

Background: The oncogenesis and developmental mechanisms of glioma must be clarified to control the disease.
Results: Migfilin relates to pathological grades, prognosis of glioma, and regulates motility of glioma cells.
Conclusion: Migfilin mediates migration and invasion through EGFR-induced PLC-γ and STAT3 pathways.
Significance: Migfilin helps us better understand the pathogenesis of glioma, and Migfilin may be a molecular marker in diagnosis and an indicator in prognosis.
Keywords: Cell Biology, Cell Migration, Cell Motility, Neurological Diseases, Pathogenesis
Abstract
Migfilin is critical for cell shape and motile regulation. However, its pathological role in glioma is unknown. Using an immunohistochemical staining assay, we demonstrate that there is a significant correlation between expression of Migfilin and pathological tumor grade in 217 clinical glioma samples. High Migfilin expression is associated with poor prognosis for patients with glioma. Investigation of the molecular mechanism shows that Migfilin promotes migration and invasion in glioma cells. Moreover, Migfilin positively modulates the expression and activity of epidermal growth factor receptor, and Migfilin-mediated migration and invasion depend on epidermal growth factor receptor-induced PLC-γ and STAT3-signaling pathways. Our results may provide significant clinical application, including use of Migfilin as a molecular marker in glioma for early diagnosis and as an indicator of prognosis.
Introduction
Gliomas are the most common primary central nervous system (CNS) tumors, accounting for more than 70% of all primary CNS neoplasms (1). According to the World Health Organization (WHO),4 gliomas are classified into four grades as follows: pilocytic astrocytoma, WHO grade I; diffuse “low grade” glioma, WHO grade II; anaplastic gliomas, WHO grade III; and glioblastoma (GBM), WHO grade IV (2). Over the past decade, great progress has been made in surgical, radioactive, and chemical anti-glioma therapies; however, the prognosis of the disease remains poor (3, 4). The survival rate of patients with anaplastic glioma and GBM are 40% at 1 year and only mildly higher (46%) after adjuvant radiochemotherapy (5). The poor clinical outcome of glioma is largely due to their widespread infiltration and recurrence at adjacent or distant regions in the brain (6). Over the last 30 years, advances in the research of molecular biology, cellular biology, and genomics have improved our understanding of glioma (7, 8). However, the molecular mechanisms are still not fully understood.
Migfilin localizes to cell-extracellular matrix (ECM) adhesion sites and cell-cell junctions and is recruited to actin cytoskeleton (9, 10). Migfilin consists of an N-terminal region, a central proline-rich (PR) region, and a C-terminal region consisting of three LIM domains. The N-terminal region binds to filamins, the PR region binds to vasodilator-stimulated phosphoprotein, and the C-terminal region binds to kindlin-2 (FERMT2) and the transcription factor CSX/NKX2–5 (9, 10). Although filamins mediate the Migfilin association with actin filaments, vasodilator-stimulated phosphoprotein mediates actin organization (11); kindlin-2 mediates the localization of Migfilin to cell-ECM adhesion sites (9), and the CSX/NKX2–5-Migfilin regulates cardiomyocyte differentiation (9, 10). In summary, Migfilin provides physical connections and transduces signals between extracellular and intracellular compartments, and it is critical for the control of a variety of fundamental cellular processes, including cell adhesion, shape of modulation, motility, and transcriptional regulation (9, 10).
In this study, we determined that Migfilin significantly correlated with pathological grades of gliomas; high Migfilin expression was associated with poor prognosis of patients with glioma; and Migfilin was an independent prognostic factor for patients with glioma. The biological significance of Migfilin revealed that it promoted migration and invasion in glioma cells. Investigation into the molecular mechanism found that Migfilin up-regulated epidermal growth factor receptor (EGFR) expression and formation of a complex with EGFR in glioma cells. Additionally, Migfilin regulated the Tyr-1173 phosphorylation of EGFR and EGFR-mediated phospholipase Cγ (PLC-γ) and signal transducer and activator of transcription 3 (STAT3)-signaling pathways. Taken together, our results suggest a role for Migfilin in the progression of glioma and identify Migfilin as a potentially important molecule for human glioma progression. Migfilin may be an ideal target for diagnosis and treatment of glioma.
EXPERIMENTAL PROCEDURES
Cell Culture
Human glioma cell lines U-87 MG, H4, and Hs 683 were purchased from the Cell Culture Center (Chinese Academy of Medical Sciences, Beijing, China) in June 2011. U-87 MG cells were cultured in minimum essential medium supplemented with 10% fetal bovine serum; H4 and Hs 683 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Cells were maintained under standard cell culture conditions at 37 °C and 5% CO2 in a humid environment.
Antibodies and Special Reagents
EGFR antibody (4267), phosphorylated (p) EGFR (pEGFR) antibodies (Tyr-1173, Tyr-845, Tyr-1068, Tyr-1148, Tyr-992, and Ser-1046/1047), protein kinase B (Akt) antibody (2920S), pAkt (serine 473) antibody (4060S), STAT3 antibody (9139S), pSTAT3 (Thr-705) antibody (9145S), extracellular signal-regulated kinases 1/2 (ERK1/2) antibody (9102S), pERK1/2 (threonine 202/tyrosine 204) antibody (4370S), PLC-γ1 antibody (5690S), pPLC-γ1 (tyrosine 783) antibody (2821S), GFP antibody (2956), GAPDH antibody (2118), and β-actin antibody (4970) were purchased from Cell Signaling Technology, Inc. Anti-Migfilin antibody was a kind gift from Dr. Cary Wu (University of Pittsburgh). Horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibody, PLC-γ1 inhibitor U-73122 (U6756), and STAT3 inhibitor cryptotanshinone (C5624) were purchased from Sigma.
Western Blotting, Immunoprecipitation, and GST Pulldown Assays
Western blot analysis was performed as follows. Cells were collected and centrifuged for harvesting. Cells were lysed on ice for 40 min in RIPA buffer (10 mm Tris, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, and 5 mm EDTA) containing Complete Protease Inhibitor Mixture (Sigma). Lysates were clarified by centrifugation at 12,000 relative centrifugal force for 20 min at 4 °C. For Western blotting, 40 μg of total protein was suspended in sample buffer. For immunoprecipitation, lysates were incubated with primary antibody followed by incubation with protein A-agarose beads (Invitrogen). The immune complexes were washed and suspended in sample buffer. In GST pulldowns, glutathione beads (Sigma) were incubated with Escherichia coli-expressed GST-Migfilin, GST mutant Migfilin, or GST alone for 4 h. Glutathione beads were then washed and incubated for 4 h with lysates of glioma cells. After washing, the protein complexes were suspended in sample buffer. All protein was loaded into each well of a 15% SDS-PAGE. Gels were transferred onto PVDF membranes (Bio-Rad), blocked with 5% milk/PBS, and incubated overnight at 4 °C with primary antibodies. Following washing and incubation with secondary antibodies in 5% milk, the membrane was washed, and the positive signals were developed with chemiluminescence reagent (Amersham). The membrane was exposed to medical x-ray film (Fuji Ltd., Tokyo, Japan).
Plasmids and Small Interfering RNA Transfection
All Migfilin plasmids were a kind gift from Dr. Cary Wu. The siRNA sequences that we used were listed as follows: Migfilin siRNA, 5′-AGGGGCAUCCACAGACAUCTT-3′, and control siRNA, 5′-UUCUCCGAACGUGUCACGU-3′. Cells were transfected with Migfilin plasmids or Migfilin siRNA using Lipofectamine2000 (Invitrogen) according to the manufacturer's instruction. Fresh medium was added 6 h after transfection.
Cell Growth Curve
Cell growth curves were assessed by adding 100 μl, containing 1 × 104 cells, to three wells of a 96-well plate. After cells had been grown for 1, 3, and 5 days, 20 μl of methanethiosulfonate reagent (Promega) was added per well, and following 1–4 h of incubation at 37 °C and 5% CO2, the absorbances were read at 490 nm with a microplate reader. The experiment was conducted in triplicate and repeated three times to draw the cell growth curve.
Transwell Migration/Invasion Assays
Migration and invasion assays were performed as follows. Migration of cells was assayed in Transwell cell culture chambers with 6.5-mm diameter polycarbonate membrane filters containing 8-μm pore size (Neuro Probe, Gaithersburg, MD). In total, 4 × 104 cells in 100 μl of serum-free medium were added to the upper chamber of the device, and the lower chamber was filled with 600 μl of culture medium with 20% fetal bovine serum. After 6 h of incubation at 37 °C, the nonmigration cells were removed from the upper surface of the membrane with a cotton swab. The filters were then fixed in methanol for 10 min, stained with Giemsa solution for 1 h, and counted. Five random microscopic fields (×100) were counted per well, and the mean was determined. For the transwell invasion assay, the membrane of the upper chamber was pre-coated with 20 ml of a 2.5 mg/ml solution of Matrigel (Falcon).
Quantitative Real Time PCR
Quantitative real time PCR analysis was performed as follows. Amplifications of 50 ng of cDNA were performed with an ABI7900HT machine (Applied Biosystems) in triplicate, in 10-μl reaction mixtures containing 1× TaqMan Universal PCR master mix (Applied Biosystems), 200 nm of primers, and 0.25 μl of dual-labeled probe (Roche ProbeLibrary). The reaction parameters were as follows: 2 min at 50 °C hold, 30 min at 60 °C hold, and 10 min at 95 °C hold, followed by 40 cycles of 15-s 95 °C melt and 1-min 60 °C anneal/extension. Measurements were performed in triplicate. Results were analyzed with an ABI sequence detector software version 2.3 using the ΔΔ cycle threshold (Ct) method for relative quantitation. A Ct <35 was used as the cutoff for estimating significantly expressed transcripts; cDNA samples with Ct values >35 were marked not expressed. Ct values between 35 and 40 were solely used for calculation of relative expression differences in treated cells versus control cells. Primers used were as follows: EGFR, forward, 5′-GGTGACCGTTTGGGAGTTGA-3′, and reverse, 5′-CCCTGAATGACAAGGTAGCG-3′; Migfilin, forward, 5′-CAGCGGAGGGACCTTCAGT-3′, and reverse, 5′-GGACACGGTCTTGTGGCAG-3′; GAPDH, forward, 5′-TGTTGCCATCAATGACCCCTT-3′, and reverse, 5′-CTCCACGACGTACTCAGCG-3′.
Cases and Tissue Samples
217 glioma specimens (125 males and 92 females, with age ranging from 1 to 74 years, average of 39.1 years and S.D. of 17.9 years) were obtained from patients undergoing therapeutic surgery for brain tumors at the Sanbo Brain Hospital of Beijing between 2008 and 2010. None of the tumors had been irradiated or treated by chemotherapy before the operation. All selected cases had sufficient material for evaluation, and samples were paraffin-embedded and selected on the basis of adequacy for immunohistochemical studies. According to WHO classification of brain tumors (2007) (2), the tumors were diagnosed as pilocytic astrocytoma (WHO grade I), astrocytoma/oligodendroglioma/mixed gliomas (WHO grade II), anaplastic gliomas (WHO grade III), and glioblastoma (WHO grade IV); there were 10 (4.6%) grade I, 77 (35.5%) grade II, 53 (24.4%) grade III, and 77 (35.5%) grade IV. Ten samples of normal brain (mostly medulla) tissue were taken from donations from individuals who died in traffic accidents; the samples were confirmed to be free of any detectable pathological conditions. For a follow-up study, patients were included who met the following criteria: 1) survived for more than 1 month after surgery and 2) did not die of any other cause other than gliomas after surgery. After surgery, patients with grades I/II were observed and received radiation therapy or chemotherapy (temozolomide) until tumor progression; and patients with grades III/IV received a combination of radiation therapy and temozolomide-based chemotherapy. The follow-up period was 35 months or until death. Informed consent from patients and ethics approval from the Institutional Research Ethics Committee was obtained.
Immunohistochemistry
Five-micrometer serial sections were cut and mounted on adherent glass slides. The sample sections were deparaffinized in xylene and rehydrated in graded ethanol. After antigen retrieval with sodium citrate, sections were blocked with 1.5% normal blocking serum in phosphate-buffered saline (PBS) for 1 h at room temperature and incubated with anti-Migfilin antibody diluted at 1:100 at 4 °C overnight. Secondary antibody was added for 1 h after rinsing by PBS. 3,3′-Diaminobenzidine was used for staining.
Evaluation of Staining
All sections were blindly analyzed by two experienced pathologists under a light microscope. Five high power fields (×400) were randomly observed on every slice. Necrotic tissue, blood vessels, and leukocytes were excluded from the quantification process. Based on the estimated percentages of positive cells, the samples were scored as follows: 0 = tissue specimens without staining; 1 = tissue specimens with <25% stained cells; 2 = tissue specimens with 25–50% stained cells; 3 = tissue specimens with 50–75% stained cells; and 4 = tissue specimens with >75% stained cells. Samples were also scored for immunostaining intensity, determined by comparing the immunoreactivity of three positive control samples that were included in each experiment as follows: 0 = none; 1 = light yellow; 2 = yellow brown; and 3 = brown. The scores for percentage of positive cells were multiplied by the scores for immunostaining intensity; the overall scores were divided into three categories as follows: negative (−), 0–4; positive (1+), 4.1–8; strong positive (2+), 8.1–12.
Statistical Analysis
All experiments were performed and repeated at least three times. Data were analyzed with SPSS 11.5 software. Correlations between the degree of staining and the subgroups according to the clinico-pathological classifications were calculated by using the Pearson χ2 test. The Kaplan-Meier method was used to estimate the overall survival rate as a function of time. Survival differences were analyzed by using the log-rank test. The Cox proportional hazards model was used for univariate and multivariate analyses of prognostic factors. A p value of less than 0.05 was considered significant.
RESULTS
Migfilin Expression Significantly Correlated with Pathological Grades of Gliomas
Immunohistochemical analysis was performed in 217 paraffin-embedded, archived glioma tissue samples and the 10 normal brain samples. Migfilin immunoreactivity was detected in 217 glioma specimens, whereas normal brain tissues showed no immunostaining (Fig. 1A). Strong Migfilin reactivity was detected in endothelial cells in all glioma tissue samples (Fig. 1B). Out of 217 samples, 81 (37.3%) showed negative (−) expression, 82 (37.7%) showed positive (1+) expression, and 54 (24.9%) showed positive (2+) expression. Then we analyzed the correlation between Migfilin expression and clinico-pathological parameters, such as age, sex, tumor location (supratentorial and subtentorial), and pathological grades. There was a positive correlation between Migfilin expression and pathological grades (p < 0.001). However, there was no significant correlation between Migfilin expression and age, sex, or tumor location (p = 0.628, p = 0.713, and p = 0.660, respectively) (Table 1 and Fig. 1, C–F). We also divided supratentorial gliomas into frontal lobe, temporal lobe, parietal lobe, and occipital lobe gliomas, and there was also no significant correlation between glioma locations and Migfilin expression levels (data not shown). Taken together, we show that expression of Migfilin significantly correlated with pathological grades of gliomas.
FIGURE 1.

Expression of Migfilin in normal brain tissue and glioma tissue (×400). A, normal brain cells exhibit no immunoreactivity for Migfilin. B, high levels of Migfilin were detected in blood vessel endothelial cells. C and D, Migfilin exhibits weak immunoreactivity in grade I and grade II gliomas. E, Migfilin exhibits moderate immunoreactivity in grade III glioma. F, Migfilin exhibits strong immunoreactivity in grade IV glioma.
TABLE 1.
Correlation between clinicopathologic parameters and migfilin expression in 217 cases of glioma
| Clinicopathologic parameters | No. of cases | Migfilin expression |
p | ||
|---|---|---|---|---|---|
| − | + | 2+ | |||
| Age (years) | 0.628 | ||||
| ≤Mean (39.1) | 104 | 44 | 31 | 29 | |
| >Mean | 113 | 37 | 51 | 25 | |
| Sex | 0.713 | ||||
| Male | 125 | 46 | 51 | 28 | |
| Female | 92 | 35 | 31 | 26 | |
| Tumor location | 0.660 | ||||
| Supratentorial | 191 | 71 | 71 | 49 | |
| Subtentorial | 26 | 10 | 11 | 5 | |
| Pathological grade | <0.001 | ||||
| I | 10 | 6 | 4 | 0 | |
| II | 77 | 43 | 24 | 10 | |
| III | 53 | 15 | 23 | 15 | |
| IV | 77 | 17 | 31 | 29 | |
High Migfilin Expression Is Associated with Poor Prognosis
Next, we analyzed the correlation between expression of Migfilin and prognosis factors in glioma. The average follow-up for surviving patients was 15 months (range, 1–35 months), and during the entire period there were 115 cancer-related deaths (53.0% of all patients). Of 81 patients with Migfilin (−) tumors, 29 (25.8%) died; of 82 patients with Migfilin (+) tumors, 45 (54.9%) died; and of 54 patients with strong positive (2+) Migfilin tumors, 41 (75.9%) died. The median follow-up time was 16 months in positive (+) Migfilin expression cases and 8 months in strong positive (2+) Migfilin expression cases (Table 2).
TABLE 2.
Effect of migfilin status on outcome for the entire cohort with follow-up data available
| Prognosis | No. of cases | Migfilin expression |
p | ||
|---|---|---|---|---|---|
| − (n = 81) | + (n = 82) | 2+ (n = 54) | |||
| Alive | 102 | 52 (64.2) | 37 (45.1) | 13 (24.1) | |
| Dead | 115 | 29 (25.8) | 45 (51.9) | 41 (75.9) | <0.001 |
| Median follow-up | − | 16 | 8 | <0.001 | |
To compare with other clinico-pathological variables, the impact of age, sex, tumor location, pathological grades, and Migfilin status on survival was analyzed. Only pathological grades and Migfilin status were significant prognostic factors on the overall survival rate (Table 3). Kaplan-Meier analysis displayed that patients with low Migfilin expression had longer survival times, whereas those with high Migfilin expression had shorter survival times (Fig. 2, log-rank, p < 0.001).
TABLE 3.
Kaplan-Meier estimates of overall survival rate dependent on age, sex, migfilin status, location, and histological grade in a follow-up period of 1–35 months for 217 patients with glioma
| Variable | Survival rate | Log-rank test |
|---|---|---|
| % | ||
| Age (years) | 0.181 | |
| ≤39.1 (n = 104) | 59 (57) | |
| >40 (n = 113) | 43 (38) | |
| Sex | 0.438 | |
| Male (n = 125) | 55 (44) | |
| Female (n = 92) | 47 (51) | |
| Tumor location | 0.992 | |
| Supratentorial (n = 191) | 87 (46) | |
| Subtentorial (n = 26) | 15 (58) | |
| Pathological grade | 0.000 | |
| I (n = 10) | 10 (100) | |
| II (n = 77) | 60 (78) | |
| III (n = 53) | 20 (38) | |
| IV (n = 77) | 12 (16) | |
| Migfilin Status | 0.003 | |
| − (n = 81) | 52 (64) | |
| + (n = 82) | 37 (45) | |
| 2+ (n = 54) | 13 (24) | |
FIGURE 2.

Kaplan-Meier curves with univariate analyses (log-rank) for patients with negative Migfilin expression (blue line, n = 81), positive Migfilin expression (green line, n = 82), and strong Migfilin expression tumors (brown line, n = 54).
We also assessed any possible prognostic impact of clinico-pathological variables in a uni- and multivariate Cox proportional survival analysis. In univariate analyses, pathological grades and Migfilin status were significant risk factors for overall mortality. We then further entered these significant variables into a multivariate Cox proportional hazard model, which identified pathological grades and Migfilin status as two independent parameters predicting higher mortality following surgery (Table 4). All of these results suggest that Migfilin expression is an independent prognostic factor for patients with glioma (relative risk, 1.344; 95% confidence interval, 1.043–1.733; p = 0.023).
TABLE 4.
Univariate and multivariate analysis by the Cox proportional hazards model
| Variable | Univariate analysis, p | Multivariate analysis |
|||
|---|---|---|---|---|---|
| B | HR | 95% confidence interval | p | ||
| Age (≥39.1 versus >39.1) | 0.504 | ||||
| Sex (male versus female) | 0.400 | ||||
| Tumor location (supratentorial versus subtentorial) | 0.079 | ||||
| Pathological grade (I or II versus III or IV) | 0.000 | 0.790 | 2.203 | 1.732–2.80 | <0.0001 |
| Migfilin status (negative versus positive) | 0.000 | 0.296 | 1.344 | 1.043–1.733 | 0.023 |
Migfilin Promotes Cell Migration and Invasion in Vitro
To clarify the underlying mechanism of Migfilin in the role of pathological grades and prognosis in glioma, we investigated the biological significance of Migfilin in glioma cells with overexpression or knockdown Migfilin. We transiently transfected FLAG-Migfilin plasmid, control plasmid, Migfilin siRNA, and control siRNA in U-87 MG, H4, and Hs 683 cells. The expression of Migfilin was confirmed by Western blotting (Fig. 3A). We first examined the effect of Migfilin on cell proliferation and found that growth of high-Migfilin cells was not different from the vector control-transfected cells, whereas Migfilin siRNA also did not affect growth compared with control siRNA cells in U-87 MG, H4, and Hs 683 cells (Fig. 3B). We then investigated the impact of Migfilin on cell migration and invasion. Migfilin overexpression in U-87 MG, H4, and Hs 683 glioma cells resulted in markedly increased migration and invasion compared with the vector control-transfected cells, as analyzed by transwell assay. In contrast, silencing endogenous Migfilin expression sharply reduced the ability of glioma cells to migrate and invade (Fig. 3, C and D). These results strongly suggest Migfilin participates in the regulation of migration and invasion of glioma cells.
FIGURE 3.

Migfilin could not induce glioma cell proliferation but could modulate the migrative and invasive ability of glioma cells in vitro. A, ectopic expression and knockdown of Migfilin in glioma cell lines U-87 MG, H4, and Hs 683 were analyzed by Western blotting using an anti-Migfilin antibody. B, growth curves reveal that Migfilin could not induce glioma cells proliferation. C and D, representative pictures (panel a) and quantification (panel b) of penetrated cells were analyzed using the Transwell migrative or invasive assay. The quantification of penetrated cells was represented as the mean of three different experiments. *, p < 0.05 versus FLAG or Control-siRNA.
C-terminal Region Is Essential for Migfilin Up-regulated EGFR Expression and Migfilin-mediated Migration and Invasion in Glioma Cells
The migrative and invasive ability of glioma cells is biologically and clinically linked to expression and activation of EGFR (12), and we were prompted to examine whether the migrative and invasive phenotype enhanced by Migfilin was associated with a change in EGFR expression. In U-87 MG cells, ectopically overexpressing Migfilin increased the expression of EGFR, and knockdown Migfilin drastically repressed EGFR expression (Fig. 4A). Real time PCR analysis was also performed to determine the expression levels of EGFR messenger ribonucleic acid (mRNA). However, ectopically overexpressing Migfilin could not increase expression of EGFR mRNA, whereas knockdown Migfilin also could not repress EGFR mRNA expression (Fig. 4B). These results suggest that Migfilin positively regulates the expression of EGFR protein but not the levels of EGFR mRNA.
FIGURE 4.

Migfilin up-regulates the expression of EGFR and forms a complex with EGFR in glioma cells. A, panel a, expression of EGFR was analyzed by Western blotting in Migfilin-transfected and Migfilin siRNA-transfected cells. Protein expression levels were normalized with actin. A, panel b, bar chart shows the relative expression of EGFR. Protein quantification was obtained by densitometric analysis of the protein absorbance × resulting band area. Protein quantified was relative to the actin internal control. All experiments were performed at least three times with consistent and repeatable results. Each value is expressed as mean ± S.D. (n = 3). *, p < 0.05 versus FLAG or Control-siRNA. B, quantification of changes of EGFR mRNA levels in Migfilin-transfected and Migfilin siRNA-transfected cells. mRNA expression levels were normalized with GAPDH. *, p < 0.05 versus FLAG or Control-siRNA. C, panels a and b, immunoprecipitation of Migfilin and EGFR. Lysates of human U-87 MG cells were mixed with rabbit anti-EGFR antibody (panel a) or mouse anti-Migfilin mAb (panel b). The immunoprecipitates or the control precipitates were analyzed by Western blotting with anti-Migfilin and EGFR antibodies, respectively. Panel c, GST fusion protein pulldown assay. Human U-87 MG cell lysates were incubated with GST-Migfilin fusion protein or GST. GST-Migfilin and GST were precipitated with glutathione beads. EGFR was detected by Western blotting with an anti-EGFR mAb. D, GST fusion protein pulldown assay. Human U-87 MG cell lysates were incubated with GST-Migfilin mutant fusion proteins or GST. GST-Migfilin mutant fusion proteins and GST were precipitated with glutathione beads. EGFR was detected by Western blotting with an anti-EGFR mAb. E, panel a, ectopic expression of mutant Migfilin in U-87 MG cells lines was analyzed by Western blotting using an anti-GFP antibody; The expression of EGFR was analyzed by Western blotting in U-87 MG cells transfected Migfilin mutant plasmids. Protein expression levels were normalized with actin; panel b, bar chart shows the relative expression of GFP and EGFR. Protein quantification was obtained by densitometric analysis of the protein absorbance × resulting band area. Protein quantified was relative to the actin internal control. All experiments were performed at least three times with consistent and repeatable results. Each value is expressed as mean ± S.D. (n = 3). *, p < 0.05 versus GFP.
We next investigated whether Migfilin and EGFR form a complex in glioma cells. We precipitated cell lysates from glioma cells with anti-EGFR mAb. Migfilin was immunoprecipitated with anti-EGFR mAb, and the immunoprecipitated material was probed by Western blotting with an anti-Migfilin antibody (Fig. 4C, panel a). At the same time, EGFR was immunoprecipitated with anti-Migfilin mAb, and the immunoprecipitated material was probed by Western blotting with an anti-EGFR antibody (Fig. 4C, panel b). In control assays, neither anti-EGFR nor anti-Migfilin was precipitated with IgG. We also precipitated cell lysates with GST-Migfilin fusion protein, and we found that EGFR was readily precipitated with GST-Migfilin. In a control experiment, EGFR was not precipitated with GST (Fig. 4C, panel c). To facilitate structure-function analyses, we attempted to identify the EGFR-binding site on Migfilin. GST fusion proteins containing different regions of Migfilin pulldown experiments were performed. GST-Migfilin(s) lacks the central PR region; GST-N-ter (residues 1–85) only contains the N-terminal region, whereas GST-PR-C-ter (residues 85-373) lacks the N-terminal region; GST-C-ter (residues 176–373) lacks the N-terminal and PR regions. As shown in Fig. 4D, EGFR was precipitated with GST-Migfilin(s) and GST-PR-C-ter, whereas the GST-N-ter was not detected, indicating that the EGFR-binding site is not on the N-terminal region or the central PR region but is on the C-terminal region. According to the results, we hypothesized that the function of the C-terminal interaction with EGFR may be required for Migfilin-positive EGFR expression. Then, the GFP-tagged Migfilin mutant plasmids were transiently transfected in U-87 MG cells. Migfilin(s) and PR-C-ter, just as Migfilin, could up-regulate the expression of EGFR protein, whereas N-terminal plasmid could not affect the expression of EGFR protein (Fig. 4E), indicating that the C-terminal region, but not the N-terminal and the central PR regions, is required for Migfilin-regulated expression of EGFR protein.
To determine whether C-terminal region also was required for Migfilin to mediate migration and invasion in glioma cells, the GFP-tagged Migfilin mutant plasmids were transiently transfected in U-87 MG cells (Fig. 5A). In transwell assays, it is showed that GFP-PR-C-ter, like GFP-Migfilin, promoted migration and invasion in U-87 MG cells, indicating that the N terminus is not required for Migfilin-mediated migration and invasion. GFP-Migfilin(s) and GFP-C-ter also promoted migration and invasion. These results suggest that the C terminus is sufficient for Migfilin to modulate migration and invasion in glioma cells.
FIGURE 5.

C-terminal region is required for Migfilin modulation of migration and invasion in glioma cells. A, ectopic expression of mutant Migfilin in glioma cells lines U-87 MG was analyzed by Western blotting using an anti-GFP antibody. B and C, representative pictures (panel a) and quantification (panel b) of penetrated cells were analyzed using the Transwell migrative or invasive assay. The quantification of penetrated cells was represented as the mean of three different experiments. *, p < 0.05 versus control.
Migfilin-mediated Migration and Invasion Is through EGFR-mediated PLC-γ and STAT3 Signaling Pathways
EGFR is a transmembrane cell-surface receptor of the ErbB receptor tyrosine kinase family. The biological activity of EGFR is extensively regulated by ligand-induced tyrosine autophosphorylation (13). Then we analyzed tyrosine phosphorylation of EGFR with antibodies against phospho-Tyr-1173, Tyr-845, Tyr-1148, Tyr-1068, and Tyr-992. Intriguingly, Migfilin could not efficiently induce EGFR phosphorylation at all tyrosine residues tested except Tyr-1173, and Migfilin exhibited a positive modulation on Tyr-1173 phosphorylation (Fig. 6A). We subsequently identified which region was required for Migfilin regulation of Tyr-1173 phosphorylation of EGFR. Only the GFP-N-ter could not induce Tyr-1173 phosphorylation, suggesting that the C terminus is essential for Migfilin regulation of Tyr-1173 phosphorylation of EGFR (Fig. 6B).
FIGURE 6.

C-terminal region is essential for Migfilin regulation of the EGFR phosphorylation. A, panel a, endogenous phosphorylation of EGFR was analyzed by Western blotting using anti-pEGFR antibodies in Migfilin-transfected and Migfilin siRNA-transfected cells. Protein expression levels were normalized with GAPDH; A, panel b, bar chart shows the relative expression of proteins. *, p < 0.05 versus FLAG or control-siRNA. B, panel a, ectopic expression of mutant Migfilin in U-87 MG cells lines was analyzed by Western blotting using an anti-GFP antibody (top panel). The expression of Tyr-1173 phosphorylation of EGFR was analyzed by Western blotting in U-87 MG cells transfected with Migfilin mutant plasmids (bottom panel). Protein expression levels were normalized with GAPDH; B, panel b, bar chart shows the relative expression of proteins. Protein quantification was obtained by densitometric analysis of the protein absorbance × resulting band area. Protein quantified was relative to the GAPDH internal control. All experiments were performed at least three times with consistent and repeatable results. Each value is expressed as mean ± S.D. (n = 3). *, p < 0.05 versus GFP.
Migfilin overexpression specifically induces Tyr-1173 phosphorylation of EGFR; therefore, we expected that EGFR-induced downstream-signaling pathways may also be affected. To test this, we investigated the effect of Migfilin on the active status of important molecules within four major EGFR-mediated signaling pathways. Migfilin plasmid or siRNA was transiently transfected in U-87 MG cells. We detected that overexpression or suppression of Migfilin could not directly affect the expression of PLC-γ, STAT3, AKT, and ERK. However, Migfilin positively affected EGFR-mediated activation of PLC-γ and STAT3 but not AKT or ERK (Fig. 7A). By transfecting GFP-tagged Migfilin mutant plasmids in U-87 MG cells, GFP-Migfilin(s), GFP-PR-C-ter, and GFP-C-ter impacted the activation of PLC-γ and STAT3, indicating that the C-terminal region is required for Migfilin modulation of EGFR-mediated activation of PLC-γ and STAT3 (Fig. 7B). To identify whether Migfilin-mediated migration and invasion depend on PLC-γ- and (or) STAT3-signaling pathways, we incubated cells with the PLC-γ inhibitor (U-73122, 2 μmol/liter) for 30 min or STAT3 inhibitor (cryptotanshinone, 5 μmol/liter) for 24 h. U-73122 and cryptotanshinone blocked Migfilin-induced migration and invasion in glioma cells (Fig. 7, C–E). These results suggest Migfilin-promoted migration and invasion depend on EGFR-mediated PLC-γ- and STAT3-signaling pathways in glioma cells.
FIGURE 7.
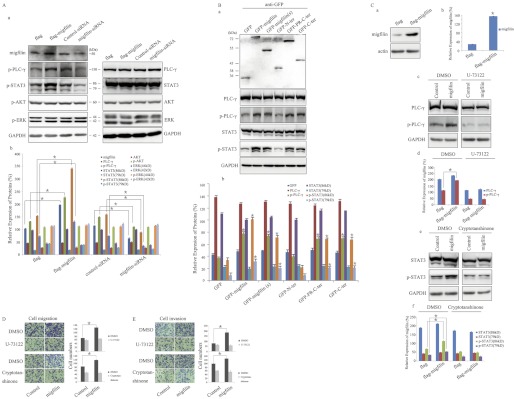
C-terminal region is essential for Migfilin regulation of EGFR-mediated PLC-γ and STAT3-signaling pathways in glioma cells. A, panel a, Western blotting analysis of endogenous ERK, PLC-γ1, STAT3, and AKT in Migfilin-transfected and Migfilin siRNA-transfected cells. Protein expression levels were normalized with GAPDH; A, panel b, bar chart shows the relative expression of proteins. *, p < 0.05 versus FLAG or control-siRNA. B, panel a, ectopic expression of mutant Migfilin in U-87 MG cells lines was analyzed by Western blotting using an anti-GFP antibody (top panel). The expression of PLC-γ1, p-PLC-γ1, STAT3, and p-STAT3 were analyzed by Western blotting in U-87 MG cells transfected with Migfilin mutant plasmids (bottom panel). Protein expression levels were normalized with GAPDH. B, panel b, bar chart shows the relative expression of proteins. *, p < 0.05 versus GFP. C, inhibitor of PLC-γ (U-73122) reduces the Migfilin-induced phosphorylation of PLC-γ, whereas inhibitor of STAT3 (cryptotanshinone) reduces the Migfilin-induced phosphorylation of STAT3. Migfilin-transfected cells were incubated with U-73122 (2 μmol/liter) for 30 min or cryptotanshinone (5 μmol/liter) for 24 h. Panels a, c, and e, Western blotting analysis of Migfilin, PLC-γ1, p-PLC-γ1, STAT3, and p-STAT3; and panels b, d, and f, bar chart shows the relative expression of proteins, and proteins expression levels were normalized with actin or GAPDH. Protein quantification was obtained by densitometric analysis of the protein absorbance × resulting band area. Protein quantified was relative to the actin or GAPDH internal control. All experiments were performed at least three times with consistent and repeatable results. Each value is expressed as mean ± S.D. (n = 3). *, p < 0.05 versus FLAG. D and E, effects of U-73122 and cryptotanshinone on Migfilin-mediated migration and invasion in glioma cells. Migfilin-transfected cells were incubated with U-73122 (2 μmol/liter) for 30 min or cryptotanshinone (5 μmol/liter) for 24 h. Representative pictures (left panel) and quantification (right panel) of penetrated cells were analyzed using the Transwell migrative or invasive assay. The quantification of penetrated cells was represented as the mean of three different experiments. *, p < 0.05 versus control.
DISCUSSION
Key findings from this study provide a significant correlation between the expression of Migfilin and pathological grades in 217 clinical glioma samples. The follow-up analysis showed that Migfilin is an independent prognostic factor for patients with glioma, with high Migfilin expression associated with poor prognosis. The biological function analysis revealed that Migfilin positively mediates migration and invasion of glioma cells. At the molecular level, we display that the C terminus is required for Migfilin to positively modulate EGFR expression and activity. In Migfilin-mediated EGFR-signaling pathways, Migfilin-mediated migration and invasion in glioma cells is through EGFR-induced PLC-γ and STAT3 pathways.
Migfilin is a widely expressed focal adhesion protein of the zyxin family (9). The expression of Migfilin is observed in the heart, lung, intestine, and uterus, whereas little transcription is detected in the brain, kidney, liver, skeletal muscle, and thymus (10, 14). Moik et al. (14) has revealed that loss of Migfilin expression permits normal development and normal postnatal aging in the Migfilin-null mouse. However, many molecules often exhibit vastly different characteristics in various cells. Papachristou et al. (15) have observed that increased cytoplasmic levels of Migfilin is associated with higher grades of human leiomyosarcoma; He et al. (16) have found that Migfilin inhibits esophageal squamous cell carcinoma cells invasion, and Xiao et al. (17) have detected that Migfilin can positively mediate bone marrow stromal cells, ECM adhesion, and migration. Therefore, we should indicate whether Migfilin contributes to genesis and the development of gliomas. We determined that Migfilin immunoreactivity was not detected in normal brain tissues but was detected in 217 glioma specimens, indicating that Migfilin might be involved in the pathogenesis of glioma. GBM is the highest grade glioma, and it is the most aggressive and deadliest form in which patients have a mean survival of 12 months after diagnosis (18, 19). Pathological grades are regarded as one of the most important predictors of outcome in patients with glioma. Our results showed that Migfilin expression significantly correlated with the WHO pathological grades, strongly suggesting that Migfilin may be involved in the progression of gliomas. During the relationship between Migfilin expression and prognosis of patients with glioma, high Migfilin expression is associated with poor prognosis of patients with glioma. Just as pathological grade is an independent prognostic factor for patients with glioma, our findings suggest that Migfilin could be used as a marker for the prognosis of glioma, providing objectivity to the pathological grades.
Gliomas, especially GBM, are characterized by extensive invasion and rapid growth. Its abilities to proliferate and widely infiltrate surrounding neuronal tissues are the leading causes of tumor recurrence and treatment failure (6, 20, 21). Therefore, it is necessary to determine the mechanism of the malignant phenotype in glioma to be able to control the disease. Migfilin, through interactions with filamin and vasodilator-stimulated phosphoprotein, orchestrates actin cytoskeleton remodeling, cell shape, and motility modulation (9, 11). In addition, Migfilin can traffic into the nucleus and is involved in gene expression (22). The abilities of proliferation, migration, and invasion play an important role in pathological grades and prognosis in gliomas, and our data presented here showed that Migfilin-overexpressing cells markedly increased migration and invasion compared with the vector control-transfected cells, whereas silencing endogenous Migfilin expression sharply decreased migration and invasion. However, Migfilin could not impact glioma cell growth. These results indicate that the role of Migfilin in pathological grades and prognosis may depend on Migfilin-mediated migration and invasion in glioma cells.
EGFR-mediated signaling pathways play an important role in the oncogenesis and development of glioma (23). Genetic analysis has shown aberrant function of p53, PTEN, and EGFR in GBM cells (24, 25). Amplification and subsequent overexpression of the EGFR gene are present in ∼30–50% of GBM (24–26). The ectopic function of EGFR has been studied as a potential prognostic indicator in glioma (27). Migration and invasion of glioma cells is a multistep process that is coordinately modulated by cancer cell attachment to ECM, degradation of ECM components, and subsequent actin cytoskeleton organization (28). Migfilin functions as an important component of cell-ECM adhesion networks and orchestrates actin cytoskeleton organization (9). Additionally, EGFR can be involved in the degradation of ECM components (29). Therefore, EGFR is a likely candidate for participation in Migfilin-mediated migration and invasion in glioma cells. Nonetheless, whether enhanced Migfilin expression in gliomas is mechanistically associated with up-regulated EGFR remains unknown. Our study first demonstrated that up-regulation of Migfilin can increase EGFR protein expression in glioma cells, and depletion of endogenous Migfilin represses EGFR expression, whereas Migfilin does not impact the levels of EGFR mRNA. Suggesting that Migfilin does not affect EGFR transcription may impact the degradation of EGFR, which needs future studies for clarification. Additionally, EGFR could form a physical interaction with the C-terminal region of Migfilin. The EGFR-binding C terminus is necessary and sufficient for Migfilin to mediate migration and invasion in glioma cells. The biological activity of EGFR not only relies on the expression of EGFR but also depends on its tyrosine phosphorylation. Ligand-induced phosphorylation of multiple tyrosine residues recruits cytoplasmic signaling molecules and triggers an intracellular signaling pathway, which is consequential in migration and invasion (30, 31). As expected, Migfilin positively mediated EGFR phosphorylation at Tyr-1173 but not at other tyrosine residues, and the C terminus also is necessary for the process. We can conclude that the EGFR-binding C terminus is required for Migfilin to mediate the expression and activation of EGFR.
There are four major EGFR downstream-signaling pathways, including the MEK-ERK pathway, the phosphatidylinositol-3-OH kinase (PI3K)-AKT pathway, the PLC-γ pathway, and the STATs pathway (32). However, Xiao et al. (17) have found that Migfilin negatively regulates ERK1/2 activation in bone marrow stromal cells. We need to identify which EGFR-mediated signaling pathways are involved in Migfilin-mediated migrative and invasive modulation in glioma cells. Among the activations of key molecules within the four signaling pathways, overexpression of Migfilin up-regulated both EGFR-mediated activations of PLC-γ1 and STAT3 but not ERK and AKT. Consistently, down-regulated expression of Migfilin repressed activation of PLC-γ and STAT3. Furthermore, the C-terminal region is required for Migfilin-mediated activation of PLC-γ and STAT3, and inhibitors of PLC-γ and STAT3 can block Migfilin-induced migration and invasion. Therefore, the EGF-binding C terminus is necessary and sufficient for Migfilin to mediate migration and invasion in glioma, and the mechanism is through PLC-γ- and STAT3-signaling pathways. Tyr-1173 phosphorylation of EGFR can be recognized by the Src homology 2 domains of PLC-γ1 and sequentially it mediates the activation of PLC-γ1 (33–35). PLC-γ1 is a key signaling molecule that catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate to yield the important second messengers inositol 1,4,5-trisphosphate and diacylglycerol. Inositol 1,4,5-trisphosphate stimulates the release of Ca2+ from intracellular stores, affecting multitudinous Ca2+-dependent enzymes, whereas diacylglycerol activates protein kinase C (PKC) and RasGRP-dependent signaling pathways (36, 37). Ca2+ plays an important role in regulating actin polymerization and the stability and formation of the actin cytoskeleton (38). Accordingly, high levels of intracellular Ca2+ in tumor cells are associated with increased cell migration and invasion (37). Thus, through the EGFR-PLC-γ-signaling pathway, Migfilin might activate Ca2+-dependent pathways to rapidly reorganize the actin cytoskeleton, and this might result in promoting migration and invasiveness in glioma cells. STAT3, a potential transcription factor, is a member of the JAK-STAT-signaling pathway (39). Phosphorylated EGFR can recruit cytoplasmic STAT3 protein to the membrane; STAT3 is subsequently phosphorylated by JAK. Phosphorylated STAT3 forms dimers and translocates to the nucleus, consequentially driving the transcription of target genes critical for cell motility and other cellular processes (40). Therefore, through the EGFR-STAT3-signaling pathway, Migfilin might promote the transcription of motility-associated genes and initiate the migration and invasiveness in glioma cells. EGFR-mediated signaling pathways are often functionally interlinked and actually should not be considered in isolation. PLC-γ1 also is a significant downstream mediator of the STAT3-signaling pathway. Phosphorylated STAT3 can mediate the activation of PLC-γ1. Furthermore, the activation of PLC-γ1 by STAT3 requires physical interaction between Tyr-705 phosphorylation of STAT3 and the Src homology 2 domains on PLC-γ1 (41). Hence, the cross-talk between pSTAT3 and pPLC-γ1 may also play a critical role in Migfilin-mediated migration and invasion in glioma cells. It is clear that Migfilin can be recruited to cell-ECM adhesions by MIG-2, which interacts with integrin-linked kinase and integrins for precise control actin remodeling (9). This raises the possibility that the transmembrane receptor integrins may also play an important role in Migfilin-EGFR-mediated migration and invasion in glioma. Therefore, future studies are required to identify this possibility.
In summary, our study identifies the important role of Migfilin in pathological grade diagnosis and prognosis in glioma, and it may indicate that Migfilin mediates the migration and invasion of glioma. Our study also elucidates the molecular mechanisms underlying Migfilin-mediated migration and invasion. In addition to providing a better understanding of the molecular and biological mechanisms of glioma malignant progression, these findings may provide significant clinical application, including the use of Migfilin as a molecular marker in the early diagnosis of glioma and as an indicator of prognosis. Furthermore, little transcript of Migfilin is detected in the normal brain tissues. Therefore, Migfilin presents a novel molecular candidate for developing and anti-glioma therapy, which may effectively and specifically reduce tumor cells without toxic effect on normal tissues.
This work was supported by 973 National Key Fundamental Research Program of China Grants 2011CB911004 and 2011CB910801 and National Natural Science Foundation of China Grant 81071633.
- WHO
- World Health Organization
- EGFR
- epidermal growth factor receptor
- GBM
- glioblastoma
- ECM
- extracellular matrix
- PR
- proline-rich
- PLC-γ
- phospholipase Cγ.
REFERENCES
- 1. Furnari F. B., Fenton T., Bachoo R. M., Mukasa A., Stommel J. M., Stegh A., Hahn W. C., Ligon K. L., Louis D. N., Brennan C., Chin L., DePinho R. A., Cavenee W. K. (2007) Malignant astrocytic glioma. Genetics, biology, and paths to treatment. Genes Dev. 21, 2683–2710 [DOI] [PubMed] [Google Scholar]
- 2. Louis D. N., Ohgaki H., Wiestler O. D., Cavenee W. K., Burger P. C., Jouvet A., Scheithauer B. W., Kleihues P. (2007) The 2007 WHO classification of tumors of the central nervous system. Acta Neuropathol. 114, 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wick W., Platten M., Weller M. (2009) New (alternative) temozolomide regimens for the treatment of glioma. Neuro. Oncol. 11, 69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang L., Ng K. Y., Lillehei K. O. (2003) Cell-mediated immunotherapy. A new approach to the treatment of malignant glioma. Cancer Control 10, 138–147 [DOI] [PubMed] [Google Scholar]
- 5. Stewart L. A. (2002) Chemotherapy in adult high grade glioma. A systematic review and meta-analysis of individual patient data from 12 randomized trials. Lancet 359, 1011–1018 [DOI] [PubMed] [Google Scholar]
- 6. Giese A., Bjerkvig R., Berens M. E., Westphal M. (2003) Cost of migration. Invasion of malignant gliomas and implications for treatment. J. Clin. Oncol. 21, 1624–1636 [DOI] [PubMed] [Google Scholar]
- 7. Poot D. H., Van Meir V., Sijbers J. (2010) General and efficient super-resolution method for multislice MRI. Med. Image Comput. Comput. Assist. Interv. 13, 615–622 [DOI] [PubMed] [Google Scholar]
- 8. Fine H. A. (2007) Promising new therapies for malignant gliomas. Cancer J. 13, 349–354 [DOI] [PubMed] [Google Scholar]
- 9. Tu Y., Wu S., Shi X., Chen K., Wu C. (2003) Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell 113, 37–47 [DOI] [PubMed] [Google Scholar]
- 10. Akazawa H., Kudoh S., Mochizuki N., Takekoshi N., Takano H., Nagai T., Komuro I. (2004) A novel LIM protein Cal promotes cardiac differentiation by association with CSX/NKX2–5. J. Cell Biol. 164, 395–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gertler F. B., Niebuhr K., Reinhard M., Wehland J., Soriano P. (1996) Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell 87, 227–239 [DOI] [PubMed] [Google Scholar]
- 12. Huang P. H., Xu A. M., White F. M. (2009) Oncogenic EGFR signaling networks in glioma. Sci. Signal. 2, re6. [DOI] [PubMed] [Google Scholar]
- 13. Bogdan S., Klämbt C. (2001) Epidermal growth factor receptor signaling. Curr. Biol. 11, R292–R295 [DOI] [PubMed] [Google Scholar]
- 14. Moik D. V., Janbandhu V. C., Fässler R. (2011) Loss of Migfilin expression has no overt consequences on murine development and homeostasis. J. Cell Sci. 124, 414–421 [DOI] [PubMed] [Google Scholar]
- 15. Papachristou D. J., Gkretsi V., Tu Y., Shi X., Chen K., Larjava H., Rao U. N., Wu C. (2007) Increased cytoplasmic level of Migfilin is associated with higher grades of human leiomyosarcoma. Histopathology 51, 499–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. He H., Ding F., Li Y., Luo A., Chen H., Wu C., Liu Z. (2012) Migfilin regulates esophageal cancer cell motility through promoting GSK-3β-mediated degradation of β-catenin. Mol. Cancer Res. 10, 273–281 [DOI] [PubMed] [Google Scholar]
- 17. Xiao G., Cheng H., Cao H., Chen K., Tu Y., Yu S., Jiao H., Yang S., Im H. J., Chen D., Chen J., Wu C. (2012) Critical role of Filamin-binding LIM protein 1 (FBLP-1)/Migfilin in regulation of bone remodeling. J. Biol. Chem. 287, 21450–21460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ohgaki H., Kleihues P. (2007) Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 170, 1445–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Louis D. N. (2006) Molecular pathology of malignant gliomas. Annu. Rev. Pathol. 1, 97–117 [DOI] [PubMed] [Google Scholar]
- 20. Wen P. Y., Kesari S. (2008) Malignant gliomas in adults. N. Engl. J. Med. 359, 492–507 [DOI] [PubMed] [Google Scholar]
- 21. Hwang J. H., Smith C. A., Salhia B., Rutka J. T. (2008) The role of fascin in the migration and invasiveness of malignant glioma cells. Neoplasia 10, 149–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu C. (2005) Migfilin and its binding partners. From cell biology to human diseases. J. Cell Sci. 118, 659–664 [DOI] [PubMed] [Google Scholar]
- 23. Mikeska T., Bock C., El-Maarri O., Hübner A., Ehrentraut D., Schramm J., Felsberg J., Kahl P., Büttner R., Pietsch T., Waha A. (2007) Optimization of quantitative MGMT promoter methylation analysis using pyrosequencing and combined bisulfite restriction analysis. J. Mol. Diagn. 9, 368–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wechsler-Reya R., Scott M. P. (2001) The developmental biology of brain tumors. Annu. Rev. Neurosci. 24, 385–428 [DOI] [PubMed] [Google Scholar]
- 25. Maher E. A., Furnari F. B., Bachoo R. M., Rowitch D. H., Louis D. N., Cavenee W. K., DePinho R. A. (2001) Malignant glioma. Genetics and biology of a grave matter. Genes Dev. 15, 1311–1333 [DOI] [PubMed] [Google Scholar]
- 26. Aldape K. D., Ballman K., Furth A., Buckner J. C., Giannini C., Burger P. C., Scheithauer B. W., Jenkins R. B., James C. D. (2004) Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J. Neuropathol. Exp. Neurol. 63, 700–707 [DOI] [PubMed] [Google Scholar]
- 27. Layfield L. J., Willmore C., Tripp S., Jones C., Jensen R. L. (2006) Epidermal growth factor receptor gene amplification and protein expression in glioblastoma multiforme. Prognostic significance and relationship to other prognostic factors. Appl. Immunohistochem. Mol. Morphol. 14, 91–96 [DOI] [PubMed] [Google Scholar]
- 28. Hoelzinger D. B., Demuth T., Berens M. E. (2007) Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J. Natl. Cancer Inst. 99, 1583–1593 [DOI] [PubMed] [Google Scholar]
- 29. van der Zee E., Jansen I., Hoeben K., Beertsen W., Everts V. (1998) EGF and IL-1α modulate the release of collagenase, gelatinase, and TIMP-1 as well as the release of calcium by rabbit calvarial bone explants. J. Periodontal. Res. 33, 65–72 [DOI] [PubMed] [Google Scholar]
- 30. Hynes N. E., Lane H. A. (2005) ERBB receptors and cancer. The complexity of targeted inhibitors. Nat. Rev. Cancer 5, 341–354 [DOI] [PubMed] [Google Scholar]
- 31. Citri A., Yarden Y. (2006) EGF-ERBB signaling. Toward the systems level. Nat. Rev. Mol. Cell Biol. 7, 505–516 [DOI] [PubMed] [Google Scholar]
- 32. Hsu J. M., Chen C. T., Chou C. K., Kuo H. P., Li L. Y., Lin C. Y., Lee H. J., Wang Y. N., Liu M., Liao H. W., Shi B., Lai C. C., Bedford M. T., Tsai C. H., Hung M. C. (2011) Cross-talk between Arg-1175 methylation and Tyr-1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat. Cell Biol. 13, 174–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chattopadhyay A., Vecchi M., Ji Q., Mernaugh R., Carpenter G. (1999) The role of individual SH2 domains in mediating association of phospholipase C-γ1 with the activated EGF receptor. J. Biol. Chem. 274, 26091–26097 [DOI] [PubMed] [Google Scholar]
- 34. Meisenhelder J., Suh P. G., Rhee S. G., Hunter T. (1989) Phospholipase C-γ is a substrate for the PDGF and EGF receptor protein-tyrosine kinases in vivo and in vitro. Cell 57, 1109–1122 [DOI] [PubMed] [Google Scholar]
- 35. Wang X. J., Liao H. J., Chattopadhyay A., Carpenter G. (2001) EGF-dependent translocation of green fluorescent protein-tagged PLC-γ1 to the plasma membrane and endosomes. Exp. Cell Res. 267, 28–36 [DOI] [PubMed] [Google Scholar]
- 36. Kim M. J., Kim E., Ryu S. H., Suh P. G. (2000) The mechanism of phospholipase C-γ1 regulation. Exp. Mol. Med. 32, 101–109 [DOI] [PubMed] [Google Scholar]
- 37. Sun L., Carpenter G. (1998) Epidermal growth factor activation of NF-κB is mediated through IκBα degradation and intracellular free calcium. Oncogene 16, 2095–2102 [DOI] [PubMed] [Google Scholar]
- 38. Janmey P. A. (1994) Phosphoinositides and calcium as regulators of cellular actin assembly and disassembly. Annu. Rev. Physiol. 56, 169–191 [DOI] [PubMed] [Google Scholar]
- 39. Darnell J. E., Jr. (1997) STATs and gene regulation. Science 277, 1630–1635 [DOI] [PubMed] [Google Scholar]
- 40. Brivanlou A. H., Darnell J. E., Jr. (2002) Signal transduction and the control of gene expression. Science 295, 813–818 [DOI] [PubMed] [Google Scholar]
- 41. Zhang P., Zhao Y., Zhu X., Sedwick D., Zhang X., Wang Z. (2011) Cross-talk between phospho-STAT3 and PLCγ1 plays a critical role in colorectal tumorigenesis. Mol. Cancer Res. 9, 1418–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]