

Background: Biosynthesis and metabolism of nicotinic acid adenine dinucleotide phosphate (NAADP) are unclear.
Results: Alkaline phosphatase (AP) was identified and characterized as enzyme involved in NAADP degradation.
Conclusion: In cells not expressing CD38, AP is a valid candidate enzyme for degradation of NAADP to nicotinic acid adenine dinucleotide.
Significance: This is the first evidence for identity of the enzyme degrading NAADP in cells not expressing CD38.
Keywords: Calcium, Calcium Signaling, NAADP, Phosphatase, Signal Transduction
Abstract
Nicotinic acid adenine dinucleotide phosphate (NAADP) is a ubiquitous second messenger providing a Ca2+ trigger in a wide range of cell types. However, its metabolism is not well understood. Here, we demonstrate the presence of endogenous NAADP in HeLa cells. CD38, a promiscuous enzyme described to be involved in NAADP metabolism, was not detectable in HeLa cells. In cell-free extracts of HeLa cells, NAADP was degraded to nicotinic acid adenine dinucleotide (NAAD). The enzyme was enriched in membranes (10,000 × g pellet) and displayed characteristics typical of alkaline phosphatase (AP), e.g. pH optimum at 8–9 and sensitivity to the inhibitors l-homoarginine and l-leucine. Importantly, NAADP at physiological concentrations (50–100 nm) was degraded to NAAD. Expression of AP isoenzymes was analyzed in HeLa cells. Based on the results together with inhibitor studies, the placental AP isoform emerged as the best candidate for NAADP degradation in HeLa cells. In contrast to HeLa cells, Jurkat T cells or HEK293 cells did not express any AP isoenzymes and did not display any NAADP 2′-phosphatase activity. Finally, the placental AP isoform was expressed heterologously in HEK293 cells, resulting in reconstitution of NAADP 2′-phosphatase activity in cell-free extracts. On the basis of the results, we provide evidence for AP as the metabolizing enzyme of NAADP in cells that do not express CD38.
Introduction
Nicotinic acid adenine dinucleotide phosphate (NAADP)2 is a ubiquitous second messenger involved in Ca2+ signaling in a wide range of cell types (reviewed in Ref. 1). NAADP acts at low nanomolar concentrations and is produced endogenously upon stimulation by several agonists (2–5). The intracellular target of NAADP is still unclear because several ion channels have been proposed to respond to NAADP, e.g. ryanodine receptors (6, 7), TRPML1 (transient receptor potential mucolipin 1) (8), and two-pore channels (9–11). The recent identification of small molecular weight soluble proteins specifically binding NAADP along with lack of direct labeling of two-pore channels (12) is in favor of a model in which such soluble NAADP-binding proteins mediate the effect of NAADP at different Ca2+ channels, as proposed originally by Gerasimenko et al. (13).
Similar to its unclear mechanism of action, the metabolism of NAADP is poorly understood. Although NAADP can be formed in vitro by CD38 via the base exchange mechanism, we and others have demonstrated that knock-out or gene silencing of CD38 either does not affect or even increases endogenous NAADP levels in cells and tissues (14, 15). However, the situation is more complex because in some cell types from CD38 knock-out mice, e.g. pancreatic acinar cells, NAADP levels are indeed lower compared with wild-type mice (16). Interestingly, increased NAADP contents in immunologically relevant tissues from CD38 knock-out mice, e.g. spleen and thymus, indicate a role for CD38 in NAADP degradation. Indeed, we and others have shown that metabolism of NAADP to 2′-phosphoadenosine diphosphoribose (2′-phospho-ADPR) takes place under physiological conditions in vitro (14, 17).
In this study, we investigated the degradation of NAADP in HeLa cells. Expression of CD38 was not detectable in these cells. The degradation product was different from the product obtained in CD38-expressing cells (2′-phospho-ADPR) and was identified by HPLC as nicotinic acid adenine dinucleotide (NAAD). The NAADP phosphatase activity was characterized and identified, and heterologous expression restored its activity.
EXPERIMENTAL PROCEDURES
Reagents
NAADP and 1,N6-etheno-nucleotides were obtained from BioLog (Bremen, Germany). Other compounds and chemicals were purchased from Sigma-Aldrich.
Cell Culture
Jurkat T lymphocytes (subclone JMP) were cultured in RPMI 1640 medium supplemented with GlutaMAX, 25 mm HEPES, 7.5% (v/v) newborn calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. HeLa and HEK293 cells were cultured in DMEM supplemented with GlutaMAX, 10% (v/v) fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin.
Analysis of Endogenous NAADP
Endogenous NAADP was extracted and quantified by the enzymatic NAADP cycling assay (5). Briefly, 2 × 108 HeLa cells were divided into identical twin samples and centrifuged at 500 × g for 5 min. One part was supplemented with 15 pmol of NAADP to calculate recovery. Cells pellets were then resuspended in 1 ml of ice-cold 20% (w/v) trichloroacetic acid and frozen in liquid N2. After two freeze-thaw cycles, samples were centrifuged at 4400 × g for 10 min at 4 °C. The supernatant was neutralized by extraction with water-saturated diethyl ether. Samples were purified by anion exchange chromatography. NAADP was quantified by the enzymatic NAADP cycling assay (5). Increase in fluorescence was measured at λex = 530 nm and λem = 590 nm on a fluorescence microplate reader (Infinite M200 with Software i-control, Tecan, Grödig, Austria). The mean recovery of NAADP in control samples was 66%. Data were subjected to Grubbs' outlier test (significance level of <0.01) and then tested using a one-sample t test versus the hypothetical value of 0.
Analysis of mRNA
Total RNA was extracted from 1 × 107 cells using an RNeasy minikit with on-column DNase digest (Qiagen). RT-PCR was performed with a Titan one-tube RT-PCR kit (Roche Applied Science) according to the manufacturer's protocol. Primers for the amplification are listed in Table 1. Nested PCR was performed with the diluted product from RT-PCR, primers (Table 1), and Taq DNA polymerase (Fermentas, St. Leon-Rot, Germany).
TABLE 1.
Primers used for RT-PCR and nested PCR
Primers specific for AP isoenzymes were adapted from Schär et al. (45).
| Forward primer | Reverse primer | |
|---|---|---|
| CD38 | 5′-GGC TCT CTA GGA GAG CCC AAC-3′ | 5′-CAC ACT CCC AAA AGT GCT GTT T-3′ |
| CD38 (nested) | 5′-CAC CAA GCG CTT TCC CCG-3′ | 5′-GAA TAC TGA AAC AGG GTT G-3′ |
| GCAP | 5′-AGC TCA TAC TCC ATA CCT G-3′ | 5′-CAC CCC CAT CCC GTC A-3′ |
| PLAP | 5′-CTC ATA CTC CAT GCC CA-3′ | 5′-CAC CCC CAT CCC ATC G-3′ |
| IAP | 5′-CTG CAG CCG GTT CCT GG-3′ | 5′-GCA CCC CCA ACC CAT CG-3′ |
| TNAP | 5′-ACA TCT GAC CAC TGC CA-3′ | 5′-GAG ACA CCC ATC CCA TC-3′ |
Preparation of Subcellular Fractions
Subcellular fractions were obtained by differential centrifugation as described previously (14, 18). Briefly, cells were lysed with a Potter-Elvehjem homogenizer, and cell debris and nuclei were discarded. Membrane protein fractions were obtained as pellets after centrifugation at 10,000 × g (P10 membranes) or at 100,000 × g (P100 membranes) and dissolved in buffer containing 140 mm NaCl, 20 mm HEPES, and EGTA-free Complete protease inhibitor (Roche Applied Science) at pH 7.4. The supernatant obtained after centrifugation at 100,000 × g was termed S100.
Western Blot Analysis of CD38
For determination of CD38 expression, P10 membrane protein (15 μg) was separated under nonreducing conditions by SDS-PAGE using a 12.5% gel and transferred onto a PVDF membrane. One part of the membrane was incubated with anti-CD38 antibody AT1 (1 μg/ml, 1 h; Santa Cruz Biotechnology, Heidelberg, Germany), and the second part was incubated with anti-Hsc70 antibody (0.04 μg/μl, 1 h; Santa Cruz Biotechnology) as a loading control. The membranes were then incubated with horseradish peroxidase-conjugated goat anti-mouse secondary antibody (0.2 μg/ml, 1 h; Dianova, Hamburg, Germany). Membranes were incubated for 1 min with ECL solution (Amersham Biosciences). Chemiluminescence was detected with a LAS-3000 Intelligent Dark Box (Fujifilm, Tokyo, Japan).
Western Blot Analysis of Alkaline Phosphatase
To analyze expression of the different isoenzymes of alkaline phosphatase (AP), 15 μg of membrane protein (P10) was separated under reducing conditions by SDS-PAGE using a 10% gel and transferred onto a PVDF membrane. One part of the membrane was incubated with antibody specific for each isoenzyme for 1 h (mouse anti-human germ cell alkaline phosphatase (GCAP; 2.5 μg/ml; AbD Serotec, Düsseldorf, Germany), rabbit anti-human placental alkaline phosphatase (PLAP; 1:800; Abcam, Cambridge, UK), mouse anti-human intestinal alkaline phosphatase (IAP; 0.6 μg/ml; Abcam), and rabbit anti-human tissue-nonspecific alkaline phosphatase (TNAP; 0.6 μg/ml; Abcam)), and the other part was incubated with mouse anti-human β-actin antibody (1:1200; Sigma-Aldrich) as a loading control. The membranes were then incubated for 1 h with the corresponding HRP-conjugated goat anti-mouse (1:2500) or goat ant-rabbit (1:5000) secondary antibody (Dianova). After incubation with ECL solution for 1 min, chemiluminescence was detected with the CAS-3000 Intelligent Dark Box.
Analysis of NAD Glycohydrolase Activity
The NAD glycohydrolase activity of CD38 was tested with a fluorescence assay. P10 membrane protein (100 μg/ml) from Jurkat and HeLa cells was incubated with 100 μm 1,N6-etheno-NAD for 30 min. NAD glycohydrolase converts the weakly fluorescent 1,N6-etheno-NAD into highly fluorescent 1,N6-etheno-ADPR. Increase in fluorescence was measured at λex = 300 nm and λem = 410 nm on the Infinite M200 fluorescence microplate reader.
Determination of Phosphate Release with Malachite Green
P10 membrane protein (1 μg/ml) from HeLa cells were incubated with 10 μm NAADP in TEA buffer (50 mm triethanolamine and 2 mm MgCl2, pH 8.0) for 30 min. The release of free phosphate was then detected by complexation with molybdate and malachite green (malachite green phosphate assay kit, BioAssay Systems, Hayward, CA) according to the manufacturer's protocol. Absorption was measured at λ = 600 nm on the Infinite M200 microplate reader.
Determination of Phosphatase Activity with para-Nitrophenyl Phosphate
P10 membrane protein (1 μg/ml) from HeLa cells were incubated with 5.5 mm para-nitrophenyl phosphate (pNPP) in DEA buffer (1 m diethanolamine and 1 mm MgCl2, pH 9.8) for 20 min. AP dephosphorylates the colorless substrate pNPP to yellow para-nitrophenol. Absorption of the dephosphorylated product was measured at λ = 405 nm on the Infinite M200 microplate reader.
Determination of Endogenous Intracellular NAADP 2′-Phosphatase Activity in HeLa Cells
HeLa cells were detached, washed twice with PBS, resuspended in either PBS (140 mm NaCl, 3 mm KCl, 8 mm Na2HPO4, and 1.5 mm KH2PO4, pH 7.4) alone or supplemented with 1 mg/ml bromelain (Sigma-Aldrich), and incubated for 30 min at 37 °C. Subsequently, cells from both conditions (with and without bromelain) were washed twice with assay buffer (140 mm NaCl, 5 mm KCl, 1 mm MgSO4, 1 mm CaCl2, 20 mm HEPES, 1 mm NaH2PO4, and 5.5 mm glucose, pH 7.4), resuspended in either assay buffer alone or containing 60 μg/ml saponin, and incubated for 5 min at 37 °C. Cells were then again washed twice and resuspended in assay buffer. Protein content was determined using the Bio-Rad protein assay. The different cell preparations were incubated with 100 μm NAADP for 1, 3, and 10 min. The reaction was stopped by addition of ice-cold TCA to a final concentration of 5% (v/v). TCA was removed by extraction with water-saturated diethyl ether. After neutralization of the samples by addition of phosphate buffer and removal of traces of residual TCA in a centrifugal evaporator, samples were analyzed for NAAD and NAADP content by HPLC.
HPLC Analysis of Nucleotides
Reaction products were filtered through a 10-kDa cutoff filter (Microcon, Millipore, Billerica, MA), diluted in elution buffer containing 20 mm KH2PO4 and 5 mm tetrabutylammonium dihydrogen phosphate at pH 6, and analyzed by ion pair reverse-phase HPLC performed on a 250 × 4.6-mm C18–5μ HyPURITY ADVANCE column (Fisher Scientific GmbH, Schwerte, Germany) or Multohyp BDS column (Chromatographie Service GmbH, Langerwehe, Germany) equipped with a 10 × 4-mm guard cartridge filled with the same material (Fisher Scientific GmbH or Chromatographie Service GmbH). Nucleotides were eluted with a nonlinear gradient with elution buffer and increasing amounts of methanol. Analysis of metabolism of 1,N6-etheno-NAD was performed at 25 °C at a flow rate of 1 ml/min with the following gradient: 0 min (0% methanol), 3 min (0% methanol), 30 min (30% methanol), 32 min (30% methanol), 34 min (0% methanol), and 40 min (0% methanol). NAADP 2′-phosphatase assays were analyzed at 25 °C at a flow rate of 0.8 ml/ml with the following gradient: 0 min (15% methanol), 3.5 min (15% methanol), 17 min (40% methanol), 27 min (45% methanol), 31 min (50% methanol), 33 min (50% methanol), 35 min (15% methanol), and 40 min (15% methanol). Nucleotides were detected by UV absorption at λ = 260 nm with a diode array detector (1200 series, Agilent Technologies, Böblingen, Germany), whereas fluorescent nucleotides were detected at λex = 300 nm and λem = 410 nm with a fluorescence detector (1200 series, Agilent Technologies). Product peaks were analyzed with ChemStation software (Agilent Technologies). Quantification was performed with external standards.
Recombinant Expression of AP in HEK293 Cells
PLAP cDNA was obtained from imaGenes (Berlin, Germany) and cloned into the vector pIRES2-EGFP (Clontech), resulting in the expression vector pIRES2-EGFP-PLAP. HEK293 cells were transfected at ∼80% confluency with Lipofectamine LTX (Invitrogen) according to the manufacturer's protocol. Expression of EGFP was verified 24 h after transfection by fluorescence microscopy. AP activity was analyzed 24 h post-transfection.
RESULTS
Analysis of endogenous NAADP levels in WT HeLa cells using the enzymatic cycling assay revealed a basal NAADP concentration of 9.9 ± 2.2 nm (mean ± S.E. (n = 13), p = 0.0007 versus hypothetical value of 0) (see also supplemental Fig. 1). Recently, CD38 has been implicated in both NAADP formation (17, 19) and degradation (14, 17). However, expression of CD38, as analyzed at the mRNA and protein levels (Fig. 1, A and B), was not detectable in HeLa cells. In comparison with Jurkat T cells, which express CD38 (Fig. 1, A and B), in protein extracts of HeLa cells only, a minor degradation of 1,N6-etheno-NAD was observed (Fig. 1C). HPLC analysis of the reaction products revealed negligible pyrophosphatase activity and lack of detectable NAD glycohydrolase activity in HeLa cells.
FIGURE 1.

Expression of CD38 is not detectable in HeLa cells. A, total RNA was extracted from Jurkat and HeLa cells. RT-PCR was performed with CD38-specific intron-spanning primers, followed by nested PCR. The PCR product was expected to be 400 bp in size. M, marker (gene ladder of 100 bp). B, P10 membrane protein from Jurkat or HeLa cells (15 μg) was separated by SDS-PAGE under nonreducing conditions and transferred onto a PVDF membrane. One part of the membrane was incubated with an antibody detecting the heat shock protein Hsc70 as a loading control. CD38 was detected using anti-CD38 antibody AT1 on the second part of the membrane. A representative experiment of three is shown. C, P10 membrane protein from Jurkat and HeLa cells (100 μg/ml) was incubated with 100 μm etheno-NAD for 30 min. Increase in fluorescence was measured at λex = 300 nm and λem = 410 nm. The background fluorescence signal obtained with HeLa cells is due to pyrophosphatase producing etheno-AMP and nicotinamide ribose phosphate (19). Data shown are means ± S.D. (n = 7–14). ***, p < 0.01 (Student's t test). AU, arbitrary units. D, shown are representative chromatograms obtained with HeLa or Jurkat membranes incubated with 100 μm etheno-NAD for 30 min. The fluorescence signal was measured at λex = 300 nm and λem = 410 nm. e-, etheno-; RU, relative units.
When NAADP was incubated with either membranes or cytosolic protein extracts from HeLa cells, the major reaction product was identified as NAAD, indicating a phosphatase activity degrading NAADP (Fig. 2A). This enzymatic activity was prominent in P10 membrane fractions and weak in P100 membranes and the S100 cytosolic fraction (Fig. 2).
FIGURE 2.
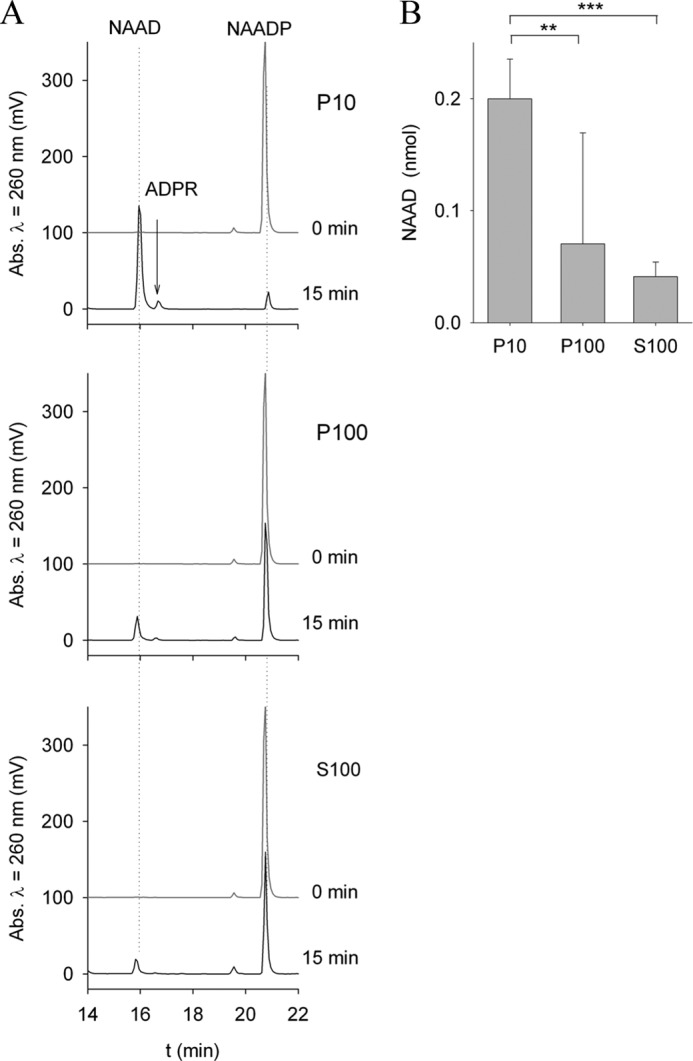
NAADP is degraded to NAAD in HeLa cells. Cytosolic protein (S100), the light membrane fraction (P100), and the heavy membrane fraction (P10), each at 1 mg/ml, were incubated with 20 μm NAADP at 37 °C for 15 min. Reaction products were analyzed by HPLC. A, shown are representative chromatograms before and 15 min after incubation with substrate. Incubation of NAADP with membrane fractions from HeLa cells resulted in dephosphorylation of NAADP and formation of NAAD. B, the reaction product NAAD was quantified after 15 min of incubation. NAAD production by P10 membrane fractions was much higher than that by P100 membrane fractions. Incubation of NAADP with cytosolic fractions (S100) resulted in relatively little NAAD formation. Data shown are means ± S.D. (n = 7–14). **, p < 0.01; ***, p < 0.001 (analysis of variance).
The NAADP 2′-phosphatase activity revealed a pH optimum between 8 and 9 (Fig. 3A). Different buffers used at the same pH showed only minor effects on NAAD formation (supplemental Fig. 2). Michaelis-Menten kinetics resulted in Km = 37 μm. The specific activity in P10 membranes was 0.8 μmol/mg/min. Because endogenous NAADP levels are in the range of ∼10–100 nm (5, 20), a Km of 37 μm appeared somewhat large and raised the question of whether this enzymatic activity might be of physiological relevance. Thus, in the next set of experiments, NAADP at physiological concentrations was used as substrate. In fact, incubation of NAADP at concentrations as low as 50 nm with P10 membranes from HeLa cells resulted in NAAD formation (Fig. 4, A and B). Furthermore, even at pH 7.2, the 2′-phosphatase reaction was observed; here, the fluorescent 1,N6-etheno-NAADP was used as substrate, and metabolism to 1,N6-etheno-NAAD was monitored by HPLC and fluorescence detection (Fig. 5). The second metabolic product observed at pH 9 only (1,N6-etheno-ADPR) also was formed in the absence of protein, obviously due to non-enzymatic degradation of 1,N6-etheno-NAADP via 1,N6-etheno-2′-phospho-ADPR, visible as a shoulder of the 1,N6-etheno-NAADP peak.
FIGURE 3.

Characterization of NAADP-degrading activity in HeLa cells. A, pH dependence of NAADP-degrading activity. Membrane protein from HeLa cells (1 mg/ml) was incubated with 20 μm NAADP at 37 °C for 5 min at different pH values. Reaction products were analyzed by HPLC. Buffers used were as follows: pH 5–6, MES; pH 7–9, TEA buffer; and pH 10–11, DEA buffer. Data shown are means ± S.D. (n = 2–4; p < 0.001). B, kinetic characterization of NAADP-degrading activity. Membrane protein from HeLa cells (0.5 mg/ml) was incubated with increasing concentrations of NAADP in TEA buffer at pH 9. Reaction products were analyzed by HPLC. Data shown are means ± S.D. (n = 1–6).
FIGURE 4.

NAADP at physiological concentrations is metabolized by NAADP-degrading activity. The physiological concentrations of NAADP in HeLa cells are 10–100 nm. Membrane protein from HeLa cells (0.5 mg/ml) was incubated with nanomolar concentrations of NAADP at 37 °C and pH 9. Samples were freeze-dried and reconstituted in water. NAAD formation was analyzed by HPLC. A, shown are representative chromatograms at 0 min and at 5 and 15 min after incubation with 50 nm NAADP. B, formation of NAAD was quantified after incubation with varying concentrations of NAADP.
FIGURE 5.

1,N6-etheno-NAADP is metabolized by NAADP-degrading activity at neutral pH. Membrane protein from HeLa cells (0.05 μg/ml) was incubated with 100 nm 1,N6-etheno-NAADP (e-NAADP) at 37 °C and pH 7.2 and 9. Samples were deproteinized using 10-kDa cutoff filters (Vivaspin). Product formation was analyzed by HPLC. Shown are representative chromatograms from samples incubated with 100 nm 1,N6-etheno-NAADP for 0, 5, 15, 40, and 60 min. Peak areas for 1,N6-etheno-2-phospho-ADPR (e-ADPRP) and 1,N6-etheno-ADPR (e-ADPR) are considerably larger due to the higher fluorescence intensity upon cleavage of the intramolecular quencher, the nicotinic acid moiety. FU, fluorescence units.
Because degradation of NAADP to NAAD preferentially occurred at alkaline pH, AP appeared to be a suitable candidate for the NAADP 2′-phosphatase. Thus, expression of isoenzymes of AP was analyzed in HeLa cells. At both the mRNA and protein levels, the AP isoenzymes PLAP, IAP, and TNAP were detected (Fig. 6, A and B).
FIGURE 6.

Expression and isoform-specific inhibition of AP isoenzymes in HeLa cells. A and B, expression analysis. A, expression analysis of AP at the mRNA level. Total RNA was extracted from HeLa cells and subjected to RT-PCR using primers specific for the different isoenzymes. Sizes of PCR products were analyzed by gel electrophoresis. Sequences of PCR products were verified by TA cloning and sequencing (not shown). mRNAs coding for PLAP (259 bp), IAP (250 bp), and TNAP (281 bp) but not GCAP (251 bp) were detected. M, marker (pUC (MspI)). B, expression analysis of AP at the protein level. Membrane protein from HeLa cells (10 μg) was separated by SDS-PAGE and transferred onto a PVDF membrane. One part of the membrane was incubated with an antibody detecting β-actin as a loading control. AP was detected with antibodies specific for the different isoenzymes. AP is highly glycosylated, and therefore, the apparent molecular mass is larger than the predicted molecular mass (57 kDa). C and D, isoform-specific inhibition. C, membrane protein from HeLa cells (1 μg/ml) was preincubated with 10 mm inhibitor for 10 min on ice. pNPP (5.5 mm) was then added and incubated for 20 min. Absorption of the dephosphorylated reaction product was measured at λ = 405 nm. D, membrane protein from HeLa cells (1 μg/ml) was preincubated with 10 mm inhibitor for 10 min on ice. NAADP (10 μm) was then added and incubated for 30 min. Release of phosphate was assessed with the malachite green assay. Data shown are means ± S.D. (n = 4–8). ***, p < 0.001 (analysis of variance).
To assess which isoform is responsible for the degradation of NAADP, isoform-specific inhibitors were tested. l-Homoarginine has been reported to inhibit mainly TNAP and has very little effect on the other isoforms (21). In contrast, l-phenylalanine especially inhibits the tissue-specific isoforms and shows little impact on TNAP (22). A third inhibitor, l-leucine, does not affect TNAP activity but strongly reduces the activity of PLAP and GCAP (21, 23). Using the nonspecific AP substrate pNPP, we observed that degradation was sensitive mainly to l-homoarginine and l-leucine, confirming expression of TNAP and PLAP in HeLa cells (Fig. 6C). Interestingly, l-homoarginine did not inhibit the degradation of NAADP by AP, suggesting that TNAP is not involved in this process (Fig. 6D). However, because l-leucine was the best inhibitor of NAADP degradation to NAAD (Fig. 6D) and because GCAP was not expressed in HeLa cells (Fig. 6B), PLAP appeared to be the best candidate for the enzymatic activity observed.
HeLa cells are known for their high expression of PLAP (22, 24, 25). Furthermore, the enzymatic characteristics observed in HeLa cells in our study indicated that PLAP is the most prominent isoform in these cells. Phosphodiesterase activity has been reported for TNAP, but not for PLAP (26, 27). Importantly, in this study, phosphodiesterase activity using cAMP as substrate was not detected in HeLa cells (data not shown). In addition, IAP and TNAP show lower specific activity in the presence of highly phosphorylated substrates (28, 29). On the contrary, no correlation between the degree of phosphorylation and phosphatase activity was seen with PLAP (30). In this study, the degree of substrate phosphorylation had no influence on the phosphatase activity (data not shown), indicating that the observed NAADP 2′-phosphatase activity is mainly due to expression of PLAP.
In Jurkat T cells, NAADP is metabolized to 2′-phospho-ADPR (14), whereas degradation of NAADP to NAAD was never observed in those cells (Fig. 7, C and D). Consistently, expression of the AP isoenzymes was not detectable in Jurkat T cells (Fig. 7, A and B).
FIGURE 7.

AP expression and activity are detectable in Jurkat cells. A, expression analysis of AP at the mRNA level. Total RNA was extracted from HeLa and Jurkat cells and subjected to RT-PCR using primers specific for the different isoenzymes. M, marker (pUG′(MspI)). B, expression analysis of AP at the protein level. Membrane protein from HeLa (H) or Jurkat (J) cells (10 μg) was separated by SDS-PAGE and transferred onto a PVDF membrane. One part of the membrane was incubated with an antibody detecting β-actin as a loading control. AP was detected with antibodies specific for the different isoenzymes. C, membrane protein from HeLa or Jurkat cells (5 μg/ml) was incubated with 5.5 mm pNPP. Absorption of the dephosphorylated reaction product was measured at λ = 405 nm. No AP activity could be detected in Jurkat cells. D, membrane protein from HeLa or Jurkat cells (5 μg/ml) was incubated with 40 μm NAADP. Release of phosphate was then assessed with the malachite green assay. Data shown are means ± S.D. (n = 4–8). n.s., not significant; ***, p < 0.001 (analysis of variance).
Taken together, our data indicate the specific degradation of NAADP at physiological concentrations to NAAD by AP, likely by the isoenzyme PLAP. To confirm these results, we aimed to recombinantly express AP in a cell in which expression of endogenous AP is not detectable. Thus, the expression and enzymatic activity of AP isoenzymes were characterized in HEK293 cells (Fig. 8). No endogenous expression of any of the AP isoenzymes (Fig. 8, A and B) and no AP activity (Fig. 8, C and D) were detected. Thus, HEK293 cells appeared to be a suitable cell system to express AP and to analyze protein extracts for recombinant AP activity toward the substrate NAADP. Indeed, expression of PLAP in HEK293 cells resulted in dephosphorylation of NAADP to NAAD, whereas no such enzymatic activity was observed in EGFP-transfected HEK293 cells (Fig. 9).
FIGURE 8.

AP expression and activity are not detectable in HEK293 cells. A, expression analysis of AP at the mRNA level. Total RNA was extracted from HeLa and HEK293 cells and subjected to RT-PCR using primers specific for the different isoenzymes. Sizes of PCR products were analyzed by gel electrophoresis. M, marker (pUG′(MspI)). B, expression analysis of AP at the protein level. Membrane protein from HeLa and HEK293 cells (10 μg) was separated by SDS-PAGE and transferred onto a PVDF membrane. One part of the membrane was incubated with an antibody detecting β-actin as a loading control. AP was detected with antibodies specific for the different isoenzymes. C, membrane protein from HeLa or HEK293 cells (5 μg/ml) was incubated with 5.5 mm pNPP. Absorption of the dephosphorylated reaction product was measured at λ = 405 nm. D, membrane protein from HeLa or HEK293 cells (5 μg/ml) was incubated with 40 μm NAADP. Release of phosphate was then assessed with the malachite green assay. Data shown are means ± S.D. (n = 4–8). n.s., not significant; ***, p < 0.001 (analysis of variance).
FIGURE 9.
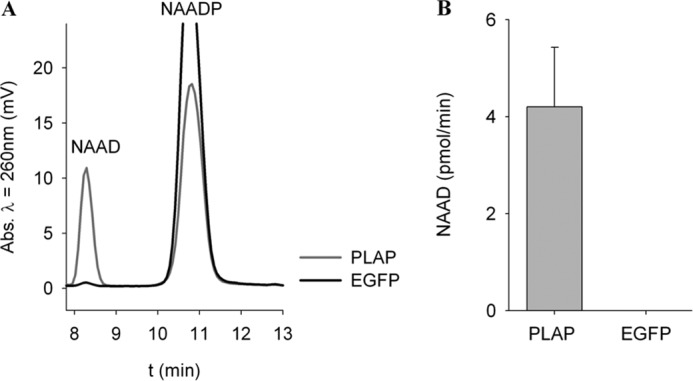
Recombinant expression of AP in HEK293 cells results in NAADP degradation. HEK293 cells were transiently transfected with pIRES2-EGFP-PLAP and control plasmid pIRES2-EGFP. 24 h after transfection, cells were incubated with 100 μm NAADP, and formation of NAAD was analyzed by HPLC. A, shown are representative chromatograms before and 2 h after incubation with substrate. Transient expression of PLAP in HEK293 cells led to dephosphorylation of NAADP and formation of NAAD. B, formation of NAAD was quantified after 2 h. Data shown are means ± S.D. (n = 5).
AP is localized on the cell surface as a glycosylphosphatidylinositol-anchored ectoenzyme (31), but expression in intracellular membranes, e.g. endoplasmic reticulum, Golgi, lysosomes, and mitochondria (32–36), has been described. Interestingly, cytosolic AP enzymatic activity was detected in saponin-permeabilized HeLa cells (35), indicating correct localization for potential involvement in cytosolic signal transduction. To test for intracellular NAADP 2′-phosphatase activity in HeLa cells, bromelain was used to proteolytically cleave surface protein. NAADP 2′-phosphatase activity decreased under such conditions (Fig. 10). Subsequent permeabilization by saponin in the absence of bromelain significantly increased NAADP 2′-phosphatase activity (Fig. 10), confirming the intracellular localization of PLAP, as described for AP 31 years ago (35). Similar results were obtained in HEK293 cells overexpressing PLAP; however, PLAP was removed from the cell surface using phosphoinositide-specific phospholipase C instead of bromelain (data not shown).
FIGURE 10.

Intracellular NAADP-degrading activity in wild-type HeLa cells. A, HeLa cells were incubated for 30 min with the protease bromelain (1 mg/ml) to release AP from the cell surface. The cells were then washed to remove both released AP and bromelain. Afterward, the cells were permeabilized using saponin. The protein content of the cells was determined, and portions equivalent to 0.2–1.0 μg of total protein were incubated with 100 μm NAADP. B, NAAD content was analyzed by HPLC after 1, 3, and 10 min to determine NAADP 2′-phosphatase activity. Data shown are means ± S.D. (n = 4). *, p < 0.05 (Student's t test).
DISCUSSION
In this study, we have demonstrated (i) the presence of endogenous NAADP in the HeLa cell line, a cell type in which expression of CD38 is not detectable; (ii) degradation of NAADP to NAAD in HeLa cell extracts; (iii) identification of AP as an NAADP-degrading enzyme; and (iv) intracellular localization of the NAADP-degrading enzyme. Although NAADP appears to be a very important and probably ubiquitous second messenger, several aspects of its metabolism are unknown. Although the base exchange reaction, catalyzed mainly (if not exclusively) by CD38, is the only reaction shown in vitro to produce NAADP (19), a growing number of reports suggest that endogenous NAADP is detected in cells or tissues even when the gene for CD38 is silenced, e.g. using an shRNA approach, or in CD38 knock-out mice (14, 15). Our study adds another example, the human cell line HeLa. Although expression of CD38 is not detectable in HeLa cells, endogenous NAADP at a similar concentration range compared with other cell types (5, 20) was clearly detected. To better understand NAADP synthesis in HeLa cells, the potential phosphorylation of NAAD was tested in our laboratory. Attempts to generate NAADP by incubation of NAAD and ATP with NAD kinase failed. Furthermore, no NAAD-phosphorylating activity could be detected using protein extracts from HeLa cells.3 Another possibility, deamidation of NADP, was also assayed. Various experiments with protein fractions from HeLa cells did not reveal an NADP deamidase activity.3 However, the presence of endogenous NAADP suggests that there must be an enzymatic activity for formation of NAADP other than CD38. The fact that neither kinase nor deamidase activity has been detectable so far indicates a more complex situation than hitherto acknowledged. It might well be that coenzyme(s) or co-substrate(s) necessary for these reactions have not been added in the right way in the in vitro assays carried out so far.
Regarding the degradation of NAADP, our enzymatic assays combined with HPLC analysis of reaction products revealed two main reactions: (i) release of nicotinic acid to produce 2′-phospho-ADPR by CD38 or (ii) release of inorganic phosphate to generate NAAD. The latter reaction was catalyzed by the PLAP isoenzyme in cell extracts from HeLa cells. An involvement of AP enzymatic activity has not yet been implicated in NAADP metabolism, but a NAADP phosphatase activity from brain membranes has been described (37). This NAADP 2′-phosphatase was characterized by a Km of 7 μm, a specific activity of ∼160 pmol/min/mg of protein, and a dependence on free [Ca2+]. Free Ca2+ at concentrations of ≥0.1 μm increased enzymatic activity substantially (37). Interestingly, Berridge et al. (37) compared their NAADP 2′-phosphatase activity with commercially available AP activity, but instead of analyzing any pH dependence, they rather focused on the Ca2+ dependence of their enzymatic activity. In our study, cells were homogenized in the presence of 1 mm Ca2+. However, although membranes obtained after two centrifugation steps were dissolved in a nominally Ca2+-free buffer (containing 110 mm KCl, 20 mm HEPES, pH 7.2, and Complete EGTA-free protease inhibitor), it is very likely that the free [Ca2+] was ≥0.1 μm in our experiments. Reports from the literature indicate a Ca2+ dependence of AP. First, AP prepared from chicken femurs was stimulated by calmodulin and 10 μm Ca2+ (30). Second, AP from basolateral membranes of rat mucosal cells was dependent on Ca2+ with optimum enzymatic activity at 40 μm Ca2+(38). However, whether the enzyme initially characterized by Berridge et al. (37) is an AP remains to be investigated.
In this study, we aimed far beyond characterization of the enzymatic activity. In fact, we used the characterization, e.g. pH optimum between 8 and 9 and specific inhibition by l-leucine, to narrow down to a suitable candidate enzyme. Most importantly, although the Km for NAADP was in the micromolar range, we demonstrated sufficient 2′-phosphatase activity also for NAADP at concentrations of 50 and 100 nm, even at pH 7.2. Finally, expression analysis of AP isoenzymes in HeLa, Jurkat, and HEK293 cells allowed heterologous expression of the AP isoenzyme PLAP in HEK293 cells, resulting in NAADP degradation to NAAD in cell extracts.
Is anything known about the involvement of PLAP in (NAADP) signaling? In vitro, PLAP is a promiscuous enzyme that degrades different adenine and uridine nucleotides, glucose 6-phosphate, β-glycerophosphate, and others (39). Phosphatidates with long fatty acid chains have been described as substrates also (40). However, the physiological substrate of AP isoenzymes has remained unclear. In this study, we have presented evidence that endogenous NAADP at very low concentrations is degraded by AP and that AP might be involved in this process under physiological conditions. However, PLAP is expressed on the cell surface as a glycosylphosphatidylinositol-anchored enzyme (31). Thus, in this sense, PLAP resembles the ectoenzyme CD38, which can be detected on the surface of many cell types (reviewed in Ref. 41). This “topological paradox” has been solved for CD38 and its ADP-ribosyl cyclase enzymatic activity since De Flora and co-workers demonstrated transport pathways for both the substrate NAD via connexin 43 and for the product cyclic ADP-ribose via nucleoside transporters, respectively (42–44). For AP, expression in the cytosol and intracellular membranes was described in earlier publications (32–36). Here, we successfully reproduced an increase in AP activity upon permeabilization of HeLa cells using NAADP as substrate, confirming the correct localization for potential involvement in cytosolic signal transduction, as reported previously (35).
Here, we present the AP isoenzyme PLAP as a candidate enzyme for degradation of NAADP to NAAD. In cells that lack CD38, such as the HeLa cell line, this degradation pathway may be important to terminate NAADP responses.
This work was supported by Deutsche Forschungsgemeinschaft Grant GU 360/15-1 (to A. H. G.).

This article contains supplemental Figs. 1 and 2.
F. Schmid and A. H. Guse, unpublished data.
- NAADP
- nicotinic acid adenine dinucleotide phosphate
- ADPR
- adenosine diphosphoribose
- NAAD
- nicotinic acid adenine dinucleotide
- AP
- alkaline phosphatase
- GCAP
- germ cell alkaline phosphatase
- PLAP
- placental alkaline phosphatase
- IAP
- intestinal alkaline phosphatase
- TNAP
- tissue-nonspecific alkaline phosphatase
- pNPP
- para-nitrophenyl phosphate
- EGFP
- enhanced GFP.
REFERENCES
- 1. Guse A. H., Lee H. C. (2008) NAADP: a universal Ca2+ trigger. Sci. Signal. 1, re10. [DOI] [PubMed] [Google Scholar]
- 2. Masgrau R., Churchill G. C., Morgan A. J., Ashcroft S. J.., Galione A. (2003) NAADP: a new second messenger for glucose-induced Ca2+ responses in clonal pancreatic β cells. Curr. Biol. 13, 247–251 [DOI] [PubMed] [Google Scholar]
- 3. Kim B. J., Park K. H., Yim C. Y., Takasawa S., Okamoto H., Im M. J., Kim U. H. (2008) Generation of nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose by glucagon-like peptide-1 evokes Ca2+ signal that is essential for insulin secretion in mouse pancreatic islets. Diabetes 57, 868–878 [DOI] [PubMed] [Google Scholar]
- 4. Yamasaki M., Thomas J. M., Churchill G. C., Garnham C., Lewis A. M., Cancela J. M., Patel S., Galione A. (2005) Role of NAADP and cADPR in the induction and maintenance of agonist-evoked Ca2+ spiking in mouse pancreatic acinar cells. Curr. Biol. 15, 874–878 [DOI] [PubMed] [Google Scholar]
- 5. Gasser A., Bruhn S., Guse A. H. (2006) Second messenger function of nicotinic acid adenine dinucleotide phosphate revealed by an improved enzymatic cycling assay. J. Biol. Chem. 281, 16906–16913 [DOI] [PubMed] [Google Scholar]
- 6. Hohenegger M., Suko J., Gscheidlinger R., Drobny H., Zidar A. (2002) Nicotinic acid adenine dinucleotide phosphate activates the skeletal muscle ryanodine receptor. Biochem. J. 367, 423–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dammermann W., Zhang B., Nebel M., Cordiglieri C., Odoardi F., Kirchberger T., Kawakami N., Dowden J., Schmid F., Dornmair K., Hohenegger M., Flügel A., Guse A. H., Potter B. V. (2009) NAADP-mediated Ca2+ signaling via type 1 ryanodine receptor in T cells revealed by a synthetic NAADP antagonist. Proc. Natl. Acad. Sci. U.S.A. 106, 10678–10683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang F., Li P. (2007) Reconstitution and characterization of a nicotinic acid adenine dinucleotide phosphate (NAADP)-sensitive Ca2+ release channel from liver lysosomes of rats. J. Biol. Chem. 282, 25259–25269 [DOI] [PubMed] [Google Scholar]
- 9. Brailoiu E., Churamani D., Cai X., Schrlau M. G., Brailoiu G. C., Gao X., Hooper R., Boulware M. J., Dun N. J., Marchant J. S., Patel S. (2009) Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J. Cell Biol. 186, 201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Calcraft P. J., Ruas M., Pan Z., Cheng X., Arredouani A., Hao X., Tang J., Rietdorf K., Teboul L., Chuang K. T., Lin P., Xiao R., Wang C., Zhu Y., Lin Y., Wyatt C. N., Parrington J., Ma J., Evans A. M., Galione A., Zhu M. X. (2009) NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459, 596–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zong X., Schieder M., Cuny H., Fenske S., Gruner C., Rötzer K., Griesbeck O., Harz H., Biel M., Wahl-Schott C. (2009) The two-pore channel TPCN2 mediates NAADP-dependent Ca2+-release from lysosomal stores. Pflugers Arch. 458, 891–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin-Moshier Y., Walseth T. F., Churamani D., Davidson S. M., Slama J. T., Hooper R., Brailoiu E., Patel S., Marchant J. S. (2012) Photoaffinity labeling of nicotinic acid adenine dinucleotide phosphate (NAADP) targets in mammalian cells. J. Biol. Chem. 287, 2296–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gerasimenko J. V., Maruyama Y., Yano K., Dolman N. J., Tepikin A. V., Petersen O. H., Gerasimenko O. V. (2003) NAADP mobilizes Ca2+ from a thapsigargin-sensitive store in the nuclear envelope by activating ryanodine receptors. J. Cell Biol. 163, 271–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmid F., Bruhn S., Weber K., Mittrücker H. W., Guse A. H. (2011) CD38: a NAADP-degrading enzyme. FEBS Lett. 585, 3544–3548 [DOI] [PubMed] [Google Scholar]
- 15. Soares S., Thompson M., White T., Isbell A., Yamasaki M., Prakash Y., Lund F. E., Galione A., Chini E. N. (2007) NAADP as a second messenger: neither CD38 nor base exchange reaction are necessary for in vivo generation of NAADP in myometrial cells. Am. J. Physiol. Cell Physiol. 292, C227–C239 [DOI] [PubMed] [Google Scholar]
- 16. Cosker F., Cheviron N., Yamasaki M., Menteyne A., Lund F. E., Moutin M. J., Galione A., Cancela J. M. (2010) The ectoenzyme CD38 is a nicotinic acid adenine dinucleotide phosphate (NAADP) synthase that couples receptor activation to Ca2+ mobilization from lysosomes in pancreatic acinar cells. J. Biol. Chem. 285, 38251–38259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Graeff R., Liu Q., Kriksunov I. A., Hao Q., Lee H. C. (2006) Acidic residues at the active sites of CD38 and ADP-ribosyl cyclase determine nicotinic acid adenine dinucleotide phosphate (NAADP) synthesis and hydrolysis activities. J. Biol. Chem. 281, 28951–28957 [DOI] [PubMed] [Google Scholar]
- 18. Schweitzer K., Mayr G. W., Guse A. H. (2001) Assay for ADP-ribosyl cyclase by reverse-phase high-performance liquid chromatography. Anal. Biochem. 299, 218–226 [DOI] [PubMed] [Google Scholar]
- 19. Aarhus R., Graeff R. M., Dickey D. M., Walseth T. F., Lee H. C. (1995) ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem. 270, 30327–30333 [DOI] [PubMed] [Google Scholar]
- 20. Churamani D., Carrey E. A., Dickinson G. D., Patel S. (2004) Determination of cellular nicotinic acid adenine dinucleotide phosphate (NAADP) levels. Biochem. J. 380, 449–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fishman W., Sie H. G. (1970) l-homoarginine; an inhibitor of serum “bone and liver” alkaline phosphatase. Clin. Chim. Acta 29, 339–341 [DOI] [PubMed] [Google Scholar]
- 22. Herz F. (1985) Alkaline phosphatase isoenzymes in cultured human cancer cells. Experientia 41, 1357–1361 [DOI] [PubMed] [Google Scholar]
- 23. Hoylaerts M. F., Manes T., Millán J. L. (1992) Molecular mechanism of uncompetitive inhibition of human placental and germ cell alkaline phosphatase. Biochem. J. 286, 23–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Singer R. M., Fishman W. H. (1974) Characterization of two HeLa sublines: TCRC-1 produces Regan isoenzyme and TCRC-2, non-Regan isoenzyme. J. Cell Biol. 60, 777–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elson N. A., Cox R. P. (1969) Production of fetal-like alkaline phosphatase by HeLa cells. Biochem. Genet. 3, 549–561 [DOI] [PubMed] [Google Scholar]
- 26. Rezende A. A., Pizauro J. M., Ciancaglini P., Leone F. A. (1994) Phosphodiesterase activity is a novel property of alkaline phosphatase from osseous plate. Biochem. J. 301, 517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang L., Balcerzak M., Radisson J., Thouverey C., Pikula S., Azzar G., Buchet R. (2005) Phosphodiesterase activity of alkaline phosphatase in ATP-initiated Ca2+ and phosphate deposition in isolated chicken matrix vesicles. J. Biol. Chem. 280, 37289–37296 [DOI] [PubMed] [Google Scholar]
- 28. Demenis M. A., Leone F. A. (2000) Kinetic characteristics of ATP hydrolysis by a detergent-solubilized alkaline phosphatase from rat osseous plate. IUBMB Life 49, 113–119 [DOI] [PubMed] [Google Scholar]
- 29. Say J. C., Ciuffi K., Furriel R. P., Ciancaglini P., Leone F. A. (1991) Alkaline phosphatase from rat osseous plates: purification and biochemical characterization of a soluble form. Biochim. Biophys. Acta 1074, 256–262 [DOI] [PubMed] [Google Scholar]
- 30. Sivanaesan L., Kwan T. K., Perumal R. (1991) The activation of chick alkaline phosphatase by calmodulin. Biochem. Int. 25, 561–570 [PubMed] [Google Scholar]
- 31. Lehto M. T., Sharom F. J. (2002) Proximity of the protein moiety of a GPI-anchored protein to the membrane surface: a FRET study. Biochemistry 41, 8368–8376 [DOI] [PubMed] [Google Scholar]
- 32. Sasaki M., Fishman W. H. (1973) Ultrastructural studies on Regan and non-Regan isoenzymes of alkaline phosphatase in human ovarian cancer cells. Cancer Res. 33, 3008–3018 [PubMed] [Google Scholar]
- 33. Lin C. W., Sasaki M., Orcutt M. L., Miyayama H., Singer R. M. (1976) Plasma membrane localization of alkaline phosphatase in HeLa cells. J. Histochem. Cytochem. 24, 659–667 [DOI] [PubMed] [Google Scholar]
- 34. Benham F., Cottell D. C., Franks L. M., Wilson P. D. (1977) Alkaline phosphatase activity in human bladder tumor cell lines. J. Histochem. Cytochem. 25, 266–274 [DOI] [PubMed] [Google Scholar]
- 35. Tokumitsu S., Tokumitsu K., Fishman W. H. (1981) Immunocytochemical demonstration of intracytoplasmic alkaline phosphatase in HeLa TCRC-1 cells. J. Histochem. Cytochem. 29, 1080–1087 [DOI] [PubMed] [Google Scholar]
- 36. Wilson P. D., Rustin G. J., Peters T. J. (1981) The ultrastructural localization of human neutrophil alkaline phosphatase in normal individuals during pregnancy and in patients with chronic granulocytic leukemia. Histochem. J. 13, 31–43 [DOI] [PubMed] [Google Scholar]
- 37. Berridge G., Cramer R., Galione A., Patel S. (2002) Metabolism of the novel Ca2+-mobilizing messenger nicotinic acid adenine dinucleotide phosphate via a 2′-specific Ca2+-dependent phosphatase. Biochem. J. 365, 295–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Birge S. J., Gilbert H. R. (1974) Identification of an intestinal sodium- and calcium-dependent phosphatase stimulated by parathyroid hormone. J. Clin. Invest. 54, 710–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Harkness D. R. (1968) Studies on human placental alkaline phosphatase. II. Kinetic properties and studies on the apoenzyme. Arch. Biochem. Biophys. 126, 513–523 [DOI] [PubMed] [Google Scholar]
- 40. Sumikawa K., Okochi T., Adachi K. (1990) Differences in phosphatidate hydrolytic activity of human alkaline phosphatase isoenzymes. Biochim. Biophys. Acta 1046, 27–31 [DOI] [PubMed] [Google Scholar]
- 41. Malavasi F., Deaglio S., Funaro A., Ferrero E., Horenstein A. L., Ortolan E., Vaisitti T., Aydin S. (2008) Evolution and function of the ADP-ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 88, 841–886 [DOI] [PubMed] [Google Scholar]
- 42. Bruzzone S., Guida L., Zocchi E., Franco L., De Flora A. (2001) Connexin 43 hemichannels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 15, 10–12 [DOI] [PubMed] [Google Scholar]
- 43. Guida L., Bruzzone S., Sturla L., Franco L., Zocchi E., De Flora A. (2002) Equilibrative and concentrative nucleoside transporters mediate influx of extracellular cyclic ADP-ribose into 3T3 murine fibroblasts. J. Biol. Chem. 277, 47097–47105 [DOI] [PubMed] [Google Scholar]
- 44. Guida L., Franco L., Bruzzone S., Sturla L., Zocchi E., Basile G., Usai C., De Flora A. (2004) Concentrative influx of functionally active cyclic ADP-ribose in dimethyl sulfoxide-differentiated HL-60 cells. J. Biol. Chem. 279, 22066–22075 [DOI] [PubMed] [Google Scholar]
- 45. Schär B. K., Otto V. I., Hänseler E. (1997) Simultaneous detection of all four alkaline phosphatase isoenzymes in human germ cell tumors using reverse transcription-PCR. Cancer Res. 57, 3841–3846 [PubMed] [Google Scholar]