

Background: Pseudomonas aeruginosa alkaline protease is a virulence factor that contributes to cystic fibrosis pathology.
Results: The ENaC channel is activated by alkaline protease.
Conclusion: Alkaline protease secretion may contribute to bacterial virulence by altering ENaC activity.
Significance: The results provide insight into patho-mechanisms associated with alkaline protease secretion.
Keywords: Cystic Fibrosis, ENaC, Physiology, Protease, Pseudomonas aeruginosa, Sodium Channels, Virulence Factors, Proteolysis
Abstract
Pseudomonas aeruginosa is an opportunistic pathogen that significantly contributes to the mortality of patients with cystic fibrosis. Chronic infection by Pseudomonas induces sustained immune and inflammatory responses and damage to the airway. The ability of Pseudomonas to resist host defenses is aided, in part, by secreted proteases, which act as virulence factors in multiple modes of infection. Recent studies suggest that misregulation of protease activity in the cystic fibrosis lung may alter fluid secretion and pathogen clearance by proteolytic activation of the epithelial sodium channel (ENaC). To evaluate the possibility that proteolytic activation of ENaC may contribute to the virulence of Pseudomonas, primary human bronchial epithelial cells were exposed to P. aeruginosa and ENaC function was assessed by short circuit current measurements. Apical treatment with a strain known to express high levels of alkaline protease (AP) resulted in an increase in basal ENaC current and a loss of trypsin-inducible ENaC current, consistent with sustained activation of ENaC. To further characterize this AP-induced ENaC activation, AP was purified, and its folding, activity, and ability to activate ENaC were assessed. AP folding was efficient under pH and calcium conditions thought to exist in the airway surface liquid of normal and cystic fibrosis (CF) lungs. Short circuit measurements of ENaC in polarized monolayers indicated that AP activated ENaC in immortalized cell lines as well as post-transplant, primary human bronchial epithelial cells from both CF and non-CF patients. This activation was mapped to the γ-subunit of ENaC. Based on these data, patho-mechanisms associated with AP in the CF lung are proposed wherein secretion of AP leads to decreased airway surface liquid volume and a corresponding decrease in mucocilliary clearance of pulmonary pathogens.
Introduction
Pseudomonas aeruginosa is an opportunistic pathogen that contributes significantly to the mortality of immune-compromised individuals, burn victims, and patients with cystic fibrosis (CF)3 (1–3). P. aeruginosa is a highly adaptable pathogen, and infection is clinically problematic due to its ability to develop antimicrobial drug resistance, form biofilms, and secrete a large number of functionally diverse virulence factors (4, 5). Secretion of virulence factors and production of biofilms contribute to bacterial defense by modulating specific host responses and establishing a physical barrier between the bacterial cells and their surrounding environment.
Among the virulence factors, Pseudomonas secretes exoproteases that putatively alter host immune signaling and scavenge nutrients from the environment (3, 6–9). Previous studies have implicated alkaline protease (AP) in multiple modes of Pseudomonas infection (10). In CF, increased AP titer in patient sputum has been correlated with decreased clinical prognosis for patients experiencing Pseudomonas exacerbation (11–15). Consistent with this, biochemical studies have demonstrated cleavage and inactivation of a variety of signaling, inflammatory, and regulatory molecules by AP (16–19). Alteration of these response pathways putatively contributes to the difficulty in controlling and clearing Pseudomonas infections. Although the pathophysiological roles of AP have yet to be fully elucidated in the CF lung, the association between AP expression and negative prognosis demonstrate that AP activity is relevant to Pseudomonas infection and exacerbation in CF patients.
Although the molecular pathophysiology underlying CF is associated with loss of cystic fibrosis transmembrane conductance regulator (CFTR) function, a putative role for the epithelial sodium channel (ENaC) regulation of pulmonary physiology and CF pathophysiology has been suggested by multiple studies (20–25). Recent work suggests that the protease-protease inhibitor balance is critical for the balance of fluid secretion in the CF lung (20, 26–29). Imbalances in protease activities, as a result of increased protease production by the host or a pathogen, impact multiple physiological events. A growing body of literature demonstrates that ENaC is regulated by protease cleavage of its large extracellular domains and may be directly affected by changes in the antiprotease-protease balance (30, 31).
ENaC is formed by a putative heterotrimer of an α-, β-, and γ-subunit (32). Structures of homologous bacterial proteins and extensive biochemical studies suggest that the subunits of ENaC are composed of two transmembrane helices separated by a large extracellular domain, with both N and C termini located intracellularly (33). The extracellular domains of ENaC are cleaved by proteases, resulting in channel activation. This activation results from release of inhibitory peptides in the extracellular domains of either or both of the α- and γ-subunits (31, 34). Cleavage of these subunits results in an increase in channel open probability (Po) and a corresponding increase in Na+ transport. Originally identified in in vitro electrophysiological studies using Xenopus oocytes, channel-activating proteases were first shown to activate ENaC through these cleavage events (35). Subsequent studies have shown that a variety of proteases regulate ENaC function in multiple mammalian epithelial tissues (36, 37).
To evaluate the potential role of protease activation of ENaC, P. aeruginosa was cultured on the apical surface of primary human bronchial epithelial (HBE) cells for electrophysiological and biochemical analyses. Growth of a protease-secreting strain of Pseudomonas, FRD1, resulted in an increase in basal ENaC current and a corresponding loss of exogenous protease-inducible current. Zymographic analyses demonstrated that AP was the predominant secreted protease and was active in the apical washes of the FRD1-treated HBE cells but not in controls. To further characterize the role of AP in activating ENaC, the protease was purified for functional and electrophysiological studies. The purified AP efficiently folded and was highly active against protease substrates at physiological conditions. Electrophysiological experiments demonstrated that the purified AP activated human and mouse ENaC in multiple cell lines. This activation was largely the result of cleavage in the γ-subunit. Similar AP activation of ENaC was seen in primary HBE cells isolated from CF and non-CF lung explant tissue. Together these data demonstrate that AP from P. aeruginosa is capable of activating ENaC. Based on these data, implications for the roles of AP in Pseudomonas pathogenesis in the CF lung are discussed.
EXPERIMENTAL PROCEDURES
Cell Culture
HBE cells were cultured from excess pathologic tissue after lung transplantation and organ donation, as described previously (38). After initial isolation, passage 0 cells were seeded onto human placental collagen-coated Costar 0.33-cm2 Transwell filters (Corning) at a density of ∼2 × 105/cm2. Reaching confluence, the apical medium was aspirated, and cultures were maintained at an air-liquid interface. Cells were fed basolaterally with differentiation medium three times weekly. Cells were studied after 3–6 weeks of culture and considered differentiated when a mucociliary phenotype was apparent by phase-contrast microscopy.
mCCDcl-11 cells (provided by Bernard Rossier and Laurent Schild, Université de Lausanne, Switzerland) were grown in 75-cm flasks (passages 28–35) in defined medium, as described previously (39). Cells were then subcultured onto 0.33-cm2 Costar Transwell filters. Twenty-four hours prior to recording, the cells were treated with a 50 μm concentration of a cell-permeant chloromethylketone furin convertase inhibitor (Enzo Life Sciences) to block the action of intracellular proprotein convertases and facilitate the delivery of uncleaved ENaC to the cell surface, as described previously (27, 38).
Fisher rat thyroid (FRT) cells were maintained in DMEM/F-12 medium supplemented with 5% FBS in flasks. Polarized FRT cultures expressing human ENaC were established by seeding 3 × 105 cells onto 0.33-cm2 Costar Transwell filters and transiently transfecting with 0.25 μg each of α-, β-, and γ-ENaC using Lipofectamine 2000 per the manufacturer's instructions. Following transfection, 50 nm dexamethasone was included in the base medium to stimulate Na+ transport, and the cells were used for short circuit current measurement 3 days later. Human α-, β-, and γ-ENaC constructs were kindly provided by Dr. Doug Eaton and subcloned into pcDNA3.1. The following mutations were introduced by site-directed mutagenesis (QuikChange): αmut (R175A/R177A/R178A/R181A/R201A/R204A), αdel (del Asp179–Arg204), γmut (R135KRR → AKAA, R178KRK → QQQQ), and γdel (del Glu139–Lys181).
Protein Expression and Purification
P. aeruginosa PAO1 genomic DNA was used as a template for PCR cloning of the aprA and aprI genes. The full-length AprA and AprI proteins were cloned into the Escherichia coli pET-DUET vector for T7-regulated expression, purification, and in vitro refolding, as described previously (40). Briefly, donor cultures were grown overnight in the presence of antibiotic and used to inoculate expression cultures. Expression cultures were grown to an A600 of ∼0.8 prior to induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside. Protein was expressed at 37 °C for 4–6 h, and the majority of overexpressed protein was insoluble. Cells were lysed by sonication (50 mm Tris-HCl, 150 mm NaCl, pH 6.8). Inclusion bodies were isolated by centrifugation and resuspended in binding buffer (50 mm Tris-HCl, 150 mm NaCl, 6 m guanidine HCl, pH 6.8), loaded onto a nickel-nitrilotriacetic acid column, washed (50 mm Tris-HCl, 150 mm NaCl, 6 m guanidine HCl, 40 mm imidazole, pH 6.8), and eluted (50 mm Tris-HCl, 150 mm NaCl, 6 m guanidine HCl, 400 mm imidazole, pH 6.8). The eluted protein was concentrated in 6 m guanidine HCl and stored at 4 °C prior to refolding.
Protease Refolding and Analysis
Refolding was performed by rapid dilution to 1–10 μm final protease concentration into 50 mm Bistris propane, 150 mm NaCl, and 2 mm CaCl2 for 15 min at 4 °C. Protease was then concentrated (Ultracon, Millipore) to 30 μm and cleared by centrifugation or filtration prior to use. Protease refolding was assessed by Western blotting and analytical gel filtration, as described previously (40). Refolded protein was separated from unfolded or aggregated protein by centrifugation at 21,000 × g relative centrifugal force for 15 min at 4 °C. The supernatant fraction was evaluated by Western blotting using a rabbit polyclonal antibody raised against the purified RTX (repeats in toxin) Ca2+-binding domain from alkaline protease using standard procedures (40). Analytical gel filtration was accomplished by injection of the protein onto a Superdex S75GL 10/300 column (GE Healthcare) pre-equilibrated in refolding buffer, described above.
Protease Activity
Protease activity was measured using fluorescence-based protease activity assays. A casein-based assay (EnzChek, Invitrogen) was used previously to assess the activity of refolded AP in vitro. The intact BODIPY-conjugated casein is quenched via intramolecular interactions, and substrate cleavage results in fluorophore dequenching. A peptide-based fluorescence assay was also used to evaluate AP activity. Based on the structural similarity to several of the human matrix metalloproteases, a fluorescent MMP substrate peptide (Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2 (where Mca represents (7-methoxycoumarin-4-yl)acetyl, and Dpa is N-3-(2,4-dinitrophenyl)-l-α,β-diaminopropionyl; R&D Systems) was used to evaluate AP activity. For both activity assays, protease was diluted into reaction buffer (50 mm Bistris propane, 150 mm NaCl, 2 mm CaCl2, and 1 μm ZnCl2) and read using a BioTek Synergy 4 multimode plate reader. Emission intensities were collected in kinetic mode, and steady state activities were fit using linear regression. Data shown were collected from at least two independent protease purifications. Data are presented as mean ± S.D. from n > 4 experiments.
Pseudomonas Growth and Protease Secretion on HBE Cells
P. aeruginosa strains FRD1 (a generous gift from Dennis Ohman, Virginia Commonwealth University Medical Center) and PAO1 (ATCC) were used to evaluate protease expression, secretion, and impact on HBE cells. Pseudomonas strains were grown in tryptic soy broth at 37 °C. Cells were harvested by centrifugation and washed five times in Ringer's solution (see below). The cell pellet was then resuspended in Ringer's solution, and cell density was normalized by A600 measurement. Optical measurements for FRD1 and PAO1 cells were standardized by colony counting after serial dilution onto LB agar plates.
Protease secretion and activity was assessed after application of Pseudomonas to the apical surface of HBE cells. Cell densities were empirically chosen to maintain HBE cell viability and high monolayer resistance after overnight co-culture. For electrophysiological recordings, HBE cells were treated with 4 × 103 cfu resuspended in 10 μl of Ringer's solution for 16 h under standard HBE culture conditions (37 °C, 5% CO2). Filters were mounted in Ussing chambers and assessed as described below. For zymography, 4 × 105 cfu of FRD1 and PAO1 cells were cultured at 37 °C overnight on the apical surface of HBE cells. After 16 h, the apical surface was washed with Ringer's solution, and the supernatant was analyzed by zymography. Samples were electrophoresed on Tris-glycine casein zymogram gels (Invitrogen), renatured, and developed following the manufacturer's protocols. The gels were incubated overnight at 37 °C and stained with Coomassie Blue. Experiments were performed on multiple independent HBE cell lines.
Short Circuit Current Measurements
Cultured cells were grown on Transwell inserts until a confluent, high resistance monolayer was obtained. Monolayer resistances were greater than 150 ohms/cm2 for all cells tested. Inserts were mounted in modified Ussing chambers (P2300, Physiological Instruments) and continuously short circuited with an automatic voltage clamp (VCC MC8, Physiological Instruments) as described previously (38). The apical and basolateral chambers each contained 3 ml of Ringer's solution (120 mm NaCl, 25 mm NaHCO3, 3.3 mm KH2PO4, 0.8 mm K2HPO4, 1.2 mm MgCl2, 1.2 mm CaCl2, and 10 mm glucose). Chambers were constantly gassed with a mixture of 95% O2, 5% CO2 at 37 °C, which maintained the pH at 7.4 and established a circulating perfusion bath within the Ussing chamber. Simultaneous transepithelial resistance was recorded by applying a 2-mV pulse per second via an automated pulse generator. Recordings were digitized and analyzed using either Acquire and Analyze 2.3 (Physiological Instruments) or PowerLab (AD Instruments, Colorado Springs, CO). A typical ISC recording included a 10-min equilibration period, followed by stimulation with trypsin or AP. INa was determined by the addition of 10 μm amiloride to the apical cell chamber at the end of each recording.
For short circuit studies, AP concentrations were chosen to match the kinetic activities of trypsin controls based on these in vitro fluorescence assays described above. Steady state reaction kinetics were measured using trypsin under concentrations known to fully activate ENaC. Following refolding and prior to short circuit measurements, both trypsin and AP activities were verified using the enzymatic assays described above.
Statistics and Curve Fitting
All data were analyzed using SigmaPlot (Systat, Chicago, IL). Time constants were calculated from curves fit using non-linear regression analysis for the exponential rise to a maximum. Differences in summarized data were evaluated by t tests, with p < 0.05 considered statistically significant.
RESULTS
ENaC Activation by P. aeruginosa
Previous studies have shown that the ENaC channel is regulated by extracellular proteolytic cleavage (30). Specific cleavage events lead to the activation of ENaC and an increase in Na+ current. P. aeruginosa secretes proteases that may cleave and activate Na+ channels, thereby decreasing airway surface liquid volume and impairing mucociliary clearance. Thus, secretion of ENaC-activating proteases may be a novel virulence mechanism used by pathological strains of Pseudomonas to manipulate the local airway environment and facilitate bacterial adherence and colonization.
To test the hypothesis that proteases secreted by P. aeruginosa manipulate ENaC activity, the apical surfaces of polarized primary HBE cells were inoculated with Pseudomonas, and the electrophysiologic effects were assessed in Ussing chambers. Two strains of P. aeruginosa were chosen for studies on HBE cells. A CF patient isolate, FRD1, was chosen based on previous reports of high expression and secretion of proteases (5). A second strain, PAO1, was chosen due to its lower virulence and reduced protease expression.
Following 16 h of HBE-bacterial co-culture, the apical medium was collected for biochemistry, and the HBE cells were used for electrophysiological studies. HBE cells exposed to FRD1 cells showed elevated basal ENaC currents when compared with the PAO1 and Ringer's solution controls (Fig. 1, A and B). To maximally activate uncleaved ENaC at the apical surface, exogenous trypsin (1 μm) was added to the apical chamber once steady-state current was achieved. No additional trypsin-activatable current was observed in FRD1-treated cells. In contrast, the PAO1 and Ringer's controls showed an increase in amiloride-sensitive current when treated with trypsin (Fig. 1A). The trypsin-induced ENaC current in the PAO1 and Ringer's control-treated cells increased to levels similar to the basal currents seen in the FRD1-treated cells. The relative changes in both basal and trypsin-stimulated currents were seen in multiple independent HBE cell lines with varying levels of ENaC current. Although the absolute currents varied between lines, the relative increase in basal activity and loss of trypsin-activation was evident across all cell lines tested.
FIGURE 1.

Pseudomonas FRD1 alteration of HBE cell physiology. The effects of Pseudomonas grown on the apical face of HBE cells were assessed biochemically and electrophysiologically. Primary HBE cells were co-cultured with Pseudomonas. A, representative traces of HBE cells exposed to Ringer's solution (black), PAO1 (light gray), or FRD1 (dark gray) Pseudomonas on the apical surface. FRD1 cells showed an increase in basal current and a loss of trypsin-activatable current in multiple HBE cell lines. Trypsin addition to the apical bath was used to evaluate maximal ENaC current and is shown as a solid line. Amiloride addition was used to evaluate ENaC-specific current and is shown as a dashed line. B, summary of basal currents shown as a function of maximal trypsin-activated current. Basal currents in the PAO1 and Ringer's controls were increased greater than 2-fold after the trypsin addition. In contrast, the FRD1-treated cells showed higher basal current and no observable activation by trypsin. C, gel zymogram of fluids washed from the apical surface of FRD1-, PAO1-, and Ringer's solution-treated cells. Proteolytic digestion of casein in the gels results in decreased Coomassie Blue staining. The ∼50-kDa white band in the FRD1-treated sample corresponds to AP secreted by the FRD1 cells. Data are presented as mean ± S.E. (error bars) from n > 3 experiments with at least 8 filters/condition.
The increase in basal current could be accounted for by at least two mechanisms. ENaC channel expression might be up-regulated in response to Pseudomonas, or the population of channels at the cell surface might be activated in the presence of the bacterial cells. The increase in basal activity and loss of trypsin-activatable current suggested that the proteolytic state of ENaC was changed in the presence of the FRD1 cells. To evaluate the differential expression and secretion of proteases by the FRD1 and PAO1 strains, gel zymography was used to evaluate protease activities. As with the electrophysiological experiments, bacterial cells were applied to the apical surface of HBE cells and grown overnight. The apical surfaces of the HBE cells were washed with Ringer's solution, and the wash was collected. Zymogram analyses demonstrated a single predominant protease band of ∼50 kDa present in the FRD1-treated HBE cell wash that was not present in the PAO1 and Ringer's controls (Fig. 1C). This band was confirmed to be alkaline protease by comparison with purified protein and Western blotting (data not shown), consistent with previous reports of high levels of alkaline protease expression and secretion by the FRD1 Pseudomonas strain.
AP Folding and Activity
To evaluate the potential role of AP in the regulation of ENaC, AP was expressed and purified for functional and structural studies. Previously, we have shown that AP refolding and activity is tightly regulated by Ca2+ binding to the RTX domain (40). To evaluate the activity of AP under conditions of physiological pH and Ca2+, purified AP was refolded in 2 mm Ca2+ across a range of pH values to include those found in normal and CF lungs (Fig. 2). Alkaline protease refolding was initially assessed by Western blot as the fraction of protein soluble after high speed centrifugation or filtration. Previous studies have shown that AP misfolding results in the formation of high molecular weight aggregates that can be removed by filtration and that folding to a native, soluble form is dependent on Ca2+ binding (40). Refolding was efficient over a broad pH range (pH 6.5–9.5), with more than 85% of refolded protein recovered in the soluble fractions (Fig. 2A). Analytical gel filtration chromatography analyses of these fractions indicated that the refolded proteins were monomeric and showed no discernible changes in hydrodynamic properties as a result of changing refolding pH (Fig. 2B). Elution chromatograms showed no signs of peak tailing or splitting, consistent with a structurally uniform population of refolded protein. Analysis of fractions collected after gel filtration chromatography showed protease activity to be found exclusively in the elution peak corresponding to monomeric protease.
FIGURE 2.

pH dependence of AP refolding and activity. The pH dependence of AP refolding and activity was assessed in vitro using purified AP. AP refolding and activity were assessed from pH 6.5 to 9.5. A, AP solubility was assessed by Western blotting after refolding and centrifugal filtration. The filtrate fraction is shown after refolding at varying pH (top) in a representative blot. Total protein, representing potentially folded and misfolded species, is shown before filtration for each refolding reaction (bottom). Molecular weight markers are shown on the left of each panel and represent apparent mass in kDa. B, representative analytical gel filtration chromatographs for AP refolded at each pH condition. The refolded protein eluted as a single major peak at ∼11.2 ml under each pH condition tested, consistent with monodisperse AP. C, AP activity was assessed using fluorescent protease substrates after refolding. Protease activity was evaluated using a casein substrate (filled circles) and a peptide substrate (open circles) at varying pH. Data are presented as mean ± S.D. (error bars) from n > 4 experiments.
Protease activity was subsequently assessed using two fluorescence-based assays. In both assays, initial substrate fluorescence is quenched by intramolecular proximity of the conjugated fluorophores. Substrate cleavage releases peptide fragments, dequenching the fluorophores. Substrates were chosen to evaluate protease activity against a globular protein and a peptide substrate. BODIPY-conjugated casein was used to evaluate AP and trypsin activities against a globular protein substrate. A fluorescent matrix metalloproteinase substrate was chosen based on the structural similarity between the AP proteolytic domain and the catalytic domains of proMMP-1, MMP-7, and MMP-12 (41–43).
Refolded AP cleaved both substrates across a range of pH conditions in the presence of Ca2+ (Fig. 2C). Cleavage of the casein substrate showed maximal activity at near neutral pH with decreasing activity as pH increased. Protease activity was reduced by ∼50–60% at pH 8.5 and 9.2 (Fig. 2C, filled circles). In contrast, proteolysis of the peptide substrate showed minimal changes across the range of pH values tested. Nearly maximal activity was seen between pH 6.5 and 8.5, with a 15% decrease in apparent activity at pH 9.2 (Fig. 2C, open circles). The difference in the pH dependence of substrate proteolysis suggests that solution properties of the substrate and/or substrate binding may be altered across the conditions tested.
AP Activation of Murine ENaC in Cortical Collecting Duct (CCD) Cells
To evaluate the activation of ENaC by AP, mouse CCD cells endogenously expressing ENaC were used to evaluate ENaC-mediated short circuit currents (INa) in Ussing chambers. Mouse CCD cells were chosen due to robust expression of ENaC and previous reports characterizing ENaC cleavage in these cells (44). Cells were pretreated (16 h) with a proprotein furin convertase inhibitor to prevent full ENaC processing along the biosynthetic pathway. The presence of uncleaved ENaC in the apical membrane of CCD cells was verified with the addition of 1 μm trypsin to the apical hemichamber (Fig. 3A, black trace). Trypsin induced an immediate activation of ISC, and amiloride addition verified that the activated current was due to ENaC-mediated Na+ transport. INa increased by ∼44%, from 41.7 ± 3.1 to 60.3 ± 7.1 μA/cm2 after activation by trypsin (Fig. 3B). In subsequent experiments, trypsin was added at the end of each protease treatment to determine the maximum ENaC-mediated Na+ transport for each filter. Amiloride was used to assess the base-line currents and verify that Na+ currents were associated with ENaC.
FIGURE 3.
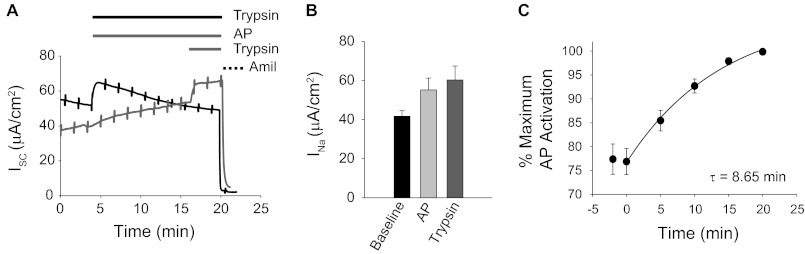
ENaC activation by AP. ENaC activation by refolded AP was evaluated by short circuit measurements in mouse CCD cells. A, representative traces of ENaC currents in CCD cells endogenously expressing ENaC. Recordings of trypsin-treated (black) and AP-treated (gray) cells are shown. Cells treated with AP were subsequently treated with trypsin to obtain maximum protease-stimulated ENaC current. Bars above the traces indicate the treatment protocol and correspond to the traces below. Amiloride treatment was assessed in each recording and used to confirm ENaC activity. B, summary of protease-stimulated ENaC currents for the CCD cells. Untreated (Baseline) and protease-induced currents are shown. C, a time course of ENaC activation by AP. ENaC activation in CCD cells occurs with a time constant of 8.65 min. Data shown are a summary of n = 14 experiments. Error bars, S.E.
To evaluate channel activation by AP, refolded protein was added to the apical bath, and INa was measured. Concentrations and volumes of AP were chosen to match the rate of substrate cleavage by trypsin using the fluorescence-based casein assay (data not shown). As with trypsin, INa increased upon the addition of the purified AP (Fig. 3A, gray trace). AP activation of ENaC resulted in a ∼39% increase in ENaC current (Fig. 3B). To establish maximal activation, trypsin was added to the apical baths after currents had plateaued in the presence of AP. Additional ENaC activation was elicited after the sequential addition of trypsin (Fig. 3A, gray trace). In contrast, the sequential addition of AP after current plateau did not result in additional ENaC activation (data not shown). AP channel activation occurred with a time constant (τ) of 8.65 min (Fig. 3C). The increase in ENaC current plateaued and remained constant after AP treatment. In contrast, trypsin treatment resulted in a peak activation of ENaC after ∼2–3 min, followed by a gradual decrease in current over the course of the experiment. This decrease in current after trypsin treatment was likely the result of persistent channel cleavage by the active protease.
AP Activation of Human ENaC
To evaluate the activation of human ENaC by AP and characterize the locations of AP cleavage, FRT cells were transfected with wild type and mutant human α-, β-, and γ-ENaC, and INa was measured. Protease activation of human ENaC in FRT cells does not require pretreatment with proprotein convertase inhibitors, thus allowing for further characterization of the cell surface proteolytic regulation. Previous studies have demonstrated that proteases activate ENaC by cleaving the extracellular loop of the α- and γ-subunits (31, 45, 46). Inhibitory peptide domains are released from the channel following limited proteolysis. ENaC open probability increases dramatically following removal of the α and/or γ inhibitory domain.
To determine which subunits are responsible for the AP-mediated ENaC activation, mutant α- and γ-subunits containing point mutations at the consensus cleavage sites or deletions of the inhibitory domain were assayed (Fig. 4A). For the α-subunit mutants, two furin sites were altered by site-directed mutagenesis to preclude cleavage at those sites (α-mut). For the γ-subunit, the furin and prostasin sites were similarly altered (γ-mut). Additionally, for both α- and γ-subunits, the inhibitory peptide that is removed following full proteolytic cleavage was deleted (α-del or γ-del) to assess the role of these sequences in AP activation (Fig. 4A, dashed segments).
FIGURE 4.

Subunit specificity of ENaC activation by AP. To further evaluate the mechanisms of ENaC activation by AP, wild type and mutant α-, β-, and γ-ENaC constructs were transfected into FRT cells and evaluated by short circuit measurements. A, schematic of the cleavage sites and deletion mutants for both α- and γ-ENaC. Constructs labeled -mut contained site-specific substitutions that alter known protease cleavage sites. Constructs labeled -del contained deletions that remove inhibitory peptide sequences. B, representative traces of mutant α-ENaC subunits, co-transfected with wild type β- and γ-ENaC subunits. Protease and amiloride treatment protocols are shown above the traces and were unchanged for each subunit combination. ENaC activation by AP was unaffected by changes in the tested α-ENaC cleavage sites. C, representative traces of mutant γ-ENaC subunits, co-transfected with wild type α- and β-ENaC subunits. ENaC activation by AP was reduced by the deletion of the γ-subunit inhibitory peptide. D, summary data for wild type and mutant ENaC channels. Untreated (Baseline) and protease-induced currents are shown for n = 8 filters/condition. Error bars, S.E.
As described previously (45), amiloride-sensitive currents could be measured after transfection of human ENaC in FRT cells (Fig. 4, B and C). The currents from wild type human ENaC increased by 50.4 ± 2.6%, from 5.8 to 8.8 μA/cm2, in response to the addition of trypsin. These currents were fully inhibited by the addition of amiloride, consistent with currents arising from ENaC activity. The addition of AP to the apical bath resulted in a 31.9 ± 1.9% increase in amiloride-sensitive current, from 5.8 to 7.9 μA/cm2. This increase in INa was ∼67% of the increase elicited by trypsin (2.1 versus 3.1 μA/cm2). The increases in ENaC activity as a result of AP and trypsin treatment were qualitatively similar in both CCD and FRT cells. (Figs. 3B and 4B).
Removal of the furin sites in the α-subunit and removal of the α-subunit inhibitory peptide had little effect on ENaC activation after the AP addition (Fig. 4, B and D). In contrast, AP activation of ENaC was altered in the channels containing mutant γ-subunits (Fig. 4, C and D). The furin-prostasin double mutant in the γ-subunit (γ-mut) showed a significantly lower base-line current that was less sensitive to AP addition than wild type. Moreover, deletion of the inhibitory domain from the γ-subunit (γ-del) resulted in a complete loss of protease activation by trypsin and AP. These data are consistent with the γ-subunit being the target of the AP-associated proteolytic activation and with previous reports that ENaC proteolytic activation is primarily determined by processing of the γ-subunit (Fig. 4D) (47).
ENaC Activation in Primary Human Bronchial Epithelial Cells
To evaluate the activation of ENaC by AP in human lung tissue, primary HBE cells were evaluated electrophysiologically. Both CF- and non-CF HBE tissues were pretreated with furin convertase inhibitor and incubated with AP as described above (Fig. 5A). Primary HBE cells from CF and non-CF tissues both showed similar ENaC activation in response to AP addition. AP treatment of non-CF HBE cells increased ENaC current by ∼120%, from 8.3 ± 0.4 to 18.0 ± 1.6 μA/cm2. Similarly, HBE cells from CF patients showed a ∼70% increase in ENaC current, from 12.2 ± 1.2 to 20.1 ± 1.4 μA/cm2. Observable differences in the absolute (Fig. 5A) or relative (Fig. 5B) activation of ENaC from HBE cells from CF and non-CF donors were seen. These changes resulted from altered base-line and maximal currents.
FIGURE 5.

ENaC activation of non-CF and CF primary HBE cells. Primary human bronchial epithelial cells were evaluated by short circuit recording to assess ENaC activation by AP. A, changes in ENaC current for CF and non-CF HBE cells. Untreated (Baseline) and protease-induced currents are shown for n > 6 recordings. B, relative currents from CF and non-CF primary HBE cells. Maximal currents were established as peak current after trypsin activation. Untreated (base line) and AP-treated currents are shown for n > 6 recordings. Error bars, S.D.
Because both CF and non-CF HBE cells showed qualitatively similar responses to AP and trypsin addition, non-CF HBE cells were utilized to evaluate the time course and dose dependence of the AP-induced ENaC activation. As with the CCD and FRT cells, the addition of AP to the apical bath resulted in a slow activation and plateau of ENaC current (Fig. 6A). AP activation was submaximal when compared with that elicited by the addition of trypsin. Subsequent trypsin addition resulted in increased ENaC current under the conditions measured. AP-induced ENaC activation was kinetically slower than that seen with trypsin, as seen in both the CCD and FRT cells. The calculated time constants for AP ranged between τ = 11.9 and 13.5 min (Fig. 6, B and C). In contrast, the time constant for trypsin was τ = 0.33 min. To evaluate the concentration dependence of the AP activation, dose-response experiments were performed. Increasing AP concentrations from 0.3 to 1.75 μm resulted in a ∼40% increase in ENaC activation. This activation appeared saturated at 1.75 μm AP because further increases in AP concentration to 3.5 μm resulted in no changes in the rates or magnitudes of ENaC activation (Fig. 6C). At saturation, AP-induced ENaC currents were submaximal when compared with those elicited by trypsin. Qualitatively, the kinetic differences seen with AP and trypsin treatment were similar between the cell types tested and ENaC orthologs evaluated.
FIGURE 6.

ENaC activation in primary HBE cells. Primary human bronchial epithelial cells were used to evaluate ENaC activation by AP. A, representative traces of ENaC currents in HBE cells after protease treatment. HBE cells were treated with either trypsin or the sequential addition of AP and trypsin and monitored by short circuit recording, as described above. Protease and amiloride treatment protocols are shown above the traces and correspond to the traces below. B, time course of ENaC activation by AP. ENaC activation in HBE cells occurs with a time constant of ∼13 min. Data shown are a summary of n = 19 experiments. C, dose response for ENaC activation by AP. AP-induced ENaC activation was assessed at 0.3 μm (open squares), 1.5 μm (filled circles), and 3.75 μm (cross-hatches). The time constants for each concentration of protease treatment are shown. Data are from n > 12 recordings. Error bars, S.E.
DISCUSSION
Alkaline protease has been implicated in multiple modes of P. aeruginosa infection, yet little is known about its specific involvement in many of these pathologies (5, 17, 19). Previous studies have demonstrated that AP expression is correlated with Pseudomonas virulence and increased resistance in CF patients, suggesting that AP may play a role in processes related to bacterial colonization and/or exacerbation in the CF lung (11, 13, 14). Current models for the regulation of ASL in CF suggest that the protease-antiprotease balance may regulate water secretion and mucus viscosity (15, 27, 38). As such, secretion of bacterial proteases may contribute to virulence by altering this balance. This increased protease activity would putatively reduce mucociliary clearance, thereby facilitating bacterial colonization.
To assess the potential role of bacterial protease on ENaC activation, primary HBE cells were exposed apically to Pseudomonas strains. Electrophysiological recordings demonstrated that ENaC currents were modulated by exposure to Pseudomonas under culture conditions (Fig. 1). Importantly, the changes in basal and activatable ENaC current suggested that the FRD1 Pseudomonas strain could modulate the electrical properties of the HBE cells. The increase in basal current could be accounted for by at least two mechanisms. ENaC might be up-regulated in response to Pseudomonas, or the population of channels at the cell surface might be activated in the presence of the bacterial cells. Trypsin treatment of these cells resulted in increased ENaC current in PAO1 and Ringer's solution, with activated ENaC current rising to that of the FRD1 basal current. The loss of trypsin-activatable current in the FRD1-treated cells suggested that the ENaC population had already been activated by proteases on the apical surface in a treatment-dependent manner. Importantly, these data demonstrate that a pool of cell surface ENaC channels is available for proteolytic activation, suggesting that at least some channels are not fully cleaved while transiting the secretory pathway.
To evaluate its role in activating ENaC in airway epithelia, AP was purified, and its activity was further assessed biochemically and electrophysiologically. Biochemical studies of the folding and activation of AP demonstrate that the protease folds efficiently and is highly active under pH conditions similar to those found in the lung (Fig. 2). Thus, the AP enzyme can be functional under conditions mimicking those of the ASL. Consistent with this, recent reports of flagellar degradation by AP have shown that the enzyme has significant activity within a physiological pH range (48). To evaluate ENaC activation by the protease, AP was added to the apical baths of Ussing chambers, and INa was measured (Figs. 3–6). Cells expressing both endogenous and heterologous ENaC responded to this treatment with an increase in amiloride-sensitive Na+ current. Both human and murine ENaC responded similarly, with respect to kinetics and magnitude, to treatment with AP. These data demonstrate that ENaC current can be activated by AP. These data also provide potential mechanistic insight into previous reports that AP is expressed highly in early stages of Pseudomonas colonization and is correlated with the virulence and degree of exacerbation of Pseudomonas in CF patients (10–12). Specifically, activation of ENaC by AP may facilitate Pseudomonas colonization and infection by modulating ENaC activity.
Differences in activation were seen between trypsin, used as a control, and AP. Maximal activation was decreased, and activation kinetics were slowed when comparing AP with trypsin controls. The changes in activation kinetics could potentially stem from differences in protease processivity and/or accessibility of specific protease sites and ENaC channel dynamics. Alternatively, the slow kinetics could suggest that ENaC activation occurs via an indirect mechanism. The submaximal activation of ENaC by AP is potentially consistent with either a direct or indirect mechanism. The inability to maximally activate ENaC by the serial addition of AP suggests that a population of channels in the membrane is highly resistant to AP stimulation. The exact nature of this protection, be it the result of direct ENaC site accessibility or an indirect activation through a cascade, warrants further investigation.
Based on these observations, we propose a model for ENaC activation by Pseudomonas during bacterial colonization. Secretion of a channel-activating protease would result in local changes to the host environment, mediated by the ENaC activation shown in this study. In the CF lung, this increased Na+ absorbance would putatively result in local dehydration of the ASL. Reduction in ASL volume and the resulting decreased mucociliary clearance capacity would then facilitate Pseudomonas adhesion to and colonization of the airway. This remodeling would also be consistent with the localization of AP to and near Pseudomonas biofilms because widespread distribution of the enzyme would not be required to elicit such proximal effects (5). Such a model is consistent with protease expression and activity profiles seen in CF patients because previous reports suggest that protease activities are differentially modulated during initial colonization and chronic infection (14).
These data suggest that the underlying molecular pathology of CF may be complicated by host-pathogen interactions. Recent studies of porcine and human CF tissues suggest that, under basal conditions, the loss of CFTR conductance underlies CF pathophysiology (49, 50). In contrast, studies of human and murine airway suggest that up-regulated ENaC current may contribute to CF pathology (51–55). Our data do not specifically address the differences in these competing hypotheses but can be accommodated within either hypothesis. The increase in ENaC current seen in both non-CF and CF HBE cells (Figs. 1 and 5) suggests that proteolytic cleavage of ENaC results in an stimulated state in both systems and is independent of CFTR expression. In both cases, the activation of ENaC would putatively result in a decrease in fluid secretion by increasing Na+ absorption, although it is not clear how CFTR function may be coregulated in such a scenario.
These data provide evidence for the activation of ENaC by the AP from Pseudomonas and suggest that ENaC may be regulated by additional proteases found in the airway. The identification and characterization of a Zn2+-metalloproteinase as a regulator of ENaC provides evidence for an additional functional and structural class of proteinases that contributes to the regulation of ENaC. In addition, the characterization of basal changes in ENaC activity, resulting from exposure to Pseudomonas, suggests that ENaC proteolysis in the airway cells may be exploited by pathogens in airway colonization. Specifically, the proteolytic activation of ENaC by AP suggests a novel mechanism by which Pseudomonas remodels its host to facilitate its colonization and modulate its virulence in the CF lung.
Acknowledgments
We thank the University of Pittsburgh Cystic Fibrosis Center Cell Core facility (supported, in part, by National Institutes of Health Grant DK072506) for primary human bronchial cells.
This work was supported, in whole or in part, by National Institutes of Health Grants DK083284 (to P. H. T.), DK078917 (to M. B. B.), and HL087932 (to M. M. M.).
- CF
- cystic fibrosis
- AP
- alkaline protease
- CFTR
- cystic fibrosis transmembrane conductance regulator
- ENaC
- epithelial sodium channel
- HBE
- human bronchial epithelial
- FRT
- Fisher rat thyroid
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- MMP
- matrix metalloprotease
- CCD
- cortical collecting duct
- ASL
- airway surface liquid.
REFERENCES
- 1. Lyczak J. B., Cannon C. L., Pier G. B. (2000) Establishment of Pseudomonas aeruginosa infection. Lessons from a versatile opportunist. Microbes Infect. 2, 1051–1060 [DOI] [PubMed] [Google Scholar]
- 2. Lyczak J. B., Cannon C. L., Pier G. B. (2002) Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 15, 194–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hobden J. A. (2002) Pseudomonas aeruginosa proteases and corneal virulence. DNA Cell Biol. 21, 391–396 [DOI] [PubMed] [Google Scholar]
- 4. Jaffar-Bandjee M. C., Lazdunski A., Bally M., Carrère J., Chazalette J. P., Galabert C. (1995) Production of elastase, exotoxin A, and alkaline protease in sputa during pulmonary exacerbation of cystic fibrosis in patients chronically infected by Pseudomonas aeruginosa. J. Clin. Microbiol. 33, 924–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sarkisova S., Patrauchan M. A., Berglund D., Nivens D. E., Franklin M. J. (2005) Calcium-induced virulence factors associated with the extracellular matrix of mucoid Pseudomonas aeruginosa biofilms. J. Bacteriol. 187, 4327–4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lazdunski A., Guzzo J., Filloux A., Bally M., Murgier M. (1990) Secretion of extracellular proteins by Pseudomonas aeruginosa. Biochimie 72, 147–156 [DOI] [PubMed] [Google Scholar]
- 7. Parmely M., Gale A., Clabaugh M., Horvat R., Zhou W. W. (1990) Proteolytic inactivation of cytokines by Pseudomonas aeruginosa. Infect. Immun. 58, 3009–3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Horvat R. T., Clabaugh M., Duval-Jobe C., Parmely M. J. (1989) Inactivation of human γ interferon by Pseudomonas aeruginosa proteases. Elastase augments the effects of alkaline protease despite the presence of α2-macroglobulin. Infect. Immun. 57, 1668–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Horvat R. T., Parmely M. J. (1988) Pseudomonas aeruginosa alkaline protease degrades human γ interferon and inhibits its bioactivity. Infect. Immun. 56, 2925–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jagger K. S., Robinson D. L., Franz M. N., Warren R. L. (1982) Detection by enzyme-linked immunosorbent assays of antibody specific for Pseudomonas proteases and exotoxin A in sera from cystic fibrosis patients. J. Clin. Microbiol. 15, 1054–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burke V., Robinson J. O., Richardson C. J., Bundell C. S. (1991) Longitudinal studies of virulence factors of Pseudomonas aeruginosa in cystic fibrosis. Pathology 23, 145–148 [DOI] [PubMed] [Google Scholar]
- 12. Tingpej P., Smith L., Rose B., Zhu H., Conibear T., Al Nassafi K., Manos J., Elkins M., Bye P., Willcox M., Bell S., Wainwright C., Harbour C. (2007) Phenotypic characterization of clonal and nonclonal Pseudomonas aeruginosa strains isolated from lungs of adults with cystic fibrosis. J. Clin. Microbiol. 45, 1697–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Granström M., Ericsson A., Strandvik B., Wretlind B., Pavlovskis O. R., Berka R., Vasil M. L. (1984) Relation between antibody response to Pseudomonas aeruginosa exoproteins and colonization/infection in patients with cystic fibrosis. Acta Paediatr. Scand. 73, 772–777 [DOI] [PubMed] [Google Scholar]
- 14. Jagger K. S., Bahner D. R., Warren R. L. (1983) Protease phenotypes of Pseudomonas aeruginosa isolated from patients with cystic fibrosis. J. Clin. Microbiol. 17, 55–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suter S. (1994) The role of bacterial proteases in the pathogenesis of cystic fibrosis. Am. J. Respir. Crit. Care Med. 150, S118–S122 [DOI] [PubMed] [Google Scholar]
- 16. Kharazmi A., Döring G., Høiby N., Valerius N. H. (1984) Interaction of Pseudomonas aeruginosa alkaline protease and elastase with human polymorphonuclear leukocytes in vitro. Infect. Immun. 43, 161–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kharazmi A., Høiby N., Döring G., Valerius N. H. (1984) Pseudomonas aeruginosa exoproteases inhibit human neutrophil chemiluminescence. Infect. Immun. 44, 587–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guyot N., Bergsson G., Butler M. W., Greene C. M., Weldon S., Kessler E., Levine R. L., O'Neill S. J., Taggart C. C., McElvaney N. G. (2010) Functional study of elafin cleaved by Pseudomonas aeruginosa metalloproteinases. Biol. Chem. 391, 705–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leidal K. G., Munson K. L., Johnson M. C., Denning G. M. (2003) Metalloproteases from Pseudomonas aeruginosa degrade human RANTES, MCP-1, and ENA-78. J. Interferon Cytokine Res. 23, 307–318 [DOI] [PubMed] [Google Scholar]
- 20. Rauh R., Diakov A., Tzschoppe A., Korbmacher J., Azad A. K., Cuppens H., Cassiman J. J., Dötsch J., Sticht H., Korbmacher C. (2010) A mutation of the epithelial sodium channel associated with atypical cystic fibrosis increases channel open probability and reduces Na+ self-inhibition. J. Physiol. 588, 1211–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Azad A. K., Rauh R., Vermeulen F., Jaspers M., Korbmacher J., Boissier B., Bassinet L., Fichou Y., des Georges M., Stanke F., De Boeck K., Dupont L., Balascáková M., Hjelte L., Lebecque P., Radojkovic D., Castellani C., Schwartz M., Stuhrmann M., Schwarz M., Skalicka V., de Monestrol I., Girodon E., Férec C., Claustres M., Tümmler B., Cassiman J. J., Korbmacher C., Cuppens H. (2009) Mutations in the amiloride-sensitive epithelial sodium channel in patients with cystic fibrosis-like disease. Hum. Mutat. 30, 1093–1103 [DOI] [PubMed] [Google Scholar]
- 22. Mall M., Bleich M., Kuehr J., Brandis M., Greger R., Kunzelmann K. (1999) CFTR-mediated inhibition of epithelial Na+ conductance in human colon is defective in cystic fibrosis. Am. J. Physiol. 277, G709–G716 [DOI] [PubMed] [Google Scholar]
- 23. Garcia-Caballero A., Rasmussen J. E., Gaillard E., Watson M. J., Olsen J. C., Donaldson S. H., Stutts M. J., Tarran R. (2009) SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc. Natl. Acad. Sci. U.S.A. 106, 11412–11417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J. L. (1989) Identification of the cystic fibrosis gene. Cloning and characterization of complementary DNA. Science 245, 1066–1073 [DOI] [PubMed] [Google Scholar]
- 25. Thibodeau P. H., Richardson J. M., 3rd, Wang W., Millen L., Watson J., Mendoza J. L., Du K., Fischman S., Senderowitz H., Lukacs G. L., Kirk K., Thomas P. J. (2010) The cystic fibrosis-causing mutation ΔF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J. Biol. Chem. 285, 35825–35835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Myerburg M. M., McKenna E. E., Luke C. J., Frizzell R. A., Kleyman T. R., Pilewski J. M. (2008) Prostasin expression is regulated by airway surface liquid volume and is increased in cystic fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 294, L932–L941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Myerburg M. M., Butterworth M. B., McKenna E. E., Peters K. W., Frizzell R. A., Kleyman T. R., Pilewski J. M. (2006) Airway surface liquid volume regulates ENaC by altering the serine protease-protease inhibitor balance. A mechanism for sodium hyperabsorption in cystic fibrosis. J. Biol. Chem. 281, 27942–27949 [DOI] [PubMed] [Google Scholar]
- 28. Tarran R., Trout L., Donaldson S. H., Boucher R. C. (2006) Soluble mediators, not cilia, determine airway surface liquid volume in normal and cystic fibrosis superficial airway epithelia. J. Gen. Physiol. 127, 591–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Caldwell R. A., Boucher R. C., Stutts M. J. (2005) Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am. J. Physiol. Lung Cell Mol. Physiol. 288, L813–L819 [DOI] [PubMed] [Google Scholar]
- 30. Kleyman T. R., Carattino M. D., Hughey R. P. (2009) ENaC at the cutting edge. Regulation of epithelial sodium channels by proteases. J. Biol. Chem. 284, 20447–20451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tan C. D., Selvanathar I. A., Baines D. L. (2011) Cleavage of endogenous γENaC and elevated abundance of αENaC are associated with increased Na transport in response to apical fluid volume expansion in human H441 airway epithelial cells. Pflugers Arch. 462, 431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stewart A. P., Haerteis S., Diakov A., Korbmacher C., Edwardson J. M. (2011) Atomic force microscopy reveals the architecture of the epithelial sodium channel (ENaC). J. Biol. Chem. 286, 31944–31952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jasti J., Furukawa H., Gonzales E. B., Gouaux E. (2007) Structure of acid-sensing ion channel 1 at 1.9 Å resolution and low pH. Nature 449, 316–323 [DOI] [PubMed] [Google Scholar]
- 34. Kashlan O. B., Boyd C. R., Argyropoulos C., Okumura S., Hughey R. P., Grabe M., Kleyman T. R. (2010) Allosteric inhibition of the epithelial Na+ channel through peptide binding at peripheral finger and thumb domains. J. Biol. Chem. 285, 35216–35223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vuagniaux G., Vallet V., Jaeger N. F., Hummler E., Rossier B. C. (2002) Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum- and glucocorticoid-regulated kinase (Sgk1) in Xenopus oocytes. J. Gen. Physiol. 120, 191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guipponi M., Vuagniaux G., Wattenhofer M., Shibuya K., Vazquez M., Dougherty L., Scamuffa N., Guida E., Okui M., Rossier C., Hancock M., Buchet K., Reymond A., Hummler E., Marzella P. L., Kudoh J., Shimizu N., Scott H. S., Antonarakis S. E., Rossier B. C. (2002) The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum. Mol. Genet. 11, 2829–2836 [DOI] [PubMed] [Google Scholar]
- 37. Planes C., Leyvraz C., Uchida T., Angelova M. A., Vuagniaux G., Hummler E., Matthay M., Clerici C., Rossier B. (2005) In vitro and in vivo regulation of transepithelial lung alveolar sodium transport by serine proteases. Am. J. Physiol. Lung Cell Mol. Physiol. 288, L1099–L1109 [DOI] [PubMed] [Google Scholar]
- 38. Myerburg M. M., Harvey P. R., Heidrich E. M., Pilewski J. M., Butterworth M. B. (2010) Acute regulation of the epithelial sodium channel in airway epithelia by proteases and trafficking. Am. J. Respir. Cell Mol. Biol. 43, 712–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bens M., Vallet V., Cluzeaud F., Pascual-Letallec L., Kahn A., Rafestin-Oblin M. E., Rossier B. C., Vandewalle A. (1999) Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J. Am. Soc. Nephrol. 10, 923–934 [DOI] [PubMed] [Google Scholar]
- 40. Zhang L., Conway J. F., Thibodeau P. H. (2012) Calcium-induced folding and stabilization of the Pseudomonas aeruginosa alkaline protease. J. Biol. Chem. 287, 4311–4322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jozic D., Bourenkov G., Lim N. H., Visse R., Nagase H., Bode W., Maskos K. (2005) X-ray structure of human proMMP-1. New insights into procollagenase activation and collagen binding. J. Biol. Chem. 280, 9578–9585 [DOI] [PubMed] [Google Scholar]
- 42. Edman K., Furber M., Hemsley P., Johansson C., Pairaudeau G., Petersen J., Stocks M., Tervo A., Ward A., Wells E., Wissler L. (2011) The discovery of MMP7 inhibitors exploiting a novel selectivity trigger. ChemMedChem 6, 769–773 [DOI] [PubMed] [Google Scholar]
- 43. Bertini I., Calderone V., Fragai M., Jaiswal R., Luchinat C., Melikian M., Mylonas E., Svergun D. I. (2008) Evidence of reciprocal reorientation of the catalytic and hemopexin-like domains of full-length MMP-12. J. Am. Chem. Soc. 130, 7011–7021 [DOI] [PubMed] [Google Scholar]
- 44. Butterworth M. B., Frizzell R. A., Johnson J. P., Peters K. W., Edinger R. S. (2005) PKA-dependent ENaC trafficking requires the SNARE-binding protein complexin. Am. J. Physiol. Renal Physiol. 289, F969–F977 [DOI] [PubMed] [Google Scholar]
- 45. Passero C. J., Carattino M. D., Kashlan O. B., Myerburg M. M., Hughey R. P., Kleyman T. R. (2010) Defining an inhibitory domain in the γ subunit of the epithelial sodium channel. Am. J. Physiol. Renal Physiol. 299, F854–F861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Passero C. J., Mueller G. M., Myerburg M. M., Carattino M. D., Hughey R. P., Kleyman T. R. (2012) TMPRSS4-dependent activation of the epithelial sodium channel requires cleavage of the γ-subunit distal to the furin cleavage site. Am. J. Physiol. Renal Physiol. 302, F1–F8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Carattino M. D., Hughey R. P., Kleyman T. R. (2008) Proteolytic processing of the epithelial sodium channel γ subunit has a dominant role in channel activation. J. Biol. Chem. 283, 25290–25295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bardoel B. W., van der Ent S., Pel M. J., Tommassen J., Pieterse C. M., van Kessel K. P., van Strijp J. A. (2011) Pseudomonas evades immune recognition of flagellin in both mammals and plants. PLoS Pathog. 7, e1002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xiong L., Montplaisir J., Desautels A., Barhdadi A., Turecki G., Levchenko A., Thibodeau P., Dube M. P., Gaspar C., Rouleau G. A. (2010) Family study of restless legs syndrome in Quebec, Canada. Clinical characterization of 671 familial cases. Arch. Neurol. 67, 617–622 [DOI] [PubMed] [Google Scholar]
- 50. Itani O. A., Chen J. H., Karp P. H., Ernst S., Keshavjee S., Parekh K., Klesney-Tait J., Zabner J., Welsh M. J. (2011) Human cystic fibrosis airway epithelia have reduced Cl− conductance but not increased Na+ conductance. Proc. Natl. Acad. Sci. U.S.A. 108, 10260–10265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boucher R. C., Stutts M. J., Knowles M. R., Cantley L., Gatzy J. T. (1986) Na+ transport in cystic fibrosis respiratory epithelia. Abnormal basal rate and response to adenylate cyclase activation. J. Clin. Invest. 78, 1245–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gowen C. W., Lawson E. E., Gingras-Leatherman J., Gatzy J. T., Boucher R. C., Knowles M. R. (1986) Increased nasal potential difference and amiloride sensitivity in neonates with cystic fibrosis. J. Pediatr. 108, 517–521 [DOI] [PubMed] [Google Scholar]
- 53. Catalan M. A., Nakamoto T., Gonzalez-Begne M., Camden J. M., Wall S. M., Clarke L. L., Melvin J. E. (2010) Cftr and ENaC ion channels mediate NaCl absorption in the mouse submandibular gland. J. Physiol. 588, 713–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhou Z., Duerr J., Johannesson B., Schubert S. C., Treis D., Harm M., Graeber S. Y., Dalpke A., Schultz C., Mall M. A. (2011) The ENaC-overexpressing mouse as a model of cystic fibrosis lung disease. J. Cyst. Fibros. 10, S172–S182 [DOI] [PubMed] [Google Scholar]
- 55. Knowles M., Gatzy J., Boucher R. (1981) Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N. Engl. J. Med. 305, 1489–1495 [DOI] [PubMed] [Google Scholar]