

Background: SERCA and ATP7A/B are P-type ATPases entailing phosphoenzyme intermediate (E-P) formation.
Results: Biochemical characterization, however, reveals distinctive features.
Conclusion: A H+-gated Ca2+ pathway coupled to E1-P to E2-P transition is SERCA-specific. Headpiece interactions with an N-metal binding extension (NMBD) and related conformational constraints are ATP7A/B-specific.
Significance: SERCA and ATP7A/B mechanisms present significant differences even though they are both P-type.
Keywords: ATPases, Calcium ATPase, Calcium Transport, Copper Transport, Enzyme Mechanisms, Membrane Transport, Recombinant Protein Expression, H+ Gating, Charge Movements, Phosphoenzyme Intermediate
Abstract
Ca2+ (sarco-endoplasmic reticulum Ca2+ ATPase (SERCA)) and Cu+ (ATP7A/B) ATPases utilize ATP through formation of a phosphoenzyme intermediate (E-P) whereby phosphorylation potential affects affinity and orientation of bound cation. SERCA E-P formation is rate-limited by enzyme activation by Ca2+, demonstrated by the addition of ATP and Ca2+ to SERCA deprived of Ca2+ (E2) as compared with ATP to Ca2+-activated enzyme (E1·2Ca2+). Activation by Ca2+ is slower at low pH (2H+·E2 to E1·2Ca2+) and little sensitive to temperature-dependent activation energy. On the other hand, subsequent (forward or reverse) phosphoenzyme processing is sensitive to activation energy, which relieves conformational constraints limiting Ca2+ translocation. A “H+-gated pathway,” demonstrated by experiments on pH variations, charge transfer, and Glu-309 mutation allows luminal Ca2+ release by H+/Ca2+ exchange. As compared with SERCA, initial utilization of ATP by ATP7A/B is much slower and highly sensitive to temperature-dependent activation energy, suggesting conformational constraints of the headpiece domains. Contrary to SERCA, ATP7B phosphoenzyme cleavage shows much lower temperature dependence than EP formation. ATP-dependent charge transfer in ATP7A and -B is observed, with no variation of net charge upon pH changes and no evidence of Cu+/H+ exchange. As opposed to SERCA after Ca2+ chelation, ATP7A/B does not undergo reverse phosphorylation with Pi after copper chelation unless a large N-metal binding extension segment is deleted. This is attributed to the inactivating interaction of the copper-deprived N-metal binding extension with the headpiece domains. We conclude that in addition to common (P-type) phosphoenzyme intermediate formation, SERCA and ATP7A/B possess distinctive features of catalytic and transport mechanisms.
Introduction
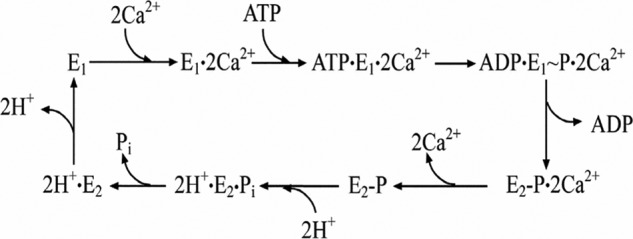
Early studies of P-type ATPases (1–3) suggested a common mechanistic feature of cation transport based on an E1 to E2 conformational transition of the ATPase protein whereby affinity and orientation of cation binding sites are changed to promote vectorial translocation. This appears clearly in the Ca2+ ATPase, where the free energy of ATP is utilized to destabilize the E1·2Ca2+ state through formation of a phosphorylated intermediate that undergoes sequential processing as proposed in Scheme 1.
SCHEME 1.
The Ca2+ ATPase (SERCA)3 is a mammalian membrane-bound protein sustaining Ca2+ transport and involved in cell Ca2+ signaling and homeostasis (4). It is made of a single polypeptide chain of 994 amino acid residues (5) distributed in 10 transmembrane segments (M1–M10) and a cytosolic headpiece including three distinct domains (A, N, and P) that are directly involved in catalytic activity (Fig. 1). The N domain contains residues (such as Phe-487) interacting with the adenosine moiety of ATP whereby the ATP substrate is cross-linked to the P domain. The P domain contains a residue (Asp-351) undergoing phosphorylation to yield a phosphoenzyme intermediate, a residue (Asp-703) coordinating Mg2+, and other features characteristic of P-type ATPases (6, 7). The A domain includes the signature sequence 181TGES that provides catalytic assistance for final hydrolytic cleavage of phosphoenzyme (8–10). Cooperative and sequential binding (11) of two Ca2+ involved in catalytic activation and transport occur on sites (I and II) located within the transmembrane region rather close to the cytoplasmic surface of the lipid bilayer (12).
FIGURE 1.

Two-dimensional folding models of the SERCA (upper panel) and ATP7B (lower panel) sequences. The diagrams show 10 (SERCA) or 8 (ATP7B) transmembrane segments including the calcium or copper binding sites (TMBS) involved in enzyme activation and transport. The extramembranous region of both enzymes comprises a nucleotide binding domain (N), the P domain, with several residues (in yellow) conserved in P-type ATPases, including the aspartate (Asp-351 or Asp-1027) that undergoes phosphorylation to form the catalytic phosphoenzyme intermediate (EP), and the A domain with the TGE conserved sequence involved in catalytic assistance of EP hydrolytic cleavage. The His-1069 residue whose mutation is frequently found in Wilson disease is shown on the N domain of ATP7B. Specific features of ATP7B are the N-metal binding domain (NMBD) extension with six putative copper binding sites and serine residues undergoing kinase-assisted phosphorylation (Ser-478, Ser-481, Ser-1211, Ser-1453).
Interesting comparative questions relate to the mammalian copper ATPases ATP7A/B (13–15), which are also P-type ATPases but sustain active transport of copper by utilization of ATP and are involved in copper homeostasis (16–20). It is shown in Fig. 1 that the ATP7B (as well as ATP7A) isoform comprises eight (rather than 10) transmembrane segments including a copper binding site (TMBS) for catalytic activation and transport and a headpiece including the N, P, and A domains with conserved catalytic motifs analogous to SERCA. A specific feature of mammalian copper ATPases is an amino-terminal extension (NMBD) that includes six copper binding sites in addition to the TMBS (19). An additional feature is the presence of serine residues (Ser-478, Ser-481, Ser-1211, Ser-1453 in ATP7B) undergoing kinase-assisted phosphorylation (21).
Several analogies listed above suggest functional similarity in calcium and copper ATPases. However, we report here that experimental characterization of partial reactions reveals that even though both ATPases include P-type phosphoenzyme formation, the two enzymes present significant diversities in energy transduction, sequence of conformational transitions, and H+ involvement.
EXPERIMENTAL PROCEDURES
Protein Production and Biochemical Characterization
Native calcium ATPase (SERCA) was obtained with the microsomal fraction of rabbit skeletal muscle (22), which is referred to as sarcoplasmic reticulum (SR) vesicles. WT and mutant recombinant calcium (SERCA1) and copper ATPases (ATP7A and ATP7B) were obtained from COS1 cells infected with adenovirus vectors containing CMV promoter-driven cDNA fused with a 3′ c-Myc tag as described previously (21). The content of specific expressed protein was evaluated in the microsomal fraction of the infected COS1 cells by SDS gel electrophoresis and Western blotting (21).
Steady state ATPase hydrolytic activity was determined after Pi production by colorimetry (21). The reaction mixture contained 50 mm MES (pH 6.0) or HEPES (pH 7.5), 80 mm KCl (or as specified in figure legends), 3 mm MgCl2, 0.2 mm EGTA, 0.2 mm CaCl2, 30 μg of SR protein/ml, and 1 μm A23187 Ca2+ ionophore. The reaction was started by the addition of 2.5 mm ATP at 30 °C temperature.
[32P]Phosphoenzyme formation was determined by incubating microsomes (50 μg of microsomal protein/ml) derived from rabbit skeletal muscle (native SERCA) with 50 μm [γ-32P]ATP at 10 or 30 °C in a reaction mixture containing 50 mm MES (pH 6.0) or HEPES (pH 7.5), 80 mm KCl, 3 mm MgCl2, and 20 μm CaCl2 or otherwise as stated in figure legends. Substrate addition and quenching at serial times with 5% trichloroacetic acid were done on continuously vortexed reaction mixtures. The quenched samples were filtered through 0.45-μm Millipore nitrocellulose filters that were then washed 3 times with cold 0.125 m perchloric acid and once with water and processed for determination of radioactivity by scintillation counting.
In most cases, however, microsomes derived from COS-1 cells expressing WT or mutant SERCA (rather than muscle microsomes containing native SERCA) were used. The microsomes containing recombinant SERCA1 (50 μg of microsomal protein/ml) were incubated with 50 μm [γ-32P]ATP at 10 or 30 °C in reaction mixtures analogous to those described above. Samples were quenched at serial times with 5% trichloroacetic acid. In this case, the quenched samples were pelleted by centrifugation at 5000 rpm for 5 min, washed with 0.125 n perchloric acid, and finally resuspended in pH 8.3 (half of the sample) or pH 6.3 loading buffer (remaining half of the sample) and separated by alkaline (23) or acid buffer (24) gel electrophoresis. The gels were dried and exposed to a phosphor screen followed by scanning on a Typhoon scanner (Amersham Biosciences) for stoichiometric determination of phosphoprotein relative to three [γ-32P]ATP standards placed on the gels.
For characterization of copper ATPase, microsomes derived from COS-1 cells expressing recombinant enzyme were preincubated for 30 min in a medium containing 5.0 mm MES triethanolamine (pH 6.0), 300 mm KCl, 10 mm DTT, 3 mm MgCl2, and 5 μm CuCl2. The low MES concentration facilitated pH changes if required by the experimental protocol.
Formation of [32P]phosphoenzyme was obtained by incubating ATP7B (50 μg of microsomal protein/ml) with 50 μm [γ-32P]ATP at 10 or 30 °C in a reaction mixture containing 50 mm MES triethanolamine (pH 6.0) or HEPES (pH 7.5), 300 mm KCl, 10 mm DTT, 3 mm MgCl2, and 5 μm CuCl2. The samples were quenched and processed for electrophoresis as explained above.
Decay of the phosphorylated enzyme intermediate in the forward direction of the catalytic cycle was determined by first reaching steady state levels of radioactive phosphoenzyme by incubation with [γ-32P]ATP for 15 s at which time 1 mm nonradioactive ATP was added, and the samples were acid-quenched at various times. The quenched samples were dissolved in detergent at acidic or alkaline pH and separated by electrophoresis at acidic or alkaline pH for determination of residual phosphoenzyme.
For reverse phosphorylation with Pi, microsomes derived from muscle or microsomes derived from COS1 cells expressing SERCA or mutant proteins were incubated with 50 mm MES (pH 6.0) or HEPES (pH 7.5), 20% Me2SO4, 10 mm MgCl2, and 2 mm EGTA. 100 mm KCl and 1 mm CaCl2 (in the absence of EGTA) were added when indicated. The reaction was started by the addition of 50 μm [32P]Pi, quenched at different times, and processed for determination of radioactive protein either on filters or electrophoresis as described for phosphorylation with ATP.
Charge Measurements
Charge movements were measured by adsorbing native SR vesicles containing SERCA or microsomes from COS-1 cells containing recombinant SERCA or Cu+ ATPase (ATP7A or ATP7B) on a solid supported membrane (SSM). The SSM consists of an alkanethiol monolayer covalently bound to a gold electrode via the sulfur atom and a phospholipid monolayer on top of it (25). After adsorption, the protein (calcium or copper ATPase) was activated by a concentration jump of a suitable substrate, i.e. ATP, in the presence of buffer solutions of various compositions. If at least one electrogenic step is involved in the relaxation process after activation, a current transient can be recorded along the external circuit (26, 27). Numerical integration of each transient is related to a net charge movement that depends upon the particular electrogenic event. In addition, kinetic information can be obtained by fitting a sum of exponentially decaying terms to the current versus time curves.
In experiments with native or recombinant SERCA, the solution contained 100 mm choline chloride (or 100 mm KCl), 25 mm MOPS, pH 7.0, 1 mm MgCl2, 0.25 mm EGTA, 0.25 mm CaCl2 (10 μm free Ca2+), and 0.2 mm DTT. Activation was obtained by the addition of ATP (100 μm) contained in a solution of the same composition. Free Ca2+ concentration was calculated with the computer program WinMAXC (28). Unless otherwise stated, 1 μm calcium ionophore A23187 was used to prevent formation of a Ca2+ concentration gradient across the SR vesicles (29). In ATP concentration-jump experiments with recombinant ATP7A or ATP7B, the non-activating solution contained 300 mm KCl (or 300 mm choline chloride), 50 mm MES triethanolamine, pH 6.0, 5.0 mm MgCl2, 0.1 mm CaCl2, 10 mm DTT, and 5 μm CuCl2; the activating solution contained, in addition, 100 μm ATP.
The concentration jump experiments were carried out by employing the SURFE2ROne device (Scientific Devices Heidelberg, Germany). The temperature was maintained at 22–23 °C for all the experiments.
To verify the reproducibility of the current transients generated within the same set of measurements on the same SSM, each single measurement of the set was repeated six times and then averaged to improve the signal to noise ratio. Standard deviations were usually found to be no greater than ±5%. Moreover, each set of measurements was usually reproduced using 3–5 different SSM electrodes. Therefore, the data points reported in Fig. 5 represent the mean of 3–5 independent normalized values. S.E. are given by individual error bars in the figure.
FIGURE 5.

Charge measurements on native SR Ca2+ ATPase (SERCA) and recombinant Cu+ ATPase (ATP7A and ATP7B). A, current transients were obtained after 100 μm ATP concentration jumps on SERCA in the presence of 10 μm free Ca2+ and 100 mm KCl at pH 7 (solid line) and 7.8 (dotted line). B, current transients were obtained after 100 μm ATP concentration jumps on SERCA in the presence of 10 μm free Ca2+ and 100 mm choline chloride (Chol.Cl) at pH 7 (solid line) and 7.8 (dotted line). C, shown are current transients obtained after 100 μm ATP concentration jumps on ATP7B in the presence of 5 μm CuCl2 and 300 mm KCl at pH 6 (solid line) and 7.8 (dotted line). D, shown is dependence of the normalized charge after 100 μm ATP concentration jumps on pH in the case of ATP7A (▵), ATP7B (▴), and SERCA (100 mm KCl, ●; and 100 mm choline chloride, ○). For each protein, the charges are normalized with respect to the value measured at pH 7. S.E. are given by error bars in the lower right panel.
RESULTS
Steady State Activity of Calcium Transport ATPase
The effects of pH, K+, and temperature on the steady state ATP utilization by native SERCA (SR vesicles) are shown in Fig. 2. In these experiments a Ca2+ ionophore (30) was added to the reaction mixture to prevent Ca2+ accumulation and back inhibition of ATPase activity, thereby obtaining constant velocity of ATP consumption. It is clear that the overall turnover is dependent on the reaction temperature, which is consistent with a 22–25 kcal mol−1 energy of activation previously reported (31). Under all conditions, the ATPase velocity is significantly increased by the presence of K+. Furthermore, the ATPase velocity is much higher when the pH is increased from 6.0 to 7.5, indicating a strong pH dependence of enzyme turnover. In contrast, it was previously found (in the absence of ionophore) that ATP-dependent Ca2+ load into SR vesicles is lower at alkaline than acid pH (29, 32).
FIGURE 2.

Steady state Ca2+ ATPase activity of native Ca2+ ATPase. ATP hydrolysis was measured at pH 6.0 or 7.5 in the presence of a Ca2+ ionophore (A23187) to prevent Ca2+ accumulation in SR vesicles and ATPase “back inhibition.” The reaction mixture contained 50 mm MES (pH 6.0) or HEPES (pH 7.5), 80 mm KCl (when indicated), 3 mm MgCl2, 20 μm CaCl2, 30 μg of SR protein/ml, and 1 μm A23187. A low rate of Ca2+-independent ATPase activity, measured in the presence of EGTA and no added Ca2+, was subtracted from the total. The reaction was started by the addition of 2.5 mm ATP at 10 or 30 °C temperature, and samples were collected at time intervals. ATPase velocity was calculated from linear slopes of Pi production.
Sequential Reactions of the SERCA Cycle
In early kinetic studies performed at 25 °C temperature and at neutral pH, we found that the addition of ATP to SERCA already activated with Ca2+ is followed by rapid phosphoenzyme formation, with a rate constant of ∼100 s−1, which was significantly slower if ATP and Ca2+ were added to enzyme deprived of Ca2+ (11). Here, as a base line to experiments on pH and temperature dependence of native and recombinant calcium and copper ATPases, we obtained measurements at pH 6.0 and 10 °C and found that even under these conditions phosphorylation after the addition of ATP and Ca2+ to enzyme deprived of Ca2+ is significantly slower than phosphorylation after the addition of ATP to enzyme preincubated with Ca2+ (Fig. 3A). This demonstrates that SERCA activation by Ca2+ requires a rate-limiting transition that is slower at acid than at alkaline pH (Fig. 3B). Therefore, activation by Ca2+ is easier if the enzyme resides in the state E1 rather than in the state 2H+·E2 (see Scheme 1 in the Introduction). Furthermore, we observed a requirement for ionic strength and a specific activation by K+ when we compared K+ to NH4+ (not shown).
FIGURE 3.

Time course of phosphoenzyme intermediate formation (A, B, C, and D) after the addition of ATP to Ca2+ ATPase and subsequent phosphoenzyme decay (E and F). Phosphorylation was obtained by the addition of 50 μm [γ-32P]ATP to ATPase preincubated with Ca2+ (● in A) or the addition of 50 μm [γ-32P]ATP and Ca2+ to ATPase deprived of Ca2+ (○ in A and all variables in B–D) at 10 or 30 °C as indicated. The reaction mixture contained 50 mm HEPES (pH 7.5) or 50 mm MES (pH 6.0), 80 mm KCl, and 3 mm MgCl2. The CaCl2 concentration was 20 μm in A. Alternatively, the reaction mixture contained 1 mm (at pH 7.5) or 5 mm EGTA (at pH 6.0) before the addition of ATP and then either 1 mm (at pH 7.5) or 5 mm (at pH 6.0) CaCl2 was added with the [γ-32P]ATP. The reaction was quenched at serial times with 5% trichloroacetic acid, and the quenched samples were filtered through 0.45-μm Millipore nitrocellulose filters that were then washed 3 times with cold 0.125 m perchloric acid and once with water and processed for determination of radioactivity by scintillation counting. Decay of phosphoenzyme intermediate (E and F) was obtained by first incubating ATPase with [γ-32P]ATP for 10 s at pH 6 and 10 °C temperature followed by a 4-fold dilution with an identical medium but containing 1 mm nonradioactive ATP. Temperature and pH are as indicated in the figure. Quenching and collection of serial samples are as described above. The experimental points are averages of values obtained in three to four different experiments.
The initial velocity of SERCA activation by Ca2+ (required for ATP utilization) is approximately the same at low and high temperature (Fig. 3, C and D). On the other hand, a higher steady state level of phosphoenzyme is reached at lower temperature, suggesting significant temperature dependence of subsequent steps leading to hydrolytic cleavage of the phosphoenzyme. In fact, it is shown in Fig. 3, E and F, that decay of radioactive phosphoenzyme intermediate after a chase with excess nonradioactive ATP is clearly temperature-dependent and is favored by alkaline pH. Therefore, completion of enzyme turnover involves transitions of conformational states that are sensitive to temperature-dependent energy of activation, as also indicated by measurements of steady state ATPase velocity (Fig. 2).
ATP Utilization by Copper ATPase (ATP7B)
In comparative studies we examined the mammalian copper ATPase. Functional characterization of copper ATPases requires heterologous expression, as native sources are not sufficiently abundant. For this reason we used microsomes derived from COS-1 cells infected with adenovirus vector and expressing recombinant ATP7A/B (or SERCA for comparison).
When phosphoprotein formation by recombinant calcium and copper ATPases are compared, we find that ATP utilization by SERCA yields only autophosphorylation of a conserved aspartate serving as phosphoenzyme intermediate. On the other hand, ATP7B yields autophosphorylation of the conserved aspartate as well as kinase-assisted phosphorylation of serine residues (21). Aspartate and serine phosphorylation can be distinguished by running samples at acid and alkaline electrophoresis. Although alkali-resistant phosphoprotein is not obtained with SERCA (Fig. 4A), phosphorylation of ATP7B includes alkali resistant and alkali labile fractions (Fig. 4B). The difference between total and alkali resistant phosphoprotein (i.e. alkali labile) corresponds to aspartyl phosphate intermediate. Alkali labile phosphorylation of ATP7B does not occur after mutations at the transmembrane copper binding site (33) and is, therefore, linked specifically to enzyme activation (E2 to E1 transition) by occupancy of the cation transport site. It is noteworthy that ATP7A undergoes aspartate phosphorylation with kinetics analogous to ATP7B, but a lower level of protein kinase assisted phosphorylation (34).
FIGURE 4.

Phosphoprotein formation after the addition of ATP to recombinant SERCA (A) or ATP7B (B–D) and subsequent decay of ATP7B phosphoenzyme (E and F). Microsomes obtained from COS-1 cells sustaining heterologous expression were incubated with 50 μm [γ-32P]ATP at 10 or 30 °C at various pH levels as explained under “Experimental Procedures.” Electrophoresis in acid buffer or alkaline buffer was then performed to distinguish total [32P]phosphoprotein (■) from alkali-resistant [32P]phosphoprotein (serine and/or threonine, □). The difference is considered alkali-labile [32P]phosphoprotein (aspartate, ●) and attributed to formation of phosphorylated enzyme intermediate. acid and alkaline refer to the media used for resuspension of samples and electrophoresis. Electrophoretic gel images correspond to sequential samples obtained within the time scale shown in the horizontal axis. Note that no alkaline-resistant phosphoprotein was obtained with SERCA, whereas both alkaline-resistant and alkaline-labile phosphorylation were obtained with ATP7B. Only the difference (phosphorylated enzyme intermediate) is shown in the lower panels (C–F). Decay of phosphoenzyme was measured as explained under “Experimental Procedures” and in the legend to Fig. 3. The experimental points are averages of values obtained in three to four different experiments. The electrophoretic images in A and B are examples of data repeated in all experiments.
We now find that even though a copper binding requirement for the E2 to the E1 transition is demonstrated by copper site mutations (33), experiments analogous to those with SERCA, in which Ca2+ is first chelated with EGTA and then Ca2+ is added to produce activation, cannot be easily done with ATP7A/B. In fact copper chelation with BCS produces inactivation due to removal of copper from the NMBD in addition to the TMBS, and very low phosphoenzyme formation is then obtained after the addition of copper and ATP.
When ATP is added to ATP7B already activated by copper, formation of phosphoenzyme intermediate is much slower than observed with SERCA and strongly temperature-dependent (Fig. 4C). In fact, not only the rate of phosphorylation but (contrary to SERCA) even the steady state level of phosphoenzyme is lower at low temperature. On the other hand, decay of the radioactive phosphoenzyme obtained after a chase with excess nonradioactive ATP shows rather low temperature dependence (Fig. 4E). This indicates that higher activation energy is required for initial formation of phosphoenzyme intermediate than for its subsequent processing. EP formation and its subsequent decay are faster at alkaline than at acid pH (Fig. 4, D and F).
Charge Transfer upon the Addition of ATP to Calcium and Copper ATPases
As previously reported (29, 35, 36), the addition of ATP to microsomal vesicles (containing SERCA or ATP7B) adsorbed on a SSM is followed by an electrogenic event recorded as a current transient. The current transient is due to the flow of electrons along the external circuit toward the electrode surface and is required to compensate for the potential difference across the vesicular membrane produced by displacement of positive charge (attributed to vectorial translocation of bound Ca2+ or Cu+) in the direction of the SSM electrode after phosphoenzyme intermediate formation by utilization of ATP (29, 36). The electrical current recorded by the SSM method is a measure of the rate of change of the transmembrane potential and is not sensitive to stationary currents. Therefore, only electrogenic steps within the first catalytic cycle are measured, whereas steady state events after the first cycle are not detected.
It is shown in Fig. 5A that in experimenting with SERCA, the total net charge decreases as the pH is raised if the main monovalent cation in the reaction mixture is provided by KCl. On the other hand, if the main cation in the reaction mixture is provided by choline chloride, the total net charge displaced increases as the pH is raised (Fig. 5B). This suggests that when lack of H+ limits H+/Ca2+ exchange (i.e. alkaline pH), conformational transitions due to alkaline pH render possible K+/Ca2+ exchange and neutralization of charge, whereas choline+/Ca2+ exchange does not occur.
Net charge displacement is also observed with microsomal vesicles containing recombinant ATP7A or ATP7B (Fig. 5, C and D), which is attributed to movement of bound Cu+ in the direction of the SSM electrode after phosphoenzyme intermediate formation by utilization of ATP (36). Contrary to SERCA, however, net charge transfer by ATP7A or -B is not modified by raising the pH even though the experiments are performed in the presence of KCl (Fig. 5C). This suggests that H+ exchange upon release of bound cation is specific for SERCA. This is even better demonstrated (Fig. 5D) when the pH is stepwise raised in experiments with SERCA in the presence of KCl (●) or choline chloride (○) or ATP7A (▵) or ATP7B (▴) in the presence of KCl.
Phosphoenzyme Formation by Utilization of Pi
A very interesting feature of the Ca2+ ATPase is its ability to form phosphorylated intermediate by utilization of Pi in the absence of Ca2+. Under the conditions (i.e. pH 6, no Ca2+ or K+) originally described (37), this reaction corresponds to E2-P formation by utilization of Pi in the reverse direction of phosphoenzyme cleavage (see Scheme 1). It is noteworthy that, contrary to enzyme activation by Ca2+ and phosphorylation by utilization of ATP (Fig. 3), the rates of enzyme phosphorylation with Pi in the absence of Ca2+ are significantly temperature-dependent (Fig. 6A).
FIGURE 6.

Time course of EP formation through utilization of [32P]Pi by WT SERCA (A) and E309Q mutant (B) and decay (C and D) of [32P]phosphoenzyme after a chase with nonradioactive Pi. Microsomes derived from COS1 cells expressing SERCA or E309Q mutant were incubated with 50 mm MES, pH 6.0, 20% Me2SO4, 10 mm MgCl2, and 2 mm EGTA. The reaction was started by the addition of 50 μm [32P]Pi, quenched at different times, and processed for determination of radioactive protein by electrophoretic analysis and detection of radioactivity (see “Experimental Procedures”). For determination of decay, the protein was incubated with 50 μm [32P]Pi as described above at 10 °C for 2 min, and then a 20-fold dilution was obtained with a medium containing 50 mm MES, pH 6.0, 10 mm MgCl2, 2 mm EGTA, and 1 mm nonradioactive Pi at 10 or 30 °C. Serial samples were taken at sequential times for acid quenching and determination of residual radioactive protein by electrophoretic analysis and detection of radioactivity. The experimental points are averages of values obtained in three to four different experiments.
Although utilization of ATP ultimately yields phosphoenzyme steady state levels determined by turnover of the catalytic cycle, experiments with Pi yield equilibrium levels of phosphoenzyme due to the absence of ATP substrate whose potential energy is required to sustain steady state activity. In the Pi reaction, the potential energy is provided by the comparative stability of the protein conformation in the non-phosphorylated versus phosphorylated state. Because in the presence of saturating Pi, equilibrium levels of phosphoenzyme are reached within 60 s (Fig. 6), it is possible to determine the dependence of phosphoenzyme equilibrium levels on experimental variables.
For the experiments shown in Figs. 6 and 7, we used both native and recombinant WT SERCA and obtained analogous results. The phosphoenzyme levels shown in the figures were derived from experiments conducted with microsomes obtained from COS1 cells expressing recombinant SERCA (see also Fig. 4A) to have a stoichiometric comparison with mutant SERCA (see below). We found that when WT SERCA is used under optimal conditions (i.e. pH 6, no Ca2+ or K+), maximal phosphoenzyme levels reach nearly half the stoichiometry of available enzyme, indicating an apparent equilibrium constant of ∼1. However, the phosphoenzyme levels are lower if the pH is raised from 6.0 to 7.5 (Fig. 7). Considering that H+ dissociation favors the E1 over the 2H+·E2 postulated in the reaction scheme, it is apparent that E1 has a lower equilibrium constant for the phosphorylation reaction with Pi. Furthermore, in the presence of Ca2+ (either at pH 6.0 or 7.5), no significant levels of phosphoenzyme were obtained (Fig. 7). Therefore, the E1·2Ca2+ state is unable to utilize Pi for formation of phosphoenzyme. Formation of phosphoenzyme by utilization of Pi is also reduced when KCl (and to a lesser extent NH4Cl) is present, likely due to enhancement of E2-P cleavage by K+ coordinated to 711–715 carbonyls and the Glu-732 side chain in the P domain (38).
FIGURE 7.

Equilibrium levels of phosphoenzyme obtained through utilization of [32P]Pi by recombinant SERCA and E309Q mutant at acid or alkaline pH. Effects of K+ and Ca2+ are shown. Microsomes derived from COS1 cells expressing SERCA or mutant (E309Q) protein were diluted (50 μg/ml) in medium containing 50 mm MES (pH 6.0) or HEPES (pH 7.5), 20% Me2SO4, and 10 mm MgCl2. 2 mm EGTA, 100 mm KCl, and 1 mm CaCl2 (in the absence of EGTA) were added as indicated in the figure. The reaction was started by the addition of 50 μm [32P]P, at 30 °C temperature and acid-quenched after 2 min. The quenched samples were processed for determination of radioactive protein by electrophoretic analysis and detection of radioactivity.
Functional Characterization of the E309Q SERCA Mutant
Whether the pH dependence of the Pi reaction is regulated specifically by ionization of acidic residues involved in Ca2+ binding at the TMBS can be further investigated by characterization of the E309Q SERCA mutant (39). In fact, Glu-309 (Fig. 1) contributes two oxygen atoms to binding of the second Ca2+ in the SERCA transmembrane region (12). It was previously reported that a single E309Q mutation interferes with binding of the second Ca2+, rendering the enzyme unable to form phosphorylated intermediate by utilization of ATP in the presence of Ca2+ even though still able to bind the first Ca2+ (40).
Taking advantage of high yield expression by the use of adenovirus vectors, we are now able to compare the phosphorylation levels of various recombinant proteins. We find that phosphorylation of E309Q mutant protein with Pi occurs with lower rates but similar temperature dependence to that observed with WT SERCA (Fig. 6, A and B). The phosphoenzyme formed at pH 6.0 in the absence of Ca2+ or K+ reached approximately the same levels when the E309Q was used, as compared with WT Ca2+ ATPase (Figs. 6 and 7). In addition, no inhibition by Ca2+ is observed (39), and a K+ effect opposite that observed with WT protein (Fig. 7) suggests that the effect of bound K+ in the P domain (38) results in different effects in the E309Q mutant, depending on the pH.
These findings indicate that, at acid pH, the E309Q mutant resides in a state that is still reactive to Pi with a favorable equilibrium constant but, most importantly, is not sensitive to Ca2+. This mutant conformational state is likely produced by a hydrogen bonding pattern that is established at pH 6.0 by the remaining acidic residues at the calcium sites when the Glu-309 acidic chain is absent. When the pH is raised to 7.5, strong inhibition by Ca2+ and minor inhibition by K+ are observed with the E309Q, in analogy to the WT protein (Fig. 7). However, phosphorylation with Pi reaches a high level (contrary to the effect of high pH on the WT protein), likely related to the absence of Glu-309 acidic chain ionization. Furthermore, a most important difference is the inability of the E309Q mutant to be activated by Ca2+ to allow utilization of ATP. This is evidently due to interference by the single E309Q mutation with binding of the second Ca2+ that is required for enzyme activation (40). On the other hand, binding of the first calcium (at alkaline pH) is sufficient to interfere with phosphorylation of the E309Q mutant by Pi.
Experiments on Copper ATPase Phosphorylation with Pi
We were not able to obtain significant levels of reverse phosphorylation of ATP7A or -B with Pi when, in analogy to calcium chelation with EGTA, we added BCS to chelate copper in our experiments with ATP7A or -B (Fig. 8). A most interesting result was obtained when the ATP7B Δ5 mutant (in which a large NMBD segment, including the first five copper binding sites, had been deleted) was used. The Δ5 construct yields low expression levels in COS1 cells unless proteasome-mediated degradation is inhibited pharmacologically. However, the Δ5 protein obtained without pharmacologic protection is still able to form EP by utilization of ATP and to sustain its hydrolytic cleavage, similarly to WT ATP7B or -A (Fig. 8) (33). Furthermore, EP formation by the Δ5 protein shows less temperature dependence than EP formation by ATP7B.
FIGURE 8.

Functional behavior of Δ5 ATP7B (large NMBD segment deleted). A and B, formation of [γ-32P]phosphoenzyme after [γ-32P]ATP utilization by Δ5 ATP7B and decay of [γ-32P]phosphoenzyme after a chase with excess nonradioactive ATP. The experiments were performed at pH 6.0 and 10 °C temperature as described in the legend to Fig. 4. The low phosphoenzyme levels are due to low concentration of expressed protein in the microsomes of the infected COS-1 cells. C, electrophoretic demonstration of phosphoenzyme formation through utilization of [γ-32P]ATP or [32P]Pi by SERCA, ATP7A, ATP7B, and Δ5 ATP7B is shown. Microsomes derived from COS1 cells expressing SERCA were incubated with 50 μm [γ-32P]ATP for 5 s at 30 °C in a reaction mixture containing 50 mm MES (pH 6.0), 3 mm MgCl2, 100 mm KCl, and 10 μm CaCl2 or 2 mm EGTA as indicated. Microsomes derived from COS1 cells expressing ATP7A, ATP7B, or Δ5 ATP7B mutant protein were incubated with 50 μm [γ-32P]ATP for 5 s at 30 °C in a reaction mixture containing 50 mm MES, pH 6.0, 300 mm KCl, 10 mm DTT, 3 mm MgCl2, and 5 μm CuCl2 or 2 mm BCS as indicated. Alternatively, microsomes derived from COS1 cells expressing SERCA were incubated with 50 μm [32P]Pi for 2 min at 30 °C in a reaction mixture containing 50 mm MES, pH 6.0, 10 mm MgCl2, 20% Me2SO4, and 1 mm CaCl2 or 2 mm EGTA as indicated. Microsomes derived from COS1 cells expressing ATP7A, ATP7B, or Δ5 ATP7B mutant protein were incubated with 50 μm [γ-32P]Pi for 2 min at 30 °C in a reaction mixture containing 50 mm MES (pH 6.0), 10 mm DTT, 10 mm MgCl2, 20% Me2SO4, and 5 μm CuCl2 or 2 mm BCS as indicated. The reactions were acid-quenched, and the samples were processed for determination of radioactive protein by electrophoretic analysis and detection of radioactivity. It is noteworthy that no kinase-mediated Ser/Thr phosphorylation was noted when Pi rather than ATP was used as a substrate.
Contrary to WT ATP7A and -B, the Δ5 protein undergoes reverse phosphorylation by Pi both in the presence and in the absence of copper (Fig. 8). This demonstrates that the NMBD has a key role in determining orientation and flexibility of the ATP7A/B headpiece domains, with a strong influence on the P domain specific reactivity to ATP or Pi. The lack of copper sensitivity for the Pi reaction is evidently due to removal of the five NMBD copper sites, whereas the copper dependence of the ATP reaction is due to the remaining TMBS copper binding. It is also interesting that ATP utilization by the Δ5 protein yields negligible alkali stable phosphorylation (Fig. 8A). This may be partly due to deletion of some of the serine residues undergoing phosphorylation (Fig. 1) but also to interference by the Δ5 deletion with docking of protein kinase C.
DISCUSSION
The experiments reported above offer a comparative characterization of the SERCA and ATP7A/B mechanisms, which we relate here to known structural features (available in greater detail for SERCA) and may also be helpful to explain further structural information on WT and mutant proteins as it will be obtained.
SERCA crystal structures show that the headpiece domains are widely separated in the transition from E2 to E1·2Ca2+ and approximate again upon nucleotide binding and formation of E1-P (12, 41, 42). Bending and rotation of the headpieces occur in conjunction with related displacement of membrane-bound segments. Our experiments demonstrate that the rate of SERCA activation by Ca2+ is rate-limiting for phosphorylation by ATP. The activation rate is slower at acid than at alkaline pH but is affected very little by raising the temperature (Fig. 3). This indicates that, in the native membrane environment, the rate of 2H+·E2 transition to E1 has rather low dependence on activation energy.
On the other hand, the high temperature dependence of phosphoenzyme decay (Fig. 3) and reverse phosphorylation by Pi (Fig. 6) indicate that conformational constraints limiting related transitions are sensitive to temperature-dependent activation energy in addition to being driven by phosphorylation-related potential energy as E1-P converts to E2-P. Diverse conformations of intermediate states (or analogs thereof) have been revealed by crystallography, indicating structural features involved in isomerization of E1-P to E2-P, cleavage of Pi, and return of E2 to E1 (8, 9). From the functional point of view, sequential transitions of these intermediate conformations yield vectorial translocation and release of bound Ca2+ in exchange for luminal H+ (29, 43–45).
Relevance of acidic residues was first indicated by the pH dependence of Ca2+ binding to SERCA in the absence of ATP, with affinity increasing from pH 6 to 8 (46, 47). Mutational analysis (39, 40, 48) and high resolution crystallography (12) then demonstrated that, within the membrane-bound region, Ca2+ binding at site I (Fig. 9) involves the side chain oxygen atoms of Asn-768, Glu-771 (M5), Thr-799, Asp-800 (M6), and Glu-908 (M8), and two water molecules. Ca2+ binding at site II (Fig. 9) involves three main chain carbonyls derived from partly unwound M4 helix, Asn-796 and Asp-800 side chain oxygen atoms (M6), and two capping oxygen atoms from Glu-309 (M4). A stable conformation (E1) is thereby obtained due to high affinity Ca2+ binding.
FIGURE 9.

Diagram of the SERCA Ca2+ binding sites in the E1·2Ca2+ and in the E2 state stabilized by thapsigargin (TG) and dibutyldihydroxybenzene (BHQ) (44) based on crystallography (12, 42) and continuum of electrostatic calculations. The larger cyan spheres in the upper diagram represent bound Ca2+ (I and II), and the small red spheres represent water. Small red circles represent bound protons, dotted pink lines represent hydrogen bonds, and green dotted lines represent Ca2+ coordination. The arrows indicate possible movements upon state transition. Data are derived from Obara et al. (44).
In the absence of Ca2+, a different conformation (E2) is acquired by SERCA. For structural studies by crystallization, the E2 conformation has been stabilized by binding of inhibitors such as thapsigargin (42) and dibutyldihydroxybenzene (44). However, it is important to consider that in the native membrane and in the absence of inhibitors, orientation and protonation of acidic chains as well as related hydrogen bonding patterns provide conformational stability for the Ca2+-free state (Fig. 9 as demonstrated in Obara et al. (44). Molecular dynamics calculations yield pK values allowing protonation (44, 49, 50) of three or four acidic residues (Glu-309, Glu-771, Glu-908, and Asp-800) and permitting hydrogen bonding that would be optimal at pH 6. On the other hand, in the absence of Ca2+, alkaline pH interferes with hydrogen bonding at the calcium site and consequently yields conformational destabilization (44, 50). Experiments with fluorescent styryl dyes and analysis of pH jump-induced relaxation process have shown a conformational transition of the empty E2-P as the pH is raised, attributed to a reversible inactivated state (51).
The experiments reported here indicate that the acidic residues in the transmembrane calcium binding domain region sustain an important role in conformational transitions pertinent to enzyme activation and transduction mechanism. In fact, the faster rate of SERCA activation observed when Ca2+ and ATP are added to SERCA previously exposed to alkaline pH (Fig. 3) demonstrates that dissociation of H+ from acidic residues accelerates the transition from E2 to E1. Furthermore, after ATP utilization at acid and to some extent at neutral pH, protonation of acidic residues occurs as a consequence of Ca2+/H+ exchange upon Ca2+ release from E2-P (43, 45), as also shown by charge movement observed in single cycle experiments (Fig. 5). At alkaline pH, Ca2+/H+ exchange and protonation of acidic residues occurs less or not at all, whereas Ca2+ may exchange with K+ as long as the luminal Ca2+ concentration is low (Fig. 5; see also Musgaard et al. (50) or may remain bound to the transport sites when the luminal Ca2+ concentration rises. Alteration of Ca2+/H+ exchange results in low transport efficiency.
It is noteworthy that although pH and concentrations of various ligands may be optimized to favor crystallization or biochemical events under experimental conditions, pH and concentrations of ligands (except Ca2+) do not change under physiological conditions. Protonation and ionization are then determined by changes in pK as acidic residues of the pathway face the lumenal or the cytosolic surface of the membrane (52). Therefore, Ca2+/H+ exchange and protonation of acidic residues sustain an important role in conformational transitions and selective ion movements within the transport cycle. In analogy to the calcium-occluded state, a proton-occluded state has been proposed (53) based on structural studies of E2-AlF4− crystals. We conclude that the transmembrane Ca2+ binding region, in addition to its linkage to phosphorylation of the cytosolic head for energy transduction, behaves as a H+-gated Ca2+ pathway for efficient translocation of Ca2+. A requirement for specific conformational transitions of this pathway was demonstrated by interference with the neighboring Cys-876–Cys-888 disulfide bond, which produces inhibition of Ca2+ transport, whereas full ATPase activity is retained (54).
Functional characterization of the E309Q mutant reveals interesting details on the conformational effects of protonated acidic residues at the calcium sites. We see that the mutant protein retains the ability to form phosphoenzyme by utilization of Pi at pH 6.0 (Figs. 6 and 7), as expected of the E2 state. Yet, using the proteolytic digestion analysis originally introduced by (55), we previously demonstrated that the E309Q mutation favors an A domain position similar to that of E1 (32) rather than E2, even in the absence of Ca2+. This does not mean that the entire conformation is E1-like, as shown by its reactivity to Pi (Fig. 7). It is apparent that this peculiar conformation of the mutant is stabilized by a hydrogen bonding pattern established by the remaining residues (Glu-771, Asp-800, and Glu-908) at acid pH. Therefore, in WT SERCA, the E2 state must be maintained specifically by the protonated Glu-309 residue in a rather strained conformation and, if the Glu-309 H+ is dissociated by raising the pH, the enzyme shifts to the E1 state. If the residual hydrogen bonding pattern of the E309Q protein is interfered with by raising the pH to 7.5, the mutant acquires Ca2+ sensitivity analogous to that of WT protein, with regard to inhibition of reverse phosphorylation with Pi (Fig. 7). However, even at alkaline pH, ATP cannot be utilized by the E309Q mutant upon the addition of Ca2+ as the second Ca2+ is not bound by the mutant at any pH. It is apparent that, in the WT protein, the acidic function of Glu-309 has conformational implications in the absence as well as in the presence of Ca2+. It is certainly involved in the E2 to E1 transition, binding the second Ca2+ required for catalytic activation, and occlusion of the bound Ca2+ upon formation of phosphorylated intermediate (56).
In comparative experiments we found that, contrary to SERCA, the rate of phosphoenzyme formation by ATP7B is highly temperature-dependent (Fig. 4C), whereas the rate of phosphoenzyme decay has a low temperature dependence (Fig. 4E). For this reason, the steady state levels of phosphoenzyme are lower at low temperature for ATP7B, whereas they are higher for SERCA. This suggests that overcoming conformational constraints, related to headpiece interactions with the NMBD (which is not present in SERCA), requires significant energy of activation to reach the phosphorylated state. These constraints pose a significant limit to initiation of the cycle as compared with SERCA, as also demonstrated by the slower kinetics of ATP-dependent charge transfer (Fig. 5).
Another interesting phenomenon is the lack of reverse ATP7B phosphorylation with Pi, suggesting that copper chelation with BCS and dissociation from the NMBD sites favors interactions of the NMBD chain with the enzyme headpiece that interfere with the phosphorylation reaction. In fact, deletion of a large segment of the NMBD allows the deleted protein (Δ5) to undergo phosphorylation by Pi (Fig. 8). This is in agreement with previous studies with Thermotoga maritima (CopA) copper ATPase (57), where the NMBD is much shorter (a single metal binding site) than that of ATP7B (six metal binding sites), and the enzyme yields high phosphorylation levels with Pi. Furthermore, the ability of the Δ5 protein to utilize ATP (Fig. 8) suggests that the deleted NMBD segment (five of six copper sites) is not necessary for catalytic activity. Its function may be related to favoring ATP7A/B interaction with other proteins involved in cellular copper homeostasis.
Finally, contrary to SERCA, net charge transfer by ATP7A or -B is not altered by raising the pH and is not reduced by K+ (Fig. 5). This indicates that cation/H+ exchange at the transport site does not play a role in copper release. In fact, structural studies of Legionella pneumophila (CopA) copper ATPase (58) do not reveal obvious involvement of acidic chains in the release phase of copper transport. Other specific mechanisms may be involved in promoting copper release from intramembrane sites of the copper ATPase, such as revealed by experiments on the Met-672–Pro-707 lumenal loop (59).
In conclusion, we find that comparative experiments with mammalian copper ATPases demonstrate distinctive mechanistic features whose knowledge complements the limited information as yet available on high resolution structure for this enzyme. Conformational constraints limiting the rate of initial phosphoenzyme intermediate formation by utilization of ATP and sensitivity to temperature-dependent activation energy are evidently related to interactions of the headpiece domains with the NMBD extension, which is not present in SERCA. These interactions are also reflected by the reverse phosphorylation reaction with Pi. On the other hand, the low temperature dependence of phosphoenzyme turnover and the lack of H+ exchange suggest that greater conformational changes are sustained in concomitance with phosphorylation by ATP and comparatively less for copper release from the phosphoenzyme.
Acknowledgment
G. I. is deeply indebted to Professor Chikashi Toyoshima for many suggestions and comments.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 HL69830. This work was also supported by Ente Cassa di Risparmio di Firenze (2009.0749) and the Italian Ministry of Education, University and Research (PRIN Project 20083YM37E).
- SERCA
- sarco-endoplasmic reticulum Ca2+ ATPase
- SR
- sarcoplasmic reticulum
- ATP7A/B
- Cu+ ATPases
- SSM
- solid supported membrane
- BCS
- bathocuproine disulfonate
- NMBD
- N-metal binding domain
- TMBS
- transmembrane binding site.
REFERENCES
- 1. Albers R. W. (1967) Biochemical aspects of active transport. Annu. Rev. Biochem. 36, 727–756 [DOI] [PubMed] [Google Scholar]
- 2. de Meis L., Vianna A. L. (1979) Energy interconversion by the Ca2+-dependent ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 48, 275–292 [DOI] [PubMed] [Google Scholar]
- 3. Post R. L., Hegyvary C., Kume S. (1972) Activation by adenosine triphosphate in the phosphorylation kinetics of sodium and potassium ion transport adenosine triphosphatase. J. Biol. Chem. 247, 6530–6540 [PubMed] [Google Scholar]
- 4. Brini M., Carafoli E. (2009) Calcium pumps in health and disease. Physiol. Rev. 89, 1341–1378 [DOI] [PubMed] [Google Scholar]
- 5. MacLennan D. H., Brandl C. J., Korczak B., Green N. M. (1985) Amino-acid sequence of a Ca2+ + Mg2+-dependent ATPase from rabbit muscle sarcoplasmic reticulum, deduced from its complementary DNA sequence. Nature 316, 696–700 [DOI] [PubMed] [Google Scholar]
- 6. Lutsenko S., Kaplan J. H. (1995) Organization of P-type ATPases. Significance of structural diversity. Biochemistry 34, 15607–15613 [DOI] [PubMed] [Google Scholar]
- 7. Pedersen P. L., Carafoli E. (1987) Ion motive ATPases. II. Energy coupling and work output. Trends Biochem. Sci. 12, 186–189 [Google Scholar]
- 8. Toyoshima C., Inesi G. (2004) Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 73, 269–292 [DOI] [PubMed] [Google Scholar]
- 9. Møller J. V., Olesen C., Winther A. M., Nissen P. (2010) The sarcoplasmic Ca2+-ATPase. Design of a perfect chemi-osmotic pump. Q. Rev. Biophys. 43, 501–566 [DOI] [PubMed] [Google Scholar]
- 10. Andersen J. P., Vilsen B. (1995) Structure-function relationships of cation translocation by Ca2+- and Na+, K+-ATPases studied by site-directed mutagenesis. FEBS Lett. 359, 101–106 [DOI] [PubMed] [Google Scholar]
- 11. Inesi G., Kurzmack M., Coan C., Lewis D. E. (1980) Cooperative calcium binding and ATPase activation in sarcoplasmic reticulum vesicles. J. Biol. Chem. 255, 3025–3031 [PubMed] [Google Scholar]
- 12. Toyoshima C., Nakasako M., Nomura H., Ogawa H. (2000) Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 405, 647–655 [DOI] [PubMed] [Google Scholar]
- 13. Dierick H. A., Ambrosini L., Spencer J., Glover T. W., Mercer J. F. (1995) Molecular structure of the Menkes disease gene (ATP7A). Genomics 28, 462–469 [DOI] [PubMed] [Google Scholar]
- 14. Tümer Z., Vural B., Tønnesen T., Chelly J., Monaco A. P., Horn N. (1995) Characterization of the exon structure of the Menkes disease gene using vectorette PCR. Genomics 26, 437–442 [DOI] [PubMed] [Google Scholar]
- 15. Petrukhin K., Lutsenko S., Chernov I., Ross B. M., Kaplan J. H., Gilliam T. C. (1994) Characterization of the Wilson disease gene encoding a P-type copper transporting ATPase. Genomic organization, alternative splicing, and structure/function predictions. Hum. Mol. Genet. 3, 1647–1656 [DOI] [PubMed] [Google Scholar]
- 16. Mercer J. F., Barnes N., Stevenson J., Strausak D., Llanos R. M. (2003) Copper-induced trafficking of the Cu-ATPases. A key mechanism for copper homeostasis. Biometals 16, 175–184 [DOI] [PubMed] [Google Scholar]
- 17. Voskoboinik I., Greenough M., La Fontaine S., Mercer J. F., Camakaris J. (2001) Functional studies on the Wilson copper P-type ATPase and toxic milk mouse mutant. Biochem. Biophys. Res. Commun. 281, 966–970 [DOI] [PubMed] [Google Scholar]
- 18. Tsivkovskii R., Purnat T., Lutsenko S. (2004) in Handbook of ATPases: Biochemistry, Cell Biology, Pathophysiology (Futai M., Kaplan J. H., Wada Y., eds) pp. 99–158, Wiley-VCH, Weinheim, Germany [Google Scholar]
- 19. Lutsenko S., Barnes N. L., Bartee M. Y., Dmitriev O. Y. (2007) Function and regulation of human copper-transporting ATPases. Physiol. Rev. 87, 1011–1046 [DOI] [PubMed] [Google Scholar]
- 20. Argüello J. M., González-Guerrero M., Raimunda D. (2011) Bacterial transition metal P1B-ATPases. Transport mechanism and roles in virulence. Biochemistry 50, 9940–9949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pilankatta R., Lewis D., Adams C. M., Inesi G. (2009) High yield heterologous expression of wild-type and mutant Cu+-ATPase (ATP7B, Wilson disease protein) for functional characterization of catalytic activity and serine residues undergoing copper-dependent phosphorylation. J. Biol. Chem. 284, 21307–21316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eletr S., Inesi G. (1972) Phase changes in the lipid moieties of sarcoplasmic reticulum membranes induced by temperature and protein conformational changes. Biochim. Biophys. Acta 290, 178–185 [DOI] [PubMed] [Google Scholar]
- 23. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 24. Weber K., Osborn M. (1969) The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J. Biol. Chem. 244, 4406–4412 [PubMed] [Google Scholar]
- 25. Pintschovius J., Fendler K. (1999) Charge translocation by the Na+/K+-ATPase investigated on solid supported membranes. Rapid solution exchange with a new technique. Biophys. J. 76, 814–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schulz P., Garcia-Celma J. J., Fendler K. (2008) SSM-based electrophysiology. Methods 46, 97–103 [DOI] [PubMed] [Google Scholar]
- 27. Tadini-Buoninsegni F., Bartolommei G., Moncelli M. R., Fendler K. (2008) Charge transfer in P-type ATPases investigated on planar membranes. Arch. Biochem. Biophys. 476, 75–86 [DOI] [PubMed] [Google Scholar]
- 28. Patton C., Thompson S., Epel D. (2004) Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium 35, 427–431 [DOI] [PubMed] [Google Scholar]
- 29. Tadini-Buoninsegni F., Bartolommei G., Moncelli M. R., Guidelli R., Inesi G. (2006) Pre-steady state electrogenic events of Ca2+/H+ exchange and transport by the Ca2+-ATPase. J. Biol. Chem. 281, 37720–37727 [DOI] [PubMed] [Google Scholar]
- 30. Scarpa A., Baldassare J., Inesi G. (1972) The effect of calcium ionophores on fragmented sarcoplasmic reticulum. J. Gen. Physiol. 60, 735–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Inesi G., Watanabe S. (1967) Temperature dependence of ATP hydrolysis and calcium uptake by fragmented sarcoplasmic membranes. Arch. Biochem. Biophys. 121, 665–671 [DOI] [PubMed] [Google Scholar]
- 32. Inesi G., Lewis D., Toyoshima C., Hirata A., de Meis L. (2008) Conformational fluctuations of the Ca2+-ATPase in the native membrane environment. Effects of pH, temperature, catalytic substrates, and thapsigargin. J. Biol. Chem. 283, 1189–1196 [DOI] [PubMed] [Google Scholar]
- 33. Pilankatta R., Lewis D., Inesi G. (2011) Involvement of protein kinase D in expression and trafficking of ATP7B (copper ATPase). J. Biol. Chem. 286, 7389–7396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Y., Pilankatta R., Hatori Y., Lewis D., Inesi G. (2010) Comparative features of copper ATPases ATP7A and ATP7B heterologously expressed in COS-1 cells. Biochemistry 49, 10006–10012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tadini Buoninsegni F., Bartolommei G., Moncelli M. R., Inesi G., Guidelli R. (2004) Time-resolved charge translocation by sarcoplasmic reticulum Ca-ATPase measured on a solid supported membrane. Biophys. J. 86, 3671–3686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tadini-Buoninsegni F., Bartolommei G., Moncelli M. R., Pilankatta R., Lewis D., Inesi G. (2010) ATP-dependent charge movement in ATP7B Cu+-ATPase is demonstrated by pre-steady state electrical measurements. FEBS Lett. 584, 4619–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Masuda H., de Meis L. (1973) Phosphorylation of the sarcoplasmic reticulum membrane by orthophosphate. Inhibition by calcium ions. Biochemistry 12, 4581–4585 [DOI] [PubMed] [Google Scholar]
- 38. Sørensen T. L., Clausen J. D., Jensen A. M., Vilsen B., Møller J. V., Andersen J. P., Nissen P. (2004) Localization of a K+-binding site involved in dephosphorylation of the sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 279, 46355–46358 [DOI] [PubMed] [Google Scholar]
- 39. Clarke D. M., Loo T. W., Inesi G., MacLennan D. H. (1989) Location of high affinity Ca2+-binding sites within the predicted transmembrane domain of the sarcoplasmic reticulum Ca2+-ATPase. Nature 339, 476–478 [DOI] [PubMed] [Google Scholar]
- 40. Strock C., Cavagna M., Peiffer W. E., Sumbilla C., Lewis D., Inesi G. (1998) Direct demonstration of Ca2+ binding defects in sarco-endoplasmic reticulum Ca2+ ATPase mutants overexpressed in COS-1 cells transfected with adenovirus vectors. J. Biol. Chem. 273, 15104–15109 [DOI] [PubMed] [Google Scholar]
- 41. Sørensen T. L., Møller J. V., Nissen P. (2004) Phosphoryl transfer and calcium ion occlusion in the calcium pump. Science 304, 1672–1675 [DOI] [PubMed] [Google Scholar]
- 42. Toyoshima C., Nomura H. (2002) Structural changes in the calcium pump accompanying the dissociation of calcium. Nature 418, 605–611 [DOI] [PubMed] [Google Scholar]
- 43. Yu X., Carroll S., Rigaud J. L., Inesi G. (1993) H+ countertransport and electrogenicity of the sarcoplasmic reticulum Ca2+ pump in reconstituted proteoliposomes. Biophys. J. 64, 1232–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Obara K., Miyashita N., Xu C., Toyoshima I., Sugita Y., Inesi G., Toyoshima C. (2005) Structural role of countertransport revealed in Ca2+ pump crystal structure in the absence of Ca2+. Proc. Natl. Acad. Sci. U.S.A. 102, 14489–14496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Levy D., Seigneuret M., Bluzat A., Rigaud J. L. (1990) Evidence for proton countertransport by the sarcoplasmic reticulum Ca2+-ATPase during calcium transport in reconstituted proteoliposomes with low ionic permeability. J. Biol. Chem. 265, 19524–19534 [PubMed] [Google Scholar]
- 46. Forge V., Mintz E., Guillain F. (1993) Ca2+ binding to sarcoplasmic reticulum ATPase revisited. I. Mechanism of affinity and cooperativity modulation by H+ and Mg2+. J. Biol. Chem. 268, 10953–10960 [PubMed] [Google Scholar]
- 47. Watanabe T., Lewis D., Nakamoto R., Kurzmack M., Fronticelli C., Inesi G. (1981) Modulation of calcium binding in sarcoplasmic reticulum adenosine triphosphatase. Biochemistry 20, 6617–6625 [DOI] [PubMed] [Google Scholar]
- 48. Andersen J. P., Vilsen B. (1998) Structure-function relationships of the calcium binding sites of the sarcoplasmic reticulum Ca2+-ATPase. Acta Physiol. Scand. Suppl. 643, 45–54 [PubMed] [Google Scholar]
- 49. Andersson J., Hauser K., Karjalainen E. L., Barth A. (2008) Protonation and hydrogen bonding of Ca2+ site residues in the E2P phosphoenzyme intermediate of sarcoplasmic reticulum Ca2+-ATPase studied by a combination of infrared spectroscopy and electrostatic calculations. Biophys. J. 94, 600–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Musgaard M., Thøgersen L., Schiøtt B. (2011) Protonation states of important acidic residues in the central Ca2+ ion binding sites of the Ca2+-ATPase. A molecular modeling study. Biochemistry 50, 11109–11120 [DOI] [PubMed] [Google Scholar]
- 51. Fibich A., Apell H. J. (2011) Kinetics of luminal proton binding to the SR Ca-ATPase. Biophys. J. 101, 1896–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yu X., Hao L., Inesi G. (1994) A pK change of acidic residues contributes to cation countertransport in the Ca-ATPase of sarcoplasmic reticulum. Role of H+ in Ca2+-ATPase countertransport. J. Biol. Chem. 269, 16656–16661 [PubMed] [Google Scholar]
- 53. Olesen C., Sørensen T. L., Nielsen R. C., Møller J. V., Nissen P. (2004) Dephosphorylation of the calcium pump coupled to counterion occlusion. Science 306, 2251–2255 [DOI] [PubMed] [Google Scholar]
- 54. Daiho T., Yamasaki K., Saino T., Kamidochi M., Satoh K., Iizuka H., Suzuki H. (2001) Mutations of either or both Cys-876 and Cys-888 residues of sarcoplasmic reticulum Ca2+-ATPase result in a complete loss of Ca2+ transport activity without a loss of Ca2+-dependent ATPase activity. Role of the Cys-876–Cys-888 disulfide bond. J. Biol. Chem. 276, 32771–32778 [DOI] [PubMed] [Google Scholar]
- 55. Danko S., Yamasaki K., Daiho T., Suzuki H., Toyoshima C. (2001) Organization of cytoplasmic domains of sarcoplasmic reticulum Ca2+-ATPase in E1P and E1ATP states. A limited proteolysis study. FEBS Lett. 505, 129–135 [DOI] [PubMed] [Google Scholar]
- 56. Inesi G., Ma H., Lewis D., Xu C. (2004) Ca2+ Occlusion and gating function of Glu-309 in the ADP-fluoroaluminate analog of the Ca2+-ATPase phosphoenzyme intermediate. J. Biol. Chem. 279, 31629–31637 [DOI] [PubMed] [Google Scholar]
- 57. Hatori Y., Hirata A., Toyoshima C., Lewis D., Pilankatta R., Inesi G. (2008) Intermediate phosphorylation reactions in the mechanism of ATP utilization by the copper ATPase (CopA) of Thermotoga maritima. J. Biol. Chem. 283, 22541–22549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gourdon P., Liu X. Y., Skjørringe T., Morth J. P., Møller L. B., Pedersen B. P., Nissen P. (2011) Crystal structure of a copper-transporting PIB-type ATPase. Nature 475, 59–64 [DOI] [PubMed] [Google Scholar]
- 59. Barry A. N., Otoikhian A., Bhatt S., Shinde U., Tsivkovskii R., Blackburn N. J., Lutsenko S. (2011) The lumenal loop Met-672–Pro-707 of copper-transporting ATPase ATP7A binds metals and facilitates copper release from the intramembrane sites. J. Biol. Chem. 286, 26585–26594 [DOI] [PMC free article] [PubMed] [Google Scholar]