

Background: OSM has the unique ability to utilize either LIFR or OSMR as a co-receptor.
Results: A unique loop in OSM reduces its affinity toward both LIFR and OSMR.
Conclusion: The loop structure in OSM is responsible for determining the affinity toward the receptor complex.
Significance: Removing the loop in OSM results in a more biologically active cytokine.
Keywords: Cell Signaling, Cytokine, Neuroprotection, Protein-Protein Interactions, Signal Transduction, STAT3, LIF, Leukemia Inhibitory Factor, Oncostatin M, gp130
Abstract
Oncostatin M (OSM) and leukemia inhibitory factor are pleiotropic cytokines that belong to the interleukin-6 (IL-6) family. These cytokines play a crucial role in diverse biological events like inflammation, neuroprotection, hematopoiesis, metabolism, and development. The family is grouped together based on structural similarities and their ability to activate the transmembrane receptor glycoprotein 130 (gp130). The common structure among these cytokines defines the spacing and the orientation of binding sites for cell surface receptors. OSM is unique in this family as it can signal using heterodimers of gp130 with either leukemia inhibitory factor receptor (LIFR) (type I) or oncostatin M receptor (OSMR) (type II). We have identified a unique helical loop on OSM between its B and C helices that is not found on other IL-6 family cytokines. This loop is located near the “FXXK” motif in active site III, which is essential for OSM's binding to both LIFR and OSMR. In this study, we show that the BC loop does not play a role in OSM's unique ability to bind OSMR. Shortening of the loop enhanced OSM's interaction with OSMR and LIFR as shown by kinetic and equilibrium binding analysis, suggesting the loop may hinder receptor interactions. As a consequence of improved binding, these structurally modified OSMs exhibited enhanced biological activity, including suppressed proliferation of A375 melanoma cells.
Introduction
IL-6 family cytokines (IL-6, IL-11, leukemia inhibitory factor (LIF),2 OSM, ciliary neurotrophic factor (CNTF), cardiotrophin-1 (CT-1), and cardiotrophin-like cytokines) possess a typical “four α-helix bundle”-like structure and act on their target cells by forming a multimeric receptor complex that includes the common receptor gp130 (1). Extensive mutagenesis studies revealed that these cytokines interact with the receptor chains through three distinct binding sites referred to as sites I, II, and III (2, 3). Cytokines requiring a co-receptor chain (IL-6, IL-11, CNTF, and CT-1) first bind to the co-receptor (IL-6R, IL-11R, or CNTF receptor) through binding site I (4–8). The glycoprotein gp130 always interacts through binding site II, and depending on the cytokine, the third binding site (site III) is used for recruitment of LIFR, OSMR, or a second gp130 molecule (9–14). Research has shown that a conserved “FXXK” motif at the core of site III is essential for all LIFR-binding proteins for their interaction with LIFR (15–17). After recruiting the required receptors, these cytokines signal via activation of the Janus kinase/signal transducer and activator of transcription (Jak/STAT) and mitogen-activated protein kinase (MAPK) pathways (18–21).
Among its family members, OSM resembles LIF most closely both in structure and function (22). The gene encoding for OSM, located on human chromosome 22q12, is only 20 kb away from LIF suggesting that these two genes evolved by gene duplication (22–25). Despite the striking similarities, OSM differs from LIF in its receptor binding. LIF first binds to leukemia inhibitory factor receptor (LIFR) and then recruits glycoprotein 130 (gp130) for its signal transduction (12, 13, 15, 26), OSM first binds gp130 and then recruits LIFR (12–16, 26–28). In addition, OSM can bind to gp130 and then recruit a unique receptor named oncostatin M receptor (OSMR) forming a distinct signaling complex (13, 14, 27). Our aim was to identify the structural features on OSM that result in its unique ability to bind OSMR and the features that result in its higher affinity toward gp130 than toward LIFR or OSMR. Based on the structural alignments, we have identified a helical loop on OSM between its B and C helices that is unique to OSM and not found on LIF or any other IL-6 family cytokine (Fig. 1). Using wild-type and mutant OSM molecules that have shortened BC loops, we show that the loop presents a steric hindrance for LIFR and OSMR, thus lowering the affinity for either receptor. Cytokines with deletions in the BC loop were able to activate LIFR-p130 and OSMR-p130 receptor complexes at 3-fold lower concentrations than the native OSM. Kinetic and equilibrium binding analysis of the ligand-receptor interactions show that improved activation is a consequence of increased affinity for LIFR and OSMR without altering the affinity for gp130. Together, these results suggest that the BC loop modulates OSM's affinity toward LIFR and OSMR by presenting a steric hindrance for their interaction. Our studies also indicate that the BC loop does not play a role in OSM's unique ability to bind OSMR.
FIGURE 1.

A and B, crystal structures of LIF (PDB 1EMR) and OSM (PDB 1EVS) with their active sites and helices A, B, C, and D identified. Both structures have an “up-up-down-down” topology with the N and C termini indicated. Also identified is the helical loop on OSM between its B and C helices. C, shown on right is the alignment of OSM structure onto LIF based on the α-carbon trace; root mean square deviation = 4.342. D, also shown on the bottom panel is the three-dimensional model for LIF in complex with LIFR and gp130 in the presence of OSM overlaid on LIF.
MATERIALS AND METHODS
Protein Design
cDNA for hOSM (NCBI Ref: NM 020530.3) was obtained from Invitrogen, and the cDNAs for hLIF and LIF05 were kindly donated by Dr. John K. Heath, University of Birmingham, UK. The gene encoding mature OSM (amino acids 26–217 of original OSM cDNA) was amplified using PCR with a FLAG tag introduced at its N terminus. The gene was then cloned into a pGEX-2T vector for protein expression as a GST-FLAG fusion protein with a thrombin cleavage site between the GST and FLAG tags. During the course of purification, we observed that the native human OSM contained a cryptic thrombin cleavage site “AGR” between its C and D helices (supplemental Fig. S1). As expected, when this fusion protein was subjected to thrombin cleavage on a glutathione-Sepharose 4B column, it resulted in the elution of two new fragments of sizes ∼17 and ∼6 kDa in addition to the native OSM, which is ∼23 kDa (supplemental Fig. S2, lane 1). To facilitate recombinant protein purification and to increase protein stability in vivo, we induced mutations at the DNA level using QuikChange® sit-directed mutagenesis kit (Stratagene, La Jolla, CA) to replace AGR with “AGA.” This modification resulted in OSM that was resistant to thrombin cleavage (supplemental Fig. S2, lane 2). The AGA modification did not alter OSM's functional activity on Müller cells (data not shown). This was expected because the modification is located in a flexible loop region away from the receptor-binding sites. From this point forward, we will refer to this AGA-modified human OSM as the wild-type OSM (OSM-WT). Recombinant proteins with modifications in the BC loop were made using the AGA-modified human OSM as the starting template. Therefore, all recombinant OSM proteins we expressed lack the thrombin cleavage site. Mutations and/or deletions of codons in the BC loop region were performed using QuikChange® mutagenesis kit (Stratagene, La Jolla, CA). Mutation of the FXXK motif on wild-type and BC loop mutant OSM molecules to AXXA was also carried out using QuikChange® mutagenesis kit (Stratagene, La Jolla, CA) (see supplemental Table S1 for list of primers used).
Expression and Purification of Proteins
Plasmids encoding wild-type or the mutant OSMs were transfected into Escherichia coli JM109 strain for protein expression. Cultures were grown in LB plus ampicillin (100 μg/ml) at 37 °C and 300 rpm until they reached midlog phase (A600 = 0.6). Isopropyl β-d-thiogalactopyranoside was then added to the culture to a final concentration of 0.1 mm, and induction was carried out for an additional 3 h at room temperature. Intracellular fusion protein was recovered from cell extracts by affinity binding to a slurry of glutathione-Sepharose 4B beads (GE Healthcare). Washes were carried out as described by the manufacturer's protocol. Isolation of the FLAG-tagged proteins was achieved by cleavage of the GST tag using human thrombin (Amersham Biosciences), at a molar ratio of 1:500 thrombin to fusion protein, in 1× PBS (pH 7.3) overnight at room temperature. Following cleavage, the elution containing wild-type or mutant OSM was pooled with an additional four batch washes (1× PBS (pH 7.3)). SDS-PAGE analysis of eluted proteins revealed that the E. coli expressed high amounts of wild type and M1 and M2 versions of OSM. The M3 mutant version of OSM was not expressible in bacteria. This could be a result of structural instability in the M3 mutant version of OSM. Cleaved proteins were further purified by fast protein liquid chromatography (FPLC) using a Mono Q anionic exchange column (Amersham Biosciences). Elution was carried out with a linear gradient of 0–1 m NaCl in 20 mm Tris buffer (pH 8.0). Eluted fractions were analyzed using SDS-PAGE. Fractions containing enriched protein were pooled and concentrated by ultrafiltration (Millipore Corp., Billerica, MA). Protein identities were confirmed by mass spectrometry, and purities were >90% as evaluated by Coomassie staining of the purified proteins run on a 4–20% gradient polyacrylamide gel (Fig. 3). Thrombin used for cleavage of the GST tag was not detected in the MS analysis. Concentration of the purified recombinant proteins was estimated using BCA assay (Pierce) and using bovine serum albumin (BSA) as the standard.
FIGURE 3.
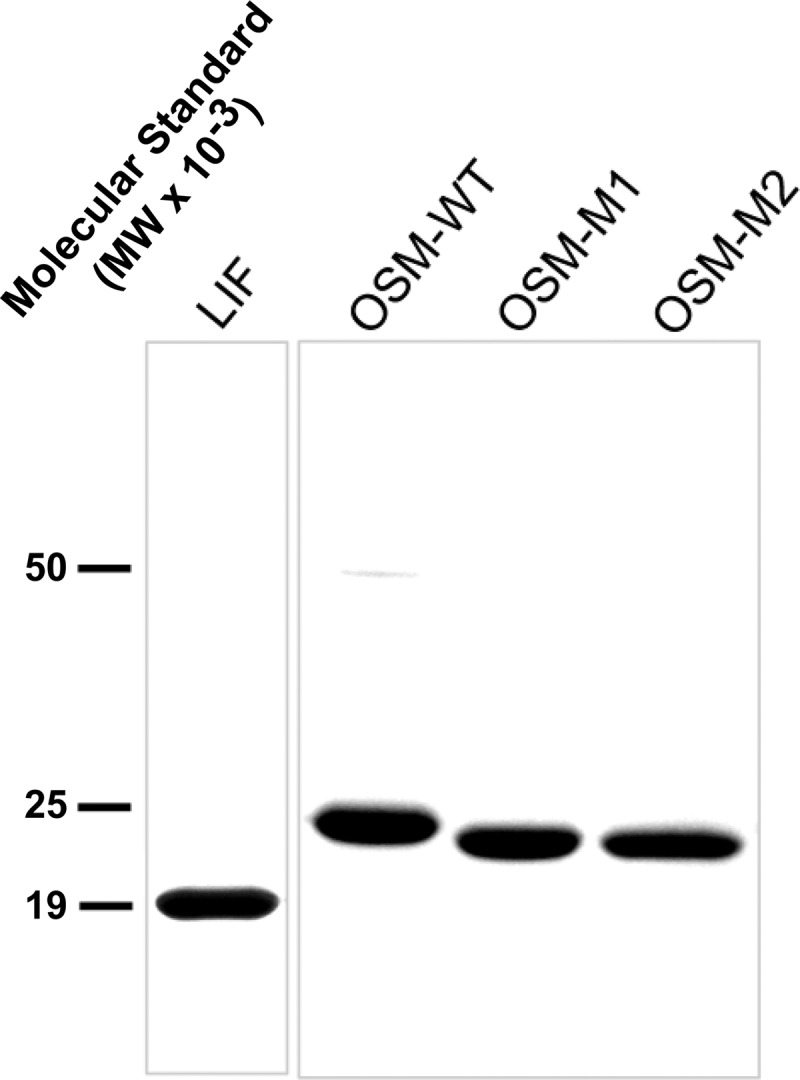
SDS-PAGE analysis of purified proteins. 8 μg of purified protein is loaded into each lane.
Circular Dichroism
CD measurements were performed on a Jasco J-715 spectropolarimeter (Jasco, Easton, MD). Steady state spectra were recorded by scanning in the wavelength region between 200 and 250 nm with 0.1-cm path length and a 1-nm bandwidth at 20 °C. Spectra of blank buffer solutions acquired under identical conditions were used for background correction. Protein concentrations were maintained at 10 μm in Dulbecco's phosphate-buffered saline (PBS) (9.33 mm potassium phosphate, 136 mm NaCl, 2.7 mm KCl, 0.6 mm MgCl2, 0.9 mm CaCl2). Estimation of the α-helical, β-sheet, and loop content in the proteins was carried out using SELCON3, CONTINNL, and CDSSTR software programs (CD Pro®, Lamar, CO).
Surface Plasmon Resonance (SPR)
Kinetic parameters of the interactions between receptor domains and the cytokines LIF, OSM-WT, OSM-M1, or OSM-M2 were analyzed by SPR using the SensiQ system (ICX Technologies, Oklahoma City, OK) as described by the manufacturer's protocol. Briefly, a carboxyl sensor with two channel, was installed in SensiQ and allowed to thermally equilibrate for about 15 min. The channels were initially cleaned with a 3-min injection of 0.1 m HCl. An activation solution of 2 mm 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride and 0.5 mm N-hydroxysulfosuccinimide was prepared in deionized water immediately before injection. Activation solution was injected over both channels for ∼3 min followed by a 10-min injection of 50 μg/ml cytokine (LIF, OSM-WT, OSM-M1, or OSM-M2) in 10 mm acetate buffer (pH 5.0) over channel 1. Channel 2 did not receive any cytokines and thus served as a reference for nonspecific binding. Unreacted N-hydroxysulfosuccinimide esters were capped with a 3-min injection of 1 m ethanolamine (pH 8.0) over both channels. Total immobilization of 500–700 response units was achieved for each of these cytokines. A concentration series of soluble LIFR (catalog no. 249-LR-050/CF, R&D Systems, Minneapolis, MN), soluble OSMR (catalog no. 4389-OR-50, R&D Systems), or soluble gp130 (catalog no. 228-GP-050/CF, R&D Systems) in running buffer (10 mm HEPES (pH 7.4), 150 mm NaCl, 3.4 mm EDTA, 0.005% Tween 20) were injected over both channels at a flow rate of 25 μl/min. Following a dissociation period of 3 min, the surfaces were regenerated by injecting 10 mm NaOH for 30 s. Rate constants for association (ka) and dissociation (kd) rates were derived by global analysis of the response curves fit to a 1:1 kinetic model (Equations 1 and 2) using QDat software (BioLogic Software, Ltd., Knoxville, TN, and Nomadics, Inc. Stillwater, OK) using 1:1 stoichiometry,
where RU is real time response units as measured by the SensiQ instrument; RUmax is the maximum response obtainable for a given concentration of the soluble receptor; t is time; and Co is concentration of the soluble receptor analyte in solution.
Cell Culture and Cytokine Stimulation
Human Müller cells and human A375 melanoma cells were grown in DMEM/F-12 and RPMI 1640 medium, respectively, supplemented with fetal bovine serum (10%) (Invitrogen), penicillin (100 units/ml), and streptomycin (100 μg/ml) (Invitrogen). Cells were seeded in a 10-cm tissue culture dish at a density of 100,000 cells/plate and allowed to grow in a 37 °C humidified atmosphere with 5% CO2. When the cells reached 80% confluency, the culture medium was changed to fresh serum-free media (DMEM/F-12 or RPMI 1640 medium supplemented with penicillin (100 units/ml) and streptomycin (50 μg/ml)). Serum starvation was carried out for 30 min before stimulation with desired doses of OSM-WT, OSM-M1, or OSM-M2 for a period of 20 min. Following stimulation, cells were harvested for measurements of STAT3 and ERK1/2 activation by Western blots.
Western Blots
Harvested cells were homogenized in a lysis buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 5 mm EDTA, 1% (v/v) Nonidet P-40, 5% (v/v) glycerol, and protease inhibitor mixture (Calbiochem). Protein content was measured using BCA protein assay (Pierce). Total protein from each sample (15 μg) was electrophoresed on 4–20% gradient SDS-polyacrylamide gels (Invitrogen) and transferred to nitrocellulose membranes (Bio-Rad). The membranes were incubated in blocking buffer (5% BSA in TBST (20 mm Tris-HCl (pH 7.5), 100 mm NaCl, and 0.1% Tween 20)) for 1 h at room temperature and then incubated overnight at 4 °C with rabbit polyclonal anti-phospho-STAT3 antibody (catalog no. 9131, Cell Signaling Technology, Beverly, MA) or anti-phospho-ERK1/2 (catalog no. 9101, Cell Signaling Technology) in blocking buffer, followed by 1-h incubation at room temperature with HRP-conjugated goat anti-rabbit secondary antibody (catalog no. NA934V, GE Healthcare). Signals were visualized using SuperSignal West Dura extended duration substrate (Pierce) and quantified by conventional digital image analysis using ImageStation 4000R (software from Kodak MI; Eastman Kodak Co.). Blots were stripped and reprobed with anti-β-actin (catalog no. ab6276-100, Abcam, Cambridge, MA) followed by appropriate secondary antibodies. Band intensities of pSTAT3 and pERK were normalized against the intensity of actin to account for loading variability.
Cell Proliferation Studies
To measure cell proliferation, ATP levels in viable cells were quantified using CellTiter-Glo Luminescent cell viability assay (Promega, Madison, WI). A375 melanoma cells were seeded in a 96-well plate at a density of 4000 cells/well in a total volume of 200 μl of RPMI 1640 medium (Invitrogen) supplemented with fetal bovine serum (10%), penicillin (50 IU/ml), and streptomycin (50 mg/ml). Cells were then treated with different doses of OSM-WT, OSM-M1, or OSM-M2 for the desired duration immediately after seeding. Control cells were treated with carrier solution, 1× PBS. Cell population in the wells was monitored using CellTiter Glo® luminescent cell viability assay (Promega, Madison, WI) according to the manufacturer's protocol.
Enzyme-linked Immunosorbent Assay (ELISA)
ELISA was used to evaluate the equilibrium binding strength of the interaction between the cytokines and their receptors. This technique has traditionally been used as a sensitive method to quantify the binding affinities between two interacting proteins. However, unlike in SPR, ELISA involves immobilization of the proteins on a flat plastic surface driven by hydrophobic and ionic interactions. This might cause some distortion in the three-dimensional structure of immobilized protein that leads to inaccuracies in the estimation of dissociation constants. It is thus important to bear in mind that the values estimated using this technique are to be used only for comparison between species but not as true equilibrium binding constants. The results obtained using this technique will thus be presented as apparent equilibrium dissociation constants (KD, app). For direct interaction studies, cytokines (LIF, OSM-WT, OSM-M1, or OSM-M2) were immobilized on the 96-well ELISA plate by incubating the wells with 200 μl of 5 nm cytokine solution in PBS (pH 7.4) overnight at 4 °C. The wells were then blocked with blocking buffer (4% BSA in PBS) for 1 h at room temperature. After washing with 250 μl of washing buffer (0.05% Tween 20 in PBS) three times, the cytokines were treated with a series of concentrations of soluble human LIFR (catalog no. 249-LR-050/CF, R&D Systems) or OSMR (catalog no. 4389-OR-050, R&D Systems) in 150 μl of blocking buffer for 2 h. The wells were then incubated with 150 μl of polyclonal anti-hLIFR (catalog no. AF249-NA, R&D Systems) or anti-hOSMR (catalog no. AF662, R&D Systems) in blocking buffer for 1 h followed by incubation with 150 μl of HRP-conjugated anti-mouse antibody (GE Healthcare) in blocking buffer for 30 min. The wells were then washed three times with washing buffer and treated with 100 μl of chromogenic Slow-TMB® HRP substrate (catalog 34024, Thermo Scientific Fisher) for 15 min. The reaction was then stopped by adding 100 μl of 2 m H2SO4, and the absorbance of each well at 450 nm was read immediately using a UV detector (iMark® Microplate Reader, Bio-Rad). For interactions of a higher order, soluble human gp130 (catalog no. 671-GP-100, R&D Systems) was immobilized on 96-well ELISA microplates by incubating the wells with 200 μl of 1 nm gp130 solution (in PBS (pH 7.4)) overnight at 4 °C. The wells were then blocked with 150 μl of blocking buffer (4% BSA in PBS) for 1 h at room temperature. After washing with 250 μl of washing buffer three times, gp130 was treated with saturating amounts of the cytokines (500 nm hLIF, 200 nm OSM-WT*, 200 nm OSM-M1, or 200 nm OSM-M2) in a volume of 150 μl of blocking buffer for a period of 2 h. The cytokine solution was discarded, and a series of concentrations of soluble human LIFR or OSMR in a final volume of 150 μl of blocking buffer were then added to the wells, and the incubation was continued for another 1 h. After washing with 250 μl of washing buffer three times, the wells were then incubated with 150 μl of polyclonal anti-hLIFR or anti-hOSMR in blocking buffer for 1 h. After three washes with 250 μl of washing buffer, the wells were then incubated with 150 μl of HRP-conjugated anti-mouse antibody in blocking buffer for 30 min. The wells were then washed again three times with washing buffer and treated with 100 μl of chromogenic Slow-TMB® HRP substrate for 30 min. The reaction was then stopped by adding 100 μl of 2 m H2SO4, and the absorbance of each well at 450 nm was read using a UV detector. Equilibrium dissociation constants (KD) are estimated by nonlinear curve fitting to the optical density values plotted against the concentrations of soluble receptor using GraphPad Prism software (GraphPad Software, La Jolla, CA).
Statistical Analysis
All statistical analyses were done using SigmaStat 3.10 (Systat Software, Inc., Richmond, CA). Results are expressed as mean ± S.D. Differences between two groups were assessed using Student's t test. A “p value” of less than 0.05 was considered significant.
RESULTS
Molecular Modeling of LIF and OSM; Identification of the BC Loop
To determine the structural differences that might account for receptor specificity, we aligned the crystal structures of hOSM (PDB code 1EVS) onto hLIF (PDB code 1EMR) based on the trace of α-carbons using Delano Scientific PyMOL molecular viewer (Fig. 1). The alignment of the backbone structures fit well with a relatively low root mean square deviation value of 4.342. The active sites II and III on both molecules exhibited good conservation in structural orientation. Previous reports showed that the FXXK motif is essential for OSM's interaction with both LIFR and OSMR (16, 29). Despite having a similar FXXK motif, other LIFR-interacting cytokines LIF, CNTF, CT-1, and cardiotrophin-like cytokine, however, cannot activate OSMR (26). This suggested that the difference in receptor specificity between hOSM and other LIFR-activating cytokines is the result of structural differences between these ligands in the vicinity of the core FXXK motif (16). One of the obvious structural differences in the alignment is the presence of an additional helical loop between its B and C helices in OSM that is not present in LIF (Fig. 1). This BC loop is positioned in close proximity to the FXXK motif in active site III. Based on the crystal structures solved for LIF in complex with LIFR (PDB code 2Q7N) or gp130 (PDB code 1PVH), we have generated a model for the trimeric complex of LIF-LIFR-gp130 using PyMOL (Fig. 1D). When OSM was superimposed over LIF in this trimeric model, the BC loop on OSM again stands out as a unique motif at the receptor-binding interface of OSM. This suggested that the BC loop is possibly playing an essential role in recognizing OSMR. To test this hypothesis, we generated substitution mutations in OSM that either remove or shorten the length of this BC loop.
Wild-type OSM contains 12 amino acids in the BC loop region. Using site-directed mutagenesis, we have deleted or modified the amino acids in this region to generate OSM molecules that contained 7, 4, or 0 amino acids. Shown in Fig. 2 are the sequences of mutant OSM molecules with truncated BC loops (OSM-M1, OSM-M2, and OSM-M3) in comparison with the wild-type OSM (OSM-WT). Glycines were incorporated into OSM-M1 and OSM-M2 to induce flexibility into the loop region thus minimizing the impact of this BC loop modification on the overall structure of OSM. OSM-WT, OSM-M1, and OSM-M2 were expressed at high levels in bacteria (Fig. 3). However, we were not able to express OSM-M3 in bacteria suggesting that complete removal of the BC loop from OSM leads to instability in the overall structure of the protein.
FIGURE 2.

Amino acid sequences of wild-type OSM and the mutant variants of OSM with truncated BC loops. Shown in gray are the α-helices present in the secondary structure of OSM as identified in the crystal structure (PDB 1EVS). Each of the helices A, B, C, and D are identified along with the BC loop region. Also highlighted in an open box is the mutated thrombin cleavage site AGA, and shown in bold letters indicated by arrows is the active FXXK site on the wild-type and mutant OSMs required by the molecules to bind LIFR and OSMR.
Structural Characterization of OSM-M1 and OSM-M2
Minor alterations in the size and composition of BC loop could potentially induce a global change in the overall structure of OSM. To determine whether the modified proteins (OSM-M1 and OSM-M2) still retained native α-helical content, we analyzed the recombinant proteins using circular dichroism (CD). Fig. 4 shows the molar ellipticity [θ] plotted against the wavelength for LIF, OSM-WT, OSM-M1, and OSM-M2. All molecules displayed similar absorption behavior. Analysis using the software programs SELCON3, CONTINLL, and CDSSTR revealed that both LIF and OSM have ∼60% α-helical content with the remaining primarily being loop regions. This is in good agreement with the crystal structures available for LIF and OSM. Analysis also showed that both OSM-M1 and OSM-M2 have similar 60% α-helical content with the remaining being loop regions. These results suggest that shortening the length of BC loop in OSM from 12 amino acids to 7 or 4 amino acids did not induce a significant global change in the overall secondary structure of OSM.
FIGURE 4.

Modifications in the BC loop area of OSM did not induce a global change in the protein's structure. A, average of three CD spectra of the purified proteins plotted as molar ellipticity (θ) versus the wavelength. B, theoretical estimation of the secondary structural content for each protein using SELCON3, CDSSTR, and CONTINNL. Values are presented as mean of estimations given by the three programs ± S.D.
Functional Evaluation of Wild-type and Mutant OSMs in Activating OSMR-gp130 Complexes
To determine whether the BC loop on hOSM is required for OSMR binding, A375 melanoma cells were treated with increasing doses of LIF, OSM wild-type, or mutant forms of OSM. A375 melanoma cells are known to express OSMR and gp130 on their cell surface, although LIFR was not detected (14). A375 cells did not respond to LIF as expected because these cells are known not to express LIFR but did responded to OSM in a linear and dose-dependent manner as demonstrated by activation of STAT3 (Fig. 5 and supplemental Figs. S3). When treated with OSM-M1 and OSM-M2, A375 cells exhibited a 3–4-fold increase in STAT3 activation relative to wild-type OSM (OSM-WT) (supplemental Fig. S3). The mutant molecules, however, did not show any change in ERK1/2 activation compared with OSM-WT.
FIGURE 5.

A, activation of STAT3 and ERK in A375 melanoma cells in response to different doses of wild-type (OSM-WT) and the mutant forms of OSM (OSM-M1 and OSM-M2). Band intensities of phospho-STAT3 (B) and phospho-ERK (C) normalized against the band intensities of β-actin are plotted against the concentration of cytokines used for stimulation. Values are presented as mean ± S.E. n ≥ 4 where n is the number of independent cultures at each dose. (*, p < 0.01; **, p < 0.001, compared with OSM-WT treatment at same dose.) Shown for comparison is the normalized phospho-STAT3 induced by LIF as estimated by the representative data shown in supplemental Fig. S3C.
Functional Evaluation of Wild-type and Mutant OSMs in Activating LIFR-gp130 Complexes
To determine whether shortening the BC loop in OSM affected the ability of OSM to activate LIFR-gp130 receptors, we used the recombinant proteins to stimulate the human retinal Müller cell line. Müller cells respond to both LIF and OSM stimulation in a dose-dependent manner by activating STAT3 (supplemental Fig. S3). To determine the receptor expression, the cells were pretreated with recombinant LIF05 (a mutant LIF molecule that specifically antagonizes the activation of LIFR but not OSMR or gp130 (15, 30)) before addition of LIF or OSM. At doses of 50 ng/ml, LIF05 was able to completely antagonize the STAT3 activation induced by both LIF and OSM demonstrating that the STAT3 activation in Müller cells is dependent upon utilization of LIFR-gp130 and not OSMR-gp130.
Treatment of Müller cells with wild-type and mutant OSM molecules again show that OSM-M1 and OSM-M2 induce a 2–3-fold greater activation of STAT3 compared with OSM-WT at similar doses (Fig. 6). Also, OSM-M1 and OSM-M2 exhibited a similar 2–3-fold higher activation of Erk1/2 compared with wild-type OSM.
FIGURE 6.

A, activation of STAT3 and ERK in human Müller cells in response to different doses of wild-type (OSM-WT) and the mutant forms of OSM (OSM-M1 and OSM-M2). Band intensities of phospho-STAT3 (B) and phospho-ERK (C) normalized against the band intensities of β-actin are plotted against the concentration of cytokines used for stimulation. Values are presented as mean ± S.E. n ≥ 4, where n is the number of independent cultures at each dose. (*, p < 0.05; **, p < 0.01, compared with OSM-WT treatment at same dose.) Shown for comparison is the normalized phospho-STAT3 induced by LIF as estimated by the representative data shown in supplemental Fig. S3A.
Removal of BC Loop Does Not Alter the Requirement of Core FXXK Motif in Active Site III
Given its proximity to the site III, it is possible that removal of the BC loop created an alternative site III that could facilitate a stronger binding to LIFR and OSMR. To determine whether the mutant OSMs, OSM-M1 and OSM-M2, still utilize the FXXK motif to interact with LIFR or OSMR, we mutated both Phe-160 and Lys-163 to alanines changing the motif to AXXA. and we evaluated their activity on Müller cells and A375 cells. Müller cells and A375 melanoma cells were serum-starved for 30 min before addition of either the wild-type (FXXK) or the alanine mutant versions (AXXA) of OSM-WT, OSM-M1. and OSM-M2. Cells were incubated for 20 min with 1 ng/ml cytokine (Fig. 7). As expected, mutating FXXK to AXXA in OSM-WT completely abolishes its ability to activate STAT3 in both A375 melanoma and Müller cells. Similar to OSM-WT, both OSM-M1 and OSM-M2 showed complete loss of activity upon alanine substitution at the active site III.
FIGURE 7.

OSM with truncated BC loop still utilizes the FXXK motif to activate LIFR and OSMR. In both A375 melanoma cells (top blot) and human Müller cells (third blot from the top) STAT3 was activated following a 20-min incubation with 1 ng/ml of the indicated forms of OSM containing the wild-type FXXK but not the alanine-substituted (AXXA) active site III. The actin blots were used for loading controls.
Shortening the Size of BC Loop Improves OSM Affinity toward LIFR and OSMR
Compared with OSM-WT, both OSM-M1 and -M2 activated signaling to higher levels at lower concentrations suggesting that removal of the BC loop on OSM increased the cytokine's ability to form a stable complex with the receptors. To directly measure the binding kinetics of these ligand-receptor interactions, we used SPR. The cytokines (LIF, OSM-WT, OSM-M1, and OSM-M2) were immobilized on the sensor chip surface, whereas recombinant soluble receptors were used as the analytes (Fig. 8). The analysis revealed that LIF had a 23-fold higher affinity toward LIFR (KD = 3.10 nm) than gp130 (KD = 72.38 nm), although OSM had a 2-fold higher affinity toward gp130 (KD = 22.69 nm) than LIFR (KD = 43.79 nm) (Table 1). When the size of BC loop is reduced from 12 to 7 amino acids (OSM-M1), the affinity of OSM toward LIFR improved dramatically (KD = 7.62 nm). When the size of the BC loop was further reduced to four amino acids, the affinity improved even more (KD = 2.74 nm). However, changing the size of BC loop did not affect OSM's affinity toward gp130 significantly (KD = OSM-WT = 22.69 nm, OSM-M1 = 26.26 nm, and OSM-M2 = 21.49 nm) (Table 1). OSM-M1 and -M2 proteins with shorter BC loops clearly display a higher affinity for LIFR still retaining their relatively high affinity toward gp130. Together, these results suggest that the BC loop on OSM is playing a significant role in determining OSM's direct interaction with LIFR.
FIGURE 8.

Kinetic analysis of soluble LIFR and soluble gp130 interaction with LIF, OSM-WT, OSM-M1, or OSM-M2. Soluble LIFR (left panels) or soluble gp130 (right panels) at various concentrations were injected over an SPR sensor chip with immobilized ligand (LIF, OSM-WT, OSM-M1, or OSM-M2). Models are indicated by a smooth gray line overlaid over response curve traces. See Table 1 for the binding constants.
TABLE 1.
Comparison of association (ka), dissociation (kd), and equilibrium dissociation (KD) constants for LIFR and gp130 binding to LIF, OSM-WT, OSM-M1 and OSM-M2
| LIFR |
gp130 |
|||||
|---|---|---|---|---|---|---|
| ka | kd | KD | ka | kd | KD | |
| ×105m−1 s−1 | ×10−3 s−1 | nm | ×105m−1 s−1 | ×10−3 s−1 | nm | |
| LIF | 7.40 | 2.30 | 3.10 | 0.74 | 5.33 | 72.38 |
| OSM-WT | 0.91 | 4.00 | 43.79 | 2.07 | 4.70 | 22.69 |
| OSM-M1 | 3.31 | 2.52 | 7.62 | 2.03 | 5.34 | 26.26 |
| OSM-M2 | 13.0 | 3.56 | 2.74 | 2.27 | 4.87 | 21.49 |
Similar SPR binding studies with OSMR showed that neither the wild-type nor the mutant OSMs (OSM-M1 and OSM-M2) exhibit a direct interaction with OSMR (Fig. 8). This is in agreement with previous results that reported a lack of direct interaction between OSM and OSMR in the absence of gp130 (16). To evaluate the binding kinetics of OSM, we needed to first from the cytokine gp130 complex which has been demonstrated to increase the affinity for the cytokine OSMR interaction. To accomplish this, we immobilized the soluble gp130 on the sensor chip surface and treated with human OSM followed by OSMR. We did observe OSM-OSMR specific binding; however, accurate association and dissociation constants could not be determined for these interactions because there was a progressive loss in the binding capacity of gp130 immobilized on the chip. To overcome this issue, we developed an ELISA to measure binding. In these assays, gp130 or OSM cytokines were immobilized on the ELISA plate and were sequentially treated with saturating amounts of wild-type or mutant OSM. The plates were then incubated with variable concentrations of soluble OSMR. Results show that after binding to gp130, both the wild-type and the mutant OSMs exhibit strong affinity toward OSMR (Fig. 9B; Table 2). Again, as observed toward LIFR, there is a significant improvement in OSM's affinity toward OSMR when the BC loop is truncated (Table 2) (KD, app OSM-WT, 10.86 ± 1.7 nm; OSM-M1, 3.71 ± 0.67 nm; and OSM-M2, 2.19 ± 0.28 nm). This represents a 3–4-fold increase in the affinity toward OSMR upon BC loop truncation on OSM. These results demonstrate that the BC loop on OSM plays a significant role in determining its affinity for both OSMR and LIFR because its truncation results in significant improvement in affinity toward both receptors. Consistent with the previous results, none of the OSM cytokines could bind OSMR in the absence of gp130 (Fig. 9A; Table 2) This result suggests that the BC loop is not responsible for determining the need for the gp130-OSM interaction prior to binding OSMR.
FIGURE 9.

ELISA analysis of soluble OSMR and soluble LIFR binding with LIF, OSM-WT, OSM-M1, and OSM-M2 or gp130-bound LIF (gp130-LIF), OSM-WT (gp130-OSM-WT), OSM-M1 (gp130-OSM-M1), and OSM-M2 (gp130-OSM-M2). Cytokines (LIF, OSM-WT, OSM-M1, or OSM-M2) immobilized on ELISA plates in the absence (A and C) or presence (B and D) of gp130 were treated with various concentrations of soluble OSMR (A and B) or soluble LIFR (C and D). Binding of soluble OSMR or LIFR to immobilized BSA was used as a control. Values are presented as mean ± S.E. n = 3, where n is the number of independent binding assays at each dose. Equilibrium KD values were estimated using a nonlinear curve fitting to the binding data using GraphPad Prism (Graph Pad Software, La Jolla, CA) (see Table 2).
TABLE 2.
Comparison of apparent equilibrium dissociation constant (KD,app) values (nm) for direct interaction of LIFR and OSMR with LIF, OSM-WT, OSM-M1, and OSM-M2 or the interaction of LIFR and OSMR with gp130-bound LIF, OSM-WT, OSM-M1, and OSM-M2
ND means not detected.
| OSMR binding |
LIFR binding |
|||
|---|---|---|---|---|
| Direct | With gp130 | Direct | With gp130 | |
| LIF | ND | ND | 8.58 ± 0.99 | 10.33 ± 1.59 |
| OSM-WT* | ND | 10.86 ± 1.70 | 60.02 ± 17.54 | 70.29 ± 18.90 |
| OSM-M1 | ND | 3.71 ± 0.67 | 10.06 ± 1.34 | 12.56 ± 1.73 |
| OSM-M2 | ND | 2.19 ± 0.28 | 8.75 ± 1.34 | 9.13 ± 1.53 |
The gp130-induced cooperative binding toward OSMR prompted us to determine whether similar cooperativity was needed for LIFR binding. The results shown in Table 2 demonstrate that previous gp130 binding to OSM or LIF did not affect their affinities toward LIFR significantly (Table 2). The results demonstrate that both LIF and OSM bind LIFR independently of the gp130 interaction. This is in significant contrast to the cooperative binding needed for OSMR interaction with OSM. Again, as observed in SPR, the mutant OSMs with a truncated BC loop exhibited a stronger affinity toward LIFR compared with the wild type (Fig. 9, C and D, and Table 2) and was also independent of gp130. The equilibrium dissociation constants (KD, app) obtained for LIFR binding using ELISA were significantly higher than the values obtained using SPR. This could be the result of possible structural distortions caused by immobilization of receptors on ELISA plates or the difference between the kinetic analysis provided by SPR versus the steady state equilibrium analysis provided by ELISA.
Inhibition of A375 Melanoma Cell Proliferation
OSM was initially discovered by its ability to suppress proliferation of several melanoma cell lines, including A375 melanoma cells (31). As expected, treating A375 cells with wild-type OSM inhibited their proliferation in a dose-dependent manner (Fig. 10). At concentrations of 20 ng/ml, OSM-WT was able to suppress A375 melanoma cell proliferation by ∼50%, although a concentration of 50 ng/ml was able to suppress the proliferation by ∼90%. In contrast, both OSM-M1 and OSM-M2 were both able to suppress the proliferation of A375 melanoma cells at significantly lower concentrations. Although 10 ng/ml concentrations were enough for the mutant OSM molecules to inhibit the proliferation by ∼50%, 20 ng/ml concentrations suppressed their proliferation by ∼90%. These data clearly suggest that reducing the size of BC loop improves the ability of OSM to activate OSMR-gp130 and suppress the proliferation of A375 melanoma cells.
FIGURE 10.

A–C, A375 melanoma cell proliferation in the presence or absence of various doses of OSM-WT, OSM-M1, and OSM-M2. D, cell numbers on 5th day of proliferation are normalized against the control cells and plotted for comparison. Values are presented as mean ± S.D. n = 4, where n is the number of independent cultures at each dose. (*, p < 0.01; **, p < 0.001, compared with control treatment at same dose.)
DISCUSSION
IL-6 family cytokines are pleiotropic cytokines that elicit a wide variety of responses in vivo mediated by the activation of signal-transducing receptors gp130, LIFR, and OSMR. Among these cytokines, OSM is unique in terms of its ability to signal through two different receptor complexes, LIFR-gp130 (type I) and OSMR-gp130 (type II). Also, OSM is unique in the order in which it binds to its receptors, i.e. gp130 followed by LIFR or OSMR (12, 26, 28, 32). Based on the crystal structures and mutational analysis conducted, it has been proposed that the ability of OSM to interact with OSMR must result from the involvement of additional residues in the vicinity of its FXXK motif that is required for OSM's binding to LIFR and OSMR (16). In comparison with other IL-6 family cytokines, we have identified that OSM has a unique α-helical loop between its B and C helices. This BC loop lies in close proximity to site III that contains the core FXXK motif. The size and location of this loop suggested that it is possibly playing an essential role in OSM's unique ability to bind OSMR. However, contrary to our expectation, shortening this loop resulted in proteins that display higher activity as indicated by improved activation of signal transduction and inhibition of A375 melanoma cell proliferation. Stimulation studies using human Müller cells that express LIFR and gp130 showed that the truncation of the BC loop on OSM improves its ability to activate LIFR-gp130 complexes also. Kinetic and equilibrium binding analysis of ligand-receptor interaction revealed that proteins lacking the BC loop on OSM had higher affinity for LIFR and OSMR. Together, these results suggest that the BC loop is clearly not essential for OSM's unique ability to bind OSMR or in its sequential binding to gp130 prior to OSMR. The loop does appear to play a role in determining the relative affinity for OSMR-OSM-gp130 and LIFR-OSM-gp130 complexes. Wild-type OSM has a 6-fold higher affinity for OSMR-gp130 than LIFR-gp130. When the loop is shortened, the affinity for both receptor complexes is increased, but the difference in affinity is reduced to 3- or 4-fold. Thus, the loop could play a role in allowing OSM to signal preferentially through OSMR-gp130 rather than LIFR-gp130. This could play a role in the biological activity of OSM in tissues or cells that express one receptor or the other.
The biological significance for reducing receptor affinity is unknown. However, it may be related to OSM's increased affinity to gp130. It is difficult to tell which came first, reduced affinity for co-receptor or increased affinity for gp130. OSM is unique in that it has a high affinity for gp130 independent of co-receptor binding. The higher affinity for gp130 could be a compensation for the lower affinity for OSMR or LIFR.
It has been reported that LIF has strong preference for binding LIFR prior to binding gp130, although OSM has a preference for binding gp130 prior to binding LIFR (12, 13, 15). Affinity measurements by SPR suggest that the mechanism behind the unique ability of OSM to first bind gp130 can be explained by its relative affinity to each receptor subunit. OSM has a 2-fold higher affinity toward gp130 than toward LIFR. LIF, which lacks the BC loop, has a 23-fold higher affinity for LIFR than for gp130. When the BC loop on OSM is truncated, OSM starts displaying a higher affinity toward LIFR than toward gp130. Clearly, the reduced affinity of OSM toward LIFR is caused by the BC loop and is likely playing a role in the difference in sequential binding between LIF and OSM.
The inability of OSM to bind soluble OSMR directly is consistent with previous observations (16). Like IL-6 and CNTF, which require binding to their α-receptor before they can bind to their signal transducing receptors, these results suggest that OSM requires binding to gp130 before it can bind OSMR. ELISA analysis of OSMR binding toward the cytokines in the presence of gp130 showed that prior gp130 binding induces remarkable cooperativity toward OSMR binding in both the wild-type and mutant OSMs (OSM-WT, OSM-M1, and OSM-M2). Although the data clearly showed that OSM utilizes the FXXK motif for OSMR binding, binding to gp130 might expose otherwise hidden residues on OSM required for OSMR binding or alter the OSM structure to move hindering residues away from the binding interface leading to the strong binding. Solving the structure of OSM in complex with gp130 would prove valuable in identifying these changes.
Finally, a number of studies conducted over the last decade have revealed the diverse biological roles of OSM. One among them is the growth modulation of cells that include tumor cells, epithelial cells, fibroblasts, and plasmacytoma cells (31, 33–37). In agreement with earlier studies, our results show that OSM inhibits the growth of A375 melanoma cells in a dose-dependent manner (Fig. 10). Mutant OSM proteins with a shorter BC loop exhibit increased potency in suppressing their proliferation (Fig. 10). This improvement in OSM's function could prove valuable in treating diseases associated with melanoma. Previous research in our laboratory has shown that STAT3 activation induced by IL-6 family cytokines, including OSM, is neuroprotective and prevents photoreceptor cell death under oxidative stress (38, 39). The mutant OSM molecules, OSM-M1 and OSM-M2, could thus serve as potent therapeutic agents in preventing photoreceptor degeneration induced by oxidative stress, e.g. retinitis pigmentosa. Also, OSM plays a key role in inflammatory response to injury and infection. OSM secreted from activated T cells and monocytes stimulates expression of the following: 1) acute phase proteins in liver (40); 2) P-selectin and E-selectin on endothelial cells (41, 42), and 3) TIMP-1 in fibroblasts (43, 44), all of which promote wound repair. The mutant OSM proteins could thus potentially find therapeutic application in promoting wound healing also.
Supplementary Material
Acknowledgment
We are grateful to John K. Heath (School of Biosciences, University of Birmingham, Birmingham, UK) for providing us with the clones for hLIF and LIF05 expression.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 EY016459 and Core Grants P20 RR017703 and P30 EY021721. This work was also supported by the Research to Prevent Blindness.

This article contains supplemental Table S1 and Figs. S1–S4.
- LIF
- leukemia inhibitory factor
- LIFR
- leukemia inhibitory factor receptor
- OSM
- oncostatin M
- OSMR
- OSM receptor
- CNTF
- ciliary neurotrophic factor
- SPR
- surface plasmon resonance
- PDB
- Protein Data Bank
- h
- human.
REFERENCES
- 1. Heinrich P. C., Behrmann I., Müller-Newen G., Schaper F., Graeve L. (1998) Interleukin-6-type cytokine signaling through the gp130/Jak/STAT pathway. Biochem. J. 334, 297–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bravo J., Heath J. K. (2000) Receptor recognition by gp130 cytokines. EMBO J. 19, 2399–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Simpson R. J., Hammacher A., Smith D. K., Matthews J. M., Ward L. D. (1997) Interleukin-6. Structure-function relationships. Protein Sci. 6, 929–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heinrich P. C., Graeve L., Rose-John S., Schneider-Mergener J., Dittrich E., Erren A., Gerhartz C., Hemann U., Lütticken C., Wegenka U. (1995) Membrane-bound and soluble interleukin-6 receptor. Studies on structure, regulation of expression, and signal transduction. Ann. N.Y. Acad. Sci. 762, 222–236 [DOI] [PubMed] [Google Scholar]
- 5. Hilton D. J., Hilton A. A., Raicevic A., Rakar S., Harrison-Smith M., Gough N. M., Begley C. G., Metcalf D., Nicola N. A., Willson T. A. (1994) Cloning of a murine IL-11 receptor α-chain; requirement for gp130 for high affinity binding and signal transduction. EMBO J. 13, 4765–4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamasaki K., Taga T., Hirata Y., Yawata H., Kawanishi Y., Seed B., Taniguchi T., Hirano T., Kishimoto T. (1988) Cloning and expression of the human interleukin-6 (BSF-2/IFN β2) receptor. Science 241, 825–828 [DOI] [PubMed] [Google Scholar]
- 7. Davis S., Aldrich T. H., Valenzuela D. M., Wong V. V., Furth M. E., Squinto S. P., Yancopoulos G. D. (1991) The receptor for ciliary neurotrophic factor. Science 253, 59–63 [DOI] [PubMed] [Google Scholar]
- 8. Robledo O., Fourcin M., Chevalier S., Guillet C., Auguste P., Pouplard-Barthelaix A., Pennica D., Gascan H. (1997) Signaling of the cardiotrophin-1 receptor. Evidence for a third receptor component. J. Biol. Chem. 272, 4855–4863 [DOI] [PubMed] [Google Scholar]
- 9. Taga T., Hibi M., Murakami M., Saito M., Yawata H., Narazaki M., Hirata Y., Sugita T., Yasukawa K., Hirano T. (1992) Interleukin-6 receptor and signals. Chem. Immunol. 51, 181–204 [PubMed] [Google Scholar]
- 10. Murakami M., Hibi M., Nakagawa N., Nakagawa T., Yasukawa K., Yamanishi K., Taga T., Kishimoto T. (1993) IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science 260, 1808–1810 [DOI] [PubMed] [Google Scholar]
- 11. Davis S., Aldrich T. H., Stahl N., Pan L., Taga T., Kishimoto T., Ip N. Y., Yancopoulos G. D. (1993) LIFRβa and gp130 as heterodimerizing signal transducers of the tripartite CNTF receptor. Science 260, 1805–1808 [DOI] [PubMed] [Google Scholar]
- 12. Gearing D. P., Comeau M. R., Friend D. J., Gimpel S. D., Thut C. J., McGourty J., Brasher K. K., King J. A., Gillis S., Mosley B. (1992) The IL-6 signal transducer, gp130. An oncostatin M receptor and affinity converter for the LIF receptor. Science 255, 1434–1437 [DOI] [PubMed] [Google Scholar]
- 13. Mosley B., De Imus C., Friend D., Boiani N., Thoma B., Park L. S., Cosman D. (1996) Dual oncostatin M (OSM) receptors. Cloning and characterization of an alternative signaling subunit conferring OSM-specific receptor activation. J. Biol. Chem. 271, 32635–32643 [DOI] [PubMed] [Google Scholar]
- 14. Auguste P., Guillet C., Fourcin M., Olivier C., Veziers J., Pouplard-Barthelaix A., Gascan H. (1997) Signaling of type II oncostatin M receptor. J. Biol. Chem. 272, 15760–15764 [DOI] [PubMed] [Google Scholar]
- 15. Hudson K. R., Vernallis A. B., Heath J. K. (1996) Characterization of the receptor-binding sites of human leukemia inhibitory factor and creation of antagonists. J. Biol. Chem. 271, 11971–11978 [DOI] [PubMed] [Google Scholar]
- 16. Deller M. C., Hudson K. R., Ikemizu S., Bravo J., Jones E. Y., Heath J. K. (2000) Crystal structure and functional dissection of the cytostatic cytokine oncostatin M. Structure 8, 863–874 [DOI] [PubMed] [Google Scholar]
- 17. Di Marco A., Gloaguen I., Graziani R., Paonessa G., Saggio I., Hudson K. R., Laufer R. (1996) Identification of ciliary neurotrophic factor (CNTF) residues essential for leukemia inhibitory factor receptor binding and generation of CNTF receptor antagonists. Proc. Natl. Acad. Sci. U.S.A. 93, 9247–9252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lütticken C., Wegenka U. M., Yuan J., Buschmann J., Schindler C., Ziemiecki A., Harpur A. G., Wilks A. F., Yasukawa K., Taga T. (1994) Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science 263, 89–92 [DOI] [PubMed] [Google Scholar]
- 19. Stahl N., Boulton T. G., Farruggella T., Ip N. Y., Davis S., Witthuhn B. A., Quelle F. W., Silvennoinen O., Barbieri G., Pellegrini S. (1994) Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6β receptor components. Science 263, 92–95 [DOI] [PubMed] [Google Scholar]
- 20. Stahl N., Farruggella T. J., Boulton T. G., Zhong Z., Darnell J. E., Jr., Yancopoulos G. D. (1995) Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science 267, 1349–1353 [DOI] [PubMed] [Google Scholar]
- 21. Darnell J. E., Jr., Kerr I. M., Stark G. R. (1994) Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–1421 [DOI] [PubMed] [Google Scholar]
- 22. Rose T. M., Lagrou M. J., Fransson I., Werelius B., Delattre O., Thomas G., de Jong P. J., Todaro G. J., Dumanski J. P. (1993) The genes for oncostatin M (OSM) and leukemia inhibitory factor (LIF) are tightly linked on human chromosome 22. Genomics 17, 136–140 [DOI] [PubMed] [Google Scholar]
- 23. Jeffery E., Price V., Gearing D. P. (1993) Close proximity of the genes for leukemia inhibitory factor and oncostatin M. Cytokine 5, 107–111 [DOI] [PubMed] [Google Scholar]
- 24. Giovannini M., Selleri L., Hermanson G. G., Evans G. A. (1993) Localization of the human oncostatin M gene (OSM) to chromosome 22q12, distal to the Ewing sarcoma breakpoint. Cytogenet. Cell Genet. 62, 32–34 [DOI] [PubMed] [Google Scholar]
- 25. Giovannini M., Djabali M., McElligott D., Selleri L., Evans G. A. (1993) Tandem linkage of genes coding for leukemia inhibitory factor (LIF) and oncostatin M (OSM) on human chromosome 22. Cytogenet. Cell Genet. 64, 240–244 [DOI] [PubMed] [Google Scholar]
- 26. Tanaka M., Miyajima A. (2003) Oncostatin M, a multifunctional cytokine. Rev. Physiol. Biochem. Pharmacol. 149, 39–52 [DOI] [PubMed] [Google Scholar]
- 27. Ichihara M., Hara T., Kim H., Murate T., Miyajima A. (1997) Oncostatin M and leukemia inhibitory factor do not use the same functional receptor in mice. Blood 90, 165–173 [PubMed] [Google Scholar]
- 28. Sporeno E., Paonessa G., Salvati A. L., Graziani R., Delmastro P., Ciliberto G., Toniatti C. (1994) Oncostatin M binds directly to gp130 and behaves as interleukin-6 antagonist on a cell line expressing gp130 but lacking functional oncostatin M receptors. J. Biol. Chem. 269, 10991–10995 [PubMed] [Google Scholar]
- 29. Liu H., Fenollar-Ferrer C., Cao A., Anselmi C., Carloni P., Liu J. (2009) Molecular dissection of human oncostatin M-mediated signal transductions through site-directed mutagenesis. Int. J. Mol. Med. 23, 161–172 [PubMed] [Google Scholar]
- 30. Vernallis A. B., Hudson K. R., Heath J. K. (1997) An antagonist for the leukemia inhibitory factor receptor inhibits leukemia inhibitory factor, cardiotrophin-1, ciliary neurotrophic factor, and oncostatin M. J. Biol. Chem. 272, 26947–26952 [DOI] [PubMed] [Google Scholar]
- 31. Zarling J. M., Shoyab M., Marquardt H., Hanson M. B., Lioubin M. N., Todaro G. J. (1986) Oncostatin M. A growth regulator produced by differentiated histiocytic lymphoma cells. Proc. Natl. Acad. Sci. U.S.A. 83, 9739–9743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu J., Modrell B., Aruffo A., Marken J. S., Taga T., Yasukawa K., Murakami M., Kishimoto T., Shoyab M. (1992) Interleukin-6 signal transducer gp130 mediates oncostatin M signaling. J. Biol. Chem. 267, 16763–16766 [PubMed] [Google Scholar]
- 33. Liu J., Spence M. J., Wallace P. M., Forcier K., Hellström I., Vestal R. E. (1997) Oncostatin M-specific receptor mediates inhibition of breast cancer cell growth and down-regulation of the c-myc proto-oncogene. Cell Growth Differ. 8, 667–676 [PubMed] [Google Scholar]
- 34. Horn D., Fitzpatrick W. C., Gompper P. T., Ochs V., Bolton-Hansen M., Zarling J., Malik N., Todaro G. J., Linsley P. S. (1990) Regulation of cell growth by recombinant oncostatin M. Growth Factors 2, 157–165 [DOI] [PubMed] [Google Scholar]
- 35. Liu J., Hadjokas N., Mosley B., Estrov Z., Spence M. J., Vestal R. E. (1998) Oncostatin M-specific receptor expression and function in regulating cell proliferation of normal and malignant mammary epithelial cells. Cytokine 10, 295–302 [DOI] [PubMed] [Google Scholar]
- 36. Grant S. L., Douglas A. M., Goss G. A., Begley C. G. (2001) Oncostatin M and leukemia inhibitory factor regulate the growth of normal human breast epithelial cells. Growth Factors 19, 153–162 [DOI] [PubMed] [Google Scholar]
- 37. Nishimoto N., Ogata A., Shima Y., Tani Y., Ogawa H., Nakagawa M., Sugiyama H., Yoshizaki K., Kishimoto T. (1994) Oncostatin M, leukemia inhibitory factor, and interleukin 6 induce the proliferation of human plasmacytoma cells via the common signal transducer, gp130. J. Exp. Med. 179, 1343–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chollangi S., Wang J., Martin A., Quinn J., Ash J. D. (2009) Preconditioning-induced protection from oxidative injury is mediated by leukemia inhibitory factor receptor (LIFR) and its ligands in the retina. Neurobiol. Dis. 34, 535–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ueki Y., Wang J., Chollangi S., Ash J. D. (2008) STAT3 activation in photoreceptors by leukemia inhibitory factor is associated with protection from light damage. J. Neurochem. 105, 784–796 [DOI] [PubMed] [Google Scholar]
- 40. Richards C. D., Brown T. J., Shoyab M., Baumann H., Gauldie J. (1992) Recombinant oncostatin M stimulates the production of acute phase proteins in HepG2 cells and rat primary hepatocytes in vitro. J. Immunol. 148, 1731–1736 [PubMed] [Google Scholar]
- 41. Yao L., Pan J., Setiadi H., Patel K. D., McEver R. P. (1996) Interleukin 4 or oncostatin M induces a prolonged increase in P-selectin mRNA and protein in human endothelial cells. J. Exp. Med. 184, 81–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Modur V., Feldhaus M. J., Weyrich A. S., Jicha D. L., Prescott S. M., Zimmerman G. A., McIntyre T. M. (1997) Oncostatin M is a proinflammatory mediator. In vivo effects correlate with endothelial cell expression of inflammatory cytokines and adhesion molecules. J. Clin. Invest. 100, 158–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Richards C. D., Kerr C., Tanaka M., Hara T., Miyajima A., Pennica D., Botelho F., Langdon C. M. (1997) Regulation of tissue inhibitor of metalloproteinase-1 in fibroblasts and acute phase proteins in hepatocytes in vitro by mouse oncostatin M, cardiotrophin-1, and IL-6. J. Immunol. 159, 2431–2437 [PubMed] [Google Scholar]
- 44. Richards C. D., Shoyab M., Brown T. J., Gauldie J. (1993) Selective regulation of metalloproteinase inhibitor (TIMP-1) by oncostatin M in fibroblasts in culture. J. Immunol. 150, 5596–5603 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.