

Background: Whether TLR3 requires proteolytic processing is not clear.
Results: TLR3 was found to be processed by cathepsins within Loop1 of leucine-rich repeat 12. Proteolytic processing is not required for TLR3 signaling but modulated its response to ligands and affected TLR3 localization in endosomes.
Conclusion: Cathepsin processing can alter TLR3 signaling.
Significance: This work contributes to understanding of TLR3 structure and function.
Keywords: Endosomes, Innate Immunity, Protease, RNA, Signal Transduction, Toll-like Receptors (TLR), Toll-like Receptor 3
Abstract
Toll-like receptors (TLRs) 3, 7, and 9 are innate immune receptors that recognize nucleic acids from pathogens in endosomes and initiate signaling transductions that lead to cytokine production. Activation of TLR9 for signaling requires proteolytic processing within the ectodomain by endosome-associated proteases. Whether TLR3 requires similar proteolytic processing to become competent for signaling remains unclear. Herein we report that human TLR3 is proteolytically processed to form two fragments in endosomes. Unc93b1 is required for processing by transporting TLR3 through the Golgi complex and to the endosomes. Proteolytic cleavage requires the eight-amino acid Loop1 within leucine-rich repeat 12 of the TLR3 ectodomain. Proteolytic cleavage is not required for TLR3 signaling in response to poly(I:C), although processing could modulate the degree of response toward viral double-stranded RNAs, especially in mouse cells. Both the full-length and cleaved fragments of TLR3 can bind poly(I:C) and are present in endosomes. However, although the full-length TLR3 has a half-life in HEK293T cells of 3 h, the cleaved fragments have half-lives in excess of 7 h. Inhibition of TLR3 cleavage by either treatment with cathepsin inhibitor or by a mutation in Loop1 decreased the abundance of TLR3 in endosomes targeted for lysosomal degradation.
Introduction
Toll-like receptors (TLRs)2 are a family of membrane-associated receptors that bind pathogen-associated molecular patterns to active innate immune responses (1, 2). TLRs are type-I glycoproteins consisting of a ligand binding ectodomain, a single-pass helix that spans the membrane, and an intracellular Toll-interleukin-1 receptor domain (TIR domain) for signal transduction. TLRs 3, 7, 8, and 9 recognize double and single-stranded RNAs or DNAs in endosomes to activate signal transduction in the cytoplasm (1, 3).
Endolysosomes play an important role in the TLR3, -7, and -9 regulation and function (3–8). They serve as sites for the sequestration of non-self-nucleic acids. The affinity of ligand recognition by TLR3 and TLR9 increases with the acidic environment found in endosomes (5, 9, 10). Endosomes also contain pH-activated proteases such as cathepsins that have been documented to cleave TLR7 and TLR9 (11–13). Cleavage is a stepwise process mediated by redundant proteases. The first step involves cathepsins that have broad sequence recognition (14) and/or asparagine endopeptidase that cleaves C-terminal to asparagines, generating an N-terminal fragment and an intermediate, untrimmed C-terminal fragment (CTF). The second step has cathepsins further trimming the CTF to produce the mature and active form of TLR9 (15). Both full-length and the mature CTF of TLR9 can bind ligand; however, only the latter could bind TLR9 adaptor for signaling transduction (11, 12). It was also found that the TLR9 CTF binds inhibitory oliogodeoxynucleotides (ODNs) with higher affinity than full-length TLR9 (16). In addition to the processing between leucine-rich repeat (LRR) 14 and LRR15 to generate the CTF, TLR9 was also found to be cleaved at the C terminus of the ectodomain to generate a soluble fragment that negatively regulates TLR9 function (17). Avian TLR15 also requires proteolytic cleavage the ectodomain to be activated (18).
Given the important role of endolysosomes in the regulation of TLR3, -7, and -9 function, trafficking chaperones are required for signaling. Unc93b1 is an endoplasmic reticulum-resident molecular chaperone responsible for translocation of TLRs 3, 7, and 9 to endolysosomes (19). A H412R substitution (3d mutation) was found to render mice deficient in immune responses mediated by TLR3, -7, and -9 by failing to bind the TLRs and facilitate their trafficking after ligand addition (19–21). Interestingly, the N-terminal region of Unc93b1 can differentially regulate TLR9 and TLR7. The Unc93b1 mutant D34A down-regulated TLR9 trafficking and signaling while up-regulating TLR7 signaling (22). D34A did not affect TLR3 signaling, although Unc93b1 contacts TLR3 through its juxtamembrane region as well as at a region within LRRs 11 and 12 of the TLR3 ectodomain (22, 23).
Although TLR3 and TLR7 are similar to TLR9 in terms of endosomal localization, there are contradictory reports as to whether they are proteolytically processed in the same way as TLR9 (11, 12, 15). Park et al. (12) reported that TLR7 cleavage was not observed, whereas Ewald et al. (11) reported that both TLR3 and TLR7 are subject to proteolytic processing the same as TLR9. Proteolytic cleavage was claimed by Ewald et al. (15) to be required for activation of TLR3 and TLR7 in murine macrophages, based upon the detection of intracellular tumor necrosis factor after ligand stimulation.
This work examines proteolytic processing of TLR3 and its effects on TLR3 function, especially in human cell lines. We found that interaction with Unc93b1 is required for the proteolytic cleavage of TLR3 in cultured cell lines. However, unlike TLR9, proteolytic cleavage is not required for TLR3 signaling, although cleavage did modulate ligand recognition in the mouse RAW264.7 cells. Finally, we documented that the cleaved fragments have increased stability when compared with the full-length TLR3 and alter trafficking to subpopulations of endolysosomes.
MATERIALS AND METHODS
Plasmid Construct
The plasmid to express the wild-type (WT) TLR3 was previously described in Qi et al. (23). The plasmid that can express TLR3 with a C-terminal hemagglutinin tag was from Invivogen, Inc. (San Diego, CA). TLR3 with mutations in Loop1 were made by site-directed mutagenesis using oligonucleotides and the QuikChange kit following the manufacturer's instructions (Stratagene, Inc., San Diego, CA). Unc93b1-Myc-FLAG and the 3d mutant were described in Qi et al. (23). All mutations were confirmed by DNA sequencing (Applied Biosystems, Foster City, CA). Plasmid pUbAU1 that can express ubiquitin with a C-terminal AU1 epitope tag was a generous gift from Dr. Khaled Tolba of the University of Miami, Miami, FL. Construct Δ1–12-HA encodes amino acids 1–51 of TLR3, fused to amino acids 354–906 followed by the influenza virus hemagglutinin (HA) tag (YPYDVPDYA). 344-C-HA encodes amino acids 1–51 and 344–906 of TLR3 and a C-terminal HA tag. Δ1–12-HA and 344-C-HA were constructed using forward primer 5′-CTCAGGTACCCGATGATCTACCCACATGTTTGGAGCACCTTAACATGG-3′ and CTCAGGTACCCGATGATCTACCCACACCCAAGATTGATGATTTTTCTTTTCAG, respectively, and reverse primer 5′-TTTTCTAGACCTCTCCATTCCTG-3′. Both PCR products were digested with KpnI and XbaI and ligated into the pUNO-TLR3-HA digested with the same restriction enzymes.
Luciferase Reporter Assay
Luciferase reporter assays for TLR-dependent signaling were performed as described previously (23). Briefly, HEK293T cells were plated in CoStar white 96-well plates in DMEM with 10% FBS and transfected with plasmids including pUNO-TLR3/4/9 to express TLRs, pNiFty-Luc/ISRE-Luc to express firefly luciferase reporters, and phRL-TK/pCMV-TK to express Renilla luciferase as an internal control. 24 h after transfection cells were induced by adding ligands of the TLRs to the culture medium, and firefly and Renilla luciferase activities were assayed using the Duo-Glo luciferase kit from Sigma and BioTek plate reader. Unless stated otherwise, TLR3 signaling was monitored with the firefly luciferase driven from the ISRE2 promoter and induced with poly(I:C) at 2.5 ng/ml (Amersham Biosciences). TLR4 signaling was assessed in cells that coexpressed TLR4 adaptors MD-2, CD14, and LPS binding protein, with the firefly luciferase driven by the NF-κB promoter and induced with LPS at 1 μg/ml (Invivogen). TLR9 signaling was assayed using firefly luciferase driven from the NF-κB promoter and induced with ODN2006 at 2 μm added to the cell culture medium (Invivogen). In Huh7.5 cells, the CMV promoter was used to express the Renilla luciferase. In HEK293T cells, a thymidine kinase promoter was used to drive the Renilla luciferase.
Quantification of IL6 Production
IL6 production by immortalized human lung epithelial BEAS-2B and mouse macrophage RAW264.7 cell lines was used to assess activities of endogenously expressed TLRs. BEAS-2B cells were cultured in BEGM media with supplements (Lonza, Basel, Switzerland; Ref. 28). TLR3 was induced with 0.125 ng/ml poly(I:C). RAW264.7 cells were cultured in DMEM supplemented with 10% FBS. Induction of TLR3, TLR4, TLR7, and TLR9 used, respectively, final concentrations of 50 ng/ml poly(I:C), 10 ng/ml LPS, 10 μg/ml gardiquimod (Invivogen) and 1 μm ODN1826 (Invivogen). After a 12-h induction, the cell culture media were harvested and centrifuged at 2000 × g to remove intact cells, and the concentration of IL6 was measured by ELISA with BD OptEIATM human IL6 ELISA set (BD Biosciences) or the DuoSet® mouse IL6 kit (R&D Systems, Minneapolis, MN). Poly(A:U) was from Invivogen. Purified Reovirus double-stranded (ds) genomic RNAs were extracted from purified virions, and Reovirus S4 was made by in vitro transcription of the sense and antisense strands followed by annealing of a 1:1 ratio of the two RNAs as described in Lai et al. (29).
Immunoprecipitations and Western Blots
Immunoprecipitations and Western blot assays used the buffers and protocols detailed in Qi et al. (23) and protein A/G-agarose beads (Santa Cruz Biotechnology). A polyclonal antibody-recognizing epitope(s) in the TLR3 ectodomain was from R&D systems (catalog #AF1487), whereas polyclonal antibody that recognized the TLR3 TIR domain was from Abcam, Inc. (Cambridge, MA; catalog #Ab84911). Anti-HA antibodies were from either Abcam (catalog #Ab9134) or Roche Applied Science (clone 3F10). Monoclonal antibodies recognizing epitope tags FLAG® and AU1 were, respectively, from Sigma (Clone M2, catalog #F1804) and Covance (Indianapolis, IN; catalog #MMS-130R). Proteins for Western blots were separated in denaturing 4–12% Bis-Tris gel (Invitrogen). The HRP-conjugated secondary antibodies that recognized mouse, rat, or goat IgGs were from Santa Cruz Biotechnologies. Proteins were detected using the ECL-plusTM or ECLTM-advance Western blotting Detection System (Amersham Biosciences), imaged using a ChemiDocTM XRS+ system (Bio-Rad), and quantified using ImageLab software (Bio-Rad).
Inhibition of Proteases and Proteasomes
z-FA-FMK (Santa Cruz Biotechnology) and bafilomycin A1 (Sigma, Inc.) were used at final concentrations of 5 μm and 2.5 nm unless stated otherwise. Lactacystin was from Sigma. IL6 production by BEAS-2B cells and RAW264.7 cells in the presence of bafilomycin A1 or z-FA-FMK had the inhibitors added to the cell culture media at the initial seeding of the cells, and the inhibitors were replaced every 24 h, as necessary. When HEK293T cells were treated with protease inhibitors and proteasome inhibitors and transfected with plasmids, the inhibitors were added at seeding. Before transfection, media was discarded and replaced with fresh media without inhibitors. 4 h after transfection culture media were replaced with fresh media with inhibitors. The media with inhibitors were replaced every 24 h as necessary.
Glycosidase Reactions
Peptide N-glycosidase F and acetylglucosaminidase H were from New England Biolabs (Ipswich, MA). Enzymatic reactions were performed with whole cell lysates under denaturing conditions according to manufacturer's instructions. They were then analyzed by Western blotting.
Biotinylation of Cell-surface Proteins
EZ-link® Sulfo-NHS-Biotin from Thermo Scientific was used to label proteins on the cell surface as instructed by the manufacturer. Briefly, HEK293T cells transfected to express TLR3 for 36 h were washed three times with PBS, and suspended in 10 mm Sulfo-NHS-Biotin dissolved in distilled water to allow labeling at room temperature for 30 min. The cells were then washed once with 50 mm Tris-Cl, pH 8.0 to quench additional labeling reactions, then washed three times with PBS, pH 8.0. Cells were then lysed and immunoprecipitated with Streptavidin-agarose beads (Sigma Aldrich, catalog #85881) preblocked with 1% BSA in PBS. The precipitated proteins were separated on denaturing SDS-PAGE and detected by Western blots.
Poly(I:C) Binding Assay
Biotinylated poly(I:C) was prepared in the dark as follows. Poly(I:C) was dissolved in a sodium acetate buffer, pH 5.5, at 2 mg/ml. This solution was diluted with an equal volume of 20 mm sodium periodate and incubated on ice for 1 h. Free sodium periodate was removed by use of a Microcon concentrator (Millipore Inc., 100-kDa cutoff) with 3 washes at 4 °C with 20 mm sodium acetate, pH 5.5. Biotin hydrazide at 5 mm (dissolved in DMSO) was added to the mixture and incubated for 2 h at room temperature. Free biotin hydrazide was removed with a Microcon concentrator as described above. One volume of 0.2 m sodium borohydride and 2 volumes of 1 m Tris-Cl, pH 8.2, were added, and the reaction was incubated on ice for 30 min. The products were purified by precipitation with two volumes of ethanol, suspended in water, and quantified by spectrophotometry. Biotinylation of poly(I:C) was confirmed by probing the RNA with streptavidin-HRP and the ECL-plusTM Western blot detection system. Cells expressing TLR3 were lysed in buffer L (50 mm Tris-Cl, 150 mm NaCl, 5 mm EDTA, and 1% Nonidet P-40, pH 5.5), clarified by a 5000 × g centrifugation for 5 min, and incubated with 2.5 ng/ml of biotinylated poly(I:C) or unlabeled poly(I:C) at 2.5 ng/ml at 37 °C for 1 h, then precipitated with streptavidin-agarose beads that had been pre-blocked with PBS amended with 1% (W/V) of BSA at 4 °C for 3 h and washed 3 times with ice-cold PBS. The precipitates were analyzed for TLR3 with Western blots. These cells were not treated with endosome inhibitors.
Pulse-chase and Cycloheximide Chase
Pulse-chase experiments used HEK293T cells transfected with plasmids at a confluency of ∼85%. After incubation in complete medium for 16 h, the cells were switched to a pulse medium consisting of DMEM medium lacking methionine and cysteine and supplemented with 10% dialyzed FBS. The pulse medium was then removed and replaced with the pulse medium amended with 150 μCi/ml EasyTagTM Expre35S35S Protein Labeling Mix (PerkinElmer Life Sciences). After 30 min of incubation with constant mixing at 37 °C, the cells were fed a complete medium supplemented with 10 mg/liter each of methionine and cysteine. Individually prepared wells of cells were harvested at specified times after the start of the chase reaction and lysed and immunoprecipitated with anti-HA (Abcam) antibody. The precipitates were separated on 4–12% denaturing Bis-Tris gel (Invitrogen) and imaged by autoradiography.
The cyclohexamide (CHX)-chase assays used HEK293T cells grown to ∼85% confluency in a 6-well plate and transfected with 2.5 μg of plasmids expressing the desired protein. 16 h later cells were resuspended in media containing 50 μg/ml CHX (Sigma, catalog #C4859) and seeded in 24-well plates. The cells were harvested at specified times after the start of the chase reaction, lysed, and analyzed by Western blots.
Confocal Microscopy
HEK-293T cells were grown on poly-l-lysine-coated coverslips to 60% confluency. The cells were transfected with pUNO-TLR3 or pUNO-TLR3 ΔL1 complexed to Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). 24 h after transfection, the cells were fixed with 4% paraformaldehyde for 15 min at room temperature and permeabilized with T buffer (0.5% Triton X-100 in PBS with 1% normal goat serum) for 30 min on ice. The cells were then blocked with 2% BSA in TBS-T (Tris-buffered saline, pH 7.4 with 0.5% Triton X-100). Incubation with primary antibodies was at 4 °C for overnight in TBS-T containing 2% BSA. After 3 washes with TBS-T, cells were incubated with secondary antibodies for 1 h at room temperature and then washed three times with TBS-T. Nuclei were stained for 30 min at room temperature with a 1:1000 dilution of the DNA-specific dye DRAQ5TM (Abcam). The coverslips were mounted on glass slides with Gold anti-fade mounting medium (Invitrogen) and dried overnight in the dark. The micrographs were acquired with a Leica TCS SP5 confocal inverted-base microscope with a 63× oil objective at Indiana University Light Microscopy Imaging Center. Images were analyzed by Leica LAS AF and Image J software. Colocalization of fluorophores was quantified using the ImageJ plug-in tool JACoP (Bolte and Cordelieres (50)).
Primary antibodies include goat anti-TLR3 ECD (R&D Systems), rabbit anti-Rab5, Rab7, Rab11 (Cell Signaling Inc.), mouse anti-EEA1 (BD Biosciences), and mouse anti-LAMP1 (Santa Cruz Biotechnology). Fluorescence-conjugated secondary antibodies include anti-rabbit Alexa 488, anti-goat Alexa 594, and anti-mouse Alexa 488 (Invitrogen).
Endosome Fractionation
Endosomal fractionation used a 0–20% continuous iodoxanol gradient (Optiprep, Sigma) prepared in a Beckman 15 ml centrifuge tube using a BioComp Gradient Master (BioComp Instruments, Inc.). HEK293T cells were grown on 10-cm culture plates and transfected with TLR3-HA plasmid. 24 h after transfection cells were lysed in 500 μl of homogenization buffer (250 mm sucrose, 1 mm EDTA, 10 mm triethanolamine, pH 7.8, with protease inhibitors) and homogenized by passing the cells through a 25-gauge needle 20 times. Cell lysis was confirmed by microscopy. The homogenate was centrifuged at 1000 × g for 10 min at 4 °C. Post-nuclear supernatant was loaded on top of the iodoxanol gradient and centrifuged at 200,000 × g for 2 h at 4 °C in SW45 rotor (Beckman Instruments). Twenty fractions were collected, and the proteins were precipitated using trichloroacetic acid at a final concentration of 25% (w/v). Pellets were washed twice with cold acetone and dissolved in either PBS for immunoprecipitation or 2× SDS-PAGE Laemmli loading buffer for SDS-PAGE and Western blots.
RESULTS
Unc93b1 Facilitates Proteolytic Cleavage of TLR3
We seek to first establish that Unc93b1 can affect TLR3 proteolytic processing. TLR3 with a C-terminal epitope tag named TLR3-HA was made to facilitate detection of processed fragments (Fig. 1A). TLR3-HA induced production of luciferase reporter from the ISRE promoter at 60% of unmodified TLR3 in the presence of the TLR3 agonist, poly(I:C), demonstrating that it is functional (supplemental Fig 1A). In HEK293T cells that overexpressed WT Unc93b1, Western blots to detect the HA tag identified two bands, one with a molecular mass comparable with full-length TLR3 and an ∼80-kDa band that is a CTF generated from full-length TLR3 (Fig. 1B). Unc93b1 mutant 3d, which resulted in defective TLR3 signaling (21), failed to facilitate production of the CTF (Fig. 1B). The CTF was present in much lower abundance in the absence of overexpressed Unc93b1 (Fig. 1B and see below). Furthermore, Unc93b1 overexpression increased the abundance of full-length (FL) TLR3 as well as resulted in a higher mobility form of TLR3 (identified with an asterisk, Fig. 1A) that is suggestive of altered post-translational modification. These results show that Unc93b1 can affect the proteolytic processing of TLR3.
FIGURE 1.
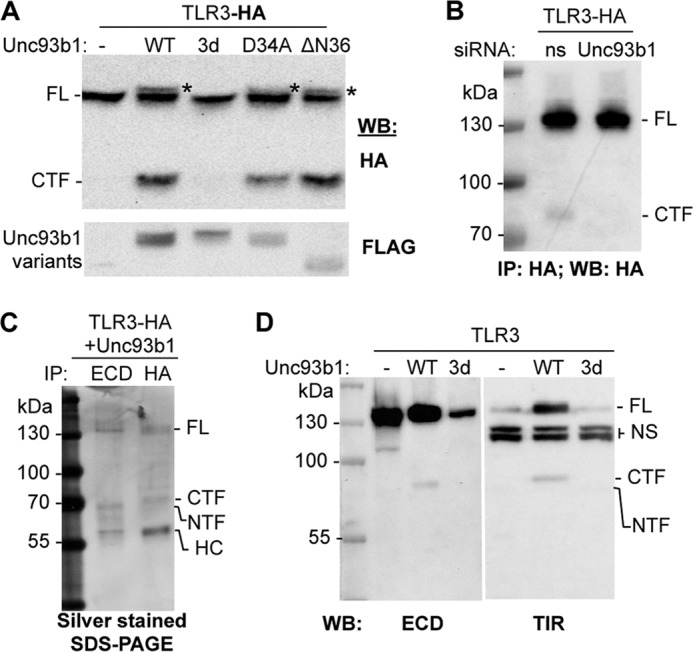
Unc93b1 promotes the proteolytic processing of TLR3 in HEK293T cells. A, shown are the effects of coexpressed Unc93b1 on proteolytic processing of TLR3. HEK293T cells were transfected to coexpress the TLR3-HA and either WT or mutant Unc93b1. All Unc93b1 contained the Myc and FLAG tag. The upper image was from a Western blot (WB) probed to detect the HA tag added to the C terminus of TLR3. The bottom image shows the abundance of the Unc93b1 proteins. The asterisks identify a higher migrating band present with Unc93b1 overexpression. B, shown are the effects of siRNA knockdown of the endogenously expressed Unc93b1 on the proteolytic cleavage of TLR3-HA. siRNA specific to Unc93b1 or a nonspecific target (ns) were transfected at 50 nm to cells expressing TLR3-HA for 72 h. The Western blot (WB) was probed to detect the HA tag as indicated in bold below the WB image. Knockdown of Unc93b1 RNA is shown in supplemental Fig 1B. IP, immunoprecipitate. C, detection of the TLR3 CTF and NTF by silver-stained SDS-PAGE is shown. The samples were immunoprecipitated from HEK293T cells that coexpressed TLR3-HA and Unc93b1. HC, heavy chain of the antibody used to immunoprecipitate TLR3. D, shown is detection of NTF and CTF in Western blots. The gel images contained Western blots of cell lysates coexpressing WT TLR3 with the empty vector (−), WT Unc93b1 (WT), and the 3d mutant (3d). The antibodies used detected epitopes present in the TLR3 ECD or the TIR domain. Nonspecific bands detected by the Abs are labeled as NS. These results were repeated twice with the same results.
To establish whether the requirements for Unc93b1-mediated TLR3 processing are related to the Unc93b1 effect on TLR7 and TLR9, we tested two mutations that differentially regulated TLR7 and TLR9 signaling and localization (22). A substitution mutant D34A and a deletion of the Unc93b1 N-terminal 36 residues (ΔN36) both resulted in the production of the CTF and the form of TLR3 with altered post-translational modification (Fig. 1B).
To determine whether proteolytic processing of TLR3 occurs in the absence of Unc93b1 overexpression, lysates from HEK293T cells transfected to express TLR3-HA were first immunoprecipitated with the antibody that recognizes the HA tag followed by a Western blot of the precipitated material. The CTF was detected in these experiments, although its abundance was low relative to the FL TLR3, suggesting that only a small portion of TLR3 was normally processed (Fig. 1C). Furthermore, siRNA knockdown of the endogenous Unc93b1 mRNA reduced the amount of TLR3 CTF relative to the amount of the full-length TLR3 (Fig. 1C). The reduction in Unc93b1 message was confirmed by quantitative RT-PCR (supplemental Fig. 1B).
We sought to identify the N-terminal fragment of the proteolyzed TLR3. Cells coexpressing TLR3-HA and Unc93b1 were subjected to immunoprecipitation with antibody recognizing TLR3 ectodomain (ECD) or HA tag. An ∼65-kDa fragment, which we will name the N-terminal fragment (NTF), was present at comparable abundance to the ∼80-kDa CTF in silver-stained SDS-PAGE of the precipitated proteins (Fig. 1D). Both the NTF and CTF were also produced by a version of TLR3 that lacks an epitope tag coexpressed with Unc93b1 in Western blots probed with the antibodies that recognized the ECD and the TIR domain. The levels of processed proteins were low relative to the detection of the HA-tagged CTF probably due to the lower affinities of the antibodies toward fragments of TLR3 (Fig. 1E). In summary, the processing of TLR3 requires Unc93b1 and leaves detectable N- and C-terminal fragments of TLR3.
TLR3 Proteolytic Cleavage Requires Glycosylation That Takes Place in the Golgi
A more accurate prediction of the proteolytic processing site(s) requires measurement of the masses of the TLR3 NTF and CTF. Because the TLR3 ECD is modified with ∼15 N-linked glycans, we treated the samples with peptide N-glycosidase F to remove N-linked glycans (24) and observed that the fragments migrated at ∼40 and ∼63 kDa, respectively, on the SDS-PAGE (Fig. 2A). The fact that the two fragments add up to the predicted mass of unglycosylated full-length TLR3 (104 kDa; Fig. 2A) suggests that the proteolytic processing was at either a discrete site in TLR3 or a series of adjacent sites. Furthermore, the estimated molecular weights of the two fragments place the cleavage site around the middle of the ECD.
FIGURE 2.
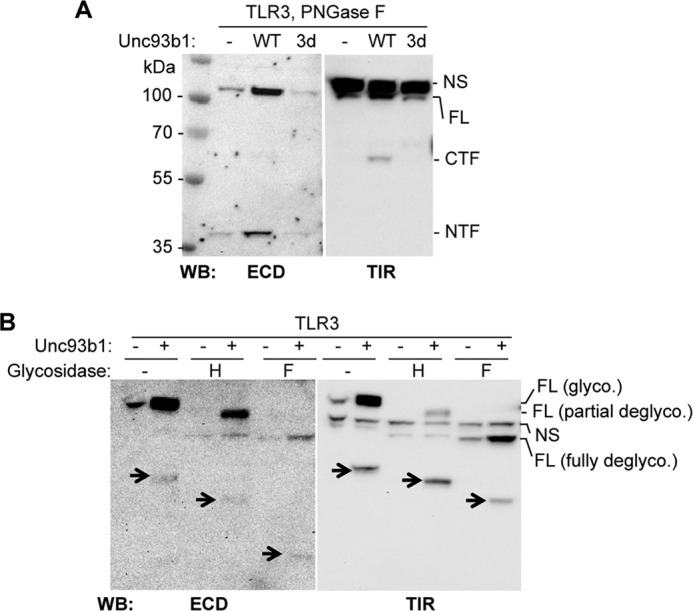
Glycosylation of TLR3 and its proteolyzed fragments. A, masses of the NTF and CTF after treatment with peptide N-glycosidase F (PNGase F) to remove N-linked glycans are shown. The NTF and CTF were detected with antibodies to ECD and TIR. NS identifies a nonspecific band that migrated above the FL TLR3. WB, Western blot. NS, nonspecific band. B, complex N-linked glycans on full-length TLR3 and the two proteolytic fragments are shown. Lysates of cells expressing TLR3 with or without overexpressed Unc93b1 were treated with acetylglucosaminidase H (H) or peptide N-glycosidase F (F). The arrows identify the NTF and CTF, as they change in electrophoretic mobilities as a result of deglycosylation. These experiments were repeated twice with the same results.
We sought to better understand the differently modified form of TLR3 that was evident upon coexpression with Unc93b1 (bands identified with asterisks in Fig. 1A). Lysates containing TLR3 and its proteolyzed fragments were treated with acetylglucosaminidase H, which cleaves high-mannose and hybrid glycans that are attached to proteins as they transit through the endoplasmic reticulum but not the complex glycans that are added in the Golgi complex (24, 25). The full-length TLR3 could be separated by SDS-PAGE to two species after acetylglucosaminidase H treatment, one that is partially deglycosylated and one fully deglycosylated, suggesting that only a fraction of TLR3 passed through the Golgi (Fig. 2B). However, treatment with acetylglucosaminidase H partially deglycosylated the NTF and CTF, suggesting that they contain both the high-mannose glycan and the hybrid glycans indicative of modification in the Golgi (Fig. 2B).
The TLR3 Cleavage Site Is Within or Adjacent to Loop1
The estimated molecular masses of the deglycosylated NTF and CTF suggest that the cleavage site is located in LRR12. LRR12 contains Loop1 (amino acids 336–343), which negatively regulates the secretion of the TLR3 ectodomain (23, 26, 27) (Fig. 3C). To examine this further, a truncation of TLR3 that lacked LRR1 through LRR12 (ΔL1–12) but retained the signal peptide and N-terminal cap of the ectodomain needed for proper localization was tested and found to migrate at a location slightly lower than the CTF (Fig. 3, A and B). Another truncation, 344-C, which lacks LRR1 through residue 343, including Loop1 (Fig. 3, A and D), had nearly identical electrophoretic mobility as the CTF (Fig. 3, A and B). To examine whether Loop1 was required for cleavage, a construct with a precise deletion of Loop1 (ΔL1) was tested and found not to produce either the NTF or the CTF (Fig. 3B). Δ64, a natural isoform of TLR3 that is expressed in human astrocytes and lacks 64 amino acids, including Loop1, also did not produce the NTF or CTF (supplemental Fig 2). These results demonstrate that the eight-amino acid sequence of Loop1 is required for the proteolytic processing of TLR3.
FIGURE 3.

Loop1 is required for proteolytic processing of TLR3. A, truncation of LRR1 to the end of Loop1 from TLR3-HA results in a molecule similar in size to the CTF generated by proteolytic cleavage. The Western blot (WB) images were from lysates of HEK293T cells transfected to express WT or truncated TLR3-HA in the absence and presence of Unc93b1, as indicated above the gel images. ΔL1–12-HA, deletion of LRR 1–12 from TLR3-HA; 344-C-HA, deletion of LRR1 up to and including 343 in LRR12 from TLR3-HA. B, shown is the effects of deleting Loop1 on TLR3 proteolytic processing in HEK293T cells. ΔL1 lacks residues 336–343 that contain Loop1. The Western blot images were probed to detect the TLR3 ECD or TIR. All images in each panel were from the same exposure of the same blot, but other samples unrelated to the results were cropped. These experiments were repeated twice, with the same results. C and D, the effect of TLR3 with mutations in or near Loop1 was on the accumulation of CTF. The presence of Unc93b1 is shown by the + symbol. The amount of CTF produced from each mutant shown on the Western blots was quantified and normalized to FL TLR3. The percentage accumulation of the CTF relative to that of wild-type TLR3 (%CTF) was shown as %CTF accumulation. L1/FLAG and L1/AU1 also contained a C-terminal HA tag, and their presence was detected the 3F10 antibody. These experiments were repeated once with reproducible results.
We mutated the residues in Loop1 in an attempt to identify the cognate residue(s) for cleavage. Five mutants that had single or multiple amino acid substitutions in Loop1 (Q336A, L337A, S340A, L342A, 4 m) all produced the CTF at quantities that are 69% or higher relative to the level from WT TLR3 (Fig. 3C). TLR9 is also cleaved by asparagine endopeptidase (12, 13, 15). Therefore, we mutated the two asparagines directly upstream of Loop1 to asparagines (Asn-323 and Asn-328). These two mutants generated the CTF at levels equal to or better than WT TLR3 (Fig. 3C). These observations suggest that TLR3 proteolytic processing does not depend on the identities of specific residues in and near Loop1. Furthermore, we do not have evidence to implicate asparagine endopeptidases in TLR3 proteolytic processing.
CTF production was examined after insertion of additional residues into Loop1 or the replacement of Loop1 with unrelated sequences. A six-residue insertion named TCM that resulted in a 14-residue loop produced CTF at 164% of the WT (Fig. 3C). Replacement of the Loop1 residues with the FLAG epitope (8 residues) or with the AU1 epitope (6 residues) resulted in CTF accumulation at, respectively, 39 and 7% that of the WT (Fig. 3, C and D). All of the mutational analysis of Loop1 showed that there is no specific sequence in Loop1 required for proteolytic cleavage. However, the length of the loop could affect efficiency of cleavage.
The Proteolytic Processing of TLR3 Takes Place in Acidic Compartments by Cathepsins
Proteolytic cleavage of TLR9 takes place in endosomes (11, 12, 15). To examine whether TLR3 processing required acidic endosomes, we treated HEK293T cells expressing TLR3 and Unc93b1 with bafilomycin A1, an inhibitor for endosome acidification, or z-FA-FMK, which inhibits cathepsins B, L, and S. CTF accumulations were significantly reduced by treatment with both bafilomycin and z-FA-FMK (Fig. 4, A and B). Comparable inhibition of TLR3-HA processing was observed in Huh7.5 cells treated with z-FA-FMK (Fig. 4C). z-FA-FMK also inhibited CTF accumulation, resulting in an increased abundance of full-length TLR3 in HEK293T cells that expressed only the endogenous Unc93b1 (Fig. 4D). These results demonstrate that proteolytic processing of TLR3 in cultured human cells requires the acidification of intracellular compartments, likely endosomes, and some combination of cathepsins B, L, and/or S. Inhibitors of asparagine endopeptidase were not tested because they are not commercially available.
FIGURE 4.
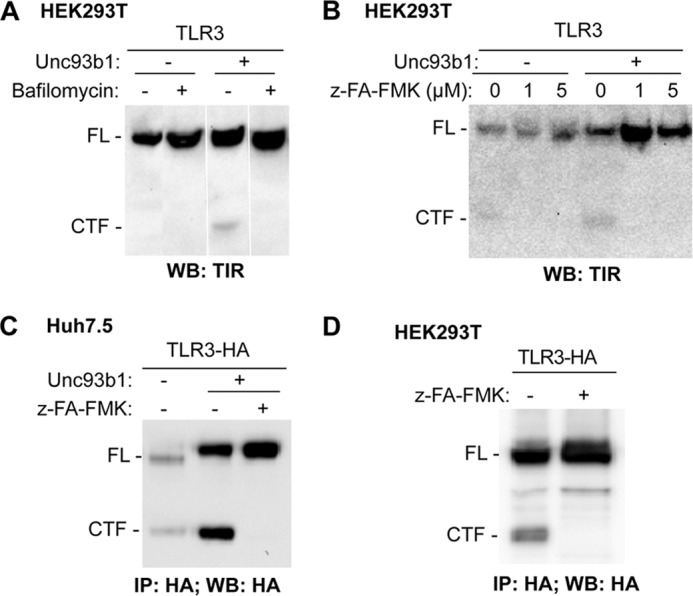
Cathepsin and endosome acidification inhibitors can inhibit TLR3 proteolytic processing. A, shown are Western blot (WB) results of TLR3 expressed in HEK293T cells mock-treated or treated with bafilomycin at a final concentration of 2.5 nm. The cotransfection of a plasmid expressing Unc93b1 is denoted with a + symbol. All of the images were from the same experiment and the same Western blot. However, unrelated results were removed. B, the cathepsin inhibitor z-FA-FMK can inhibit CTF accumulation while increasing the abundance of the full-length TLR3 in transiently transfected HEK293T cells. C, z-FA-FMK can inhibit TLR3 proteolytic processing in transiently transfected Huh7.5 cells. Transfection efficiency of Huh7.5 cells is lower than HEK293T cells; thus, TLR3-HA was immunoprecipitated (IP) to increase sensitivity of detection. D, z-FA-FMK can inhibit TLR3 proteolytic processing in HEK293T cells with endogenous level Unc93b1. Cell lysates were immunoprecipitated with anti-HA antibody and detected by Western blot for HA epitope. These experiments were repeated three times with the same results.
Proteolytic Processing Is Not Required for TLR3 Signaling
The current model for TLR9 signaling is that its cleaved CTF is the mature and active form of the receptor (11, 12, 15). Given that the D34A and ΔN36 mutants of Unc93b1 had different effects on TLR3 signaling when compared with the effect on TLR9 (22), we examined whether proteolytic processing was required for TLR3 signaling. In transiently transfected HEK293T cells, bafilomycin treatment reduced signaling by TLR3 and TLR9 but had minimal effects on TLR4 signaling (Fig. 5A, supplemental Table 1). These results are consistent with the known requirements of endosomal acidification and Unc93b1 for TLR3 and TLR9 signaling but not TLR4 (8). Treatment of HEK293T cells with z-FA-FMK resulted in reduced TLR9 signaling induced by ODN2006 (Fig. 5A, supplemental Table 1) but did not significantly affect LPS-induced TLR4 signaling. Strikingly, TLR3-dependent signaling induced by poly(I:C) was unaffected by z-FA-FMK (Fig. 5A). In Huh7.5 cells, TLR9 signaling was also reduced in the presence of z-FA-FMK, whereas the effect on TLR3 was modest and not statistically different from the control treatments (supplemental Table 1). The human lung epithelial BEAS-2B cells express endogenous TLR3 and can secrete IL6 when induced with poly(I:C) (28, 29). Poly(I:C)-activated BEAS-2B cells treated with z-FA-FMK produced IL6 at levels comparable with the control cells, whereas treatment with bafilomycin reduced IL6 production significantly (Fig. 5B, supplemental Table 1). BEAS-2B cells do not respond to TLR9 agonists (31); hence, the effects of the inhibitors on TLR9 signaling could not be examined. Nonetheless, all of these results suggest that cathepsin processing is not required for signaling by TLR3.
FIGURE 5.

Proteolysis of TLR3 is not required for TLR3 signaling. A, shown are the effects of cathepsin inhibitor z-FA-FMK on luciferase reporter levels induced by TLR3, -4, and -9 ligands in transiently transfected HEK293T cells. Where added, bafilomycin (BAF) and z-FA-FMK were dissolved in DMSO at final concentrations of 2.5 nm and 5 μm, respectively. The final concentration of DMSO was 0.5%. The ratio of the firefly luciferase reporter driven from an ISRE (TLR3 in HEK293T cells) or NF-κB (TLR4 and -9 in HEK293T cells, and TLR3, -4, and -9 in Huh7.5 cells) promoter was normalized to the Renilla luciferase expression. -Fold induction was calculated as firefly/Renilla ratio of ligand-induced cells over the ratio of mock-treated cells. B, IL6 production by human lung epithelial BEAS-2B cells induced by poly(I:C) (0.125 ng/ml) in response to inhibitors is shown. IL6 concentration was determined by ELISA. C, shown are the effects of inhibitors on IL6 production by mouse macrophage RAW264.7 cells in response to ligands for different Toll-like receptors. Poly(I:C) (50 ng/ml), LPS (10 ng/ml), gardiquimod (10 ng/μl), and ODN1826 (1 μm) are ligands, respectively, for TLR3, TLR4, TLR7, and TLR9. D, -fold induction of luciferase reporters of TLR3 mutants ΔL1 and Δ64 relative to WT TLR3 in HEK293T cells. E, -fold induction of luciferase reporters of TLR3 mutant ΔL1 compared with WT TLR3 in Huh7.5 cells is shown. F, relative -fold induction of luciferase reporters of TLR3 mutants with C-terminal HA tag and WT TLR3-HA in HEK293T cells is shown. In all graphs the error bars show the range of S.D. from three independent assays. The graphs are representative of data from three independent experiments.
We examined whether signaling of the mouse TLR3 required proteolytic processing, whereas the human TLR3 did not. RAW264.7 cells were treated with ligands for TLR3, 4, 7, or 9. IL6 production was then quantified using ELISA. In the presence of bafilomycin, IL6 production mediated by the endosomal TLRs 3, 7, and 9 was significantly reduced, whereas signaling by the cell-surface TLR4 was not, as expected. z-FA-FMK inhibited signaling of TLR7 and TLR9, induced by gardiquimod and ODN1826, respectively, to the same degree as treatment with bafilomycin. However, z-FA-FMK did not inhibit poly(I:C)-induced IL6 production (Fig. 5C, supplemental Table 1). Thus, in mouse cells, TLR3 signaling in response to poly(I:C) in mouse cells does not require processing by cathepsins, consistent with our observations with human cells.
We examined whether mutant TLR3 constructs that are not proteolytically processed remained competent for signaling. TLR3 ΔL1 was active for signaling in poly(I:C)-induced HEK293T and Huh7.5 cells (Fig. 5, D and E). A signaling-defective mutant named Δ64 served as a negative control in this experiment (Fig. 5D). Loop1 substitutions L1/FLAG and L1/AU1, which have reduced levels of CTF (Fig. 3D), signaled to the same level as WT TLR3 (Fig. 5F). Finally, we tested the ability of ΔL1–12 and 344-C to signal in the luciferase reporter assay. Although their molecular sizes are very close to the CTF, they were both deficient in signaling in response to poly(I:C) (Fig. 5F).
Our results contradict those in a recent report by Garcia-Cattaneo et al. (30), who observed that a construct expressing the CTF of TLR3 was competent for signaling in a human retinal epithelial cell line. Therefore, we sought to determine whether the N-terminal cap of the ECD or the signal peptide in our 344-C construct inhibited signaling of the fragment. We deleted the N-terminal cap, resulting a construct named S-344-C, which expressed the signal peptide (amino acid 1–25), fused with amino acid 344 until the C-terminal end of TLR3. Although this truncation expressed well and the molecular size is similar to the CTF, it is deficient in signaling transduction (supplemental Fig 3). When we further replaced the signal peptide by a methionine residue to form M-344-C, the truncation was not properly accumulated and was also deficient in signaling (supplemental Fig 3). Therefore, all of our results from cells treated with cathepsin inhibitors and with various mutated TLR3 constructs consistently show that proteolytic cleavage is not required for TLR3 to signal.
Proteolytic TLR3 Fragments Are Present in Endosomes and on the Cell surface and Can Bind Poly(I:C)
We examined whether the full-length TLR3, the NTF, and the CTF can localize to endosomes and bind poly(I:C). TLR3 ectodomain preferentially binds dsRNA at acidic pH values (10). We incubated biotinylated poly(I:C) with HEK293T cells transfected to express TLR3 for 1 h. The cells were then lysed, and the biotinylated RNAs were recovered using streptavidin resin. Western blots of the recovered proteins revealed that the NTF, the CTF, and full-length TLR3 all associated with poly(I:C) (Fig. 6A). Proteolyzed TLR3 thus remains competent to bind poly(I:C).
FIGURE 6.
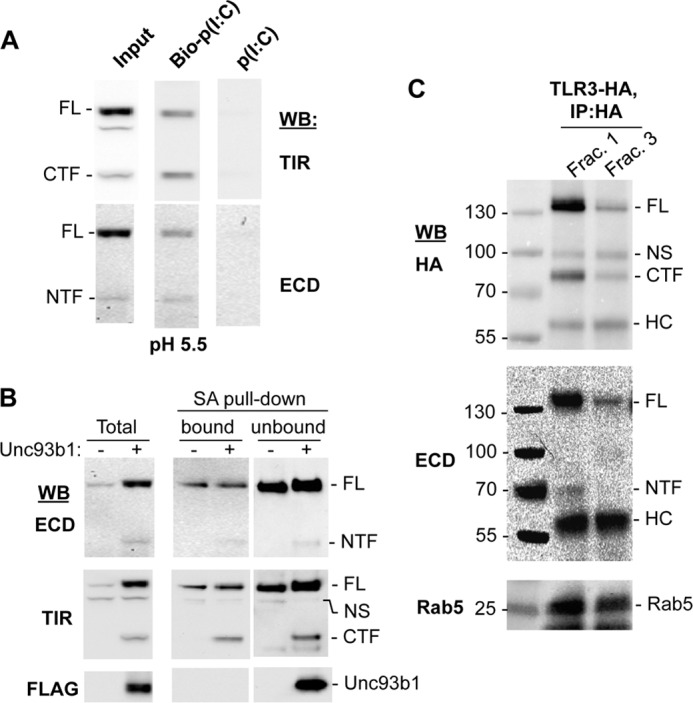
The proteolyzed TLR3 fragments are on the surface, in endosomes, and can bind poly(I:C). A, the TLR3 NTF and CTF retain the ability to bind poly(I:C). HEK293T cells transfected to express TLR3 and Unc93b1 were lysed in acidic buffer, pH 5.5. Poly(I:C) or biotinylated poly(I:C) was incubated with cell lysates and precipitated with streptavidin-agarose beads. The bound materials were detected using Western blots (WB) probed with antibodies to ECD or TIR of TLR3. B, fragments of TLR3 can be detected on the cell surface. HEK293T cells transfected to express TLR3 with or without coexpressed Unc93b1 were labeled with sulfo-NHS-Biotin, which is impermeable to cell membranes. The streptavidin-agarose beads precipitated (bound) and the remaining supernatant not bound to the beads (unbound) as well as the total cell lysate (Total) were all analyzed for TLR3 and the cleaved fragments by Western blots. C, uncleaved and cleaved TLR3 are enriched in endosomes. HEK293T cells expressing TLR3-HA were homogenized, and organelles were fractionated using an iodoxanol gradient. Fractions that are positive for the endosome marker Rab5 in Western blots (Frac. 1 and Frac. 3) were immunoprecipitated with HA antibody, and TLR3 was detected by Western blots. NS, nonspecific band; HC, heavy chain.
TLR3 can localize to both the plasma membrane and the endosome of a number of human cell lines, and localization is critical for cytokine production in epithelial and fibroblast cells (23, 32–34). To determine whether the proteolyzed TLR3 is present on the cell surface, we biotinylated the cell surface proteins, then lysed the cells and affinity-purified the biotinylated materials using streptavidin-agarose beads. The endoplasmic reticulum resident Unc93b1 was detected only in the extract that did not bind to streptavidin, indicating that it was not accessible to biotinylation. In contrast, full-length TLR3, the NTF as well as the CTF, did bind streptavidin, indicating that at least a portion of the processed TLR3 is present on the cell surface (Fig. 6B).
To determine whether proteolytically processed TLR3 is present in endosomes, cells expressing TLR3-HA were lysed, and the lysate was fractionated on an iodoxanol gradient. Immunoprecipitation with anti-HA antibody identified both the full-length TLR3 and the CTF, as would be expected (Fig. 6C, top). Notably, the NTF was also coimmunoprecipitated in this reaction, indicating that the NTF retains interaction with either TLR3 or the CTF (Fig. 6C, middle). These results indicate that proteolytically processed TLR3 remains competent for the activities associated with signaling.
TLR3 Proteolytic Processing Can Influence dsRNA Recognition in RAW264.7 Cell
Although poly(I:C) is the standard ligand to activate TLR3 signaling, it is a synthetic mimic of dsRNA, the natural ligand for TLR3 during viral infections. We sought to determine whether the proteolytic processing can affect TLR3 recognition of ligands other than poly(I:C). BEAS-2B cells and RAW264.7 cells were treated with either DMSO or z-FA-FMK then induced with poly(A:U) or dsRNA extracted from Reovirus virions (Reo-ds) (Fig. 7A) (28, 35). Treatment with z-FA-FMK did not affect poly(I:C)-induced signaling, consistent with our previous observations. However, z-FA-FMK caused a small but reproducible increase in IL6 production by BEAS-2B cells treated with polyI(A:U) and Reo-ds. The antimicrobial peptide LL37 can enhance viral dsRNA-induced signaling by TLR3 in BEAS-2B cells (28). IL6 production increased dramatically with the combination of Reo-ds and LL37. However, inhibition of cathepsins by z-FA-FMK did not differentially increase IL6 production (Fig. 7A).
FIGURE 7.

Proteolytic processing can affect TLR3 signaling to some dsRNAs. A, BEAS-2B cells treated with DMSO or z-FA-FMK (5 μm final concentration) were induced with ligands for 12 h, and the amount of IL6 secreted into the media was quantified by ELISA. TLR3 ligands include poly(I:C) (0.125 ng/ml, p(I:C)), poly(A:U) (0.3 ng/ml, p(A:U)), Reovirus dsRNA (1.25 ng/ml, Reo-ds). When added, LL37 was used at 3 μm. B, RAW264.7 cells were treated with DMSO or z-FA-FMK (5 μm) and induced with ligands for 12 h before culture media were collected and the secreted IL6 quantified by ELISA. The ligands and their concentrations used were p(I:C) (50 ng/ml), p(A:U) (5 ng/ml), Reo-ds (25 ng/ml), and ODN1826 (1 μm). In these experiments, the error bars are S.D. of three independent treatments. Graphs are representative of two repeats.
The murine RAW264.7 macrophage cells require higher concentrations of poly(I:C) to induce TLR3, and the resulting IL6 levels were lower than those induced by poly(A:U) or ODN1826, a mouse TLR9 agonist (Figs. 5C and 7B). In RAW264.7 cells, treatment with z-FA-FMK reduced signaling by TLR9, consistent with the results of Ewald et al. (15). With the TLR3 ligands, response to Reo-ds was significantly enhanced by z-FA-FMK. Unexpectedly, poly(A:U)-induced IL6 production was decreased by z-FA-FMK (Fig. 7B). These results suggest that the response to dsRNAs can differ between human and murine TLR3s. Knockdown of TLR3 with siRNAs resulted in decreased IL6 production in response to poly(I:C), poly(A:U), and Reo-ds, but not ODN1826, confirming the effects were mediated by TLR3 (supplemental Fig 2). Furthermore, proteolytic cleavage of TLR3 in human and murine cell lines could affect ligand recognition and signaling by TLR3.
Ubiquitination of TLR3
Because proteolytic processing had only a modest effect on ligand recognition by TLR3 in human cells, we sought to determine whether it would affect TLR3 turnover given that Unc93b1 increased both proteolytic processing and TLR3 accumulation (Fig. 1B). Because TLR3 turnover is not well understood, we first determined whether it could be ubiquitinated by coexpressing it with an epitope-tagged ubiquitin, ubiquitin-AU1. In the absence of overexpressed Unc93b1, the accumulation of full-length TLR3 decreased to ∼10% that in cells expressing ubiquitin-AU1 (Fig. 8A). Immunoprecipitation of TLR3-HA with an HA antibody and subsequent Western blotting to detect ubiquitin-AU1 showed that TLR3 migrated as a broad smear that ranged from ∼70 kDa to more than 250 kDa, suggesting that not only is TLR3 multiply ubiquitinated but that some of the ubiquitinated TLR3 has been proteolyzed (Fig. 8A). Upon the coexpression of both Unc93b1 and ubiquitin-AU1, the accumulation of the CTF was reduced by ∼30–50% relative to the FL TLR3 in multiple experiments, indicating that the CTF can be ubiquitinated (Fig. 8A and data not shown). The complementary experiment to immunoprecipitate ubiquitin-AU1 followed by Western blot detection of TLR3 confirmed that coexpression of Unc93b1 and TLR3 reduced TLR3 ubiquitination in the presence of ubiquitin-AU1. These results indicate that Unc93b1 protects TLR3 from ubiquitination and possibly degradation (Fig. 8A).
FIGURE 8.
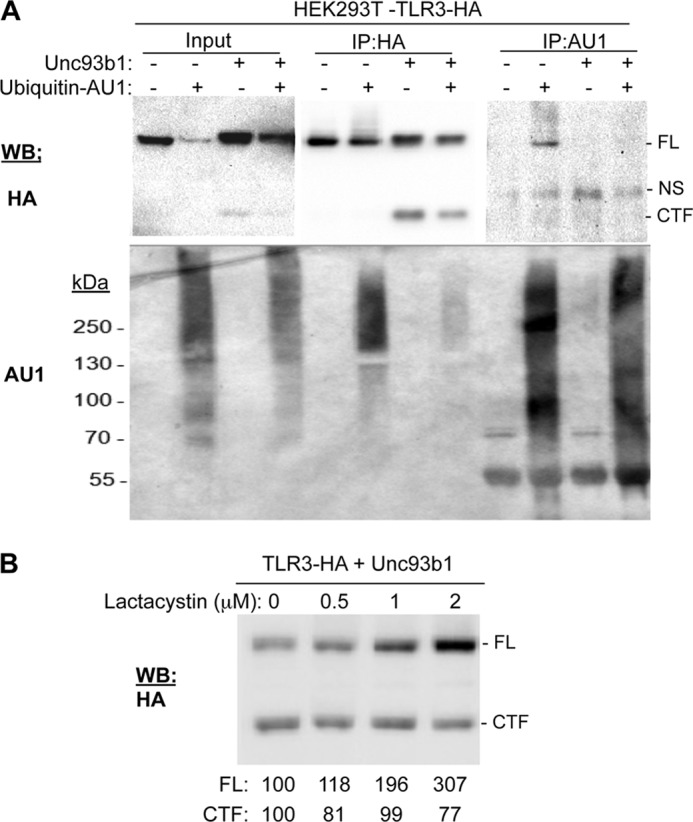
Full-length TLR3 is ubiquitinated and degraded by proteasomes. A, ubiquitination of TLR3 is shown. HEK293T cells transfected to express TLR3-HA with or without exogenous Unc93b1 and ubiquitin-AU1 as indicated were lysed and immunoprecipitated (IP) using either anti-HA antibody or anti-AU1 antibody in the presence of 1 mm de-ubiquitinase inhibitor, N-ethylmaleimide. NS, nonspecific band. TLR3-HA and Ubiquitin-AU1 were detected by Western blot (WB). B, HEK293T cells expressing TLR3-HA and Unc93b1 were treated with lactacystin for 48 h after transfection at indicated concentrations. Quantifications of TLR3 FL and CTF by Western blot were normalized to the control treatment, and the percentages are shown below the blot image. These experiments were repeated three times with highly similar results.
To address whether full-length TLR3 and cleaved products are degraded in the proteasomes, cells coexpressing TLR3-HA and Unc93b1 were treated with the proteasome-specific inhibitor lactacystin (Fig. 8B). Lactacystin increased FL TLR3 accumulation in a concentration-dependent manner by up to 3-fold, indicating that TLR3 turnover likely involves proteasomes. Intriguingly, unlike the FL TLR3, the amount of CTF did not increase along with lactacystin (Fig. 8B). These results suggest that the full-length TLR3 and the proteolytically processed TLR3 could have different fates.
The Half-lives for TLR3 and the Proteolytic Fragments
We seek to determine whether TLR3 and the CTF have different half-lives. A pulse-chase experiment with 35S-labeled proteins showed that the FL TLR3 had a half-life of ∼3 h in the absence of overexpressed Unc93b1 (Fig. 9, A and B). The coexpression of Unc93b1 increased the half-life of FL TLR3 to ∼7 h. The accumulation of the CTF in the presence of Unc93b1 increased over time, reaching a maximum at ∼7 h, and did not decrease even by 24 h (Fig. 9, A and B). The half-life of the CTF is thus in excess of 24 h. These results show that Unc93b1 can protect TLR3 from degradation. Furthermore, proteolytic processing of TLR3 will result in cleaved fragments of TLR3 that are more stable than the full-length TLR3.
FIGURE 9.

The half-lives of full-length TLR3 and CTF. A, shown is the half-life of TLR3 in radiolabeled pulse-chase assays. HEK293T cells expressing TLR3-HA with or without Unc93b1 were pulse-labeled with [35S]methionine and [35S]cysteine and then harvested after a chase period at the times in hours indicated. TLR3 was identified by immunoprecipitation (IP) with an antibody to HA tag, SDS-PAGE, and autoradiography. The signals in the autoradiogram were quantified by use of phosphorimaging. Three independent experiments yielded comparable results. B, shown is a graph quantifying the results from A. C, determination of the half-lives of TLR3, NTF, and CTF by a CHX-chase assay is shown. HEK293T cells expressing TLR3-HA with or without Unc93b1 were treated with 50 μg/ml CHX for the period of time indicated above the Western blot (WB) image. TLR3 or truncated forms of TLR3 were identified by the presence of the HA tag or an antibody that recognized the TLR3 ECD. In the blots probed to detect the TLR3 ECD, the middle panel was not treated with peptide N-glycosidase F and appeared heterogeneous due to N-linked glycans. The bottom-most panel contains the NTF treated with peptide N-glycosidase F (PNGase F) to better resolve the NTF and allowed its quantification. D, a graph quantifying the results from the CHX-chase experiment is shown. These experiments were repeated twice, and the half-lives of the TLR3 and CTF were comparable with the values shown.
A CHX-chase experiment was also used to independently examine the half-lives of TLR3-HA and its proteolyzed fragments (36). Prolonged incubation with CHX is toxic to cells; therefore, the chase was performed for only 7 h. However, because the CHX-chase does not require immunoprecipitation of the proteins before detection by Western blots, the signals for the various proteins are stronger. WT TLR3 had a half-life of ∼3 h using the CHX-chase protocol, consistent with the results from the pulse-chase experiment. The coexpression of Unc93b1 increased the half-life to ∼6 h. Notably, the CTF and NTF reached maximum accumulation at ∼4 h and remained robust up to 7 h (Fig. 9, C and D). Although the half-lives differed slightly, the pulse-chase and the CHX-chase experiments demonstrated that once processed, the proteolytic fragments generated from TLR3 have higher stabilities than the full-length TLR3.
Proteolytic Processing Affects Localization of TLR3 in Subsets of Endosomes
To further characterize the basis for the differential stabilities of TLR3 and proteolytically processed TLR3, we quantified the accumulation of TLR3 in subsets of endosomes with and without z-FA-FMK treatment using confocal fluorescence microscopy. We distinguished subsets of endosomes by markers that specify subpopulations that had been recently formed from the plasma membrane (EEA1 and Rab5) or matured endosomes (Rab7) or those that are destined to recycle to the plasma membrane (Rab11) or to fusion with lysosomes (LAMP1) (37–39). TLR3 in cells not treated with z-FA-FMK was more abundant in early endosomes marked with EEA1 or Rab5. TLR3 was present in lower abundance in endosomes marked with Rab7, Rab11, or LAMP1. With z-FA-FMK treatment to inhibit proteolytic processing, the abundance of TLR3 in endosomes marked with Rab7 and LAMP1 was significantly increased (Fig. 10A). Quantification of TLR3 colocalization with the various endosomal markers showed that z-FA-FMK treatment resulted in a significant shift of TLR3 from early endosomes to those that are marked with Rab7 and LAMP1 (Fig. 10B).
FIGURE 10.

Localization of TLR3 into endosomes with and without inhibition of proteolytic cleavage. A, confocal microscopy detecting colocalization of WT TLR3 or ΔL1 in HEK293T cells with different endosomes identified using antibodies to the marker proteins are listed on the left of the micrographs. z-FA-FMK was used at 5 μm. Endosomal markers are represented as pseudocolor green, TLR3 or ΔL1 are represented in red, and the nucleus is in blue. Locations where TLR3 colocalized with an endosomal marker are in yellow. B, quantification of the colocalization of WT TLR3 or ΔL1 with endosomal marker proteins is shown. Each bar shows the mean of at least three independent experiments, each of which contains a minimum of 25 cells for each treatment whose fluorescent spots were quantified using the Image J software plug-in colocalization tools JACoP (50). For cells transfected to express WT TLR3, groups that were mock-treated or treated with z-FA-FMK were compared. Cells expressing ΔL1 were compared with those expressing WT TLR3 in the absence of inhibitors. The p values were calculated using the Student's t test.
We also compared the localization of the WT TLR3 with the ΔL1 mutant that is deficient for CTF production but competent for signaling. ΔL1 also showed decreased colocalization with EEA1 and Rab5 in comparison to WT TLR3. In addition, even without z-FA-FMK treatment, ΔL1 preferentially accumulated in endosomes that were marked with Rab7 and LAMP1 (Fig. 10). These results suggest that proteolytic processing affects TLR3 trafficking to functionally distinct endosomes.
DISCUSSION
In this study we documented that the human TLR3 is proteolytically processed in HEK293T and Huh7.5 cells within or near Loop1 in LRR12 of the TLR3 ectodomain, leaving two fragments. Cleavage was increased by the coexpression of the Unc93b1 protein and decreased by the siRNA knockdown of the endogenous Unc93b1. The degree of proteolytic cleavage in HEK293T cells was low, likely due to the saturation of Unc93b1 and possibly other proteins required for this process by TLR3 overexpression (Fig. 1C). Analysis of the N-linked glycosylation of uncleaved and cleaved TLR3 using glycosidases revealed that the processing of TLR3 is preceded by the addition of complex glycans in the Golgi complex (Fig. 2B). These results are consistent with observations for trafficking of TLR9 (40). However, whether cleavage of TLR3 could occur before its localization in endosomes remains to be determined.
TLR3 cleavage could be inhibited by z-FA-FMK, an inhibitor of cathepsin B, L, and S, and also by the deletion of Loop1 in LRR12 of the TLR3 ectodomain. Both the uncleaved TLR3 and the N- and C- terminal fragments resulting from proteolytic cleavage can bind poly(I:C), localize to the surface of HEK293T cells, and activate innate immune signaling in human and mouse cells. However, although cleavage does not alter poly(I:C)-induced TLR3 signaling in both human and murine cell lines and only had a modest effect on TLR3 signaling to some dsRNAs in human cells, it significantly alters dsRNA-induced TLR3 signaling in murine RAW264.7 cells. Importantly, in human cells, the unprocessed TLR3 was degraded more rapidly than the processed TLR3 fragments and was preferentially accumulated in endosomes targeted for protein degradation.
Replacements of all of the residues in Loop1 still resulted in some degree of proteolytic processing, indicating that cathepsin cleavage is not particularly sequence-specific. However, the amount of processed TLR3 is correlated with the length of Loop1 (Fig. 3), perhaps due to the change of accessibility to cathepsins by length.
Proteolytic processing of TLR3 is not essential for signaling, contrary to the situation for TLR9 (11, 12, 15). When proteolytic cleavage is inhibited by either the cathepsin inhibitor z-FA-FMK or deletion of Loop1, TLR3 can still be activated with poly(I:C) in human Huh7.5 and HEK293T cell lines, which express transiently transfected TLR3, and also in the BEAS-2B cell line, which expresses endogenous TLR3. Even in the murine RAW264.7 cells, treatment with cathepsin inhibitor z-FA-FMK did not affect TLR3 signaling induced by poly(I:C). In the above cell lines where TLR9 function could be examined, TLR9 signaling was significantly decreased by z-FA-FMK (Fig. 5). TLR3 processing was not the focus of the work of Ewald et al. (15), and the difference in the interpretation of the results by them and us could be due to analysis of TLR3 signaling by different approaches. Although we used a luciferase reporter assay for HEK293T and Huh7.5 cells and the measurement of IL6 production for BEAS-2B and RAW264.7 cells, Ewald et al. (15) used the accumulation of intracellular TNF accumulation.
TLR3 may be subject to more complex regulations compared with TLR9. Depending on the cell types, TLR3 can be functional in both endosomes and at the plasma membrane. Monoclonal antibodies blocking cell-surface TLR3 can inhibit dsRNA-induced cytokine secretion by fibroblasts and lung epithelium cells (32, 41). Cell-surface TLR3s have also been reported to be activated by siRNAs in murine cell lines (42). In addition, TLR3 has two distinct binding sites for dsRNA binding, one located from the N terminus to LRR3 and the other in LRR19 through 21 of the ectodomain. Both are required for binding to poly(I:C) (43–48), in comparison to TLR9 and TLR7, which recognize ligands primarily through one binding site (49). Both cleaved and uncleaved TLR3 appear to be competent for signaling, but proteolytic cleavage may confer additional activity to the recognition of some ligands by TLR3, as we have observed for the recognition of Reovirus dsRNA and poly(A:U) RNA in RAW267.4 cells. Another means of regulation of TLR3 activity could be by the abundance of cathepsins in different cell lines. In fact, we have consistently observed different amounts of cleaved fragments in two human cell lines, with <5% in HEK293T and ∼30% in Huh7.5 cells relative to uncleaved TLR3, in the presence of only endogenously expressed Unc93b1.
Finally, the proteolyzed TLR3 appears to have distinct properties when compared with the full-length TLR3. The CTF had a longer half-life in both the pulse-chase and CHX-chase experiments (Fig. 9) compared with uncleaved TLR3, and its accumulation is not affected by treatment with lactacystin, a proteasome inhibitor (Fig. 8B). In addition, when TLR3 could be cleaved by cathepsins, it is more abundant in early endosomes, whereas inhibition of cleavage results in higher accumulation to recycling endosomes and lysosomes (Fig. 10). The differences in localization to different endosomes could affect ligand recognition by TLR3 and its cleaved fragments, as different endosomes could carry different cargos and thus different TLR3 ligands. The higher stability of the CTF (and likely the NTF) compared with the full-length TLR3 could affect the duration of signaling. Although we observed that TLR3s with a deletion of LRR1–12 or residues 1–343 were inactive for signaling in HEK293T cells, Garcia-Cattaneo et al. (30) reported that the CTF of TLR3 was active for signaling in a human retinal epithelial cell line. Whether this difference is due to the cell lines or the constructs used remains to be determined. However, all of the results on TLR3 proteolytic processing in this and other works demonstrate that TLR3 signaling is under a complex network of regulation that will include protein trafficking, cathepsin activity, and the contribution from distinct endosomes.
Supplementary Material
Acknowledgments
We thank Dr. Jarrat Jordan and Lani San Mateo of Centocor Inc. for numerous helpful discussions that shaped this study. We thank Jim Powers and the Indiana University Light Microscopy Center for help with confocal microscopy.
This work was supported in part by a grant from the Indiana Economic Development Corp. (to C. C. K.).

This article contains supplemental Table 1 and Figs. S1–S3.
- TLR
- Toll-like receptor
- TIR domain
- Toll-interleukin-1 receptor domain
- CTF
- C-terminal fragment
- NTF
- N-terminal fragment
- ODN
- oliogodeoxynucleotide
- LRR
- leucine-rich repeat
- CHX
- cyclohexamide
- ECD
- ectodomain
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- z-FA-FMK
- benzyloxycarbonyl-Phe-Ala-fluoromethyl ketone
- FL
- full-length.
REFERENCES
- 1. Kumar H., Kawai T., Akira S. (2009) Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 388, 621–625 [DOI] [PubMed] [Google Scholar]
- 2. Trinchieri G., Sher A. (2007) Cooperation of Toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 7, 179–190 [DOI] [PubMed] [Google Scholar]
- 3. Blasius A. L., Beutler B. (2010) Intracellular toll-like receptors. Immunity 32, 305–315 [DOI] [PubMed] [Google Scholar]
- 4. Barton G. M., Kagan J. C. (2009) A cell biological view of Toll-like receptor function. Regulation through compartmentalization. Nat. Rev. Immunol. 9, 535–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barton G. M., Kagan J. C., Medzhitov R. (2006) Intracellular localization of Toll-like receptor 9 prevents recognition of self-DNA but facilitates access to viral DNA. Nat. Immunol. 7, 49–56 [DOI] [PubMed] [Google Scholar]
- 6. Chaturvedi A., Pierce S. K. (2009) How location governs toll-like receptor signaling. Traffic 10, 621–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kindrachuk J., Potter J. E., Brownlie R., Ficzycz A. D., Griebel P. J., Mookherjee N., Mutwiri G. K., Babiuk L. A., Napper S. (2007) Nucleic acids exert a sequence-independent cooperative effect on sequence-dependent activation of Toll-like receptor 9. J. Biol. Chem. 282, 13944–13953 [DOI] [PubMed] [Google Scholar]
- 8. McGettrick A. F., O'Neill L. A. (2010) Localization and trafficking of Toll-like receptors. An important mode of regulation. Curr. Opin. Immunol. 22, 20–27 [DOI] [PubMed] [Google Scholar]
- 9. de Bouteiller O., Merck E., Hasan U. A., Hubac S., Benguigui B., Trinchieri G., Bates E. E., Caux C. (2005) Recognition of double-stranded RNA by human toll-like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J. Biol. Chem. 280, 38133–38145 [DOI] [PubMed] [Google Scholar]
- 10. Leonard J. N., Ghirlando R., Askins J., Bell J. K., Margulies D. H., Davies D. R., Segal D. M. (2008) The TLR3 signaling complex forms by cooperative receptor dimerization. Proc. Natl. Acad. Sci. U.S.A. 105, 258–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ewald S. E., Lee B. L., Lau L., Wickliffe K. E., Shi G. P., Chapman H. A., Barton G. M. (2008) The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature 456, 658–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Park B., Brinkmann M. M., Spooner E., Lee C. C., Kim Y. M., Ploegh H. L. (2008) Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat. Immunol. 9, 1407–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sepulveda F. E., Maschalidi S., Colisson R., Heslop L., Ghirelli C., Sakka E., Lennon-Duménil A. M., Amigorena S., Cabanie L., Manoury B. (2009) Critical role for asparagine endopeptidase in endocytic Toll-like receptor signaling in dendritic cells. Immunity 31, 737–748 [DOI] [PubMed] [Google Scholar]
- 14. Turk V., Stoka V., Vasiljeva O., Renko M., Sun T., Turk B., Turk D. (2012) Cysteine cathepsins. From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 1824, 68–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ewald S. E., Engel A., Lee J., Wang M., Bogyo M., Barton G. M. (2011) Nucleic acid recognition by Toll-like receptors is coupled to stepwise processing by cathepsins and asparagine endopeptidase. J. Exp. Med. 208, 643–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Avalos A. M., Ploegh H. L. (2011) Competition by inhibitory oligonucleotides prevents binding of CpG to C-terminal TLR9. Eur. J. Immunol. 41, 2820–2827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chockalingam A., Cameron J. L., Brooks J. C., Leifer C. A. (2011) Negative regulation of signaling by a soluble form of toll-like receptor 9. Eur. J. Immunol. 41, 2176–2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. de Zoete M. R., Bouwman L. I., Keestra A. M., van Putten J. P. (2011) Cleavage and activation of a Toll-like receptor by microbial proteases. Proc. Natl. Acad. Sci. U.S.A. 108, 4968–4973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim Y. M., Brinkmann M. M., Paquet M. E., Ploegh H. L. (2008) UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 452, 234–238 [DOI] [PubMed] [Google Scholar]
- 20. Brinkmann M. M., Spooner E., Hoebe K., Beutler B., Ploegh H. L., Kim Y. M. (2007) The interaction between the ER membrane protein UNC93B and TLR3, -7, and- 9 is crucial for TLR signaling. J. Cell Biol. 177, 265–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tabeta K., Hoebe K., Janssen E. M., Du X., Georgel P., Crozat K., Mudd S., Mann N., Sovath S., Goode J., Shamel L., Herskovits A. A., Portnoy D. A., Cooke M., Tarantino L. M., Wiltshire T., Steinberg B. E., Grinstein S., Beutler B. (2006) The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 7, 156–164 [DOI] [PubMed] [Google Scholar]
- 22. Fukui R., Saitoh S., Matsumoto F., Kozuka-Hata H., Oyama M., Tabeta K., Beutler B., Miyake K. (2009) Unc93B1 biases Toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. J. Exp. Med. 206, 1339–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qi R., Hoose S., Schreiter J., Sawant K. V., Lamb R., Ranjith-Kumar C. T., Mills J., San Mateo L., Jordan J. L., Kao C. C. (2010) Secretion of the human Toll-like receptor 3 ectodomain is affected by single nucleotide polymorphisms and regulated by Unc93b1. J. Biol. Chem. 285, 36635–36644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maley F., Trimble R. B., Tarentino A. L., Plummer T. H., Jr. (1989) Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal. Biochem. 180, 195–204 [DOI] [PubMed] [Google Scholar]
- 25. Kornfeld R., Kornfeld S. (1985) Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 54, 631–664 [DOI] [PubMed] [Google Scholar]
- 26. Bell J. K., Botos I., Hall P. R., Askins J., Shiloach J., Davies D. R., Segal D. M. (2006) The molecular structure of the TLR3 extracellular domain. J. Endotoxin. Res. 12, 375–378 [DOI] [PubMed] [Google Scholar]
- 27. Choe J., Kelker M. S., Wilson I. A. (2005) Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science 309, 581–585 [DOI] [PubMed] [Google Scholar]
- 28. Lai Y., Adhikarakunnathu S., Bhardwaj K., Ranjith-Kumar C. T., Wen Y., Jordan J. L., Wu L. H., Dragnea B., San Mateo L., Kao C. C. (2011) LL37 and cationic peptides enhance TLR3 signaling by viral double-stranded RNAs. PLoS One 6, e26632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lai Y., Yi G., Chen A., Bhardwaj K., Tragesser B. J., Rodrigo A Valverde, Zlotnick A., Mukhopadhyay S., Ranjith-Kumar C. T., Kao C. C. (2011) Viral double-strand RNA-binding proteins can enhance innate immune signaling by toll-like receptor 3. PLoS One 6, e25837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Garcia-Cattaneo A., Gobert F. X., Müller M., Toscano F., Flores M., Lescure A., Del Nery E., Benaroch P. (2012) Cleavage of Toll-like receptor 3 by cathepsins B and H is essential for signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 9053–9058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sha Q., Truong-Tran A. Q., Plitt J. R., Beck L. A., Schleimer R. P. (2004) Activation of airway epithelial cells by toll-like receptor agonists. Am. J. Respir. Cell Mol. Biol. 31, 358–364 [DOI] [PubMed] [Google Scholar]
- 32. Duffy K. E., Lamb R. J., San Mateo L. R., Jordan J. L., Canziani G., Brigham-Burke M., Korteweg J., Cunningham M., Beck H. S., Carton J., Giles-Komar J., Duchala C., Sarisky R. T., Mbow M. L. (2007) Down modulation of human TLR3 function by a monoclonal antibody. Cell. Immunol. 248, 103–114 [DOI] [PubMed] [Google Scholar]
- 33. Matsumoto M., Funami K., Tanabe M., Oshiumi H., Shingai M., Seto Y., Yamamoto A., Seya T. (2003) Subcellular localization of Toll-like receptor 3 in human dendritic cells. J. Immunol. 171, 3154–3162 [DOI] [PubMed] [Google Scholar]
- 34. Weber C., Müller C., Podszuweit A., Montino C., Vollmer J., Forsbach A. (2012) Toll-like receptor (TLR) 3 immune modulation by unformulated small interfering RNA or DNA and the role of CD14 (in TLR-mediated effects). Immunology 136, 64–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alexopoulou L., Holt A. C., Medzhitov R., Flavell R. A. (2001) Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413, 732–738 [DOI] [PubMed] [Google Scholar]
- 36. Wang H., Song P., Du L., Tian W., Yue W., Liu M., Li D., Wang B., Zhu Y., Cao C., Zhou J., Chen Q. (2011) Parkin ubiquitinates Drp1 for proteasome-dependent degradation. Implication of dysregulated mitochondrial dynamics in Parkinson disease. J. Biol. Chem. 286, 11649–11658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen J. W., Chen G. L., D'Souza M. P., Murphy T. L., August J. T. (1986) Lysosomal membrane glycoproteins. Properties of LAMP-1 and LAMP-2. Biochem. Soc. Symp. 51, 97–112 [PubMed] [Google Scholar]
- 38. Mu F. T., Callaghan J. M., Steele-Mortimer O., Stenmark H., Parton R. G., Campbell P. L., McCluskey J., Yeo J. P., Tock E. P., Toh B. H. (1995) EEA1, an early endosome-associated protein. EEA1 is a conserved α-helical peripheral membrane protein flanked by cysteine “fingers” and contains a calmodulin-binding IQ motif. J. Biol. Chem. 270, 13503–13511 [DOI] [PubMed] [Google Scholar]
- 39. Stenmark H., Olkkonen V. M. (2001) The Rab GTPase family. Genome Biol. 2, reviews3007.1–review3007.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chockalingam A., Brooks J. C., Cameron J. L., Blum L. K., Leifer C. A. (2009) TLR9 traffics through the Golgi complex to localize to endolysosomes and respond to CpG DNA. Immunol. Cell Biol. 87, 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Matsumoto M., Kikkawa S., Kohase M., Miyake K., Seya T. (2002) Establishment of a monoclonal antibody against human Toll-like receptor 3 that blocks double-stranded RNA-mediated signaling. Biochem. Biophys. Res. Commun. 293, 1364–1369 [DOI] [PubMed] [Google Scholar]
- 42. Kleinman M. E., Yamada K., Takeda A., Chandrasekaran V., Nozaki M., Baffi J. Z., Albuquerque R. J., Yamasaki S., Itaya M., Pan Y., Appukuttan B., Gibbs D., Yang Z., Karikó K., Ambati B. K., Wilgus T. A., DiPietro L. A., Sakurai E., Zhang K., Smith J. R., Taylor E. W., Ambati J. (2008) Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature 452, 591–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bell J. K., Askins J., Hall P. R., Davies D. R., Segal D. M. (2006) The dsRNA binding site of human Toll-like receptor 3. Proc. Natl. Acad. Sci. U.S.A. 103, 8792–8797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Botos I., Liu L., Wang Y., Segal D. M., Davies D. R. (2009) The toll-like receptor 3-sRNA signaling complex. Biochim. Biophys. Acta 1789, 667–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fukuda K., Watanabe T., Tokisue T., Tsujita T., Nishikawa S., Hasegawa T., Seya T., Matsumoto M. (2008) Modulation of double-stranded RNA recognition by the N-terminal histidine-rich region of the human toll-like receptor 3. J. Biol. Chem. 283, 22787–22794 [DOI] [PubMed] [Google Scholar]
- 46. Liu L., Botos I., Wang Y., Leonard J. N., Shiloach J., Segal D. M., Davies D. R. (2008) Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science 320, 379–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pirher N., Ivicak K., Pohar J., Bencina M., Jerala R. (2008) A second binding site for double-stranded RNA in TLR3 and consequences for interferon activation. Nat. Struct. Mol. Biol. 15, 761–763 [DOI] [PubMed] [Google Scholar]
- 48. Tokisue T., Watanabe T., Tsujita T., Nishikawa S., Hasegawa T., Seya T., Matsumoto M., Fukuda K. (2008) Significance of the N-terminal histidine-rich region for the function of the human toll-like receptor 3 ectodomain. Nucleic Acids Symp. Ser. (Oxf) 203–204 [DOI] [PubMed] [Google Scholar]
- 49. Wei T., Gong J., Jamitzky F., Heckl W. M., Stark R. W., Rössle S. C. (2009) Homology modeling of human Toll-like receptors TLR7, 8, and 9 ligand-binding domains. Protein Sci. 18, 1684–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bolte S., Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.