluorostyrene.
Figure 4.
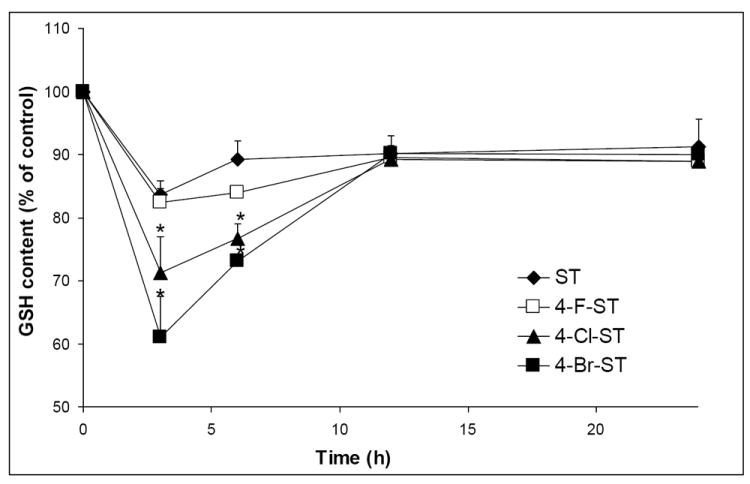
CYP2E1 cells were incubated with styrene (ST), 4-fluorostyrene (F-ST), 4-chlorostyrene (Cl-ST) or 4-bromostyrene (Br-ST) at concentration of 100 μM. Cellular GSH contents were determined at designed time points of exposure. Control: cells were treated with vehicle as 100% for the corresponding time points (* indicates significant difference from styrene or styrene oxide, p < 0.01, means ± SE, n = 4).
4. Discussion
The biochemical mechanisms of styrene-induced cytotoxicity remain unknown. As part of our mechanistic study, we developed structure-activity (toxicity) relationships of selected halogenated styrene analogues. The hypothesis for the study was that cytotoxicities induced by styrene derivatives with similar metabolic pathway are associated with their biochemical and chemical properties. The rationale of the study includes that halogens belong to group 17 of the periodic table and share similar chemical characteristics but with different reactivity. We reasoned that the halogenated styrene analogues would undergo a similar metabolic activation through cytochrome P450 oxidation at different rates. We chose styrene, 4-fluorostyrene, 4-chlorostyrene and 4-bromostyrene for our structure-toxicity relationship study and found that 4-bromostyrene was the most toxic to CYP2E1 cells, followed by 4-chlorostyrene and 4-fluorostyrene. Styrene showed a similar degree of toxicity as 4-fluorostyrene (Figure 1 and Table 1). The results indicate that the cytotoxicity of the halogenated styrene compounds increases with the increase in the periodic number of the halogens substituted at para position of styrene. Actually, we chemically synthesized 4-iodostyrene and observed its cytotoxicity (more toxic than 4-bromostyrene) in CYP2E1 cells. Unfortunately, observed unstableness of 4-iodostyrene made us to give up its further investigation.
Styrene-induced cytotoxicity has been suggested to require metabolic activation. To determine whether the epoxidation of the vinyl group is a key step to initiate styrene toxicity, we determined Vmax/Km of enzymatic epoxidation of styrene and the three halogenated styrene derivatives in a recombinant human CYP2E1. Parameter Vmax/Km represents the catalytic efficiency of a biochemical reaction. The order of Vmax/Km of styrene and the three halogenated styrene derivatives in CYP2E1 was found to be 4-bromostyrene > 4-chlorostyrene > 4-fluorostyrene ≈ styrene (Table 1), which matches the order of the cytotoxicities induced by the four compounds (Figure 1 and Table 1). It can explain that higher turnover (Vmax/Km) of bioactivation of the halogenated styrenes produces more reactive metabolites responsible for their cytotoxicities.
The electrophilicity of electrophilic species is a determining factor in SN1 (unimolecular nucleophilic substitution) and SN2 (bimolecular nucleophilic substitution) reactions. We hypothesize that the electrophilicity of oxides derived from styrene and the styrene analogues is another important element for their cytotoxicities. We evaluated the electrophilicities of styrene oxide, 4-fluorostyrene oxide, 4-chlorostyrene oxide and 4-bromostyrene oxide by determining the rates of cysteamine adductions with the corresponding oxides. The order of the electrophilicities of the oxides was 4-bromostyrene oxide > 4-chlorostyrene oxide > 4-fluorostyrene oxide ≈ styrene oxide (Table 1). Interestingly, the order of electrophilicities of the oxides was also consistent to that of cytotoxicities induced by the corresponding parent styrene analogues. This implicates a possible correlation between the electrophilicities of the oxides and the cytotoxicities induced by styrene and the halogenated styrenes.
Taken together, two factors, i.e. the total amount of the oxides generated and the electrophilicity of the oxides, appear to correlate with the cytotoxicities of styrene and the three halogenated styrene derivatives. Here we introduce a new parameter, namely bioactivation-reactivity (BR) index, which combines the efficiency of reactive metabolite formation with the electrophilicity of the reactive metabolite. This parameter is presented by the value of k × Vmax/Km, and this provides a platform to compare those metabolism-dependent electrophilic species. Parameter k stands for the rate of a reaction of an electrophilic metabolite with a nucleophilic agent. Bioactivation-reactivity indexes of 4-bromostyrene, 4-chlorostyrene, 4-fluorostyrene and styrene were calculated and are listed in Table 1. The order of the BR indexes was 4-bromostyrene > 4-chlorostyrene > 4-fluorostyrene ≈ styrene, which matches the order of cytotoxicities induced by these compounds.
As an electrophilic species, styrene oxide has been reported to react with nucleophilic biomolecules, such as glutathione (GSH) (Carlson et al., 2006; Gamer et al., 2004; Harvilchuck and Carlson, 2008; Turner et al., 2005), proteins (Christakopoulos et al., 1993; Fustinoni et al., 1998; Johanson et al.,2000; Osterman-Golkar et al., 2000; Vodicka et al., 1999; Vodicka et al., 2001; Vodicka et al., 2003) and nucleic acids (Maki-Paakkanen et al., 1991). Turner et al. observed the decrease in GSH in the liver and lung as early as one hour after styrene administration in a time- and dose-dependent manner (Turner et al., 2005). A time-course of GSH depletion was observed in CYP2E1 cells after exposure to styrene and the three halogenated styrene derivatives (Figure 4). 4-Bromostyrene was found to deplete cellular GSH the greatest, followed by 4-chlorostyrene and 4-fluorostyrene/styrene. The order in potency of GSH depletion by styrene and the halogenated styrene analogues matches that of their cytotoxicities as well as their BR indexes (Table 1).
Adduction of albumin and hemoglobin by styrene oxide has been well documented (Christakopoulos et al., 1993; Fustinoni et al., 1998; Johanson et al., 2000; Osterman-Golkar et al., 2000; Vodicka et al., 1999; Vodicka et al., 2001; Vodicka et al., 2003). Recently, our lab reported cellular protein adduction derived by styrene oxide in cultured tissues and cells after exposure to styrene (Yuan et al., 2010). In the present study, we compared the radioactivity bound to cellular proteins obtained from CYP2E1 cells after incubation with 14C-styrene, 14C-4-fluorostyrene or 14C-4-bromostyrene. Both total radioactivity tracing (Figure 2) and electrophoresis-based radioactivity imaging (Figure 3) showed similar order in degree of protein adduction induced by the individual radioactive styrene and halogenated styrene derivatives with those of their bioactivations (BR index), GSH depletion and cytotoxicity. The consistent patterns observed support our hypothesis that the cytotoxicity induced by styrene derivatives, which share a similar bioactivation process correlates their metabolic and biochemical identities. The “amplifying” effect of para-substitution of chlorine and bromine, along with the structure-toxicity relationship developed, provides us a unique approach for the mechanistic studies of styrene-induced cytotoxicity. Additionally, it appears that similar cellular proteins were modified by reactive metabolites of styrene, 4-fluorostyrene and 4-bromostyrene (Figure 3). This opens new avenues for investigation of the role of protein modification in styrene-induced cytotoxicity.
Halogens are known electron-withdrawing groups, and the inductive effects of fluorine, chlorine and bromine substituted on styrene may cause decreased electron density of the π-systems, which disfavors the oxidation of styrene. However, the unshared p-electrons of halogens can also enhance the aromatic electron density by p-π delocalization (Sykes, 1986). It is likely that the effectiveness of the substituted halogen atoms participating in the electron delocalization plays a favorable role in the observed rates of bioactivation of the halogenated styrenes. Besides, the para-substituted halogens can alter the reactivity of the epoxide group of styrene oxide through the p-π electron delocalization that stabilizes the developing cation of the epoxides of the halogenated styrenes (Scheme 2). We anticipate that the capability of the electron delocalization plays a significant role in the observed reactivity (electrophilicity) of the oxides to cysteamine. The elevated capacity of the electron delocalization increases the probability for chemical reactions of the oxides with cellular biomolecules, which possibly triggers the process of their cytotoxicities.
Scheme 2.
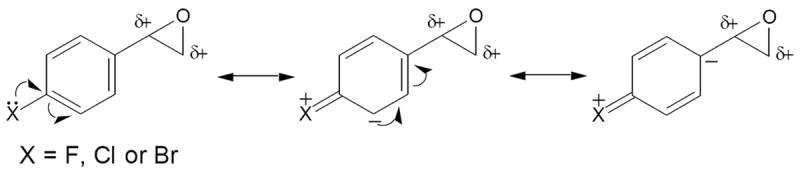
CYP2E1 has been reported to be one of cytochrome P450 enzymes responsible for the metabolic activation of styrene (Guengerich et al., 1991; Nakajima et al., 1994). Our earlier study demonstrated that CYP2E1 cells are more susceptible to styrene than the wild-type cells (Chung et al., 2006). Similar observation was obtained in CYP2E1 and wild-type cell lines after exposure to the halogenated styrenes (Figure 1). This implicates that the halogenated styrenes were metabolically activated by similar enzyme system.
The halogenated styrenes were used in the present study as model compounds to probe the correlation between metabolic activation of styrene and its cytotoxicity. Little is known about metabolisms and cytotoxicities of the halogenated styrenes. The present metabolism study demonstrated that the three halogenated styrenes were exclusively biotransformed to the corresponding oxides (6-8) by CYP2E1. The results showed the consistence with our original hypothesis that the halogenated styrenes undergo similar bioactivation pathway(s) as that of styrene.
In conclusion, the structure-toxicity relationship study showed that para-substituted halogenated styrene analogues induced cytotoxicity in an order of 4-bromostyrene > 4-chlorostyrene > 4-fluorostyrene and that styrene was as toxic as 4-fluorostyrene. CYP2E1 participated in the bioactivation and cytotoxicity of the halogenated styrenes in CYP2E1 abundant cells. The cytotoxicities induced by styrene and the halogenated styrene analogues were proportional to the efficiency of their bioactivation to oxides, electrophilicity of the oxides, potency of cellular GSH depletion, and degree of protein adduction. The results support our hypothesis that cytotoxicities induced by latent toxicants with similar metabolic pathways corellate with their biochemical and chemical properties. The novel parameter bioactivation-reactivity index we introduced provides a useful platform for the comparison of latent toxicants (metabolic activation required).
Highlights.
We investigated the structure-toxicity relationship of halogenated styrenes.
Cytotoxicity order: 4-bromostyrene > 4-chlorostyrene > 4-fluorostyrene ≈ styrene.
Bioactivation of the styrenes follows the same order of their toxicities.
GSH depletion and protein covalent binding are consistent with their toxicities.
The results indicate bioactivation of styrene is correlated with its cytotoxicity.
Acknowledgments
This work was supported by NIH Grant HL080226. We thank Mr. Andrew Lowe of Seattle Children’s Research Institute for his assistance in the preparation of the manuscript.
Footnotes
Conflict of interests
The authors declare that there are no conflicts of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bond JA. Review of the toxicology of styrene. Crit Rev Toxicol. 1989;19:227–249. doi: 10.3109/10408448909037472. [DOI] [PubMed] [Google Scholar]
- 2.Carlson GP. Comparison of mouse strains for susceptibility to styrene-induced hepatotoxicity and pneumotoxicity. J Toxicol Environ Health. 1997;51:177–187. doi: 10.1080/00984109708984020. [DOI] [PubMed] [Google Scholar]
- 3.Carlson GP. Effects of inducers and inhibitors on the microsomal metabolism of styrene to styrene oxide in mice. J Toxicol Environ Health. 1997;51:477–488. doi: 10.1080/00984109708984038. [DOI] [PubMed] [Google Scholar]
- 4.Carlson GP, Turner M, Mantick NA. Effects of styrene and styrene oxide on glutathione-related antioxidant enzymes. Toxicology. 2006;227:217–226. doi: 10.1016/j.tox.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Christakopoulos A, Bergmark E, Zorcec V, Norppa H, Maki-Paakkanen J, Osterman-Golkar S. Monitoring occupational exposure to styrene from hemoglobin adducts and metabolites in blood. Scand J Work Environ Health. 1993;19:255–263. doi: 10.5271/sjweh.1476. [DOI] [PubMed] [Google Scholar]
- 6.Chung J-K, Yuan W, Liu G, Zheng J. Investigation of styrene toxicity in CYP2E1 transgenic cells. Toxicology. 2006;226:99–106. doi: 10.1016/j.tox.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Cruzan G, Cushman JR, Andrews LS, Granville GC, Miller RR, Hardy CJ, Coombs DW, Mullins PA. Subchronic inhalation studies of styrene in CD rats and CD-1 mice. Fundam Appl Toxicol. 1997;35:152–165. doi: 10.1006/faat.1996.2273. [DOI] [PubMed] [Google Scholar]
- 8.Cruzan G, Bus J, Hotchkiss J, Harkema J, Banton M, Sarang S. CYP2F2-generated metabolites, not styrene oxide, are a key event mediating the mode of action of styrene-induced mouse lung tumors. Regul Toxicol Pharmacol. 2012;62:214–220. doi: 10.1016/j.yrtph.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Fleming-Jones ME, Smith RE. Volatile organic compounds in foods: A five-year study. J Agric Food Chem. 2003;51:8120–8217. doi: 10.1021/jf0303159. [DOI] [PubMed] [Google Scholar]
- 10.Fustinoni S, Colosio C, Colombi A, Lastrucci L, Yeowell-O’Connell K, Rappaport SM. Albumin and hemoglobin adducts as biomarkers of exposure to styrene in fiberglass-reinforced-plastics workers. Int Arch Occup Environ Health. 1998;71:35–41. doi: 10.1007/s004200050247. [DOI] [PubMed] [Google Scholar]
- 11.Gadberry MG, DeNicola DB, Carlson GP. Pneumotoxicity and hepatotoxicity of styrene and styrene oxide. J Toxicol Environ Health. 1996;48:273–294. doi: 10.1080/009841096161339. [DOI] [PubMed] [Google Scholar]
- 12.Gamer AO, Leibold E, Deckardt K, Kittel B, Kaufmann W, Tennekes HA, van Ravenzwaay B. The effects of styrene on lung cells in female mice and rats. Food Chem Toxicol. 2004;42:1655–1667. doi: 10.1016/j.fct.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Green T, Toghill A, Foster J. The role of cytochromes P-450 in styrene induced pulmonary toxicity and carcinogenicity. Toxicology. 2001;169:107–117. doi: 10.1016/s0300-483x(01)00488-7. [DOI] [PubMed] [Google Scholar]
- 14.Guengerich FP, Kim D-H, Iwasaki M. Role of human cytochrome P-450IIE1 in the oxidation of many low molecular weight cancer suspects. Chem Res Toxicol. 1991;4:168–179. doi: 10.1021/tx00020a008. [DOI] [PubMed] [Google Scholar]
- 15.Harvilchuck JA, Carlson GP. Comparison of styrene and its metabolites styrene oxide and 4-vinylphenol on cytotoxicity and glutathione depletion in Clara cells of mice and rats. Toxicol Lett. 2008;183:28–35. doi: 10.1016/j.tox.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 16.Harvilchuck JA, Pu X, Klaunig JE, Carlson GP. Indicators of oxidative stress and apoptosis in mouse whole lung and Clara cells following exposure to styrene and its metabolites. Toxicology. 2009;264:171–788. doi: 10.1016/j.tox.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 17.Hodgson AT, Wooley JD, Daisey JM. Emissions of volatile organic compounds from new carpets measured in a large-scale environmental chamber. Air Waste. 1993;43:316–324. doi: 10.1080/1073161x.1993.10467136. [DOI] [PubMed] [Google Scholar]
- 18.Huff J, Infante PF. Styrene exposure and risk of cancer. Mutagenesis. 2011;26:583–884. doi: 10.1093/mutage/ger033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.IARC. Some industrial chemicals. IARC Monogr Eval Carcinog Risks Hum. 1994;60:233–246. [PMC free article] [PubMed] [Google Scholar]
- 20.IARC. Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. IARC Monogr Eval Carcinog Risks Hum. 2002;82:437–522. [PMC free article] [PubMed] [Google Scholar]
- 21.Johanson G, Ernstgard L, Gullstrand E, Lof A, Osterman-Golkar S, Williams CC, Sumner SC. Styrene oxide in blood, hemoglobin adducts, and urinary metabolites in human volunteers exposed to (13)C(8)-styrene vapors. Toxicol Appl Pharmacol. 2000;168:36–49. doi: 10.1006/taap.2000.9007. [DOI] [PubMed] [Google Scholar]
- 22.Lanosa MJ, Willis DN, Jordt S, Morris JB. Role of metabolic activation and the TRPA1 receptor in the sensory irritation response to styrene and naphthalene. Toxicol Sci. 2010;115:589–595. doi: 10.1093/toxsci/kfq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leibman KC. Metabolism and toxicity of styrene. Environ Health Perspect. 1975;11:115–119. doi: 10.1289/ehp.7511115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maki-Paakkanen J, Walles S, Osterman-Golkar S, Norppa H. Single-strand breaks, chromosome aberrations, sister-chromatid exchanges, and micronuclei in blood lymphocytes of workers exposed to styrene during the production of reinforced plastics. Environ Mol Mutagen. 1991;17:27–31. doi: 10.1002/em.2850170105. [DOI] [PubMed] [Google Scholar]
- 25.Mörbt N, Mögel I, Kalkhof S, Feltens R, Röder-Stolinski C, Zheng J, Vogt C, Lehmann I, von Bergen M. Proteome changes in human bronchoalveolar cells following styrene exposure indicate involvement of oxidative stress in the molecular-response mechanism. Proteomics. 2009;9:4920–4933. doi: 10.1002/pmic.200800836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakajima T, Elovaara E, Gonzalez FJ, Gelboin HV, Raunio H, Pelkonen O, Vainio H, Aoyama T. Styrene metabolism by cDNA-expressed human hepatic and pulmonary cytochromes P450. Chem Res Toxicol. 1994;7:891–896. doi: 10.1021/tx00042a026. [DOI] [PubMed] [Google Scholar]
- 27.Osterman-Golkar S, Christakopoulos A, Zorcec V, Svensson K. Dosimetry of styrene 7,8-oxide in styrene- and styrene oxide-exposed mice and rats by quantification of haemoglobin adducts. Chem Biol Interact. 1995;95:79–87. doi: 10.1016/0009-2797(94)03348-x. [DOI] [PubMed] [Google Scholar]
- 28.Rappaport SM, Ting D, Jin Z, Yeowell-O’connell K, Waidyanatha S, McDonald T. Application of Raney nickel to measure adducts of styrene oxide with hemoglobin and albumin. Chem Res Toxicol. 1993;6:238–244. doi: 10.1021/tx00032a014. [DOI] [PubMed] [Google Scholar]
- 29.Scott D, Preston RJ. A critical review of the cytogenetic effects of styrene with an emphasis on human population monitoring: a synopsis. Crit Rev Toxicol. 1994;24(Suppl):S47–8. doi: 10.3109/10408449409020140. [DOI] [PubMed] [Google Scholar]
- 30.Shen S, Zheng F, Gao L, Zeng S, Zheng J. Detection of phenolic metabolites of styrene in mouse microsomal incubations. Drug Metab Disp. 2010;38:1934–1943. doi: 10.1124/dmd.110.033522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sykes P. Electrophilic and nucleophilic substitution in aromatic system. In: Sykes P, editor. A guidebook to mechanism in organic chemistry. Longman Singapore Publishers Pte Ltd; Singapore: 1986. pp. 130–177. [Google Scholar]
- 32.Tang W, Hemm I, Eisenbrand G. Estimation of human exposure to styrene and ethylbenzene. Toxicology. 2000;144:39–50. doi: 10.1016/s0300-483x(99)00188-2. [DOI] [PubMed] [Google Scholar]
- 33.Teixeira JP, Gaspar J, Coelho P, Costa C, Pinho-Silva S, Costa S, Da Silva S, Laffon B, Pásaro E, Rueff J, Farmer P. Cytogenetic and DNA damage on workers exposed to styrene. Mutagenesis. 2010;25:617–621. doi: 10.1093/mutage/geq049. [DOI] [PubMed] [Google Scholar]
- 34.Turner M, Mantick NA, Carlson GP. Comparison of the depletion of glutathione in mouse liver and lung following administration of styrene and its metabolites styrene oxide and 4-vinylphenol. Toxicology. 2005;206:383–388. doi: 10.1016/j.tox.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 35.Vodicka P, Koskinen M, Stetina R, Soucek P, Vodickova L, Matousu Z, Kuricova M, Hemminki K. The role of various biomarkers in the evaluation of styrene genotoxicity. Cancer Detect Prev. 2003;27:275–284. doi: 10.1016/s0361-090x(03)00096-5. [DOI] [PubMed] [Google Scholar]
- 36.Vodicka P, Soucek P, Tates AD, Dusinska M, Sarmanova J, Zamecnikova M, Vodickova L, Koskinen M, de Zwart FA, Natarajan AT, Hemminki K. Association between genetic polymorphisms and biomarkers in styrene-exposed workers. Mutat Res. 2001;482:89–103. doi: 10.1016/s0027-5107(01)00214-7. [DOI] [PubMed] [Google Scholar]
- 37.Vodicka P, Tvrdik T, Osterman-Golkar S, Vodickova L, Peterkova K, Soucek P, Sarmanova J, Farmer PB, Granath F, Lambert B, Hemminki K. An evaluation of styrene genotoxicity using several biomarkers in a 3-year follow-up study of hand-lamination workers. Mutat Res. 1999;445:205–224. doi: 10.1016/s1383-5718(99)00127-8. [DOI] [PubMed] [Google Scholar]
- Yuan W, Jin H, Chung J-K, Zheng J. Evidence for cellular protein covalent binding derived from styrene metabolite. Chem Biol Interact. 2010;186:323–330. doi: 10.1016/j.cbi.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]