

Background: Pyrrolysyl-tRNA synthetase attaches pyrrolysine to tRNAPyl. It is encoded by pylS in Archaea but by pylSn and pylSc in Bacteria.
Results: PylSn binds tRNAPyl specifically in an electrophoretic mobility shift assay.
Conclusion: PylSn and the PylS N terminus participate in binding the tRNA that is key to the genetic encoding of pyrrolysine.
Significance: PylSn and the homologous PylS N terminus previously had no known function.
Keywords: Amino Acid, Aminoacyl tRNA Synthetase, Archaea, Bacteria, RNA-binding Protein, Transfer RNA (tRNA), 22nd Amino Acid, Genetic Code, Pyrrolysine, Pyrrolysyl-tRNA Synthetase
Abstract
Pyrrolysine is represented by an amber codon in genes encoding proteins such as the methylamine methyltransferases present in some Archaea and Bacteria. Pyrrolysyl-tRNA synthetase (PylRS) attaches pyrrolysine to the amber-suppressing tRNAPyl. Archaeal PylRS, encoded by pylS, has a catalytic C-terminal domain but an N-terminal region of unknown function and structure. In Bacteria, homologs of the N- and C-terminal regions of archaeal PylRS are respectively encoded by pylSn and pylSc. We show here that wild type PylS from Methanosarcina barkeri and PylSn from Desulfitobacterium hafniense bind tRNAPyl in EMSA with apparent Kd values of 0.12 and 0.13 μm, respectively. Truncation of the N-terminal region of PylS eliminated detectable tRNAPyl binding as measured by EMSA, but not catalytic activity. A chimeric protein with PylSn fused to the N terminus of truncated PylS regained EMSA-detectable tRNAPyl binding. PylSn did not bind other D. hafniense tRNAs, nor did the competition by the Escherichia coli tRNA pool interfere with tRNAPyl binding. Further indicating the specificity of PylSn interaction with tRNAPyl, substitutions of conserved residues in tRNAPyl in the variable loop, D stem, and T stem and loop had significant impact in binding, whereas those having base changes in the acceptor stem or anticodon stem and loop still retained the ability to complex with PylSn. PylSn and the N terminus of PylS comprise the protein superfamily TIGR03129. The members of this family are not similar to any known RNA-binding protein, but our results suggest their common function involves specific binding of tRNAPyl.
Introduction
Pyrrolysine has been called “the 22nd amino acid” (1), because it is the most recently discovered genetically encoded protein residue found in a naturally occurring organism (2). Pyrrolysine was first described in the structure of the methyltransferase initiating monomethylamine metabolism in Methanosarcina barkeri (3), and subsequently residues with the mass of pyrrolysine were demonstrated in the dimethylamine and trimethylamine methyltransferases (4). For each of these proteins, the position of pyrrolysine corresponds in the encoding gene to an amber codon (TAG/UAG) (5, 6). Pyrrolysine is inserted into the growing monomethylamine methyltransferase protein with little detectable UAG termination product in Methanosarcina spp. Even foreign genes with in-frame amber codons are expressed in the methanogen, albeit with less efficient UAG translation (7, 8).
Underlying UAG translation in Methanosarcinaceae is the pyl gene cluster (see Fig. 1) (9, 10). The first gene in the pyl gene cluster is pylT, encoding the tRNAPyl that possesses the CUA anticodon required to translate amber codons as pyrrolysine. Deletion of pylT renders Methanosarcina acetivorans unable to produce methylamine methyltransferases but with no other apparent defect (11). The secondary structure of tRNAPyl is unusual, having a small D loop and variable loop and an elongated anticodon stem, as well as lacking the base typically found between the acceptor and D stems (9). Nonetheless, tRNAPyl assumes the expected L-shaped tertiary structure but with a more compact core relative to most tRNA species (12, 13).
FIGURE 1.

Representative microbes with sequenced genomes that contain the pyl genes. A and B, the archaeal genomes are deposited at NCBI under accession numbers NC_003552 (A) and NC_007955 (B). C and D, the bacterial genomes have accession numbers NC_011830 (C) and AASZ01001259 as well as AASZ01001367 (D). The proteobacterial example is a gut symbiont sequenced in a metagenomic study of a marine worm. In all examples but D, the pyl genes are found in an apparent single transcriptional unit. In D, the pylTScSn genes and the pylBCD genes are found on separate contigs (groups of overlapping clones). The number labeling each gene is the locus number in the annotated genome. The numbers below genes in A–C show the percentage of similarity for each gene product when aligned with the corresponding M. acetivorans pyl gene product, in D the percentage of similarity to the corresponding gene products in M. acetivorans is shown, followed by the percentage of similarity to D. hafniense pyl gene products. Genes involved in the genetic encoding of pyrrolysine are in red, and those in biosynthesis of pyrrolysine are in blue. The pylSn genes are shown in pink, which matches the shading of the homologous 5′ region of the archaeal pylS genes.
Following pylT in the archaeal pyl gene cluster is the pylS gene that encodes a pyrrolysyl-tRNA synthetase able to ligate tRNAPyl to chemically synthesized pyrrolysine (14) or pyrrolysine analogs (15). Transformation of Escherichia coli with the pylS and pylT genes allows in vivo suppression of UAG codons when supplemented with pyrrolysine (14) or its derivatives (16). Pyrrolysine is made independently of tRNAPyl by the products of the downstream pylB, pylC, and pylD genes (10, 17). Each molecule of pyrrolysine is made from two molecules of lysine (18) with (3R)-3-methyl-d-ornithine an intermediate (18–20). Further transformation of E. coli with pylB, pylC, and pylD allows E. coli to decode UAG as endogenously produced pyrrolysine (17). The ability of pylS and pylT to encode a functional orthogonal pair within E. coli has led to their application for insertion of modifiable lysine derivatives into recombinant proteins (10, 21).
Homologs of the pyl genes are also found in isolated species of bacteria, including Gram-positive organisms such as Desulfitobacterium hafniense, Desulfotomaculum acetoxidans, Acetohalobium arabaticum, and Thermincola spp. JR, as well as a δ-proteobacterial worm symbiont (9, 10, 22–25). The archaeal pylS gene is represented as split genes in bacteria (Fig. 1 and supplemental Fig. S1). The bacterial pylSn gene product is homologous to the N terminus of archaeal PylS, whereas the pylSc gene product is homologous to the C terminus of PylS (9, 26).
The structure of the Methanosarcina mazei PylS with an N-terminal truncation of 185 amino acids (Δ185PylS) revealed the catalytic site of PylS (27, 28). The deletion was necessary to eliminate the highly basic yet hydrophobic N-terminal portion of the protein, whose presence interfered with protein stability and crystallization (29). The structure of D. hafniense PylSc is similar to that of M. mazei Δ185PylS, although with an apparently tighter binding pocket for pyrrolysine (13, 30). PylSc binds tRNAPyl primarily through interactions with residues of the acceptor and D stem with no direct contact to residues of the T arm, anticodon arm, or variable loop (13).
Loss of the N-terminal portion of archaeal PylS is detrimental to in vivo activity. Truncation or mutation in the first 106 residues from M. barkeri PylS led to the loss of in vivo aminoacylation of tRNAPyl, as well as UAG codon suppression in a recombinant system (31). However, these experiments did not delineate whether protein structure or activity was compromised by the N-terminal truncation. Recombinant D. hafniense PylSc was reported to charge cognate tRNAPyl with a pyrrolysine analog in vitro, but pylSc has been reported to produce undetectable or very low level UAG translation in vivo (31). The biochemical function of the N terminus of PylS or PylSn has never been demonstrated.
Here we show using EMSA that PylS can bind with specificity to tRNAPyl, an activity that is lost when the N-terminal portion of M. barkeri PylS is deleted. Furthermore, we show that the D. hafniense PylSn is also capable of binding tRNAPyl and that PylSn recognizes tRNAPyl specifically. This binding is susceptible to mutations in the variable loop and T-arm of tRNAPyl, areas that do not interact with PylSc. Thus, PylSn and the homologous region of PylS are tRNAPyl-binding proteins, although neither has detectable homology with known families of RNA-binding proteins.
EXPERIMENTAL PROCEDURES
Expression of Genes and Purification of Products
The D. hafniense PylSc and M. barkeri PylS proteins were purified following recombinant expression of their genes as described previously (14, 30). The D. hafniense pylSn gene was amplified by PCR from genomic DNA using Ex Taq (Takara Mirus Bio, Madison, WI) and primers PylSnF and PylSnR (the sequence of these and other primers are listed in supplemental Table S1). The amplified pylSn gene was subsequently ligated into PCR2.1-Topo (Invitrogen) and then inserted into the NcoI and NotI sites of expression vector pET22b(+) (Novagen, Madison, WI) so as to produce sequence encoding PylSn with a C-terminal hexahistidine tag under the control of an inducible T7 promoter. The plasmid was then transformed into E. coli BL21 (DE3) (Stratagene, Santa Clara, CA). The desired sequence of this and the following gene constructs were confirmed by sequencing of both sense and antisense strands using a 3730 DNA sequence analyzer (Applied Biosystems, Carlsbad, CA) at the Ohio State University Plant-Microbe Genomics Facility.
The cells were grown at 37 °C in LB broth (10 g/liter tryptone, 5 g/liter yeast extract, and 10 g/liter sodium chloride) containing 100 mg/liter ampicillin. When the culture had grown to early log phase (A600 = 0.4–0.6), 1 mm isopropyl β-d-1-thiogalactopyranoside was added to induce production of PylSn. The cells were harvested by centrifugation 3.5 h later and then suspended in 500 mm NaCl, 10 mm imidazole, and 20 mm sodium phosphate buffer, pH 7.4, prior to lysis using a French pressure cell. Following centrifugation at 27,000 × g, the supernatant was applied to a 1-ml bed volume nickel-activated HiTrap HP column (GE Healthcare). Bound protein was then eluted with a 40-ml linear gradient of 10–500 mm imidazole in 500 mm NaCl and 20 mm sodium phosphate, pH 7.4. PylSn eluted at 250 mm imidazole, and the collected peak was 95% pure judged by densitometry of the preparation following separation by SDS-acrylamide gel electrophoresis and subsequent Coomassie staining. PylSn was stored in elution buffer supplemented with 20% glycerol at −20 °C.
N-terminally truncated M. barkeri MS PylS (Δ92PylS) was prepared using genomic DNA as template for PCR amplification with primers 92SF and 92SR that resulted in amplification of the gene such that it would produce PylS lacking N-terminal residues 1–92. The amplified truncated pylS gene was inserted into pET22b (+) at the NdeI and XhoI sites so as to produce Δ92PylS with a hexahistidine C terminus. The Δ92PylS protein was purified by nickel affinity column as described above and eluted at ∼250 mm imidazole.
The PylSnΔ120S protein in which PylSn replaces the N terminus of M. barkeri PylS was made as follows. The M. barkeri PylS gene was amplified from genomic DNA using FusCF and FusCR and cloned into pET15b using the NcoI and BamH1 sites. This resulted in a truncated pylS gene whose product would lack the first 120 N-terminal residues but have a C-terminal hexahistidine tag. The D. hafniense pylSn gene was then amplified from genomic DNA using primers FusNF and FusNR and then ligated into the pET15b clone bearing the inserted truncated PylS gene using the NcoI and EcoRV sites. The resultant plasmid was transformed into E. coli BL21 and used to produce the purified fusion protein designated PylSnΔ120S protein using the methods outlined above. The PylSnΔ120S eluted from the nickel affinity column again at ∼250 mm histidine.
Recombinant D. hafniense tRNAPyl was produced in E. coli cells bearing the pETDuet-1 plasmid in which the genomic D. hafniense pylT gene was inserted into the XbaI site lying directly behind the T7 promoter. The pylT gene was amplified from genomic DNA using primers PylTF and PylTR. The tRNAPyl was then isolated with the E. coli RNA pool as described below.
E. coli RNA pool isolation-E. coli BL21 (DE3) was grown in LB broth at 37 °C to an A600 of ∼0.5 and then centrifuged at 5000 × g. The cell pellet was suspended in 10 mm EDTA in 0.3 m sodium acetate, pH 4.5, before extraction with phenol:chloroform (5:1) equilibrated with the same buffer (Invitrogen). Following centrifugation at 18,000 × g for 15 min, the aqueous layer was extracted a second time. The RNA was then precipitated by addition of a three-fold volume of 100% ethanol, resuspended in 0.3 m sodium acetate, pH 4.5, and precipitated again. The final pellet was air-dried and dissolved in TE buffer (10 mm Tris-Cl, 1 mm EDTA, pH 7.0). The isolated cellular RNA pool contained 30% tRNA when examined by fluorescent intensity of agarose electrophoretic gels stained with ethidium bromide. The deacylated tRNA pool was prepared by adding equal volume of deacylation buffer (100 mm NaCl in Tris-HCl, pH 9.5) and then heating at 70 °C for 30 min. The deacylated tRNA pool was exchanged into TE buffer using Microspin G-25 columns (GE Healthcare).
In Vitro Transcription of tRNA
The tRNAs used in this work were prepared following modifications of published procedures (32, 33). An oligonucleotide corresponding to the 5′ end of the tRNA gene was made such that it also contained an upstream sequence with a T7 promoter. A second oligonucleotide corresponding to the 3′ end of the tRNA gene was made such that it overlapped the first oligonucleotide for 10–12 nucleotides centered on the sequence encoding the anticodon loop. The annealed oligonucleotides were extended using Sequenase (Affymetrix, Santa Clara, CA) to make a template for run-off transcription employing the Megashortscript T7 kit (Invitrogen) according to the manufacturer's recommendations and having 0.5 μm template incubated for a reaction time of 4 h. The resultant tRNA transcript was purified on a denaturing 14% polyacrylamide gel and recovered by elution in TE buffer for 4 h at 22 °C, followed by ethanol precipitation. The recovered tRNA transcript pellet was dissolved in TE buffer and stored at −20 °C. To produce 32P-tRNA, [32P]ATP (20 μCi; final specific radioactivity of 133 μCi/nmol; PerkinElmer Life Sciences) was added to the transcription reaction.
Assay for Amino Acid Activation and tRNA Charging
Aminoacylation of tRNAPyl was detected using acid-urea acrylamide gel electrophoresis as described previously (14). The reactions were conducted at 37 °C in 50 mm KCl, 10 mm MgCl2, 5 mm ATP, and 5 mm dithiothreitol in 10 mm Hepes buffer, pH 7.2. Additionally, assays contained as substrate 50 μm pyrrolysine (kind gift of Michael Chan) (34) and recombinant tRNAPyl present in the E. coli RNA pool (1 mg/ml total RNA). Enzyme was added as indicated in the text. All of the reactions were initiated with enzyme and ended after 30 min by the addition of 7 m urea in 0.3 m sodium acetate buffer, pH 5.0. The reaction mixture was then loaded on a 14% polyacrylamide gel containing 7 m urea in 0.3 m sodium acetate, pH 5.0, and ran at 50 V in the same acetate buffer for 16 h at 4 °C. The tRNA was then electroblotted onto a Hybond N+ membrane (GE Healthcare) and detected by hybridization with a 5′-labeled 32P-tRNAPyl probe employing a Storm phosphorimager (GE Healthcare).
Amino acid activation was assayed using ATP-pyrophosphate exchange, essentially as performed previously (14, 35). All assays were performed at 37 °C in duplicate and were repeated with at least two independent enzyme preparations. Assays contained 20 mm Hepes (pH 7.2), 10 mm MgCl2, 25 mm KCl, 4 mm DTT, 5 mm ATP, 2 mm [32P]PPi (5–15 dpms/pmol; PerkinElmer Life Sciences). In standard assays, 50 μm pyrrolysine and 0.5–1 μm enzymes were added. Identical reactions without enzyme were set up as negative controls. To determine kinetic parameters of PylSc, pyrrolysine concentrations were varied over the range of 0.1–4 Km, and the data were analyzed by nonlinear regression using Prism software (GraphPad, San Diego, CA).
Electrophoretic Mobility Shift Assay
Binding of 32P-labeled tRNA by various proteins was assayed by EMSA. Protein-tRNA complexes were formed by incubation of 17–50 nm 32P-tRNA with various amount of protein on ice for 15 min in 30 μl of binding buffer (50 mm KCl, 10 mm MgCl2, and 5 mm DTT in 10 mm Hepes, pH 7.2). As indicated, in experiments having high amounts of PylSn, 33 μg/ml heparin was also added to the reaction buffer to suppress protein aggregation. The tRNA-protein mixture was then loaded onto a 10% polyacrylamide gel containing 10% glycerol, 1 mm EDTA in 45 mm Tris borate buffer, pH 8.0, which was electrophoresed in the same buffer at 175 V for 2 h at 4 °C. The gel was then exposed to a phosphorus screen for 32P-tRNA and 32P-tRNA-protein complex detection using a Storm phosphorimager. The density of individual bands was quantified by ImageQuant (GE Healthcare). The apparent Kd values of tRNA-protein complexes were obtained by nonlinear regression of the data using Prism software. For assays with variant tRNAPyl for which PylSn had poor affinity, the apparent Kd was estimated by applying the data to following equation: θ = (protein)/((protein) + Kd), where θ is the fractional saturation of tRNA-protein complex seen at the upper end of the PylSn concentrations tested (36).
RESULTS
The N-terminal Region of PylS Influences Complex Formation with tRNAPyl
We first tested whether binding of PylS with its cognate tRNA could be detected by electrophoretic mobility shift assay, because this has not previously been documented. Increasing concentrations of M. barkeri MS PylS were incubated with in vitro transcribed M. barkeri 32P-tRNAPyl (20 nm) prior to electrophoresis in a nondenaturing 10% acrylamide gel. Migration of the tRNAPyl band was retarded in a manner allowing fit to a binding curve having an apparent Kd for the PylS-tRNAPyl complex of 0.12 ± 0.01 μm (Fig. 2A).
FIGURE 2.

Electrophoretic mobility of in vitro transcribed M. barkeri tRNAPyl incubated in the presence of M. barkeri PylS or Δ92PylS. Protein concentrations added to tRNAPyl are indicated at the bottom of each gel. A, electrophoretic mobility of 20 nm tRNAPyl following incubation with increasing concentrations of PylS. B, electrophoretic mobility of 0.5 μm tRNAPyl subsequent to incubation with PylS or Δ92PylS.
N-terminally truncated PylS was unable to form a stable complex with tRNAPyl detectable by EMSA. We made three constructs of the truncated pylS genes with deletions that would correspond to the first 92, 119, or 145 residues of the protein. Of these, only the first construct, Δ92PylS, produced a stable form of the protein that could be successfully purified. However, Δ92PylS could not form a stable complex in EMSA experiments performed under the same conditions as with intact PylS. Even with 10 μm Δ92PylS, which was the highest concentration tested, no shift of the free tRNAPyl band was apparent (Fig. 2B). The lack of activity in these assays was not due to an improperly folded protein, as evidenced by the retention of pyrrolysine-dependent PPi-ATP exchange activity, which Δ92PylS catalyzed with a kcat of 14.4 ± 0.6 min−1, relative to 6 min−1 observed with wild type PylS preparations. Δ92PylS also aminoacylated tRNAPyl in a manner indistinguishable from wild type PylS when assayed using acid-urea electrophoresis (data not shown). These results suggested that although the N terminus is not required for activity in these assays, the presence of the N terminus sufficiently stabilized interaction with tRNAPyl to form a complex detectable by EMSA.
Bacterial PylSn Binds tRNAPyl
The co-crystalization of PylSc and tRNAPyl, as well as modeling studies, revealed how the C terminus of PylS interacts with tRNAPyl (13, 27). However, the above experiments indicate that N-terminal domain might recognize tRNAPyl as well. Unfortunately, our attempts to generate a stable protein with only the N-terminal region of PylS did not succeed. Therefore, we turned to PylSn, the bacterial homolog of the N terminus of archaeal PylS domain, which is encoded as a separate gene independent of the gene encoding the catalytic PylSc domain in some Bacteria. D. hafniense PylSn aligns with residues 1–95 of M. barkeri PylS (supplemental Fig. S1).
Recombinant D. hafniense PylSn bearing a C-terminal hexahistidine tag was relatively stable if kept at low protein concentration at high ionic strength in stock solutions supplemented with sufficient glycerol to keep the solution from freezing at −20 °C. Even under these conditions, stored protein aggregated and precipitated. Therefore, fresh preparations were consistently used in studies of PylSn binding to tRNA. We noted that aggregated PylSn nonspecifically bound nucleic acid in high molecular weight complexes do not enter the gel during EMSA. However, such artifacts were minimized with fresh preparations stored as indicated.
The addition of freshly prepared PylSn to assay mixtures with in vitro transcribed D. hafniense tRNAPyl resulted in a pronounced shift in position of tRNAPyl following migration in PAGE gels (Fig. 3A), indicating that PylSn forms a distinct complex with tRNAPyl. No other band was visible in the gel other than those for tRNAPyl and PylSn-tRNAPyl complex, and significant aggregation of protein-tRNA complex was not observed at the tRNA and protein concentrations tested. Increasing amounts of PylSn were added to 17 nm tRNAPyl to determine the apparent Kd for the formation of the PylSn-tRNAPyl complex. The data best fit a nonlinear regression curve corresponding to a Kd of 0.13 ± 0.01 μm.
FIGURE 3.

Efficacy of D. hafniense PylSn or PylSc in forming electrophoretically detectable complexes with D. hafniense tRNAPyl. Protein concentrations employed are indicated at the bottom of each lane. A, electrophoretic mobility of 17 nm tRNAPyl after incubation with indicated amounts of PylSn. B, electrophoretic mobility of 0.5 μm tRNAPyl after incubation with PylSc or PylSn.
Similar to the results obtained with Δ92PylS, bacterial PylSc did not form an EMSA-detectable complex when incubated with D. hafniense tRNAPyl (Fig. 3B), nor did the addition of PylSc cause a change in the migration of the PylSn-tRNAPyl complex. This was not due to an inactive enzyme, D. hafniense PylSc carried out an PPi-ATP exchange reaction dependent on pyrrolysine (kcat = 19.5 ± 1.2 min−1, Km = 44 ± 4 μm) and charged D. hafniense tRNAPyl recombinantly produced in E. coli with pyrrolysine in a manner indistinguishable from wild PylS when assayed using acid-urea electrophoresis.
PylSn could rescue the ability of N-terminally truncated M. barkeri PylS to bind tRNAPyl as detectable by EMSA. Six different chimeric proteins were prepared in which variable lengths of the N-terminal region of PylS were replaced by PylSn. Only one of these, the fusion protein PylSnΔ120S, in which PylSn replaced the first 120 residues of PylS, could be successfully purified. PylSnΔ120S formed an EMSA-detectable complex when incubated with D. hafniense tRNAPyl, and the apparent Kd for the formation of the PylSnΔ120S-tRNAPyl complex was measured as 0.35 ± 0.03 μm (data not shown). Unlike wild type, the fusion protein was not able to detectably aminoacylate tRNAPyl, possibly because of altered spacing of the N- and C-terminal tRNAPyl-binding domains.
PylSn Binding of tRNA Is Selective for tRNAPyl
The above data indicated that PylSn binds tRNAPyl but did not reveal whether binding was specific for tRNAPyl or could be generalized to other tRNAs. Therefore, we evaluated the ability of D. hafniense PylSn to bind various in vitro transcribed tRNA species from D. hafniense including tRNAGAG (Leu) (Fig. 4), tRNAUCC (Gly), tRNAUUU (Lys), and tRNAUGC (Ala) (data not shown). We also tested Bos taurus mitochondrial tRNAUGA (Ser) (Fig. 4), which shares some of the unique features of tRNAPyl including a three-base variable loop and elongated anticodon stem (37). When assayed under conditions where PylSn readily caused a gel shift of free tRNAPyl, PylSn did not cause a shift analogous to the PylSn-tRNAPyl complex with these tRNA species.
FIGURE 4.
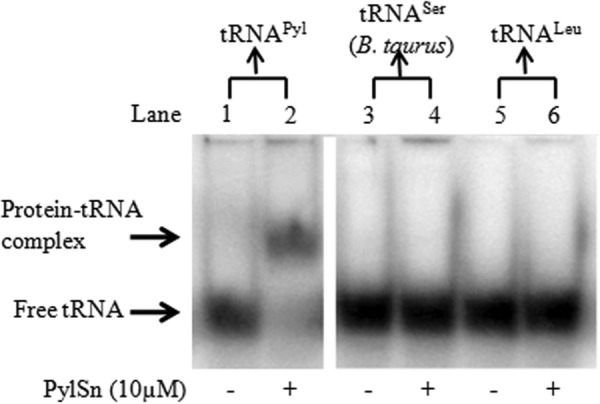
Specificity of D. hafniense PylSn for binding tRNAPyl as measured by electrophoretic shift. The panels show electrophoretic mobility shift assay of 0 μm and 10 μm PylSn with 17 nm tRNAPyl (lanes 1 and 2); 0 μm and 10 μm PylSn with 30 nm B. taurus mitochondrial tRNASer (lanes 3 and 4); and 0 μm and 10 μm PylSn with 30 nm tRNALeu (lanes 5 and 6). Both panels are from the same polyacrylamide gel.
The specificity of PylSn binding to D. hafniense tRNAPyl was further tested by competition assays in which increasing concentration of the crude deacylated tRNA pool extracted from E. coli was incubated with 17 nm of 32P-tRNAPyl transcript prior to the addition of 1 μm of PylSn. No decrease in the band representing the tRNAPyl-PylSn complex could be detected because the estimated tRNA concentration increased from 0 to 1.7 μm; nor did the free tRNAPyl concentration appear to increase (Fig. 5). These data suggest that PylSn can form a stable complex with tRNAPyl even in the presence of a 100-fold molar excess of other tRNA species.
FIGURE 5.
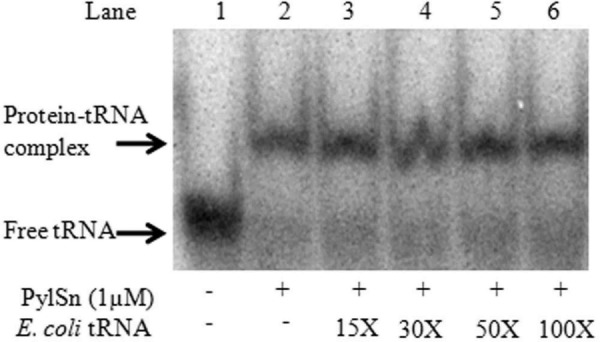
D. hafniense PylSn binds tRNAPyl even in the presence of competing RNA. The panels show electrophoretic mobility shift assay of 17 nm tRNAPyl incubated in the presence of 1 μm PylSn and the E. coli tRNA pool. The calculated concentration of E. coli tRNA added to each binding assay was 0, 0.25, 0.5, 0.85, or 1.7 μm (lanes 2–6). The tRNA pool was 30% of total RNA in the preparation.
Certain Base Substitutions in tRNAPyl Compromise Binding by PylSn
As a further test of the specificity of PylSn for tRNAPyl, we produced in vitro transcribed variants of D. hafniense tRNAPyl having base substitutions at the positions indicated in Figs. 6 and 7. The positions changed were chosen based on their high conservation in known examples of tRNAPyl because these bases might serve as determinants for PylSn binding or disrupt tertiary structure important for PylSn recognition. The variant tRNAPyl were then compared in the ability to form complexes with wild type D. hafniense tRNAPyl in EMSA. Representative EMSA gels are shown in Fig. 8. We screened mutations under conditions that would detect base changes yielding strong binding defects, i.e. the variant tRNAPyl-PylSn complex must have a Kd greater than 2.5 μm to be distinguishable from wild type.
FIGURE 6.
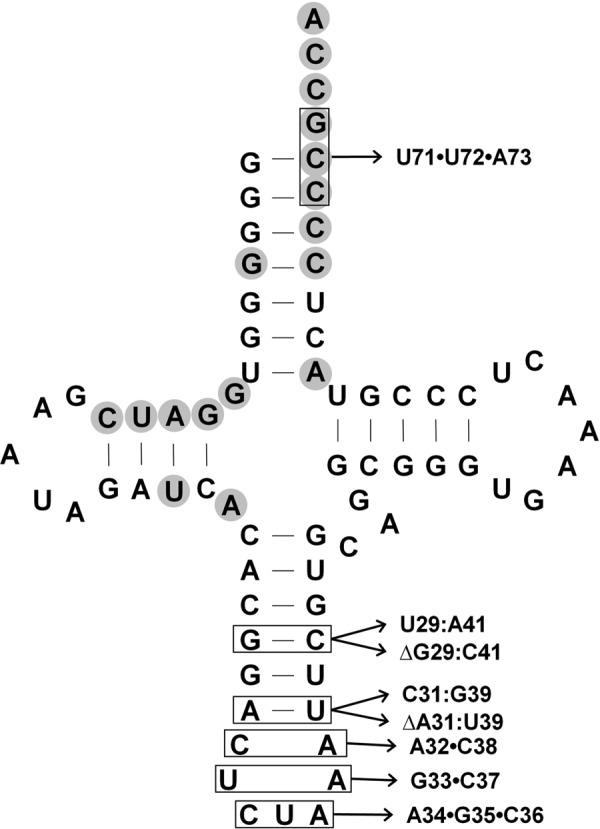
Base changes made in the D. hafniense tRNAPyl in the acceptor stem and anticodon stem and loop. Those bases known to directly contact PylSc residues (13) are shown with gray shading. Locations in which bases were changed are shown boxed in this figure. None of these bases proved essential for EMSA detectable binding by PylSn.
FIGURE 7.
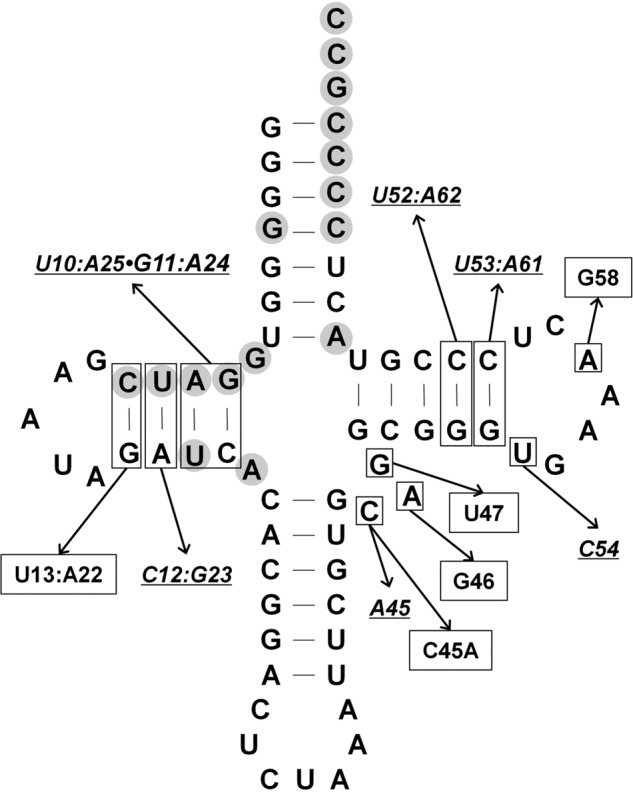
Base changes made within the D. hafniense tRNAPyld-stem and T stem and loop. Targeted base positions are indicated by the boxed bases in the tRNA. Bases that directly interact with PylSc are shown with gray shading (13). The base changes that strongly affected PylSn binding to tRNA, but still showed detectable binding in EMSA, are underlined. Base changes that eliminated the tRNA ability to form an EMSA-detectable complex with PylSn are boxed.
FIGURE 8.
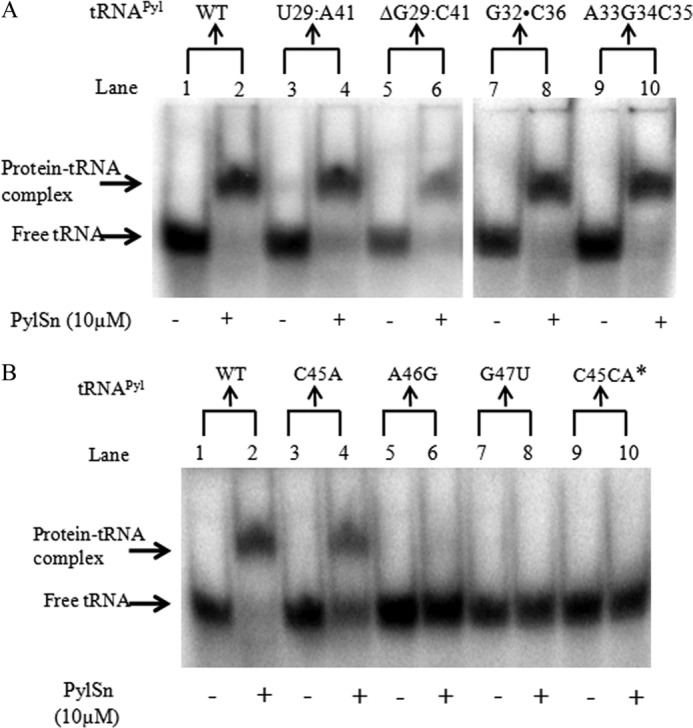
Representative EMSA gels showing the effects of several base changes in the D. hafniense tRNAPyl on PylSn binding. A, electrophoretic mobility of D. hafniense tRNAPyl incubated in the absence (odd-numbered lanes) or presence (even-numbered lanes) of 10 μm PylSn. The tRNAPyl variants carried substitutions in the anticodon stem and loop as indicated at the top of the gel for each pair of lanes. All lanes are from a single electrophoretic gel. B, the panels show electrophoretic mobility of tRNAPyl variants carrying substitutions in the variable loop when incubated in the presence of 10 μm PylSn (even-numbered lanes) or in its absence (odd-numbered lanes). C45CA*, lanes 9 and 10 contain tRNAPyl in which an additional A was inserted between C45 and A46.
No bases are universally conserved in the anticodon stem of tRNAPyl, although several base pairs are found in the majority of examples, such as G29:C41 and A31:U39 (10). However, substitution of either with an alternative base pair did not result in a severe defect in PylSn binding under our assay conditions (Figs. 6 and 8). Furthermore, outright deletion of either base pair did not affect binding of PylSn in our assays relative to wild type, indicating the elongated anticodon stem of tRNAPyl is not essential for PylSn binding. Six of the seven residues in the anticodon loop are identical in all known examples of tRNAPyl. Mutations of these residues are deleterious to the in vivo ability of tRNAPyl to act as an amber suppressor in E. coli, especially U33 and A37, the residues that flank the anticodon (38). However, none of the four residues flanking the anticodon are essential for binding of tRNAPyl by PylSn (Fig. 6). The CUA anticodon itself was changed to AGC, and this construct is still readily bound by PylSn. Similarly, a change to the acceptor stem did not detectably change PylSn binding, as shown by substitution of C71 and C72, along with the discriminator base G73, with the sequence UUA.
In contrast, changes to the D stem strongly affected PylSn binding. Three of the four base pairs in the D stem are universally conserved in tRNAPyl and the remaining pair, A11:U24, is found in 70% of bacterial tRNAPyl sequences. Mutation of the two base pairs distal to the D loop (Fig. 8) led to weak binding (Kd = ∼30 μm), whereas independent mutation of the two base pairs proximal to the D loop (Fig. 7) led to similarly weak (U12:A23 to C12:G23) or no detectable binding (C13:G22 to U13:A22). The assay conditions were set up such that a shift of 10% of the tRNA would have indicated an apparent Kd of 200 μm.
One of the most distinctive features of tRNAPyl is the very small variable loop composed of only three residues (Fig. 7). Mutations in any of these three residues significantly decreased the affinity of PylSn for tRNAPyl (Figs. 7 and 8). The least disruptive mutation was C45A, which nonetheless had only weak binding with an estimated Kd of 10 μm. In contrast, tRNAPyl having A46G or G47U substitution did not detectably interact with PylSn in EMSA. Insertion of an additional base into the variable loop also led to no detectable binding. The detrimental effect of substitution of the latter two residues may reflect their important role in stabilizing the L-shaped tertiary structure by interaction with the D stem helix (13), indicating the importance of the tRNAPyl structure for binding to PylSn.
Mutations within the T stem and T loop also strongly affected binding between PylSn and tRNAPyl. Two highly conserved residue pairs are present in the T-stem, namely G52:C62 and G53:C61. Mutation of the first or second GC pair reduced affinity of PylSn for the tRNA with apparent Kd values of ∼10 and 40 μm, respectively (Fig. 7). Mutations in the T-loop also reduced PylSn affinity for the tRNA. For example, no interaction of tRNAPyl with PylSn was detectable (Kd > 200 μm) following substitution of A58 with C. This effect may be due to the need for A58 interaction with U54 in the properly folded tRNA (13); however, alteration of U54 to G led to a more modest decrease in affinity, with a Kd of ∼20–30 μm, perhaps indicating that A58 might be involved in direct PylSn binding and recognition. Mutation of A58 was also reported to have similar detrimental effects on archaeal PylRS function in vivo (38).
DISCUSSION
PylSn has an unknown structure and has had no demonstrated function. Here we show for the first time that the bacterial PylSn forms a complex with tRNAPyl, but not noncognate tRNAs or tRNAPyl with certain bases modified. Loss of the N terminus of archaeal PylS leads to a loss of activity in EMSA, indicating that this homolog of PylSn has the same capacity to form a stable complex with tRNAPyl. D. hafniense PylSn used as a search query for a conserved domain BLAST search at the National Center for Biotechnology Information revealed only a single hit against one domain superfamily, TIGR03912, which includes the discrete pylSn gene products of Bacteria as well as the N termini of archaeal PylS proteins. A further search using either the BLAST or PSI-BLAST programs did not reveal significant homology to a known RNA-binding protein. PylSn and the N terminus of PylS thus appear to represent a novel tRNA binding domain.
A recent survey of the genomes hosted at NCBI revealed those from 12 species of Bacteria and Archaea that possessed the pyrrolysine biosynthetic genes pylBCD (10). All these genomes encode a TIGR03912 domain as the N-terminal portion of pylS or as a separate pylSn gene. Those with pylSn also possess the pylSc gene. This implies the tRNAPyl binding ability of the TIGR03912 family plays a role in the genetic encoding of tRNAPyl sufficient to lead to the retention of the independent gene or linked domain along with the other pyl genes. Given the current data, the simplest hypothesis is that the binding of tRNAPyl by PylSn, or the N terminus of PylS, plays a role in facilitating the charging of tRNAPyl by the catalytic domain, possibly by increasing its affinity for tRNAPyl. The published Kd of PylSc for D. hafniense tRNAPyl is 6.9 μm. This is remarkably high in comparison with the Kd values measured for archaeal PylS and tRNAPyl, which are typically 10-fold lower (31). The PylSn-tRNAPyl complex has a Kd in this same low range. Notably, loss of the ability of PylS to form an electrophoretically detectable complex with tRNAPyl occurs upon deletion of the N-terminal domain, and PylSc was not capable of forming a complex with tRNAPyl detectable by electromobility shift, suggesting higher stability of the PylSn and tRNAPyl complex.
The flexible linker connecting the catalytic and N-terminal domains of PylS provides a direct connection between the high affinity tRNAPyl-binding domain and the catalytic domain, whose complex with tRNAPyl is less stable. However, for PylSn to function similarly to increase the overall affinity of bacterial pyrrolysyl-tRNA synthetase for tRNAPyl, it would presumably require the formation of a ternary complex of the two proteins and the tRNA. Such a complex may or may not include direct interaction of the two proteins, because PylSn could act to change the conformation of tRNAPyl, so as to increase the binding affinity of PylSc.
Substitutions of highly conserved residues in tRNAPyl revealed portions of the tRNA whose sequence are critical for binding of tRNAPyl by PylSn. Substitution of conserved residue pairs in the D stem, residues of which directly interact with PylSc (13), strongly influence the binding of PylSn to tRNAPyl (Figs. 6 and 8). PylSc binds the D stem from the minor groove side, leaving it possible that the decreased PylSn affinity for these variants reflects PylSn interaction with the opposite side of the D stem. However, both A46 and G47 of the variable loop respectively interact with U12:A23 and C13:G22 of the D stem (13), and it is likely these mutations disrupt the tertiary structure of tRNAPyl. On the other hand, the G10:C25 and A11:U24 pairs do not directly participate in this interaction, and changes of these base pairs still strongly affected PylSn affinity. Interestingly, base substitutions within the T stem and loop also strongly influenced tRNAPyl binding to PylSn, indicating that parts of the tRNAPyl core that have no reported contact with PylSc are important for PylSn recognition. The requirement for A58 is especially interesting; given that this residue does not directly pair or stack with any other residue in the core, it may represent a determinant directly recognized by PylSn.
The cloverleaf structure of tRNAPyl is distinguished by the exceptionally small variable loop, as well as an elongated anticodon stem. Only the sequence of one of these elements appears crucial for PylSn binding. Substitution of any base within the variable loop, or insertion of additional base into this region, either eliminated or compromised interaction between tRNAPyl In contrast, the elongated anticodon stem and anticodon loop are not a strict requirement for PylSn binding; deletion of a base pair in the stem at most moderately interfered with PylSn binding. Although the effects these changes of tRNAPyl have on PylSn binding may reflect effects on structure or direct base interactions, the sensitivity of PylSn binding to changes in tRNAPyl D and T stems and T loop further illustrate the specificity of PylSn for tRNAPyl.
Although the retention of the pylSn gene (or the corresponding part of archaeal pylS) in presently sequenced genomes suggests a long term advantage to retention of this tRNAPyl-binding domain, the physiological advantage is not yet apparent. In vitro, PylSc acts as an aminoacyl-tRNA synthetase, when presented with pyrrolysine, or pyrrolysine analogs, and tRNAPyl (this work and Ref. 39). Similarly, we show here that an N-terminal truncation of archaeal PylS retains the ability to activate pyrrolysine and charge tRNAPyl. However, the current data from recombinant systems in which PylS or PylSc and tRNAPyl are employed in vivo to suppress amber codons in reporter genes yield conflicting information. PylS was reported to lose the ability to function in vivo when pylS suffers deletion of the N-terminal domain, and pylSc did not detectably function in amber suppression leading to expression of a reporter gene (31). A very sensitive genetic assay revealed weak UAG translation with pylSc, which was not enhanced by co-expression with pylSn (13). A more recent paper indicated that PylSc by itself is capable of supporting fairly high levels of amber codon translation in E. coli (40). Our own in vivo experiments agree with the latter (data not shown) and suggest that PylSc does not require PylSn to function in a recombinant system.
This suggests subtle yet important effects of PylSn on PylSc function, or of the N-terminal region of PylS on catalytic domain, have led to retention of this tRNAPyl-binding protein/domain in genomes. The tighter and more stable binding displayed by PylSn for tRNAPyl relative to PylSc may influence the kinetics of tRNAPyl acylation so that lower expression of pylT allows an adequate pool of pyrrolysyl-tRNAPyl to fuel UAG translation. Alternatively, orthogonality of PylSc and tRNAPyl has yet to be formally proven, and it is possible the specificity of tRNAPyl binding by PylSn and its homologs may increase the fidelity of pyrrolysine incorporation into protein. Even a small error rate in which pyrrolysylation of a noncognate tRNA occurs could result in a deleterious physiological effect. In any case, it is clear that the specific binding of tRNAPyl by the N terminus of PylS, or by PylSn, is of sufficient merit to render retention of their encoding genes a positive trait in those organisms that decode UAG as pyrrolysine.
Supplementary Material
Footnotes
This work was supported in part by National Institutes of Health Grant GM070663. This work was also supported by Department of Energy Grant DE-FG0202-91ER200042 (to J. A. K.).

This article contains supplemental Table S1 and Fig. S1.
REFERENCES
- 1. Atkins J. F., Gesteland R. (2002) The 22nd amino acid. Science 296, 1409–1410 [DOI] [PubMed] [Google Scholar]
- 2. Krzycki J. A. (2004) Function of genetically encoded pyrrolysine in corrinoid-dependent methylamine methyltransferases. Curr. Opin. Chem. Biol. 8, 484–491 [DOI] [PubMed] [Google Scholar]
- 3. Hao B., Gong W., Ferguson T. K., James C. M., Krzycki J. A., Chan M. K. (2002) A new UAG-encoded residue in the structure of a methanogen methyltransferase. Science 296, 1462–1466 [DOI] [PubMed] [Google Scholar]
- 4. Soares J. A., Zhang L., Pitsch R. L., Kleinholz N. M., Jones R. B., Wolff J. J., Amster J., Green-Church K. B., Krzycki J. A. (2005) The residue mass of l-pyrrolysine in three distinct methylamine methyltransferases. J. Biol. Chem. 280, 36962–36969 [DOI] [PubMed] [Google Scholar]
- 5. Burke S. A., Lo S. L., Krzycki J. A. (1998) Clustered genes encoding the methyltransferases of methanogenesis from monomethylamine. J. Bacteriol. 180, 3432–3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Paul L., Ferguson D. J., Jr., Krzycki J. A. (2000) The trimethylamine methyltransferase gene and multiple dimethylamine methyltransferase genes of Methanosarcina barkeri contain in-frame and read-through amber codons. J. Bacteriol. 182, 2520–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. James C. M., Ferguson T. K., Leykam J. F., Krzycki J. A. (2001) The amber codon in the gene encoding the monomethylamine methyltransferase isolated from Methanosarcina barkeri is translated as a sense codon. J. Biol. Chem. 276, 34252–34258 [DOI] [PubMed] [Google Scholar]
- 8. Longstaff D. G., Blight S. K., Zhang L., Green-Church K. B., Krzycki J. A. (2007) In vivo contextual requirements for UAG translation as pyrrolysine. Mol. Microbiol. 63, 229–241 [DOI] [PubMed] [Google Scholar]
- 9. Srinivasan G., James C. M., Krzycki J. A. (2002) Pyrrolysine encoded by UAG in Archaea. Charging of a UAG-decoding specialized tRNA. Science 296, 1459–1462 [DOI] [PubMed] [Google Scholar]
- 10. Gaston M. A., Jiang R., Krzycki J. A. (2011) Functional context, biosynthesis, and genetic encoding of pyrrolysine. Curr. Opin. Microbiol. 14, 342–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mahapatra A., Patel A., Soares J. A., Larue R. C., Zhang J. K., Metcalf W. W., Krzycki J. A. (2006) Characterization of a Methanosarcina acetivorans mutant unable to translate UAG as pyrrolysine. Mol. Microbiol. 59, 56–66 [DOI] [PubMed] [Google Scholar]
- 12. Théobald-Dietrich A., Frugier M., Giegé R., Rudinger-Thirion J. (2004) Atypical archaeal tRNA pyrrolysine transcript behaves towards EF-TU as a typical elongator tRNA. Nucleic Acids Res. 32, 1091–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nozawa K., O'Donoghue P., Gundllapalli S., Araiso Y., Ishitani R., Umehara T., Söll D., Nureki O. (2009) Pyrrolysyl-tRNA synthetase-tRNA(Pyl) structure reveals the molecular basis of orthogonality. Nature 457, 1163–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blight S. K., Larue R. C., Mahapatra A., Longstaff D. G., Chang E., Zhao G., Kang P. T., Green-Church K. B., Chan M., K., Krzycki J. A. (2004) Direct charging of tRNACUA with pyrrolysine in vitro and in vivo. Nature 431, 333–335 [DOI] [PubMed] [Google Scholar]
- 15. Polycarpo C., Ambrogelly A., Bérubé A., Winbush S. M., McCloskey J. A., Crain P. F., Wood J. L., Söll D. (2004) An aminoacyl-tRNA synthetase that specifically activates pyrrolysine. Proc. Natl. Acad. Sci. U.S.A. 101, 12450–12454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Polycarpo C. R., Herring S., Bérubé A., Wood J. L., Söll D., Ambrogelly A. (2006) Pyrrolysine analogues as substrates for pyrrolysyl-tRNA synthetase. FEBS Lett. 580, 6695–6700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Longstaff D. G., Larue R. C., Faust J. E., Mahapatra A., Zhang L., Green-Church K. B., Krzycki J. A. (2007) A natural genetic code expansion cassette enables transmissible biosynthesis and genetic encoding of pyrrolysine. Proc. Natl. Acad. Sci. U.S.A. 104, 1021–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gaston M. A., Zhang L., Green-Church K. B., Krzycki J. A. (2011) The complete biosynthesis of the genetically encoded amino acid pyrrolysine from lysine. Nature 471, 647–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Quitterer F., List A., Eisenreich W., Bacher A., Groll M. (2012) Crystal structure of methylornithine synthase (PylB). Insights into the pyrrolysine biosynthesis. Angew. Chem. Int. Ed. Engl. 51, 1339–1342 [DOI] [PubMed] [Google Scholar]
- 20. Cellitti S. E., Ou W., Chiu H. P., Grünewald J., Jones D. H., Hao X., Fan Q., Quinn L. L., Ng K., Anfora A. T., Lesley S. A., Uno T., Brock A., Geierstanger B. H. (2011) d-Ornithine coopts pyrrolysine biosynthesis to make and insert pyrroline-carboxy-lysine. Nat. Chem. Biol. 7, 528–530 [DOI] [PubMed] [Google Scholar]
- 21. Fekner T., Chan M. K. (2011) The pyrrolysine translational machinery as a genetic-code expansion tool. Curr. Opin. Chem. Biol. 15, 387–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Y., Gladyshev V. N. (2007) High content of proteins containing 21st and 22nd amino acids, selenocysteine and pyrrolysine, in a symbiotic deltaproteobacterium of gutless worm Olavius algarvensis. Nucleic Acids Res. 35, 4952–4963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Woyke T., Teeling H., Ivanova N. N., Huntemann M., Richter M., Gloeckner F. O., Boffelli D., Anderson I. J., Barry K. W., Shapiro H. J., Szeto E., Kyrpides N. C., Mussmann M., Amann R., Bergin C., Ruehland C., Rubin E. M., Dubilier N. (2006) Symbiosis insights through metagenomic analysis of a microbial consortium. Nature 443, 950–955 [DOI] [PubMed] [Google Scholar]
- 24. Goodchild A., Saunders N. F., Ertan H., Raftery M., Guilhaus M., Curmi P. M., Cavicchioli R. (2004) A proteomic determination of cold adaptation in the Antarctic archaeon, Methanococcoides burtonii. Mol. Microbiol. 53, 309–321 [DOI] [PubMed] [Google Scholar]
- 25. Kim S. H., Harzman C., Davis J. K., Hutcheson R., Broderick J. B., Marsh T. L., Tiedje J. M. (2012) Genome sequence of Desulfitobacterium hafniense DCB-2, a Gram-positive anaerobe capable of dehalogenation and metal reduction. BMC Microbiol. 12, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krzycki J. A. (2005) The direct genetic encoding of pyrrolysine. Curr. Opin. Microbiol. 8, 706–712 [DOI] [PubMed] [Google Scholar]
- 27. Yanagisawa T., Ishii R., Fukunaga R., Kobayashi T., Sakamoto K., Yokoyama S. (2008) Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode N(epsilon)-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem. Biol. 15, 1187–1197 [DOI] [PubMed] [Google Scholar]
- 28. Kavran J. M., Gundllapalli S., O'Donoghue P., Englert M., Söll D., Steitz T. A. (2007) Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation. Proc. Natl. Acad. Sci. U.S.A. 104, 11268–11273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yanagisawa T., Ishii R., Fukunaga R., Nureki O., Yokoyama S. (2006) Crystallization and preliminary x-ray crystallographic analysis of the catalytic domain of pyrrolysyl-tRNA synthetase from the methanogenic archaeon Methanosarcina mazei. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62, 1031–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee M. M., Jiang R., Jain R., Larue R. C., Krzycki J., Chan M. K. (2008) Structure of Desulfitobacterium hafniense PylSc, a pyrrolysyl-tRNA synthetase. Biochem. Biophys. Res. Commun. 374, 470–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Herring S., Ambrogelly A., Gundllapalli S., O'Donoghue P., Polycarpo C. R., Söll D. (2007) The amino-terminal domain of pyrrolysyl-tRNA synthetase is dispensable in vitro but required for in vivo activity. FEBS Lett. 581, 3197–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sampson J. R., Uhlenbeck O. C. (1988) Biochemical and physical characterization of an unmodified yeast phenylalanine transfer RNA transcribed in vitro. Proc. Natl. Acad. Sci. U.S.A. 85, 1033–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Francklyn C. S., First E. A., Perona J. J., Hou Y. M. (2008) Methods for kinetic and thermodynamic analysis of aminoacyl-tRNA synthetases. Methods 44, 100–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hao B., Zhao G., Kang P. T., Soares J. A., Ferguson T. K., Gallucci J., Krzycki J. A., Chan M. K. (2004) Reactivity and chemical synthesis of l-pyrrolysine. The 22nd genetically encoded amino acid. Chem. Biol. 11, 1317–1324 [DOI] [PubMed] [Google Scholar]
- 35. Li W. T., Mahapatra A., Longstaff D. G., Bechtel J., Zhao G., Kang P. T., Chan M. K., Krzycki J. A. (2009) Specificity of pyrrolysyl-tRNA synthetase for pyrrolysine and pyrrolysine analogs. J. Mol. Biol. 385, 1156–1164 [DOI] [PubMed] [Google Scholar]
- 36. Bovee M. L., Yan W., Sproat B. S., Francklyn C. S. (1999) tRNA discrimination at the binding step by a class II aminoacyl-tRNA synthetase. Biochemistry 38, 13725–13735 [DOI] [PubMed] [Google Scholar]
- 37. Watanabe Y., Kawai G., Yokogawa T., Hayashi N., Kumazawa Y., Ueda T., Nishikawa K., Hirao I., Miura K., Watanabe K. (1994) Higher-order structure of bovine mitochondrial tRNA(SerUGA). Chemical modification and computer modeling. Nucleic Acids Res. 22, 5378–5384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ambrogelly A., Gundllapalli S., Herring S., Polycarpo C., Frauer C., Söll D. (2007) Pyrrolysine is not hardwired for cotranslational insertion at UAG codons. Proc. Natl. Acad. Sci. U.S.A. 104, 3141–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Herring S., Ambrogelly A., Polycarpo C. R., Söll D. (2007) Recognition of pyrrolysine tRNA by the Desulfitobacterium hafniense pyrrolysyl-tRNA synthetase. Nucleic Acids Res. 35, 1270–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Katayama H., Nozawa K., Nureki O., Nakahara Y., Hojo H. (2012) Pyrrolysine analogs as substrates for bacterial pyrrolysyl-tRNA synthetase in vitro and in vivo. Biosci. Biotechnol. Biochem. 76, 205–208 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.