

Background: The Sulf enzymes regulate HS-dependent extracellular signaling during skeletal muscle regeneration.
Results: The skeletal muscle-specific Sulf double null mice exhibit reduced new myofiber size and defective Wnt signaling.
Conclusion: Sulfs promote canonical Wnt signaling to antagonize noncanonical signaling, thereby enhancing myoblast fusion.
Significance: Understanding the mechanisms of myoblast fusion will help to enhance the efficacy of muscle regeneration.
Keywords: Cell Signaling, Heparan Sulfate, Muscle Regeneration, Myogenesis, Wnt Pathway, Sulf, Myoblast Cell, Satellite Cell
Abstract
Skeletal muscle regeneration is mediated by satellite cells (SCs). Upon injury, SCs undergo self-renewal, proliferation, and differentiation into myoblasts followed by myoblast fusion to form new myofibers. We previously showed that the heparan sulfate (HS) 6-O-endosulfatases (Sulf1 and -2) repress FGF signaling to induce SC differentiation during muscle regeneration. Here, we identify a novel role of Sulfs in myoblast fusion using a skeletal muscle-specific Sulf double null (SulfSK-DN) mouse. Regenerating SulfSK-DN muscles exhibit reduced canonical Wnt signaling and elevated non-canonical Wnt signaling. In addition, we show that Sulfs are required to repress non-canonical Wnt signaling to promote myoblast fusion. Notably, skeletal muscle-relevant non-canonical Wnt ligands lack HS binding capacity, suggesting that Sulfs indirectly repress this pathway. Mechanistically, we show that Sulfs reduce the canonical Wnt-HS binding and regulate colocalization of the co-receptor LRP5 with caveolin3. Therefore, Sulfs may increase the bioavailability of canonical Wnts for Frizzled receptor and LRP5/6 interaction in lipid raft, which may in turn antagonize non-canonical Wnt signaling. Furthermore, changes in subcellular distribution of active focal adhesion kinase (FAK) are associated with the fusion defect of Sulf-deficient myoblasts and upon non-canonical Wnt treatment. Together, our findings uncover a critical role of Sulfs in myoblast fusion by promoting antagonizing canonical Wnt signaling activities against the noncanonical Wnt pathway during skeletal muscle regeneration.
Introduction
Satellite cells (SCs)2 are stem cells that reside between the sarcolemma and the basal lamina of myofibers in adult skeletal muscle (1, 2). In addition to unique localization, SCs also express a distinct transcription factor Pax7. Although SCs are normally quiescent, injury causes SC to exit quiescence and enter into the cell cycle. SCs proliferate to self-renew and to expand the pool of committed myoblasts. The activation, proliferation, and self-renewal of SCs are controlled by a plethora of extracellular signals, including Notch, fibroblast, and hepatocyte growth factors and Wnts (1, 2).
Myoblast fusion to form new myofibers is an integral step of skeletal muscle regeneration (3). Fusion is initiated by forming nascent myofibers with only a few nuclei followed by fusion of more myoblasts to generate a mature myotube with many nuclei. Myoblast fusion in mammals not only involves a number of proteins that are located on the cell surface or at the contact site but also requires intracellular focal adhesion kinase (FAK), Erk5, Rac1, and CDC42 (4–6). Relatively little is known about the extracellular signals that regulate myoblast fusion. Recent studies suggest a role of canonical Wnt signaling in myoblast fusion. Although new myofibers form in regenerating muscle between day 4 and day 5 post-injury, injection of canonical Wnt3a increases the size of new myofibers without changing myofiber number in vivo (7). In vitro assays also show that canonical Wnts induce myoblast fusion (8, 9).
Several extracellular signals involved in skeletal muscle regeneration are regulated by the heparan sulfate (HS) 6-O-endosulfatases (Sulfs), Sulf1&2 (10). HS is a linear carbohydrate polymer that is covalently attached to a protein core of heparan sulfate proteoglycans on the cell surface, in the basement membrane, and in the extracellular matrix (11). HS consists of tandem repeats of uronic acid and glucosamine. This disaccharide unit undergoes a variety of modifications during biosynthesis in the Golgi, including epimerization and 2-O-sulfation of uronic acid and N-, 3-O-, and 6-O-sulfation of glucosamine. These modifications are usually incomplete, thereby generating domains that have high or low levels of sulfation or lack sulfation along the length of the HS chain (11).
Sulfs are the only known HS-editing enzymes that modify sulfated HS sequences after HS biosynthesis (10). Sulfs are tethered on the cell surface after secretion (12). Both Sulf1 and Sulf2 remove a subset of 6-O-sulfate groups specifically from trisulfated IdoA2S-GlcNS6S (D2S6) and disulfated GlcA-GlcNS6S (D0S6) disaccharides (13–15). Sulf-mediated HS 6-O-desulfation disrupts the formation of HS-FGF2-FGF receptor ternary complex, leading to repressed FGF signaling (16, 17). In contrast, Sulfs promote canonical Wnt signaling by reducing Wnt binding to HS, thereby promoting Wnt bioavailability for receptor binding (13, 18). The ability of Sulfs to enhance canonical Wnt signaling requires Sulf localization into lipid rafts on the cell membrane (19).
Our previous studies in systemic Sulf double mutant mice demonstrate that Sulfs are required for timely differentiation of SCs during skeletal muscle regeneration (20). Only Sulf1 is present in uninjured skeletal muscle and quiescent SCs, whereas both Sulfs are expressed by activated satellite cells. Although Sulfs are dispensable for skeletal muscle formation during development, they redundantly repress FGF2 and hepatocyte growth factor signaling in activated SCs to induce differentiation (20). Notably, Sulf-deficient and wild type (WT) SCs differentiate similarly without the presence of FGF2 in culture, and Sulf deficiency only leads to a transient increase in the number of SCs in regenerating skeletal muscle (20). These observations support that Sulfs are regulators rather than essential components of the extracellular signaling pathways, and as a result, Sulfs function in a growth factor-dependent manner.
In addition to SCs, myoblasts and new myofibers also express both Sulf1 and Sulf2 (20), suggesting additional roles of Sulfs in myoblasts during new myofiber formation. However, Sulf1−/−;Sulf2−/− mice exhibit partial post-natal lethality, multiple tissue defects, and reduced skeletal muscle mass (21, 22). These phenotypes complicate functional studies of Sulfs in the adult using this model. To overcome the limitation, we generated a skeletal muscle-specific Sulf double null (SulfSK-DN) mouse line to investigate functions of Sulfs during skeletal muscle regeneration. SulfSK-DN mice exhibit SC phenotypes during regeneration, similar to Sulf1−/−;Sulf2−/− mice (20). In addition, SulfSK-DN mice show defects in new myofiber formation. This study focuses on mechanisms underlying Sulf-regulated myoblast fusion.
EXPERIMENTAL PROCEDURES
Mice
The skeletal muscle-specific Sulf double mutant mice (SulfSK-DN) were generated by crossing a knock-in Myf5-Cre line (23) with a mouse that carried floxed Sulf1 and Sulf2 genes (Sulff/f;Sulf2f/f) (21) followed by mating Myf5-Cre+;Sulf1f/+;Sulf2f/+ male mice with Sulff/f;Sulf2f/f female mice. After genotyping, Myf5-Cre+;Sulf1f/f;Sulf2f/f mice were then crossed with Sulff/f;Sulf2f/f mice to generate SulfSK-DN mice. Littermates that do not carry the Myf5-Cre allele were used as WT controls in all experiments. All lines were in C57BL/6 background. Details of genotyping are provided in the supplemental Methods. All animal studies were approved by the Institutional Animal Care and Use Committee of Boston University Medical Campus.
Antibodies and Other Reagents
Recombinant mouse secreted frizzled-related protein 2 (sFRP2), mouse Dkk1, mouse Wnt3a, and human Wnt7a were purchased from R&D Systems. Cardiotoxin (Naja nigricollis, catalog #217504) was purchased from Calbiochem. Primary antibodies against p-CamKiiα(Thr-286)-R (sc-12886-R), p-JNK (sc-6254), p-FAK (sc-81493), MYH3 (sc-53091), LRP5 (sc-134463), and myogenin (sc-12732) were purchased from Santa Cruz Biotechnology. Other antibodies include anti-Wnt3a antibody (catalog #09-162, Millipore), anti-active β-catenin antibody (catalog #05-665, Millipore), anti-laminin antibody (catalog #L9393, Sigma), anti-Myc antibody (9E10, Sigma), anti-Gapdh antibody (catalog #Ab8245, Abcam), and anti-MF20 and anti-Pax7 antibodies (Developmental Studies Hybridoma Bank, Iowa University).
Skeletal Muscle Injury and Regeneration
Tibialis anterior (TA) muscles of 2-month-old WT and SulfSK-DN mice were injured by injecting 100 μl of 10 μm cardiotoxin (CTX) as day 0. Muscles were collected on day 3 or day 5 post-injury. Half of the TA muscles were frozen in liquid nitrogen for protein and mRNA analysis, and the remaining half was preserved in Optimal Cutting Temperature (OCT, catalog #4583, Tissue-Tek) compound and quick-frozen in liquid nitrogen-cooled isopentane for immunohistochemical analysis. For Wnt signaling studies, TA muscles were injured with CTX for 24 h and then injected with 50 μl of sFRP2 (2 μg/ml) or Wnt7a (25 ng/ml).
Heparan Sulfate Analysis
Tissue-derived HS structural analysis was performed as described previously (24, 25). TA muscles of 2-month-old WT and SulfSK-DN mice were digested with Pronase at 37 °C for 24 h followed by heat inactivation of the Pronase and further digestion with benzonase at 37 °C for 3 h. To release glycosamino glycans from proteins, samples were treated with 0.5 m NaOH for 24 h at 4 °C. The solutions were neutralized with formic acid and centrifuged at 11,000 × g for 10 min. The supernatants were subjected to DEAE weak ion exchange and washed with 0.1 m NaCl, pH 6. GAGs were eluted with 1 m NaCl, desalted by PD-10 column, and vacuum-dried. To break down HS chains, GAGs were incubated with heparin lyase I, II, III (Ibex, Montreal, Canada) at 37 °C overnight followed by second aliquot of enzymes for 16 more hours. Samples, along with seven artificial mixtures of D2S6 and D2S0 standards, were subjected to size exclusion chromatography LC/MS analysis and run tandem MS to differentiate D0S6 and D2S0. HS disaccharide standards and all chemicals were from Sigma.
Satellite Cell Isolation, Myoblast Culture, and Immunocytochemistry
Satellite cell isolation from hind limb skeletal muscles of 2-month-old mice was prepared as described previously (20). Cells were maintained in growth medium containing 20% fetal bovine serum (Hyclone), 2% chick embryonic extract, and 1% penicillin/streptomycin (Invitrogen) in DMEM medium. All culture plates were coated with ECL Cell Attachment Matrix (1:20 dilution, catalog #08-110, Millipore) for 1 h at 37 °C. For staining, low passage primary myoblast cells (∼50,000 cells per well) were seeded at relative low ∼25% confluence on the 8-well Permanox Chamber Slide (catalog #177445, Nunc) in growth medium for 24 h before switching to differentiating medium that contained 2% horse serum and 1% penicillin/streptomycin in DMEM for 24 or 48 h. Cells in differentiating medium were treated with vehicle, rWnt3a (30 ng/ml), rWnt7a (100 ng/ml), sFRP2 (2 μg/ml), or Dkk1 (0.5 μg/ml) before cells were fixed with 4% paraformaldehyde for 30 min at room temperature. Cell immunolabeling was performed as described previously (20). Primary antibodies included anti-myogenin (1:50) and p-FAK (1:50), anti-MF20 (1:10), Myf5 (1:500), anti-caveolin3 (1:1000), and anti-LRP5 (1:500). Antigen-antibody complex was detected with fluorescence-conjugated secondary antibodies (1:250) and nuclei with Hoechst dye (1 μg/ml). Signals were visualized under a fluorescent Zeiss 100 microscope. For confocal microscopy, immunolabeled myoblast cells were imaged using a Zeiss Axiovert 100 m LSM 510 equipped with an argon and two HeNe lasers.
Immunohistochemistry
Optimal Cutting Temperature-preserved TA muscles were cut into 8-μm sections and stored at −20 °C. Sections were thaw to room temperature and fixed with 4% paraformaldehyde for 20 min followed by immunostaining procedures described previously (20). Primary antibodies included anti-laminin (1:100) and anti-eMyh (1:50) or anti-Pax7 (1:3). Secondary antibodies included Alexa-488 anti-mouse or Alexa548 anti-rabbit (1:250, Molecular Probes). Nuclei were stained by Hoechst dye (1 μg/ml). Signals were visualized under the fluorescent Zeiss 100 microscope. A minimum of three non-overlapping, representative images were taken under a 20× objective lens for further analysis. Myofiber size was quantified using AxioVision 4.8 software. At least three muscles under each treatment condition were utilized for statistical analysis.
Western Blot
Flash-frozen TA muscles were minced into small pieces in RIPA buffer containing phosphatase inhibitor (catalog #524627, Calbiochem) and proteinase inhibitor (catalog #11836153001, Roche Diagnostics). Muscle pieces were homogenized by PowerGen 125 (Fisher), sonicated, and centrifuged, and supernatants were collected with an equal volume of 2× Laemmli buffer. For assays with primary cultures, myoblasts (∼1 × 105 cells per well) in 6-well plates were induced to form myofibers by maintaining in differentiating medium for 24 or 48 h. Cells were washed once with PBS, collected in 100 μl of Laemmli Buffer (2×). Protein content in the cell and tissue samples was measured using a protein assay kit (Bio-Rad). Protein samples (20 μg) were resolved by NuPAGE® 4–12% Bis-Tris gels (catalog #NP0323BOX, Invitrogen) followed by transferring onto Hybond-ECL nitrocellulose membrane (catalog # RPN303D, Amersham Biosciences). Membranes were blocked with 5% BSA for 1 h and incubated with primary antibodies against active β-catenin (1:5,000), p-CamKii (1:2,500), p-JNK (1:2,000), p-FAK (1:10,000), caveolin3 (1:10,000) and Gapdh (1:100,000) in 2.5% BSA overnight at 4 °C. Membranes were washed 3× with phosphate buffer containing 0.1% Triton X-100 (PBST) and incubated with secondary antibody (1:10000) in 2.5% milk for 1 h. Membranes were washed 3 times with PBST and exposed to ECL reagent for ∼2 min followed by signal detection.
Real-time Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Satellite cells in 6-well plates were treated with vehicle or Wnt3a in differentiating medium for 24 h and collected for RNA isolation using RNeasy Mini Kit (catalog #74106, Qiagen). Total RNA (1 μg) was reverse-transcribed using SuperScript® III First-Strand Synthesis SuperMix (catalog #18080-400, Invitrogen). Quantitative PCR was performed using Power SYBR® Green PCR Master Mix (catalog #4367659, Applied Biosystems) and the StepOnePlusTM Real-time PCR system. The relative level of Axin 2 mRNA expression was calculated by normalizing to β-actin mRNA using ΔCt. Primers used in PCR were: Axin2_forward (5′-CGCAGGCTGGCAGAGGTGTC-3′) and Axin2_reverse (′-GGGTTGGCGAAGGGTGAGGC-3′; β-actin_forward (5′-GCAGCTCCTTCGTTGCCGGT-3′) and β-actin_reverse (5′-TACAGCCCGGGGAGCATCGT-3′).
Heparin Binding Assay
HEK 293T cells were transfected with pCMV-DyRed (empty vector), pCMV-Sulf2-Myc, and pCMV-Sulf2(C-S)-Myc plasmids using LipoD392TM in vitro transfection reagent (catalog #SL100668, SingaGen Laboratories). After 48 h, cell lysates were collected and dialyzed in Tris-buffered saline (TBS) buffer containing proteinase and phosphatase inhibitors as described previously (13). Dialyzed cell lysates were then incubated with heparin-conjugated agarose beads in TBS buffer containing 50 mm MgCl2 overnight at 37 °C with rotation. Treated heparin beads were washed 3 times with TBS, aliquoted in 20 μl per tube, and blocked with 1% BSA in TBS for 1 h at room temperature. After washing, heparin beads were incubated with rWnt3a (1 ng) or increasing amounts of rWnt7a (10, 30, or 100 ng) in 50 μl for 30 min at room temperature. Beads were washed before adding 40 μl of 2× Laemmli buffer to collect bound Wnts followed by Western blot analysis.
Statistical Analysis
Data were presented as the mean ± S.D. of a minimum of three independent assays. Statistic significance was analyzed using Student's t test as paired or equal variance where appropriate.
RESULTS
The Skeletal Muscle-specific Sulf Double Null Mice Exhibit Tissue-specific Changes in HS 6-O-Sulfation
The SulfSK-DN mice were generated by crossing a knock-in Myf5-Cre line (23) with a Sulf1f/f;Sulf2f/f line that carries loxp sites flanking the second coding exons of both Sulf1 and Sulf2 genes (supplemental Figs. 1 and 2) (21). This exon encodes for essential amino acid sequences within Sulf enzymatic domain (21). Consistent with Cre expression in the skeletal muscle and satellite cells (23, 26), recombinant null alleles of Sulf1 and Sulf2 genes were detected in the skeletal muscle of SulfSK-DN mice (supplemental Figs. 1 and 2). In addition, satellite cells isolated from SulfSK-DN mice had diminished Sulf1 and Sulf2 mRNA expression as compared with WT controls (supplemental Fig. 3).
To validate a loss of Sulf enzymatic activity in SulfSK-DN mice, we compared the sulfation level of HS isolated from the TA muscles of WT and SulfSK-DN mice at 2 months of age using mass spectrometry. The skeletal muscle expresses HS with very low levels of sulfation (24). Disaccharide analysis showed that the HS samples from both WT and SulfSK-DN mice contained a large majority of non-sulfated, N-acetylated disaccharides and similarly low average numbers of sulfate group per disaccharide in depolymerized HS samples (Fig. 1, A and B). Considering that Sulfs only remove a subset of 6-O-sulfate groups from D2S6 and D0S6 substrates (13–15) that are of low abundance in the skeletal muscle (24), our assay did not detect any significant changes in general HS sulfation in SulfSK-DN mice likely due to sensitivity issue. In addition, the skeletal muscle contains multiple cell types including fibroblast cells and endothelial cells that are not affected by Myf5-Cre expression. The complexity of cellular sources of HS provides an additional compounding factor to our disaccharide assay. Because of these limitations, we also did not detect any significant increase in the abundance of trisulfated D2S6 in SulfSK-DN mice.
FIGURE 1.

Comparison of HS structure and HS biosynthetic gene expression in TA muscles of WT and SulfSK-DN mice. Purified HS from the muscle was enzymatically digested into disaccharides followed by mass spectrometry analysis to assess levels of N-sulfated (N-S) and N-acetylated (N-Ac) disaccharides in A, average number of sulfate groups per disaccharide in B, and the abundance of disulfated Sulf substrate (D0S6) and di-sulfated non-substrate (D2S0) in C. Sulf deficiency selectively increases the abundance of D0S6 without affecting N-sulfation or general sulfation levels of HS in the skeletal muscle. D, quantitative RT-PCR analysis compares mRNA expression of genes involved in HS biosynthesis in TA muscles of WT and SulfSK-DN mice. No change in mRNA expression of Ndst1–4, Hs2Ost, and Hs6Ost1 genes was observed. Data represent the mean and S.D. of four independent assays. **, p < 0.05. GE, gene expression.
Disulfated Sulf substrate D0S6 was found to be more abundant than the trisulfated substrate D2S6. Because Sulfs enzymatically convert D2S6 into D2S0 and D0S6 into D0S0 (13–15), HS of SulfSK-DN muscles is predicted to contain less D2S0 but more D0S6 than the WT control. We thus reasoned that relative abundance of D2S0 and D0S6 would provide a sensitive assessment for changes in Sulf activity in SulfSK-DN muscles. Indeed, compared with WT controls, HS of SulfSK-DN mice exhibited an ∼50% increase in the abundance of D0S6 (22.4 ± 8.4 versus 15.4 ± 5.2%, p < 0.05) with a reciprocal reduction in the levels of D2S0 (77.6 ± 8.4 versus 84.6 ± 5.2%, p < 0.05) (Fig. 1C). The mRNA expression of genes involved in sulfation during HS biosynthesis, such as N-deacetylase/N-sulfotransferase 1–4 (Ndst 1–4), 2-O-sulfotransferase (Hs2st), and 6-O sulfotransferase (Hs6st), was not changed in SulfSK-DN muscles (Fig. 1D). Collectively, our findings indicate that SulfSK-DN mice have selectively deficient Sulf activities in the skeletal muscle.
SulfSK
DN mice appear indistinguishable from WT littermates. The skeletal muscles of SulfSK-DN mice also develop normally, consistent with our previous observation that Sulfs are not required for the formation of the skeletal muscle or satellite cells (20). Therefore, SulfSK-DN mice provide an ideal model to investigate Sulf function during skeletal muscle regeneration.
Sulfs Promote SC Differentiation and Myoblast Fusion during Skeletal Muscle Regeneration
To investigate the role of Sulfs in skeletal muscle regeneration, the TA muscles of WT and SulfSK-DN mice were injured by CTX injection followed by histological assays to assess the efficacy of regeneration. At day 5 post-injury, SulfSK-DN muscles had almost twice as many Pax7+ SCs as WT controls (comparing Fig. 2Ab with Fig. 2Ad, quantified in Fig. 2Ba). The increase was transient, as no difference in the number of SCs was identified in SulfSK-DN muscles at day 14 post-injury (supplemental Fig. 4). This observation is consistent with a previously characterized role of Sulfs in promoting SC differentiation (20).
FIGURE 2.

Sulfs promote SC differentiation and myoblast fusion during skeletal muscle regeneration. TA muscles of WT and SulfSK-DN mice were harvested at day 5 post-injury. A, Pax7+ SCs and eMyh+ newly formed myofibers were identified by immunohistochemistry using mid-belly cross-sections. The basement membrane of myofibers was labeled with a laminin antibody. Nuclei were labeled by Hoechst dye. Scale bar = 100 μm. B, shown is quantification of relative abundance of Pax7+ SCs (a), relative abundance of new myofibers with increasing number of myonuclei (b) and sizes (c), and average number and size of new myofibers within a regenerating area of 1 mm2 (d) of WT and SulfSK-DN muscles. Data presented are the mean and S.D. of more than 1000 new myofibers from 3 independent experiments. #, p < 0.01; **, p < 0.05; *, p < 0.1.
Regenerating TA muscles of SulfSK-DN mice also exhibited delayed new myofiber formation. Newly formed myofibers at day 5 post-injury were identified by their centrally located myonuclei and expression of the embryonic isoform of myosin heavy chain (eMyh) (Fig. 2A, a and c). We measured the size of mid-belly cross-sections of new myofibers. Compared with WT muscles, SulfSK-DN muscles had significantly higher percentages of small new myofibers (less than 375 μm2 in area size) and less abundant large new myofibers (more than 875 μm2 in area size) (Fig. 2, Bc), resulting in reduced average size of new myofibers in SulfSK-DN mice (498 ± 203 μm2 (SulfSK-DN) versus 967 ± 243 μm2 (WT), p < 0.01) (Fig. 2Bd).
Although a transient increase in SC number during regeneration of SulfSK-DN mice may affect the size of the myoblast pool for fusion, Sulfs may directly regulate myoblast fusion because Sulfs are highly expressed in myoblasts and new myofibers (20). If SulfSK-DN mice have a reduced number of myoblasts, the total area of new myofibers would be reduced as compared with WT controls. Alternatively, if SulfSK-DN myoblasts were defective in fusion, regenerating SulfSK-DN muscles would have smaller myofibers with less myonuclei but more myofibers, as compared with WT controls, so that the total new myofiber area would be similar between these two genotypes. To distinguish between these two possibilities, we quantified the number and area of new myofibers and myonuclei per fiber in both WT and SulfSK-DN muscles at day 5 post-injury. We found that regenerating SulfSK-DN muscles had more new myofibers than WT muscles (525 ± 232 (SulfSK-DN) versus 282 ± 96 (WT) per mm2; p < 0.05) (Fig. 2Bd). However, the total area of new myofibers was found to be similar between SulfSK-DN and WT muscles. In addition, ∼80% of new myofibers in SulfSK-DN muscles had only one myonuclei, as compared with 58% in WT muscles. Also, less than 3% of new SulfSK-DN myofibers had more than two myonuclei, as compared with 13% in WT muscles (Fig. 2Bb). These observations support a role of Sulfs in myoblast fusion.
To further test if Sulfs promote myoblast fusion, WT and SulfSK-DN SCs in low passage cultures were induced to differentiate into myoblasts by removal of FGF2. Satellite cells were seeded at relatively low density so that individual myofibers are easily identifiable. Under this condition, both WT and SulfSK-DN SCs turn off Pax7 expression, withdraw from cell cycle and similarly activate myogenin expression (Fig. 3, A and B) (20). After 48 h, we assessed myoblast fusion by double staining. Newly formed myofiber was labeled with a monoclonal MF20 antibody. Because the myogenin antibody is also monoclonal and, therefore, cannot be used in combination with the MF20 antibody, we tested a polyclonal anti-Myf5 antibody. Myf5 protein was found to be readily detectable in the myonuclei in our assay (Fig. 3, C and D), although Myf5 mRNA expression is known to be down-regulated during myoblast fusion. Double staining using this Myf5 antibody and the MF20 antibody showed that ∼50% of the WT myofibers contained more than one nucleus, as compared with less than 20% of multinucleated SulfSK-DN myofibers (Fig. 3, C and D). This observation provides direct evidence that Sulfs promote myoblast fusion.
FIGURE 3.

Sulfs promote myoblast fusion in culture. Primary cultures of WT and SulfSK-DN myoblast cells were maintained in differentiation media without the presence of FGF2 for 48 h before assessment of myogenin expression in A and B and fusion in C and D. Nuclei were labeled by Hoechst. Myofibers were labeled with the MF20 antibody in C. Percentages of myogenin+ cell in WT and SulfSK-DN cultures were shown in B. The efficacy of fusion was quantified by percentages of mono- and multi-nucleated MF20+ myofibers in D. Data represent the mean and S.D. of three independent experiments. #, p < 0.01; **, p < 0.05; *, p < 0.1. Scale bar = 100 μm.
It is important to note that no difference in new myofiber size was found between WT and SulfSK-DN muscles at day 14 post-injury (supplemental Fig. 4). This is consistent with regulatory, rather than essential, roles of Sulfs in HS-dependent signaling pathways that are involved in myoblast fusion.
Sulfs Differentially Regulate the Canonical and Non-canonical Wnt Signaling Pathways
Candidate HS-dependent extracellular signals that are involved in myoblast fusion are canonical Wnts. Myoblasts express several canonical and non-canonical Wnts (27). Canonical Wnts trigger stabilization and nuclear translocation of β-catenin, whereas non-canonical Wnts activates Ca2+-dependent PKC, CamKii, and/or small G-proteins such as Rac and RhoA that lead to activation of c-Jun N-terminal kinase (Jnk) (28, 29).
We tested whether Sulfs regulate the Wnt pathways by comparing basal and stimulated signaling levels between WT and SulfSK-DN myoblasts in culture. Canonical Wnt signaling was assayed by Western blot to measure stabilized non-phosphorylated β-catenin. SulfSK-DN cultures had lower levels of stabilized β-catenin under basal conditions and in response to increasing levels of exogenous canonical Wnt3a, as compared with WT cultures (Fig. 4, A and B). However, treatment with 6-bromoin-dirubin-3′-oxime, a specific inhibitor of GSK3β, significantly increased the level of non-phosphorylated β-catenin to a similar level in both WT and SulfSK-DN myoblast cultures (Fig. 4A). GSK3β is a downstream kinase that facilitates the degradation of β-catenin through phosphorylation (28, 29). This finding is consistent with the established role of Sulfs in promoting the bioavailability of canonical Wnts in the extracellular matrix (13, 18), and therefore, blockade of downstream inhibitors of canonical Wnt signaling rescued the signaling defect of SulfSK-DN myoblast cells.
FIGURE 4.

Sulfs differentially regulate canonical and noncanonical Wnt signaling in myoblast culture. A, primary WT and SulfSK-DN myoblast cultures were treated with increasing amounts of recombinant Wnt3a (Wnt3a; 0, 1, 3, 10, 30, 100 ng/ml) and specific glycogen synthase kinase 3β inhibitor 6-bromoin-dirubin-3′-oxime (5 μm) for 24 h and subjected to Western blot analysis to detect stabilized β-catenin. Gapdh was the loading control. Images represent one of three independent experiments. B, quantification of stabilized β-catenin levels upon Wnt3a treatment after normalizing to Gapdh is shown. Densitometry was shown as the mean ± S.D., n = 3. **, p < 0.05; *, p < 0.1. C, WT and SulfSK-DN myoblast cultures were treated with vehicle or Wnt3a (30 ng/ml) for 48 h. The induction of mRNA expression of canonical Wnt target gene Axin2 was assayed by quantitative RT-PCR. Relative gene expression (in C) and -fold induction (in D) of Axin2 gene expression were shown. Data represent the mean and S.D. of four independent experiments. #, p < 0.01. E, WT and SulfSK-DN cultures were treated with increasing amount of recombinant Wnt7a (Wnt7a; 0, 1, 3, 10, 30, 100 ng/ml) for 1 h. Western blot assays were used to detect downstream noncanonical Wnt signaling components, p-Jnk and p-CamKii. Signals were normalized to Gapdh. Results of two independent experiments was shown in F. DU, density unit.
Wnt3a increased mRNA expression of a target gene Axin2 by ∼45-fold above the basal level in WT myoblast cultures as compared with ∼14-fold in SulfSK-DN myoblast cultures (Fig. 4C). This observation indicated that Sulfs promoted canonical Wnt signaling by ∼3-fold in myoblasts (Fig. 4D), consistent with previous findings (13, 28). Interestingly, the basal level of Axin2 gene expression was higher in SulfSK-DN myoblast cultures than in WT myoblast cultures (Fig. 4C). Considering Axin2 is a negative regulator of the canonical Wnt signaling, the elevated basal Axin2 gene expression may provide a mechanism to repress basal canonical Wnt signaling in SulfSK-DN myoblasts as compared with WT myoblasts.
In contrast, downstream effectors of the non-canonical Wnt signaling pathway, phosphorylated CamKii (p-CamKii) and Jnk (p-Jnk), were elevated in SulfSK-DN cultures as compared with WT cultures under basal conditions (Fig. 4, E and F). Stimulation with a physiologically relevant noncanonical Wnt7a (27) increased the level of p-CamKii in both cultures, and the level in SulfSK-DN cultures was consistently higher than WT cultures at all tested concentrations (Fig. 4, E and F). Interestingly, Jnk activation in SulfSK-DN myoblasts was higher than WT myoblasts at 1 ng/ml Wnt7a, peaked at 3 ng/ml Wnt7a, and was reduced to levels similar or even lower than WT myoblast cells at higher concentrations of Wnt7a (Fig. 4, E and F). Notably, WT myoblast cultures exhibited a similar dose curve of p-Jnk in response to Wnt7a as SulfSK-DN myoblast cultures except that the peak in WT cultures was at 10 ng/ml Wnt7a (Fig. 4, E and F). Collectively, these observations indicate that Sulfs negatively regulate the noncanonical Wnt pathway in the myoblasts: Sulfs repress CamKii activation and blunt Jnk activation.
Wnt7a Injection Mimics Sulf Deficiency during Skeletal Muscle Regeneration
Elevated non-canonical Wnt signaling in SulfSK-DN myoblasts may block myoblast fusion. If true, WT muscles injected with Wnt7a would exhibit similar defects in new myofiber formation as SulfSK-DN muscles. Before testing this hypothesis, we characterized the kinetics of non-canonical Wnt signaling during skeletal muscle regeneration. Western blot assays showed that levels of downstream p-CamKii and p-Jnk increased upon injury, peaked around day 3 post-injury, and fully recovered 1 week after injury (Fig. 5A). Although CamKii and Jnk can be activated by multiple signals, we observed that their activation was affected by Wnt-specific inhibitors, indicating that CamKii and Jnk activation were reliable readouts for non-canonical Wnt signaling in our assays (see Fig. 7, A and B). Therefore, non-canonical Wnt signaling is activated before and during myoblast fusion between day 3 and day 5 post-injury.
FIGURE 5.

Elevated non-canonical Wnt signaling partially mimics Sulf deficiency during skeletal muscle regeneration. A, temporal activation of Camkii and Jnk, two downstream effectors of noncanonical Wnt signaling, after CTX-induced injury of TA muscles of WT mice was assayed by Western blot. Gapdh was used as the loading control. B, Sulfs repress CamKii and Jun activation during muscle regeneration. TA muscles of WT and SulfSK-DN mice were collected before injury (−CTX) and on day 3 post-injury (+CTX). Levels of p-CamKii and p-Jnk Gapdh were assayed by Western blot and normalized to Gapdh. The image represented one of three independent experiments. C, TA muscles of WT mice were treated with vehicle or Wnt7a (50 μl of 25 ng/ml) at day 1 post-injury. Muscles were collected at day 3 post-injury for immunoblot analyses of p-CamKii and p-Jnk. Gapdh was the loading control. D, vehicle- and Wnt7a-treated TA muscles of WT mice were collected on day 5 post-injury for immunohistochemical analyses to identify eMyh+ new myofibers. Scale bar = 100 μm. E, shown is quantification of the SC population (a), relative abundance of new myofibers with one and more myonuclei (b), and of increasing sizes (c and d) in vehicle and Wnt7a-treated muscles of WT mice. Data presented are the mean and S.D. of three independent experiments. #, p < 0.01; **, p < 0.05; *, p < 0.1. DU, density unit.
FIGURE 7.

Sulfs indirectly repress noncanonical Wnt signaling but directly promote opposing canonical Wnt signaling. A and B, primary myoblast cultures (in A) or regenerating TA muscles (in B) of WT and SulfSK-DN mice were treated with Wnt inhibitors (2 μg/ml sFRP2 or 0.5 μg/ml Dkk1). Downstream activators of canonical and noncanonical Wnt signaling, stabilized β-catenin and p-CamKii/p-Jnk, respectively, were assayed by Western blot. Gapdh was the loading control. Quantification was performed by normalizing signals of p-CamKii and stabilized β-catenin to Gapdh by densitometry. Images represent one of four independent experiments. C, primary WT myoblast cultures of WT and SulfSK-DN mice in growth medium were immunolabeled using an antibody against LRP5. WT myoblast cells exhibited punctuated LRP5 labeling (pointed by white arrows), whereas LRP5 labeling appeared diffusive in DKK1-treated WT myoblast cultures and in SulfSK-DN myoblast cultures. The percentage of cells that exhibited punctuated LRP5 staining was shown in images. Images shown were representative of three independent experiments. Scale bars, 50 μm. D, colocalization of LRP5 and caveolin3, a lipid raft marker, in WT myoblasts by confocal microscopy is shown. Arrows pointed to punctuated immunolabeling of LRP5 and caveolin3 around the cell surface. #, p < 0.01; **, p < 0.05; *, p < 0.1. DU, density unit.
We compared the levels of p-CamKii and p-Jnk between WT and SulfSK-DN muscles at day 3 post-injury, the peak of non-canonical Wnt signaling. Consistent with results from myoblast cultures, SulfSK-DN muscles exhibited higher levels of p-CamKii (5.5 ± 3.4 (SulfSK-DN) versus 1.25 ± 1.13 (WT) density units; p < 0.05) and p-Jnk (4.57 ± 2.67 (DN) versus 0.58 ± 0.43 (WT) density units; p < 0.05) (Fig. 5B). These observations in vivo support our in vitro data that Sulfs are negative regulators of non-canonical Wnt signaling.
To mimic elevated non-canonical Wnt signaling in SulfSK-DN muscles, we injected Wnt7a (50 μl at a concentration of 25 ng/ml) into WT muscles 24 h post-injury and assessed changes in signaling activity and the efficacy of regeneration at day 3 and day 5 post-injury. Western blot assays showed that Wnt7a treatment increased the levels of p-CamKii and p-JnK (Fig. 5C). Wnt7a also led to an increase in the Pax7+ SC population as compared with vehicle-treated controls at day 5 post-injury (360 ± 105 (Wnt7a) versus 224 ± 37 (vehicle) per mm2; p < 0.05) (Fig. 5Ea), consistent with a role of Wnt7a in promoting symmetric cell division for the expansion of SCs (27). Notably, Wnt7a treatment also increased the abundance of myofibers with one nucleus within muscle cross-sections, although the effect did not reach statistical significance (Fig. 5, D and Eb). In addition, Wnt7a increased the abundance of small new myofibers (Fig. 5Ec) and reduced the average size of new myofibers (850 ± 178 μm2 (Wnt7a) versus 935 ± 232 μm2 (vehicle); p < 0.05) (Fig. 5Ed). Overall, the effects of exogenous Wnt7a on regenerating WT muscles mimicked phenotypes of SulfSK-DN muscles. These findings provide evidence that Sulfs repress non-canonical Wnt signaling to promote myoblast fusion during regeneration.
Sulfs Indirectly Repress Non-canonical Wnt Signaling by Promoting Opposing Canonical Wnt Signaling
To identify mechanisms underlying differential effects of Sulfs on canonical and non-canonical Wnt signaling, we performed Wnt-heparin binding assays. Recombinant Wnt protein was incubated with heparin-conjugated agarose beads that were predigested by similar amounts of Sulf2 and inactive Sulf2 (C-S) (Fig. 6A) (20). Sulf2(C-S) lacks enzymatic activity due to a point mutation that converts an essential Cys to Ser in the enzymatic domain (30). The effect of Sulf on Wnt-HS binding was assessed by comparing the amount of Wnt bound to Sulf2-desulfated heparin and Sulf2(C-S)-treated heparin. The canonical Wnt3a (1 ng) bound to heparin, a highly sulfated and secreted HS derivative. In addition, Wnt3a-heparin binding was reduced by the active Sulf2 enzyme but not inactive Sulf2(C-S) (Fig. 6A). In contrast, Wnt7a did not bind to heparin even at a concentration 100 times that of Wnt3a (Fig. 6B). Consistently, HS binding Weintraub sequences (31) were identified in Wnts that have been shown to activate canonical pathway, such as Wnt3a, but not in a large majority of non-canonical Wnts, including Wnt7a and others that are expressed by regenerating skeletal muscle (Table 1) (27).
FIGURE 6.
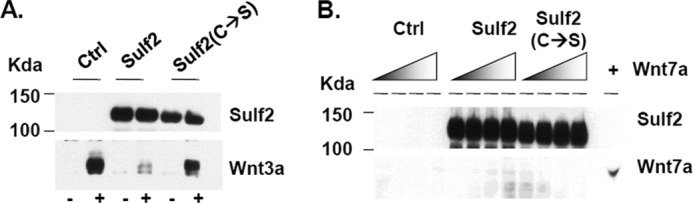
Non-canonical Wnt7a does not bind heparin. A, 293T cells transfected with empty vector (Ctrl), Sulf2-Myc, or inactive Sulf2(C-S)-myc expression vector were assayed for Sulf2 expression by Western blot using the Myc antibody. Wnt3a (1 ng) bound to heparin or Sulf2-digested heparin was assayed. Sulf2 reduced Wnt3a-heparin binding. B, increasing amounts of Wnt7a (0, 10, 30, or 100 ng) were incubated with heparin or Sulf2-digested heparin to allow binding. The amount of Wnt7a bound to heparin was assayed by Western blot. No significant binding of Wnt7a to heparin was observed. Images represent one of three independent experiments.
TABLE 1.
HS binding Weintraub sequences within Wnts
B means any basic amino acid residue. X means any amino acid residue. Weintraub sequences (in bold) are found in several Wnts, including Wnt3a that is known to induce canonical signaling in myogenic population. Wnts that have been shown to induce noncanonical signaling in myogenic cells, such as Wnt7a, lack Weintraub sequences. Note: not all Wnts listed in the table have been tested for their function during skeletal muscle regeneration or their activities in the canonical versus noncanonical signaling pathways in myogenic cells.
| Protein | GI number | Reference number | Residues | Heparan sulfate binding concensus sequence (XBBBXBX or XBBXBX) |
|---|---|---|---|---|
| Wnt1 | 4885655 | NP_005421.1 | 61–80 | LQLLSRKQRR LIRQNPGILH |
| Wnt2 | 4507927 | NP_003382.1 | 171–190 | FVDAKERKGK DARALMNLHN |
| Wnt2b | 13518017 | NP_004176.2 | 181–200 | KAFVDAKEKR LKDARALMNL |
| Wnt3 | 13540477 | NP_110380.1 | 321–340 | TRTEKRKEKC HCIFHWCCYV |
| Wnt3a | 14916475 | NP_149122.1 | 321–340 | ERRREKCRCV FHWCCYVSCQ |
| Wnt4 | 17402922 | NP_110388.2 | ||
| Wnt5a | 40806205 | NP_003383.2 | ||
| Wnt5b | 14249180 | NP_116031.1 | 251–270 | SAAAMRVTRK GRLELVNSRF |
| Wnt6 | 16507239 | NP_006513.1 | ||
| Wnt7a | 17505191 | NP_004616.2 | ||
| Wnt7b | 17505193 | NP_478679.1 | ||
| Wnt8a | 17505195 | NP_490645.1 | ||
| Wnt8b | 110735437 | NP_003384.2 | 271–290 | ECLRRGRALG RWERRSCRRL |
| Wnt9a | 15082261 | NP_003386.1 | 61–80 | RLKLERKQRR MCRRDPGVAE |
| Wnt9b | 17017976 | NP_003387.1 | 51–70 | HLKQCDLLKL SRRQKQLCRR |
| Wnt10a | 16936520 | NP_079492.2 | 161–180 | ASRRGDEEAF RRKLHRLQLD |
| Wnt10b | 16936522 | NP_003385.2 | 241–260 | ENLKRKCKCH GTSGSCQFKT |
| Wnt11 | 17017974 | NP_004617.2 | ||
| Wnt16 | 17402916 | NP_476509.1 | 261–280 | DKTKRKMRRR EKDQRKIPIH |
A lack of Wnt7a-heparin binding suggested that Sulfs repress non-canonical Wnt7a signaling through indirect mechanisms. One such mechanism may be Sulf-enhanced canonical Wnt signaling, as the canonical pathway is known to antagonize non-canonical Wnt signaling (32–34). If true, blocking canonical Wnt signaling would activate the non-canonical pathway. To test this, we treated WT myoblast cultures for 48 h with Dkk1, a specific inhibitor of the canonical Wnts (34). Dkk1 treatment blocked β-catenin stabilization while significantly increasing the level of p-CamKii by ∼2.7-fold to a level similar to that in SulfSK-DN cultures (Fig. 7A). In addition, treatment with sFRP2 also significantly increased p-CamKii levels in WT myoblasts but to a smaller extent than Dkk1 (Fig. 7A), consistent with sFRP2 interference with both canonical and non-canonical Wnts. Compared with WT myoblasts, Dkk1 and sFRP2 had little effect on non-canonical Wnt signaling in SulfSK-DN myoblasts (Fig. 7A), which may be due to already low levels of canonical Wnt signaling in SulfSK-DN myoblasts (Figs. 4A and 7A).
Treatment with sFRP2 differentially affected activation of CamKii and Jnk in regenerating WT and SulfSK-DN muscles. sFRP2 injected 24 h after injury elevated the levels of p-CamKii and p-Jnk in WT muscles as compared with vehicle-treated muscles at day 3 post-injury (Fig. 7B). In contrast, sFRP2 treatment reduced p-CamKii and p-Jnk in regenerating SulfSK-DN muscles to levels similar to vehicle-treated WT muscles (Fig. 7B). These observations in vivo further support a Sulf-dependent antagonizing role of the canonical Wnt pathway against the noncanonical pathway.
Mechanisms underlying the opposition of the noncanonical Wnt pathway by canonical Wnt signaling are complex and likely involve components on the cell surface and at intracellular compartments in the myoblasts (32–35). However, because Sulfs are HS-modifying enzymes that function in the lipid raft to potentiate canonical Wnt signaling (19), we reasoned that a lipid raft-associated mechanism may be involved in Sulf regulation of the noncanonical pathway. Indeed, it was shown previously that cell membrane localization of the Wnt co-receptors, LRP5/6, in the lipid raft and internalization with caveolin3, a lipid raft marker, activates the canonical Wnt pathway, whereas DKK1 disrupts LRP5/6 internalization with caveolin3, leading to activation of the noncanonical Wnt pathway (33, 34). To assess whether Sulfs regulated subcellular colocalization LRP5/6 with caveolin3, we performed immunostaining for LRP5/6 in WT and SulfSK-DN myoblasts. We found that more than 50% of WT myoblast cells (58 ± 8%) exhibited a punctuated LRP5 immunolabeling that was largely colocalized with caveolin3 around the cell surface (Fig. 7, C and D). In contrast, less than 15% of DKK1-treated WT myoblasts (14 ± 6%) and SulfSK-DN myoblasts (11 ± 2%) exhibited punctated LRP5 staining, and LRP5 was largely diffusively distributed in these cultures (Fig. 7C). Importantly, the level of caveolin3 expression in myoblasts was not affected by Sulf deficiency (see Fig. 10B). This observation suggests that Sulfs regulate the subcellular localization of LRP5/6, a switch between the two Wnt signaling pathways.
FIGURE 10.

Sulfs promote FAK activation and clustering. A, TA muscles of WT and SulfSK-DN mice were collected at day 3 post-injury for Western blot assays of p-FAK. Gapdh was the loading control. Images represent one of four independent experiments. B, WT and SulfSK-DN myoblasts in cultures were treated with vehicle or Wnt7a (100 ng/ml) for 48 h before Western blot assays of p-FAK and caveolin3. Gapdh was the loading control. Images represent one of three independent experiments. C, shown is immunocytochemistry to assay subcellular localization of p-FAK (green) in WT myoblast cells that were treated with vehicle or Wnt7a for 48 h and in SulfSK-DN myoblasts in differentiation medium. Nuclei were labeled with Hoechst dye (blue). Arrowheads point to punctuated p-FAK clustering. Percentages of cells with punctuated p-FAK immunolabeling are shown in d. Data are the mean and S.D. of four independent experiments. #, p < 0.01. Scale bars, 100 μm.
sFRP2 Treatment Rescued the Muscle Regeneration Defects of SulfSK-DN Mice
To establish that elevated non-canonical Wnt signaling causes the defects in skeletal muscle regeneration of SulfSK-DN mice, we tested whether blockade of over-activated non-canonical Wnt signaling by sFRP2 at 24 h post-injury rescued regeneration defects of SulfSK-DN muscles. At day 5 post-injury, compared with vehicle treatment, sFRP2 increased the size of new myofibers and abundance of multinucleated myofibers (Fig. 8, A and B, b and c). The average size of new myofibers in sFRP2-treated SulfSK-DN TA muscles was significantly larger than vehicle-treated SulfSK-DN muscles (644 ± 54 μm2 (sFRP2) versus 468 ± 53 μm2 (vehicle); p < 0.05) (Fig. 8Bd). However, the size of these new myofibers was smaller as compared with WT muscles, indicating that sFRP2 partially rescued the defect in myofiber regeneration in SulfSK-DN mice (Fig. 8Bd). sFRP2 treatment of regenerating SulfSK-DN TA muscles also led to a decrease in Pax7+ SC population (229 ± 11 (sFRP2) versus 332 ± 69 (vehicle) per mm2; p < 0.05) (Fig. 8Ba). These observations together with our results that sFRP2 represses non-canonical Wnt signaling in vivo provide strong evidence that elevated non-canonical Wnt signaling is the likely cause for defective myofiber formation in SulfSK-DN mice.
FIGURE 8.

sFRP2 treatment partially rescued defects in SCs and new myofiber size of SulfSK-DN muscles. A, TA muscles of SulfSK-DN mice were treated with vehicle or sFRP2 (50 μl of 2 μg/ml) on day 1 post-injury and collected on day 5 post-injury for immunohistochemical analyses to identify eMyh+ new myofibers. B, quantification of SC number (a), relative abundance of mono- and multinucleated new myofibers (b), and new myofiber size (c and d) in vehicle- and sFRP2-treated SulfSK-DN muscles is compared with WT muscles. Data represent the mean and S.D. of three independent experiments. **, p < 0.05; *, p < 0.1. Scale bars, 100 μm.
Canonical and Non-canonical Wnts Differentially Regulate Myoblast Fusion and Myofiber Size in Culture
To directly test that non-canonical Wnt signaling affects myoblast fusion, WT myoblast cultures were treated with Wnt7a (100 ng/ml) for 48 h before fusion was assessed. Wnt7a treatment led to higher percentages of single-nucleated myofiber and lower percentages of myofibers with more than four nuclei as compared with vehicle-treated cultures (Fig. 9A), suggesting that non-canonical Wnt signaling repressed myoblast fusion. In addition, canonical Wnt3a partially rescued fusion defects of SulfSK-DN cultures, as Wnt3a increased the abundance of multinucleated myofibers (Fig. 9B). Therefore, results of these in vitro assays indicate that the canonical and non-canonical Wnt pathways have opposing effects on myoblast fusion. Sulfs, by promoting canonical Wnt signaling to antagonize non-canonical Wnt signaling, may function to increase myoblast fusion.
FIGURE 9.

Canonical and noncanonical Wnts differentially affects myoblast fusion. A, primary myoblast cells of WT mice were treated with vehicle or Wnt7a (100 ng/ml) in differentiating media for 48 h. Myoblast fusion was assessed after immunolabeling followed by quantification of relative abundance of mono- and multinucleated myofibers. B, primary myoblast cells of SulfSK-DN mice were treated with vehicle or Wnt3a (30 ng/ml) before immunolabeling and assessment of fusion. Data represent the mean and S.D. of three independent experiments. #, p < 0.01; **p < 0.05; *, p < 0.1. Scale bars, 100 μm.
Sulfs Regulate Non-canonical Wnt-dependent FAK Activation and Distribution to Promote Myoblast Fusion
We further investigated the mechanism of non-canonical Wnt and Sulf regulation of myoblast fusion. Because myofiber phenotypes of SulfSK-DN mice are similar to those of FAK−/− mice (4), we investigated whether Sulfs and non-canonical Wnts regulate myoblast fusion through the FAK signaling pathway. We assayed both the level and subcellular distribution of activated p-FAK in WT and SulfSK-DN myoblasts with and without Wnt7a treatment. Western blot assays showed that FAK was activated at day 3 post-injury (Fig. 10A) at the peak of CamKii and Jnk activation (Fig. 5A). Compared with WT controls, levels of p-FAK were significantly reduced in regenerating SulfSK-DN muscles (0.31 ± 0.28 (SulfSK-DN) versus 2.69 ± 0.65 (WT), p < 0.01) at day 3 post-injury (Fig. 10A) and in SulfSK-DN myoblast cultures (Fig. 10B), suggesting that Sulfs are required for FAK activation. Interestingly, Wnt7a had no effect on p-FAK levels (Fig. 10B). However, Wnt7a treatment reduced myoblast fusion in culture similar to Sulf deficiency (Fig. 9). Because subcellular localization of p-FAK is crucial for FAK function, we speculated that Wnt7a and Sulfs may similarly regulate subcellular distribution of p-FAK to affect myoblast fusion.
To assess the effect of Sulfs and non-canonical Wnts on subcellular distribution of p-FAK, we assayed p-FAK immunoreactivity in low density WT and SulfSK-DN myoblast cultures. After 24 h in differentiation media, myoblasts differentiated into myocytes, and a large majority of them were mononucleated before fusion (supplemental Fig. 5). At this time point, we did not observe any difference in punctuated p-FAK immunostaining, which is indicative of clustering of p-FAK in cell surface subdomains between WT and SulfSK-DN cultures and at the presence of Wnt7a (supplemental Fig. 5). However, after 48 h in culture when myoblasts fuse to form multinucleated myofibers, WT cultures retained punctuated p-FAK immunoreactivity, whereas Wnt7a disrupted clustering of p-FAK on the cell membrane (Fig. 10C, a and b). To compare, we quantified the percentages of cells with punctuated p-FAK staining in both control and Wnt7a-treated WT cultures. The majority of vehicle-treated control myoblasts and nascent myofibers (57.5 ± 6.5%) exhibited punctuated immunolabeling of p-FAK as compared with small percentages of Wnt7a-treated cultures (18.0 ± 5.7%, p < 0.01) (Fig. 10Cd). In addition, only 13% of cells in SulfSK-DN cultures exhibited punctuated p-FAK distribution, similarly to that in Wnt7a-treated WT cultures (18.0 ± 5.7%) (Fig. 10C, c and d). Together, defective myoblast fusion caused by Sulf deficiency and Wnt7a treatment was associated with aberrant subcellular distribution of p-FAK.
DISCUSSION
In this study we investigate functions of Sulfs during skeletal muscle regeneration using a skeletal muscle-specific SulfSK-DN mouse line. SulfSK-DN mice exhibit two distinct phenotypes of muscle regeneration. One phenotype is a transient increase in the number of satellite cells, similar to that observed in systematic Sulf double null mice (20). We demonstrated previously that Sulfs repress FGF2 and hepatocyte growth factor signaling to induce differentiation of satellite cells (20). Here, we show that exogenous Wnt7a further increases the number of satellite cells during regeneration of WT skeletal muscles, whereas sFRP injection partially rescues this satellite cell phenotype in regenerating muscles of SulfSK-DN mice. These observations suggest that Sulfs may have an additional role in regulating non-canonical Wnt signaling to control the number of satellite cells. The non-canonical Wnts have been shown to induce symmetric expansion of a subset of satellite cells (27). Because this subset of satellite cells express Pax7 but not Myf5, their expression of Sulfs is not affected in SulfSK-DN mice. Therefore, non-canonical Wnts may also regulate Pax7 expression in Myf5+ SCs. If true, Sulfs may play dual inhibitory roles in the FGF/hepatocyte growth factor and non-canonical Wnt pathways to induce SC differentiation.
This study focuses on the other phenotype of SulfSK-DN mice, delayed myoblast fusion during skeletal muscle regeneration. Results of mimic and rescue assays in vivo demonstrate that Sulfs repress non-canonical signaling to promote myoblast fusion. We ruled out the possibility that Sulfs directly regulate non-canonical Wnt signaling based on a lack of non-canonical Wnt-HS binding. Instead, we proposed that Sulfs indirectly represses non-canonical Wnt signaling by promoting opposing activities of canonical Wnt signaling. We provide several lines of evidence in support of opposing interactions of the two Wnt pathways. First, Sulf deficiency is associated with a reduction in canonical Wnt signaling but an increase in non-canonical Wnt signaling in vivo and in vitro. Second, blockade of canonical Wnt signaling by Dkk1 elevates non-canonical Wnt signaling in wild type myoblast cultures. Third, the two Wnt pathways have opposite effects on myoblast fusion. Fourth, Sulfs potentiate the co-localization of LRP5 and caveolin3. Sulfs, which are in the lipid raft (19), may enhance the formation of a Wnt-frizzled receptor-LRP5/6 co-receptor complex in the lipid raft, thereby increasing the bioavailability of LRP5 to interact with caveolin3 for activation of the canonical Wnt pathway. Sulf deficiency may decrease the formation of the complex, resulting in reduced LRP5/6 internalization with caveolin3 and subsequently elevated noncanonical Wnt signaling. Collectively, we propose that Sulfs coordinate canonical and non-canonical Wnt signaling activities to promote myoblast fusion during skeletal muscle regeneration. Without Sulfs, canonical Wnt signaling is diminished, whereas non-canonical Wnt signaling is elevated, resulting in defective new myofiber formation.
Our studies on the level and subcellular distribution of p-FAK support FAK signaling as a downstream candidate of Sulf-enhanced myoblast fusion. We show that Sulfs are required for both activation and surface distribution of FAK. Activated FAK was found to cluster on the cell membrane of myoblasts. The distinct cell surface distribution of p-FAK was not affected by Sulf deficiency or non-canonical Wnt7a at 24 h in differentiation media after myoblasts differentiated before fusion. However, at 48 h, both Sulf-deficient and Wnt7a-treated myoblast cultures showed disrupted cell surface clustering of p-FAK with associated defects in myoblast fusion. These findings indicate that Sulfs and noncanonical Wnts have opposite effects on subcellular distribution of p-FAK during myoblast fusion. The clustering of p-FAK may reflect cell-cell contact that is required for myoblast fusion. The function of p-FAK clustering in myoblast fusion needs to be tested by future work using tools that allow spatially and temporally controlled FAK activation in myoblasts. Based on our finding that non-canonical Wnt signaling blocks activated FAK clustering, Sulfs may promote myoblast fusion by antagonizing non-canonical Wnt-disrupted FAK subcellular localization. Mechanisms underlying differential regulatory effects of Sulf and non-canonical Wnt on FAK activation and membrane clustering are unknown. However, we speculated that Sulfs might affect functions of syndecan-4, a heparan sulfate proteoglycan that is highly expressed in satellite cells (36). Syndecan-4 is required to activate FAK and focal adhesion formation (37) at least partially mediated through the attached HS chains (38).
Recently, noncanonical Wnts have been shown to induce skeletal muscle hypertrophy after Wnt7a was overexpressed in the skeletal muscle or myoblasts for extended periods of time (39). Whether Sulfs also regulate noncanonical Wnts-induced muscle hypertrophy require further investigation.
In summary, our findings indicate that Sulfs have multiple roles during skeletal muscle regeneration. Sulfs not only repress FGF2/hepatocyte growth factor signaling in SCs to induce differentiation but also enhance the opposition of the canonical Wnt pathway against non-canonical Wnt signaling to promote myoblast fusion. Thus, Sulfs may serve as potential therapeutic targets to increase the efficacy of skeletal muscle regeneration for the treatment of muscular dystrophy and age-related atrophy.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Training Grant 5T32AG000115–24 (to T. T.) and Grants R01AG034939 (to X. A.) and R01HL098950 and P41RR10888 (to J. Z.).

This article contains supplemental Methods and Figs. 1–5.
- SC
- satellite cell
- CamKii
- Ca2+/calmodulin-dependent protein kinase II
- p-CamKii
- phosphorylated CamKii
- FAK
- focal adhesion kinase
- FGF2
- fibroblast growth factor 2
- LRP5/6
- low density lipoprotein receptor-related protein 5 and 6
- GSK3β
- glycogen synthase kinase 3β
- HS
- heparan sulfate
- Sulf
- HS 6-O-endosulfatase
- SulfSK-DN
- skeletal muscle-specific Sulf double null mice
- TA
- tibialis anterior
- sFRP2
- secreted frizzled-related protein 2
- CTX
- cardiotoxin
- DN
- dominant negative.
REFERENCES
- 1. Dhawan J., Rando T. A. (2005) Stem cells in postnatal myogenesis. Molecular mechanisms of satellite cell quiescence, activation, and replenishment. Trends Cell Biol. 15, 666–673 [DOI] [PubMed] [Google Scholar]
- 2. Rudnicki M. A., Le Grand F., McKinnell I., Kuang S. (2008) The molecular regulation of muscle stem cell function. Cold Spring Harbor Symp. Quant. Biol. 73, 323–331 [DOI] [PubMed] [Google Scholar]
- 3. Pavlath G. K. (2010) Spatial and functional restriction of regulatory molecules during mammalian myoblast fusion. Exp. Cell Res. 316, 3067–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Quach N. L., Biressi S., Reichardt L. F., Keller C., Rando T. A. (2009) Focal adhesion kinase signaling regulates the expression of caveolin-3 and β1 integrin, genes essential for normal myoblast fusion. Mol. Biol. Cell 20, 3422–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sunadome K., Yamamoto T., Ebisuya M., Kondoh K., Sehara-Fujisawa A., Nishida E. (2011) ERK5 regulates muscle cell fusion through Klf transcription factors. Dev. Cell 20, 192–205 [DOI] [PubMed] [Google Scholar]
- 6. Vasyutina E., Martarelli B., Brakebusch C., Wende H., Birchmeier C. (2009) The small G-proteins Rac1 and Cdc42 are essential for myoblast fusion in the mouse. Proc. Natl. Acad. Sci. U.S.A. 106, 8935–8940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brack A. S., Conboy I. M., Conboy M. J., Shen J., Rando T. A. (2008) A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell 2, 50–59 [DOI] [PubMed] [Google Scholar]
- 8. Rochat A., Fernandez A., Vandromme M., Molès J. P., Bouschet T., Carnac G., Lamb N. J. (2004) Insulin and Wnt1 pathways cooperate to induce reserve cell activation in differentiation and myotube hypertrophy. Mol. Biol. Cell 15, 4544–4555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pansters N. A., van der Velden J. L., Kelders M. C., Laeremans H., Schols A. M., Langen R. C. (2011) Segregation of myoblast fusion and muscle-specific gene expression by distinct ligand-dependent inactivation of GSK-3β. Cell. Mol. Life Sci. 68, 523–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ai X., Kusche-Gullberg M., Lindahl U., Emerson C. P., Jr. (2005) in Chemistry and Biology of Heparin and Heparan Sulfate (Garg H. G., Linhardt R. J., Hales C. A., eds) pp. 245–258, Elsevier Ltd., New York [Google Scholar]
- 11. Esko J. D., Lindahl U. (2001) Molecular diversity of heparan sulfate. J. Clin. Invest. 108, 169–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dhoot G. K., Gustafsson M. K., Ai X., Sun W., Standiford D. M., Emerson C. P., Jr. (2001) Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science 293, 1663–1666 [DOI] [PubMed] [Google Scholar]
- 13. Ai X., Do A. T., Lozynska O., Kusche-Gullberg M., Lindahl U., Emerson C. P., Jr. (2003) QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfateproteoglycans to promote Wnt signaling. J. Cell Biol. 162, 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lamanna W. C., Baldwin R. J., Padva M., Kalus I., Ten Dam G., van Kuppevelt T. H., Gallagher J. T., von Figura K., Dierks T., Merry C. L. (2006) Heparan sulfate 6-O-endosulfatases. Discrete in vivo activities and functional cooperativity. Biochem. J. 400, 63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morimoto-Tomita M., Uchimura K., Werb Z., Hemmerich S., Rosen S. D. (2002) Cloning and characterization of two extracellular heparin-degrading endosulfatases in mice and humans. J. Biol. Chem. 277, 49175–49185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lai J., Chien J., Staub J., Avula R., Greene E. L., Matthews T. A., Smith D. I., Kaufmann S. H., Roberts L. R., Shridhar V. (2003) Loss of HSulf-1 up-regulates heparin binding growth factor signaling in cancer. J. Biol. Chem. 278, 23107–23117 [DOI] [PubMed] [Google Scholar]
- 17. Wang S., Ai X., Freeman S. D., Pownall M. E., Lu Q., Kessler D. S., Emerson C. P., Jr. (2004) QSulf1, a heparan sulfate 6-O-endosulfatase, inhibits fibroblast growth factor signaling in mesoderm induction and angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 101, 4833–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nawroth R., van Zante A., Cervantes S., McManus M., Hebrok M., Rosen S. D. (2007) Extracellular sulfatases, elements of the Wnt signaling pathway, positively regulate growth and tumorigenicity of human pancreatic cancer cells. PLoS One 2, e392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tang R., Rosen S. D. (2009) Functional consequences of the subdomain organization of the sulfs. J. Biol. Chem. 284, 21505–21514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Langsdorf A., Do A. T., Kusche-Gullberg M., Emerson C. P., Jr., Ai X. (2007) Sulfs are regulators of growth factor signaling for satellite cell differentiation and muscle regeneration. Dev. Biol. 311, 464–477 [DOI] [PubMed] [Google Scholar]
- 21. Ai X., Kitazawa T., Do A. T., Kusche-Gullberg M., Labosky P. A., Emerson C. P., Jr. (2007) Sulf1 and Sulf2 regulate heparan sulfate-mediated GDNF signaling for esophageal innervation. Development 134, 3327–3338 [DOI] [PubMed] [Google Scholar]
- 22. Holst C. R., Bou-Reslan H., Gore B. B., Wong K., Grant D., Chalasani S., Carano R. A., Frantz G. D., Tessier-Lavigne M., Bolon B., French D. M., Ashkenazi A. (2007) Secreted sulfatases Sulf1 and Sulf2 have overlapping yet essential roles in mouse neonatal survival. Plos One 2, e575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tallquist M. D., Weismann K. E., Hellström M., Soriano P. (2000) Early myotome specification regulates PDGFA expression and axial skeleton development. Development 127, 5059–5070 [DOI] [PubMed] [Google Scholar]
- 24. Shi X., Zaia J. (2009) Organ-specific heparan sulfate structural phenotypes. J. Biol. Chem. 284, 11806–11814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Staples G. O., Shi X., Zaia J. (2011) Glycomics analysis of mammalian heparan sulfates modified by the human extracellular sulfatase HSulf2. PLoS One 6, e16689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kuang S., Kuroda K., Le Grand F., Rudnicki M. A. (2007) Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 129, 999–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Le Grand F., Jones A. E., Seale V., Scimè A., Rudnicki M. A. (2009) Wnt7a activates the planar cell polarity pathway to drive the symmetric expansion of satellite stem cells. Cell Stem Cell 4, 535–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Logan C. Y., Nusse R. (2004) The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 [DOI] [PubMed] [Google Scholar]
- 29. Rao T. P., Kühl M. (2010) An updated overview on Wnt signaling pathways. A prelude for more. Circ. Res. 106, 1798–1806 [DOI] [PubMed] [Google Scholar]
- 30. Schmidt B., Selmer T., Ingendoh A., von Figura K. (1995) A novel amino acid modification in Sulfatases that is defective in multiple sulfatase deficiency. Cell 82, 271–278 [DOI] [PubMed] [Google Scholar]
- 31. Cardin A. D., Weintraub H. J. (1989) Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 9, 21–32 [DOI] [PubMed] [Google Scholar]
- 32. Kühl M., Geis K., Sheldahl L. C., Pukrop T., Moon R. T., Wedlich D. (2001) Antagonistic regulation of convergent extension movements in Xenopus by Wnt/β-catenin and Wnt/Ca2+ signaling. Mech. Dev. 106, 61–76 [DOI] [PubMed] [Google Scholar]
- 33. Yamamoto H., Komekado H., Kikuchi A. (2006) caveolin is necessary for Wnt-3a-dependent internalization of LRP6 and accumulation of β-catenin. Dev. Cell 11, 213–223 [DOI] [PubMed] [Google Scholar]
- 34. Yamamoto H., Sakane H., Yamamoto H., Michiue T., Kikuchi A. (2008) Wnt3a and Dkk1 regulate distinct internalization pathways of LRP6 to tune the activation of β-catenin signaling. Dev. Cell 15, 37–48 [DOI] [PubMed] [Google Scholar]
- 35. Nalesso G., Sherwood J., Bertrand J., Pap T., Ramachandran M., De Bari C., Pitzalis C., Dell'accio F. (2011) WNT-3A modulates articular chondrocyte phenotype by activating both canonical and non-canonical pathways. J. Cell Biol. 193, 551–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cornelison D. D., Filla M. S., Stanley H. M., Rapraeger A. C., Olwin B. B. (2001) Syndecan-3 and syndecan-4 specifically mark skeletal muscle satellite cells and are implicated in satellite cell maintenance and muscle regeneration. Dev. Biol. 239, 79–94 [DOI] [PubMed] [Google Scholar]
- 37. Wilcox-Adelman S. A., Denhez F., Goetinck P. F. (2002) Syndecan-4 modulates focal adhesion kinase phosphorylation. J. Biol. Chem. 277, 32970–32977 [DOI] [PubMed] [Google Scholar]
- 38. Mahalingam Y., Gallagher J. T., Couchman J. R. (2007) Cellular adhesion responses to the heparin-binding (HepII) domain of fibronectin require heparan sulfate with specific properties. J. Biol. Chem. 282, 3221–3230 [DOI] [PubMed] [Google Scholar]
- 39. von Maltzahn J., Bentzinger C. F., Rudnicki M. A. (2012) Wnt7a-Fzd7 signaling directly activates the Akt/mTOR anabolic growth pathway in skeletal muscle. Nat. Cell Biol. 14, 186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.