

Background: Hormone regulation of ion channels requires assembly of multiprotein complexes.
Results: The epithelial sodium channel is present in a hormone-dependent ∼1.1-MDa complex, which requires the scaffold protein CNK3 for assembly.
Conclusion: CNK3-dependent assembly of this regulatory complex is essential for control of epithelial sodium transport.
Significance: This is the first demonstration of scaffold-mediated assembly for a sodium channel-regulatory complex.
Keywords: Aldosterone, Hormones, Signal Transduction, Sodium Channels, Sodium Transport, ENaC Scaffold, ENaC-regulatory Complex, Aldosterone Signaling
Abstract
Hormone regulation of ion transport in the kidney tubules is essential for fluid and electrolyte homeostasis in vertebrates. A large body of evidence has suggested that transporters and channels exist in multiprotein regulatory complexes; however, relatively little is known about the composition of these complexes or their assembly. The epithelial sodium channel (ENaC) in particular is tightly regulated by the salt-regulatory hormone aldosterone, which acts at least in part by increasing expression of the serine-threonine kinase SGK1. Here we show that aldosterone induces the formation of a 1.0–1.2-MDa plasma membrane complex, which includes ENaC, SGK1, and the ENaC inhibitor Nedd4-2, a key target of SGK1. We further show that this complex contains the PDZ domain-containing protein connector enhancer of kinase suppressor of Ras isoform 3 (CNK3). CNK3 physically interacts with ENaC, Nedd4-2, and SGK1; enhances the interactions among them; and stimulates ENaC function in a PDZ domain-dependent, aldosterone-induced manner. These results strongly suggest that CNK3 is a molecular scaffold, which coordinates the assembly of a multiprotein ENaC-regulatory complex and hence plays a central role in Na+ homeostasis.
Introduction
Epithelial ion channels and transporters are regulated by hormonal and non-hormonal signals, which alter their plasma membrane abundance and activity (1–6). A large body of evidence has suggested that these transporters and channels exist in multiprotein regulatory complexes, which influence their steady state levels and net activity (7–12). However, in only a few cases have intact complexes been identified (7, 13–16), and very little is known about the existence and function of validated regulators within such complexes. The epithelial Na+ channel (ENaC)3 in particular is a heterotrimeric highly selective Na+ channel, which is essential for the control of Na+ and K+ balance, extracellular fluid volume, and blood pressure in most vertebrates. Animals or humans with defects in this channel (or upstream regulators) have severe disorders of Na+ wasting and K+ excess (17, 18). Its activity and cell surface expression are tightly regulated by a variety of hormonal and non-hormonal signals, most notably the salt-regulating hormone aldosterone. This steroid hormone acts through an intracellular receptor, the mineralocorticoid receptor, to trigger coordinated changes in the expression of several mediators, which increase plasma membrane ENaC numbers (and to a lesser extent open probability) and hence Na+ transport.
Recent evidence has suggested that several ENaC-regulatory proteins function within a multiprotein complex, which governs the net cell surface expression and activity of the channel (1, 12, 19). In the absence of aldosterone, Raf-1 and the ubiquitin ligase Nedd4-2 are constitutively expressed ENaC inhibitors, which associate with the channel, enhance its internalization and, if unabated lead to its degradation. SGK1 is an aldosterone-regulated serine-threonine kinase, which selectively increases ENaC plasma membrane abundance by phosphorylating and inhibiting Nedd4-2 (20, 21) and possibly other targets, including Raf-1. SGK1 has been shown to interact with both ENaC and Nedd4-2 and to be present at the plasma membrane (12, 21–24); however, there has been no direct evidence for an intact multiprotein complex containing ENaC and its validated regulators at the plasma membrane. Furthermore, how SGK1 and other regulatory factors are recruited to ENaC and organized into a regulatory complex at the plasma membrane has remained obscure. Importantly, none of the previously identified ENaC-interacting proteins has been shown to function as a scaffold protein for this putative plasma membrane complex.
Here we report the identification of a plasma membrane complex of ∼1.0–1.2 MDa containing ENaC and some of its established regulators, including SGK1 and Nedd4-2, in mouse distal nephron cells treated with aldosterone. We further identify in the same complex, connector enhancer of kinase suppressor of Ras isoform 3 (CNK3), a putative scaffold protein recently shown to be a functionally important aldosterone-regulated protein in the distal nephron (25). Unlike other aldosterone-regulated gene products, CNK3 has the classic structural features of a scaffold protein, and its close relative, CNK1, has been shown to function as a scaffold (26, 27). Notably, like CNK1, CNK3 contains a PSD-95/DLG-1/ZO-1 (PDZ) domain, which is found in many proteins implicated in the plasma membrane expression and function of membrane transporters. In particular, members of the NHERF family have been shown to enhance the plasma membrane stability of the epithelial ion transporters NaPi-IIa and sodium-hydrogen exchanger 3 channel (NHE3) (28–31). These observations suggested the hypothesis that CNK3 serves as an aldosterone-induced scaffold, which enhances the recruitment of SGK1 and potentially other stimulatory factors to ENaC. We therefore characterized the functional effects and physical interactions of CNK3. We found that CNK3 directly interacts with ENaC at the cell surface as well as with other ENaC-interacting proteins, including Nedd4-2, Raf-1, and SGK1. Importantly, we demonstrated that CNK3 stimulates interactions of regulatory proteins with ENaC and with each other. CNK3 also increases ENaC cell surface expression as well as endogenous ENaC activity in renal epithelial cells. These effects of CNK3 are aldosterone-dependent and require an intact PDZ domain. Taken together, the functional and biochemical data support the idea that CNK3 is an aldosterone-induced scaffold protein, which coordinates the dynamic assembly of a ∼1.0–1.2-MDa plasma membrane ENaC-regulatory complex.
EXPERIMENTAL PROCEDURES
Cloning of Expression Constructs
Complementary synthetic DNA oligonucleotides for RNA interference (RNAi) targeting mouse CNK3 (5′-GGAGCAGGTGCTACATCAACTTTCAAGAGAAGTTGATGTAGCACCTGCTCC-3′) and a non-target control (5′-TTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAA-3′) were hybridized and inserted into pENTR/U6 vector (Invitrogen). shRNA cassettes were recombined by Gateway cloning into a modified pLenti-6 destination vector (pGT3) to generate lentiviral shRNA expression constructs. The ORF for mouse wild-type CNK3 was amplified by RT-PCR from mouse total kidney mRNA and inserted into pcDNA3.1 directional TOPO vector (Invitrogen) according to the manufacturer's recommendations. A V5 epitope tag was added in-frame at the C terminus to allow detection of expression using anti-V5 antibody. The pcDNA3.1-human CNK3 and pcDNA3.1-human SGK1 constructs were generated similarly from total HEK 293T cell mRNA. A C-terminal FLAG or V5 epitope tag was added to facilitate analyses of expression using anti-FLAG or anti-V5 antibody. pcDNA3.1-ΔPDZ-mCNK3-V5 was generated by PCR amplification using a primer set flanking the PDZ domain and introducing EcoRI restriction sites. The construct was recircularized by ligating EcoRI-digested compatible ends. Expression constructs mCNK3-V5 and mCNK3ΔPDZ-V5 resistant to shRNA targeting CNK3 were generated by introducing multiple silent mutations in the region of the CNK3 ORF targeted by the shRNA. The generation/source of pMO-mSGK1(S422D)-HA, pMO-mGILZ1, pMO-FLAG-mRaf-1, and pMO-mNedd4-2-FLAG constructs has been described previously (12, 19, 32). pMO-FLAG-α/β/γ-mouse ENaC constructs were generated using pcDNA-α/β/γ-ENaC plasmids as templates and inserting the FLAG epitope in the extracellular region of α-, β-, and γ-ENaC. All constructs were confirmed by DNA sequencing.
Production of Lentiviruses Encoding shRNA Targeting Mouse CNK3 and Generation of mpkCCDc14 Cells Stably Silencing Endogenous CNK3 Expression
Lentivirus production and infection of cells were essentially carried out according to the Invitrogen ViraPower Lentiviral Expression Systems User Manual (33). Briefly, 6 × 106 HEK 293T cells were seeded in 75-cm2 cell culture flasks. Cells were then transfected with lentiviral vectors using Lipofectamine 2000 (Invitrogen) and incubated in 10 ml of culture medium. Every 24 h, the medium was replaced, and supernatants containing viruses were collected, centrifuged (3,000 rpm for 5 min at 4 °C), and stored at 4 °C. Lentiviral particles were concentrated by using PEG-itTM (System Bioscience) according to the manufacturer's instructions and resuspended in 500 μl of residual medium. Viral stocks were stored in 100-μl aliquots at −80 °C. Viral titers were determined by performing a human immunodeficiency virus, type 1 p24 ELISA (PerkinElmer Life Sciences).
mpkCCDc14 cells were grown in 12-well plates until they reached ∼60% confluence. Cells were transduced overnight with viral supernatants at a multiplicity of infection of 1, washed, and seeded in antibiotic-free medium in 10-cm dishes. 48 h post-transduction, recombinants were selected with blasticidin (5 μg/ml) for 10 days. Polyclonal cells were expanded and tested for relative CNK3 knockdown efficiency by quantitative PCR as described previously (25). Recombinant cells were grown in the presence of 1 μg/ml blasticidin to maintain selection pressure. The CNK3 knockdown efficiency (as analyzed by quantitative PCR) was >70%.
Cell Culture and Electrophysiological Measurements
HEK 293T cells were routinely maintained in high glucose DMEM supplemented with 100 units/ml penicillin, 100 mg/ml streptomycin, and 10% FBS. mpkCCDc14 cells were regularly maintained and grown on Transwell filters as described previously (32, 34). Cells were transfected with 4 μg of appropriate expression vector (wild-type as well as shRNA-resistant mCNK3-V5 or ΔPDZ-mCNK3-V5 constructs or empty vector as a control) using a high efficiency electroporation protocol (nucleofection, Amaxa Biosystems, Inc.) as described previously (32). Electrophysiological measurements were performed prior to and after aldosterone stimulation also as described previously (12, 32). All experiments were repeated at least three independent times with similar results.
Immunoprecipitations and Immunoblotting
3 × 106 HEK 293T cells were seeded in 10-cm cell culture dishes and allowed to attach overnight. Cells were then transfected with appropriate expression vectors as specified in the figures using Lipofectamine (Invitrogen) according to the manufacturer's instructions. The amounts of DNA transfected were as follows: 2 μg of mSGK1/S422D-HA, 2 μg of hSGK1-V5, 500 ng of FLAG-mRaf-1, 100 ng of FLAG-mNedd4-2, 750 ng of mCNK3-V5, 4 μg of ΔPDZ-mCNK3-V5, 750 ng of hCNK3-FLAG, and 300 ng of each FLAG-mENaC subunit per dish. 24 h post-transfection, cells were harvested, and lysates were processed for protein analyses. CNK3 interaction with SGK1 could only be detected in the presence of co-transfected GILZ1 (1.5 μg of untagged mGILZ1/10-cm dish). Immunoprecipitations (IPs) of the V5 epitope were carried out with Dynabeads Protein G (Invitrogen) according to the manufacturer's suggestions. In brief, total cell lysates were incubated for 30 min with 4 μg of anti-V5 antibody (Ab) (Invitrogen). Ab-antigen complexes were then captured for 20 min at 4 °C by adding 1 mg of Dynabeads Protein G. The lysate-Ab-bead mixture was kept at 4 °C under rotary agitation at all times. Captured immune complexes were separated magnetically and washed at least three times. IPs of the HA and FLAG epitopes were performed as described previously (12, 19). Immunoblotting was also performed as described previously (19, 25).
The following antibodies were used for immunoblotting: anti-FLAG-HRP Ab (Sigma), anti-HA-HRP Ab (Roche Applied Science), anti V5-HRP Ab (Invitrogen), anti-α-ENaC Ab (kindly provided by Johannes Loffing, University of Lausanne), and anti-β-ENaC Ab (kindly provided by Mark Knepper, National Institutes of Health, Bethesda, MD). Detection of endogenous aldosterone-induced ENaC-regulatory proteins in mpkCCDc14 cells was performed using the commercially available anti-SGK1 Ab (Sigma), anti-CNK3 Ab (Protein Tech Group Inc., Chicago, IL), anti-Nedd4-2 Ab (Abcam), and anti-Raf-1 Ab (Cell Signaling Technology). Total GAPDH or α-tubulin was used as a loading control. Appropriate secondary light chain-recognizing Abs were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA).
For densitometric analyses of immunoblots, signal intensities of specified bands were determined using NIH ImageJ software and normalized to that of the internal control as described previously (12, 19). For quantification of CNK3 signals, signal intensities from both bands were added to provide one value. Values so obtained were used to determine mean ± S.E. for a graphical representation.
Two-dimensional Blue Native (BN) PAGE Analyses of Multiprotein Complexes Containing ENaC and Key ENaC-regulatory Proteins
mpkCCDc14 cells were grown on 15-cm dishes and treated with aldosterone (1 μm for 2 h) or vehicle (equal volume of ethanol). HEK 293T cells were transfected with ENaC (α-, β-, γ-FLAG-ENaC subunits; 750 ng/subunit/15-cm dish) either alone or in combination with mSGK1/S422D-HA (5 μg/15-cm dish), mGILZ1 (3.75 μg/15-cm dish), and mCNK3-V5 (5 μg/15-cm dish) using Lipofectamine (Invitrogen) according to the manufacturer's instructions. Transfection was allowed to proceed for 48 h. At the end of the treatment period (mpkCCDc14 cells) or transfection (HEK 293T cells), cells layers were exposed to a water-soluble membrane-permeable cross-linking agent, dimethyl-3,3′-dithiobispropionimidate·2 HCl (2 mm) (Thermo Fisher Scientific, Hanover Park, IL) at room temperature as described previously (19). We used dimethyl-3,3′-dithiobispropionimidate·2 HCl to help preserve possible transient protein-protein interactions that are likely to be disrupted during the process of plasma membrane isolation. Following an hour of cross-linking, plasma membrane fractions were isolated from these cells using a plasma membrane protein extraction kit (BioVision, Mountain View, CA) according to the manufacturer's instructions. Extracts were solubilized in 2.5% dodecyl maltoside. Plasma membrane purity was confirmed by the absence of calnexin (an integral endoplasmic reticulum membrane marker; antibody purchased from Cell Signaling Technology) or GAPDH (a cytosolic marker) and by the independent enrichment of Na+-K+-ATPase (a plasma membrane marker; antibody purchased from Abcam) in the same fractions (supplemental Fig. 1B). BN gel electrophoresis was performed with these extracts using 3–12% native PAGE Novex Bis-Tris gels and the XCell SureLock MiniCell electrophoresis system (both from Invitrogen). For further separation by second dimension SDS-PAGE, the vertical lanes from first dimension BN-PAGE were cut out, and the gel strips were processed using an Invitrogen two-dimensional BN-PAGE kit according to the manufacturer's instructions. They were then placed into a 4–20% Tris-glycine two-dimensional SDS-polyacrylamide gel (Invitrogen) and overlaid with 1× SDS Laemmli loading buffer. The second dimension run and immunoblotting were performed according to standard procedures.
Cell Surface Biotinylation Assays and Double Pulldowns Using Iminobiotin
Cell surface biotinylation assays were essentially performed as described previously (12). Briefly, HEK 293T cells were co-transfected as specified in the figures. 48 h after transfection, cells were treated with EZ-Link sulfo-NHS-biotin (Pierce) for an hour prior to extraction and recovery of biotinylated proteins with immobilized NeutrAvidin beads (Thermo Scientific). Immunoprecipitates were analyzed for cell surface (biotinylated) β-ENaC or CNK3-V5 by immunoblotting using anti-β-ENaC antibody (kindly provided by Mark Knepper, National Institutes of Health) or anti-V5 antibody (Invitrogen). As a control for integrity of cells during the biotinylation process, the blot was stripped and reprobed for GAPDH. Total cellular GAPDH was used as a loading control. Densitometric analyses were performed using NIH ImageJ software as described above. Endogenous surface SGK1 and CNK3 proteins in mpkCCDc14 cells were detected using anti-SGK1 Ab (Sigma) and anti-CNK3 Ab (Protein Tech Group Inc.) respectively, following NeutrAvidin pulldowns.
For the double pulldown assays, transfected cells were treated with EZ-Link NHS-iminobiotin (Pierce) instead of biotin for an hour prior to extraction and recovery of iminobiotinylated proteins with immobilized NeutrAvidin beads according to the manufacturer's instructions (Thermo Scientific). Iminobiotinylated proteins immobilized on NeutrAvidin beads were recovered in a minimal volume of elution buffer for 5 min and then subjected to a second pulldown using EZview anti-FLAG affinity matrix (Sigma) as described previously (12).
GST Pulldown Assays
In vitro transcription and translation of full-length wild-type mCNK3 was performed using the TnT T7 Quick Coupled Transcription and Translation rabbit reticulocyte system (Promega) in the presence of [35S]methionine according to the manufacturer's instructions. Glutathione S-transferase (GST)-ENaC fusion proteins were expressed using the GST purification system (Amersham Biosciences) as described previously (24) with minor modifications as follows. The C-terminal cytoplasmic domains of mouse α- (amino acids 613–700) and β-ENaC (amino acids 557–638) were subcloned from the respective full-length mouse pMO-ENaC plasmids into the GST-containing prokaryotic expression vector pGEX-4T1 (Amersham Biosciences) using PCR. The plasmids were confirmed by sequencing and then transformed into BL-21 Escherichia coli for high level expression followed by purification on a glutathione-Sepharose column according to the manufacturer's protocol. GST alone, purified simultaneously using the pGEX-4T3 plasmid, served as a negative control. Binding experiments and signal visualization were performed as described previously (24).
Statistical Analyses
Data are represented as mean ± S.E. In all experiments involving statistical analyses, three to six samples were tested, and at least three independent experiments were performed with the same treatment protocol. Unless otherwise specified, all statistical comparisons were evaluated using a Student's unpaired two-tailed t test, and significance was defined as a p value <0.05.
RESULTS
ENaC, SGK1, Nedd4-2, and CNK3 Are Part of a Large ∼1.0–1.2-MDa Regulatory Complex
We used the two-dimensional BN-PAGE technique (35–38) to characterize the size and composition of ENaC-containing multiprotein complexes isolated from plasma membrane preparations of mpkCCDc14 cells. These cells, which express endogenous ENaC and respond to aldosterone with an increase in ENaC-dependent Na+ current, provide a faithful model of aldosterone-regulated Na+ transport in the kidney tubule. Cells were treated with vehicle (Fig. 1A) or aldosterone (Fig. 1B), and plasma membranes were isolated and subjected to two-dimensional electrophoresis as described under “Experimental Procedures.” In the first dimension (“blue native” PAGE), intact protein complexes were separated by size, and in the second dimension (SDS-PAGE), they were denatured and separated into their separate components. Thus, proteins that are components of the same multiprotein complex co-migrate in the first (horizontal) dimension and are separated into distinct spots in the same vertical line in the second dimension (35). In the absence of aldosterone, we observed that ENaC was present at levels slightly above background (detected by anti-α-ENaC antibody) and distributed along the horizontal axis between ∼720 kDa and 1.2 MDa. A separate spot at ∼600 kDa was also observed. The presence of ENaC complexes in the range of 600–720 kDa is consistent with previous reports (14, 39). Under these conditions, we were unable to detect clear plasma membrane signals for previously identified ENaC regulators, SGK1, Nedd4-2, GILZ1, and CNK3.
FIGURE 1.

Detection of multiprotein complexes containing ENaC and some of its key regulatory proteins. Solubilized plasma membrane lysates from mpkCCDc14 cells treated with vehicle (A) or aldosterone (1 μm for 2 h) (B) were subjected to two-dimensional BN/SDS-PAGE to separate native multiprotein complexes. Immunoblotting (IB) was performed using specific antibodies as described under “Experimental Procedures.” HEK 293T cells were transfected with ENaC either alone (C) or with key regulatory proteins (D) as described under “Experimental Procedures.” Solubilized plasma membrane lysates from these cells were subjected to BN-PAGE followed by subsequent two-dimensional SDS-PAGE to separate out the components of the multiprotein complexes. Immunoblotting was performed using specific antibodies as described under “Experimental Procedures.” These blots are representative of at least four similar immunoblots.
Upon aldosterone treatment, plasma membrane ENaC increased markedly and was detected in a much tighter distribution primarily between 1.0 and 1.2 MDa (in the native dimension). A weaker but distinct signal was detected between 600 and 720 kDa (Fig. 1B, see α-ENaC panel). We next probed the blots for previously characterized ENaC regulators, some of which had been shown to interact with ENaC and with each other (12) but which had not been shown to be present in a single native complex. As shown in the lower panels of Fig. 1B, we detected the negative regulator, Nedd4-2, as well as the positive regulator, SGK1, in the same distribution as ENaC in the native dimension (see arrow). Consistent with earlier observations (12), we also detected Raf-1 at modest levels in the native complex (first dimension) (see supplemental Fig. 1); however, it did not appear in the subsequent SDS-PAGE blots because expression was below the limit of detection for the two-dimensional blot. Interestingly, despite its effect on ENaC plasma membrane localization (12, 32), we could not detect GILZ1 in this membrane complex. Based on a recent report identifying the putative scaffold protein CNK3 as an aldosterone-stimulated regulator of ENaC (25), we also probed for this protein. As shown in Fig. 1B, bottom panel, CNK3 was found entirely in two distinct spots, largely co-migrating with ENaC between 1.0 and 1.2 MDa. Together with previous data (12) and data below, these findings strongly support the conclusion that we have identified a native plasma membrane complex containing ENaC and key regulators, Nedd4-2, SGK1, and CNK3. We have previously used the term ENaC-regulatory complex (ERC) to describe this multiprotein complex consisting of ENaC and its regulators.
To examine directly whether these regulators are sufficient to cause the increase in plasma membrane ENaC, we transiently transfected HEK 293T cells with ENaC either alone (Fig. 1C) or with the regulators (Fig. 1D). These mammalian human embryonic kidney cells are known to reliably express and traffic to the plasma membrane a variety of integral membrane proteins, including ion transporters and channels like ENaC (12, 32, 40, 41). When expressed alone in these cells, ENaC (again detected using anti-α-ENaC antibody) was found at levels slightly above background in the region between 700 kDa and 1.1 MDa (Fig. 1C), similar to native ENaC in mpkCCDc14 cells in the absence of aldosterone. In the presence of co-transfected regulators, ENaC plasma membrane expression was markedly induced with a distinct signal detected at 1.0–1.2 MDa (Fig. 1D). A strong ENaC signal was also detected more diffusely between 480 kDa and 1.0 MDa. We next probed the blots for the various regulators and as in the native complex identified SGK1 and CNK3, but not GILZ1, in the 1.0–1.2-MDa complex. CNK3 in particular was strikingly localized to the 1.0–1.2-MDa region; SGK1 was found in the same location as well as more diffusely between ∼60 and 750 kDa. The signal at around 60 kDa, which was particularly intense, is consistent with SGK1 alone, which runs at slightly above 50 kDa on denaturing gels (as seen in the second dimension of the present blots). This observation is consistent with the idea that SGK1 is not tightly associated with the ERC and tends to dissociate during electrophoresis (see also below). GILZ1 was not in any of these membrane blots.
CNK3 and SGK1 Are Functionally Important Stimulators of ENaC
It is well established in multiple cultured cell types and in vivo that SGK1 is a physiologically important regulator of vertebrate Na+ homeostasis primarily through effects to increase ENaC plasma membrane abundance in a subset of epithelial cells in the kidney tubules (42–48). By phosphorylating and inhibiting Nedd4-2, SGK1 inhibits ENaC ubiquitinylation and thereby decreases channel internalization from the plasma membrane and subsequent degradation (20, 49, 50). The full range of SGK1 effects and the subcellular locations for these effects have not been determined. However, increasing recent evidence supports the idea that a significant locus of SGK1 action is at the plasma membrane (Fig. 1 and Refs. 22 and 51). Importantly, SGK1 and Nedd4-2 have been shown to interact with ENaC at the plasma membrane as well as with each other (12, 23, 52–55).
Less is known about CNK3; however, one recent report demonstrated that CNK3 is a mineralocorticoid receptor target gene highly expressed in native collecting duct and that it stimulates ENaC-mediated Na+ transport in M1 kidney collecting duct cells (25). Furthermore, in this same report, CNK3 was detected in both cytosolic and membrane fractions. To study CNK3 function in the context of the present experiments, we first examined the effect of shRNA-mediated knockdown of endogenous CNK3 on aldosterone-induced ENaC activity in mpkCCDc14 cortical collecting duct principal cells, which recapitulate key features of aldosterone-regulated Na+ transport in native distal nephron more closely than M1 cells (56). The cells were stably transduced with lentiviruses that code for either shRNA targeting mouse CNK3 or a non-target control shRNA. Cells were then analyzed on Transwell filters for aldosterone-stimulated transepithelial Na+ transport as described previously (32, 34). Fig. 2A depicts the -fold change in aldosterone-induced Na+ current over basal. CNK3 depletion resulted in a dramatic decrease in aldosterone-stimulated transepithelial Na+ transport, confirming the functional role of CNK3 in ENaC stimulation in this cell line. We next assessed the effects of heterologous CNK3 expression on ENaC cell surface expression using biotinylation assays in ENaC-transfected HEK 293T cells as described previously (12). The abundance of plasma membrane ENaC was analyzed by immunoblotting using an antibody specific to β-ENaC. As shown in Fig. 2B, CNK3 significantly stimulated ENaC surface expression. Thus, together with prior literature, these experiments confirm that SGK1 and CNK3 are functionally important in regulating ENaC activity through their effects on channel cell surface expression.
FIGURE 2.

CNK3 positively regulates ENaC surface expression and activity. A, knockdown of endogenous CNK3 decreases ENaC activity in mpkCCDc14 cells. mpkCCDc14 cells stably expressing CNK3 or control (non-target) shRNA were plated on Transwell filters and allowed to grow to high resistance. The cell monolayers were then treated with aldosterone (1 μm for 3 h) prior to determination of ENaC activity. Shown is the -fold change in aldosterone-induced equivalent short circuit current in CNK3 shRNA-expressing cells. Also shown is a representative blot demonstrating knockdown of endogenous CNK3. ***, p < 0.001 compared with control (n = 6). B, cell surface biotinylation assay for ENaC in HEK 293T cells transiently transfected with FLAG-tagged α-, β-, and γ-mENaC subunits. After transfection, cells were treated with EZ-Link sulfo-NHS-biotin before protein extraction and recovered by affinity immobilization using NeutrAvidin-conjugated agarose beads followed by immunoblot (IB) analysis with anti-β-ENaC antibody. Shown is a graphical representation of CNK3-dependent augmentation of surface ENaC based on multiple repeats of biotinylation assays. Also shown is a representative blot demonstrating CNK3 overexpression. ***, p < 0.001 compared with control (n = 4). Error bars represent S.E. WCL, whole-cell lysate.
ERC Components Nedd4-2, Raf-1, SGK1, and CNK3 Physically Interact with ENaC and with Each Other
The two-dimensional gel experiments support the conclusion that in the presence of aldosterone plasma membrane ENaC exists in a 1.0–1.2-MDa complex that contains the regulatory proteins Nedd4-2, SGK1, and CNK3 (Fig. 1). To further examine the physical interactions among these complex components, we performed a series of co-IP experiments in transfected HEK 293T cells. In the absence of CNK3, SGK1 showed weak but significant interaction with ENaC (Fig. 3), consistent with previous results (24). Interestingly, this interaction was markedly stimulated by CNK3 (Fig. 3A, top panel, compare lanes 2 and 3; Fig. 3B shows a graphical representation of the same). SGK1 also physically interacted with Nedd4-2 (Fig. 4A, top panel) although weakly, consistent with previous reports (12, 52). As for ENaC, the interaction of SGK1 with Nedd4-2 was markedly stimulated by co-expression of CNK3 (Fig. 4A, top panel, compare lanes 2 and 3; Fig. 4B shows a graphical representation of the same). We also found that SGK1 physically interacted with CNK3 itself (Fig. 5A). Finally, we examined the physical interactions of CNK3 with ENaC, Nedd4-2, and Raf-1, finding robust specific interactions (Fig. 5, B and C). Together, these findings confirm the physical interactions among components of the 1.0–1.2-MDa plasma membrane ERC and begin to suggest that CNK3 functions as a molecular scaffold that stimulates formation of this complex.
FIGURE 3.
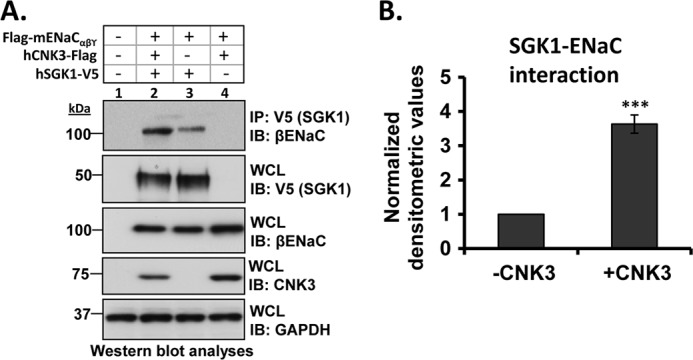
CNK3 markedly stimulates interaction of SGK1 with ENaC. A, co-immunoprecipitation assays in HEK 293T cells revealing the ability of CNK3 to markedly augment interaction of SGK1 with ENaC. Blots showing total cellular expression of the various proteins specified are also depicted. GAPDH was used as a loading control. IB, immunoblot; WCL, whole-cell lysate. Also shown is a graphical representation of the above data (B) demonstrating significantly increased SGK1-ENaC interaction in the presence of CNK3. ***, p < 0.001 compared with control (n = 4). Error bars represent S.E.
FIGURE 4.

CNK3 significantly stimulates interaction of SGK1 with its target substrate, Nedd4-2. A, co-immunoprecipitation assays in transiently transfected (as specified) HEK 293T cells demonstrating that CNK3 stimulates the interaction of SGK1 with Nedd4-2. Blots showing total cellular expression of the various proteins specified are also depicted. GAPDH was used as a loading control. IB, immunoblot; WCL, whole-cell lysate. Also shown is a graphical representation of the above data (B) clearly demonstrating that CNK3 markedly augments the SGK1-Nedd4-2 interaction. ***, p < 0.001 (n = 4). Error bars represent S.E.
FIGURE 5.

CNK3 is an integral component of the ERC. A–C, co-immunoprecipitation assays in HEK 293T cells demonstrating the ability of CNK3 to physically interact with SGK1 (A), ENaC (B), and the ENaC-inhibitory proteins Raf-1 and Nedd4-2 (C). Blots depicted are representative of at least four independent experiments each, performed with the same treatment protocol. Blots showing total cellular expression of the various proteins specified are also depicted. GAPDH was used as a loading control. IB, immunoblot; WCL, whole-cell lysate.
Role of the PDZ Domain in CNK3 Interactions
CNK3 possesses multiple protein-protein interaction motifs, which are typical of a molecular scaffold (57), and its close relative, CNK1, has been shown to be a scaffold (58). In particular, CNK3, like CNK1, has a PDZ domain; PDZ domains have been implicated in the assembly of large multiprotein complexes at the cell membrane, linking cytosolic signaling proteins to membrane transporters (59–62). To begin to explore the function of the CNK3 PDZ domain in ERC formation and ENaC regulation, we generated a CNK3 mutant that lacks the PDZ domain (referred to as ΔPDZ-CNK3) and examined the interaction of wild-type and mutant CNK3 with key ERC components in whole-cell lysates. As shown in Fig. 6A (top panel), ΔPDZ-CNK3 failed to co-immunoprecipitate ENaC, whereas full-length/wild-type (WT) CNK3 showed robust interaction (also shown in Fig. 5B). ΔPDZ-CNK3 also failed to interact with SGK1 (Fig. 6B, top panel). In contrast, ΔPDZ-CNK3 was able to interact with both Nedd4-2 and Raf-1 (supplemental Fig. 2), the ENaC-inhibitory proteins.
FIGURE 6.

The PDZ domain in CNK3 is essential for its ability to interact with ENaC and the key ENaC-regulatory kinase, SGK1. HEK 293T cells were transiently transfected with FLAG-tagged α-, β-, and γ-mENaC subunits and V5-tagged WT- or ΔPDZ-CNK3 with or without SGK1 as specified. Shown are co-immunoprecipitation assays demonstrating the drastically reduced ability of ΔPDZ-CNK3 to interact with ENaC (A) (n = 4) or SGK1 (B) (n = 5). Blots showing total cellular expression of the various proteins specified are also depicted. GAPDH was used as a loading control. IB, immunoblot; WCL, whole-cell lysate.
The Role of the CNK3 PDZ Domain in ENaC Regulation
To further explore the functional role of CNK3 and its PDZ domain in ENaC regulation and formation of the plasma membrane complex, a series of surface biotinylation experiments were performed with wild-type CNK3 and the PDZ domain-deficient mutant (Fig. 7A). Consistent with the two-dimensional gel experiments (Fig. 1D), in transfected HEK 293T cells, wild-type CNK3 significantly increased surface biotinylated ENaC (Fig. 7A, top panel, Pulldown: N, IB: β-ENaC, compare lanes 3 and 4). Also consistent with the two-dimensional gel experiments as well as with the co-IP experiments (Fig. 5B), CNK3 was present in the surface biotinylated fraction only when ENaC was co-expressed (Fig. 7A, top panel, Pulldown: N, IB: V5, compare lanes 2 and 4), supporting the idea that ENaC brings CNK3 to the cell surface. In contrast to wild-type CNK3, we were not able to detect any cell surface-associated ΔPDZ-CNK3 (Fig. 7A, top panel, Pulldown: N, IB: V5, lane 5), nor did ΔPDZ-CNK3 stimulate ENaC surface expression (Fig. 7A, top panel, Pulldown: N, IB: β-ENaC, lane 5). In fact, ΔPDZ-CNK3 appeared to have a dominant negative effect.
FIGURE 7.

CNK3 interacts with ENaC at the cell surface and significantly stimulates its plasma membrane abundance. A, cell surface biotinylation assays in HEK 293T cells showing a marked reduction in the ability of ΔPDZ-CNK3 to stimulate surface expression of ENaC in contrast to that of WT-CNK3. Also shown is the dependence of CNK3 on ENaC for its surface recruitment. GAPDH served as a negative control for NeutrAvidin IP (“N”) blots that primarily contained surface proteins (n = 4). Sequential cell surface biotinylation and co-immunoprecipitation assays confirm that WT-CNK3 interacts with cell surface ENaC and stimulates its plasma membrane abundance (see “Experimental Procedures” for experimental details). Blots showing total cellular expression of the various proteins specified are also depicted. In whole-cell lysates (WCL), GAPDH was used as a loading control (n = 4). B, cell surface biotinylation assays for endogenous ENaC, CNK3, and SGK1 proteins in mpkCCDc14 kidney epithelial cells. Aldosterone stimulation (1 μm for 3 h) markedly augments co-localization of ENaC and associated CNK3 and SGK1 proteins on the cell surface. Blots showing total cellular expression of the various proteins specified are also depicted. In whole-cell lysates, α-tubulin was used as a loading control (n = 4). C, graphical representation of the -fold increase in total (left bar) versus plasma membrane-associated (right bar) CNK3 in response to 3-h treatment with aldosterone. Surface expression (NeutrAvidin pulldown) and whole-cell lysate data from B were quantitated by densitometry. Bars represent signal ratios in the presence versus absence of aldosterone. Total CNK3 continued to increase at later time points, reaching a peak of ∼3-fold stimulation at 6–8 h of aldosterone treatment (not shown; see text for details). ***, p < 0.001 (n = 4). Error bars represent S.E. IB, immunoblot.
To confirm that the capture of CNK3 in the surface biotin-labeled fraction was due to its interaction with ENaC, double pulldown experiments were performed. Eluates of the cell surface proteins isolated by biotinylation as above were subjected to immunoprecipitation using anti-FLAG antibody (to capture FLAG-ENaC), and then these immunoprecipitates were probed for CNK3 (V5) by Western blot. This sequential pulldown confirms that WT-CNK3 physically interacts with cell surface ENaC and that it significantly stimulates ENaC surface expression (Fig. 7A, middle panel of blots). Consistent with results shown thus far, ΔPDZ-CNK3 failed to co-fractionate with plasma membrane ENaC and inhibited rather than stimulated ENaC cell surface expression. The observed changes were restricted to surface ENaC in that total cellular ENaC levels were not affected by CNK3 expression (Fig. 7A, whole cell lysate panel of blots, IB: β-ENaC).
Next we examined the effects of aldosterone on cell surface recruitment of endogenous ENaC, SGK1, and CNK3 by performing cell surface biotinylation assays in untransfected mpkCCDc14 renal epithelial cells (Fig. 7B). As expected, upon aldosterone stimulation, we found enhanced levels of endogenous surface ENaC (Fig. 7B, WCL panel, IB: CNK3). In the same fraction, we could also detect robust aldosterone-stimulated surface localization of CNK3 as well as SGK1. Interestingly, the increase in plasma membrane abundance of CNK3 after 3-h treatment with aldosterone was substantially more robust than the increase in total cellular levels (Fig. 7C). At later time points, CNK3 expression increased further (data not shown). Double pulldown of endogenous ENaC (as in Fig. 7A) was not technically feasible; however, taken together with the data in Fig. 7A, these results strongly support the conclusion that endogenous CNK3 physically interacts with ENaC at the plasma membrane in a PDZ domain-dependent, aldosterone-stimulated fashion.
Finally, to confirm the functional importance of the PDZ domain in aldosterone-regulated transepithelial Na+ transport, we performed rescue experiments in mpkCCDc14 cells in which expression of endogenous CNK3 was knocked down (Fig. 8). mpkCCDc14 “shRNA4” cells stably silencing the CNK3 gene or “control” parental mpkCCDc14 cells were transiently transfected with shRNA-resistant expression constructs (res-WT-CNK3 and res-ΔPDZ-CNK3, which harbor silent mutations that confer shRNA resistance) or empty vector as specified. Cells were grown on Transwell filters and analyzed for aldosterone-induced ENaC activity as described previously (32). We found that res-WT-CNK3 restored transepithelial Na+ transport in cells lacking endogenous CNK3, but res-ΔPDZ-CNK3 did not (Fig. 8), confirming the functional importance of CNK3 and strongly supporting the conclusion that its PDZ domain is required for its ENaC-stimulatory activity.
FIGURE 8.

The PDZ domain in CNK3 is essential for its ability to stimulate ENaC activity. In contrast to full-length-CNK3, ΔPDZ-CNK3 cannot restore aldosterone-induced ENaC activity in mpkCCDc14 cells stably silencing the expression of endogenous CNK3. mpkCCDc14 cells stably silencing the expression of endogenous CNK3 (shRNA4) were transiently transfected with shRNA-resistant full-length (“res-CNK3”) or ΔPDZ-CNK3 (“res-ΔPDZ-CNK3”) constructs using nucleofection. Parental mpkCCDc14 cells nucleofected with an empty vector (VC) were used as a positive control (control). Unmodified wild-type CNK3 expression construct was used as an additional control (“WT-CNK3”). Shown are a graphical representation of the Ieq measured 3 h after aldosterone stimulation (1 μm) and a representative Western blot demonstrating the rescue of the exogenously expressed shRNA-resistant WT- and ΔPDZ-CNK3-V5 constructs. *, p < 0.05; ***, p < 0.001 (n = 6). Error bars represent S.E. IB, immunoblot; WCL, whole-cell lysate.
CNK3 Physically Interacts with ENaC C-terminal Tails
The above experiments demonstrate that CNK3 interacts with ENaC at the plasma membrane, but they do not assess whether this represents a direct physical interaction. We therefore examined whether CNK3 physically interacts with ENaC in vitro. The C-terminal tails of ENaC subunits are important sites of regulation and have been shown to interact with other regulators, including SGK1, ERK1/2, and Nedd4-2. We therefore expressed α- and β-ENaC C-terminal tails in E. coli as glutathione S-transferase fusion proteins, captured them on glutathione-Sepharose beads, and purified them to near homogeneity. The immobilized fusion proteins were then incubated with 35S-labeled CNK3 expressed in reticulocyte lysate as described previously (24). We observed that in vitro translated full-length CNK3 specifically interacted with α- and β-ENaC (Fig. 9, lanes 2 and 3) but exhibited negligible binding to GST alone (Fig. 9, lane 1). These data provide strong support for the idea that CNK3 physically interacts with the α- and β-ENaC tails with the caveat that a generic component of reticulocyte lysate could function as an intermediary.
FIGURE 9.
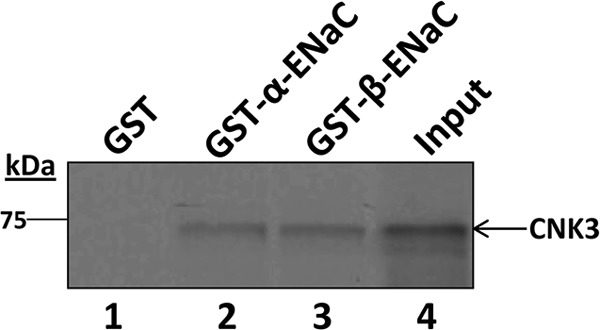
CNK3 physically interacts with α- and β-ENaC in vitro. The [35S]Met-labeled in vitro translation product for wild-type CNK3 was incubated with GST alone, GST-α-ENaC, or GST-β-ENaC as indicated. After recovery of fusion proteins on glutathione-Sepharose beads, the bound fraction was analyzed by SDS-PAGE and visualized by autoradiography. 10% of in vitro translated product used in the binding assays is represented as “Input.” Shown is a representative autoradiograph from five independent GST pulldown assays performed. Similar results were obtained in all the experiments performed.
DISCUSSION
A variety of evidence over the last several years has suggested that ion channels and transporters are contained within multiprotein complexes, which are required for their regulation. However, direct characterization of intact complexes with identification of validated regulators has been scarce. In the case of ENaC, several reports have addressed the potential role of ENaC-interacting proteins (9, 63, 64) and aldosterone-induced proteins (65, 66) in the control of plasma membrane channel levels and activity, and a few have isolated intact multiprotein complexes. In at least two reports (14, 15), complexes >600 kDa were identified, and one of these was followed up with identification of some components of the intact complex, including a 41-kDa pertussis toxin-sensitive Gαi-3 (67). However, functional roles of associated proteins in ENaC regulation were not established. A functional picture only began to emerge subsequently with the identification of ENaC-interacting proteins (54) and aldosterone-induced proteins using recombinant DNA methodologies (25, 32, 68–72). In particular, the ubiquitin ligase Nedd4-2 was identified in a yeast two-hybrid interaction screen as an ENaC-interacting protein (54), whereas SGK1 was identified as an aldosterone-induced mRNA using a subtractive library approach (68). Together with the subsequent demonstration that SGK1 phosphorylates and inhibits Nedd4-2 (20), these findings provided a new paradigm in ion channel regulation through kinase-dependent modulation of a channel-inhibitory ubiquitin ligase. More recently, it has become clear that SGK1 interacts with ENaC at the plasma membrane (22, 24) as well as with Nedd4-2 and Raf-1 (12, 21, 23). Indeed, based on co-IP data, it appeared that inhibitory and stimulatory ENaC factors are present together in a multiprotein ENaC-associated complex, which physically interacts with the channel and controls its subcellular localization and stability (12). However, the demonstration of an intact complex containing ENaC along with key established regulators at the cell surface has been challenging. More importantly, the basis of assembly of this putative multiprotein complex has also remained elusive.
Here we report for the first time the identification of an ∼1.0–1.2 MDa native complex containing ENaC and key regulatory factors, including SGK1, Nedd4-2, c-Raf, and CNK3, at the plasma membrane in mpkCCDc14 cells treated with aldosterone (Fig. 1B). A complex of the same size and containing these same regulators was also identified in plasma membrane isolated from HEK 293T cells transiently transfected with ENaC and a select set of regulators (Fig. 1D). Together with previous data, our present findings support the idea that in the absence of aldosterone Nedd4-2 and Raf-1 (and likely additional proteins) associate with the channel and trigger channel internalization and degradation (73–77). In the presence of aldosterone, the levels of stimulatory factors (notably SGK1 and CNK3) rise, are recruited into the ERC, and inhibit Nedd4-2 and the Raf-MEK-ERK signaling module, thereby increasing ENaC retention at the plasma membrane and hence steady state levels (Figs. 3–9).
Our data further suggest that CNK3 functions as a scaffold for this complex. Scaffold proteins play key roles in facilitating specific signal transduction through potentially pleiotropic networks by interacting with multiple components of signaling cascades, tethering them into complexes, and localizing them at particular subcellular compartments, thereby enhancing signaling specificity. Our data demonstrate that CNK3 interacts with ENaC itself (Figs. 5B and 7A), that this interaction is direct (Fig. 9), and that it occurs at the cell surface in an aldosterone-dependent manner (Fig. 7B). Interestingly, the effect of aldosterone on CNK3 association with ENaC at the plasma membrane is more robust and occurs more rapidly than stimulation of CNK3 expression (Fig. 7B). The basis for this effect is uncertain at this time, but it seems likely that it reflects the actions of more rapidly induced aldosterone target genes on preformed CNK3. CNK3 also physically interacts with SGK1 and Nedd4-2 (Fig. 5, A and C) and stimulates interactions of SGK1 with both ENaC (Fig. 3) and Nedd4-2 (Fig. 4). The interactions of CNK3 with ENaC and with SGK1, but not with Nedd4-2 or Raf-1, were dependent on an intact PDZ domain (Fig. 6 and supplemental Fig. 2). Importantly, the ability of CNK3 to stimulate interaction of SGK1 and ENaC was dependent on an intact PDZ domain as was its ability to promote ENaC cell surface expression (Fig. 7A) or stimulate Na+ current (Fig. 8). Indeed, Na+ transport in polarized mpkCCDc14 cells in which endogenous CNK3 was suppressed by shRNA could be restored by exogenous expression of shRNA-resistant WT-CNK3 but not by shRNA-resistant ΔPDZ-CNK3 (Fig. 8).
It is notable that PDZ domain-containing proteins have been found previously to regulate membrane transporters in polarized epithelial cell membranes (78, 79). For instance, the stimulatory effects of glucocorticoids on the NHE3 in proximal tubule cells are at least in part dependent on SGK1, which is recruited in a PDZ domain-dependent interaction by NHERF2 to the plasma membrane (60). Similarly, the ability of the PDZ protein NHERF1 to interact dynamically with the parathyroid hormone receptor (a G protein-coupled membrane receptor) and cognate adaptor proteins regulates receptor trafficking and signaling in a spatially and temporally coordinated manner (61). A detailed analysis of precise residues within the PDZ domain important for the association of CNK3 with its interacting ENaC-regulatory proteins is an important issue and remains to be elucidated. It will also be of great interest to identify the structural motifs involved in CNK3 interaction with Nedd4-2 and Raf-1 and to determine how these motifs are utilized in conjunction with the PDZ domain to effect the dynamic assembly and disassembly of the ERC.
In conclusion, we demonstrate the presence of an aldosterone-induced ∼1.0–1.2-MDa ENaC-containing native complex at the plasma membrane and also provide evidence that CNK3 is a hormone-induced scaffolding platform that orchestrates the assembly of this complex, recruits SGK1, and thereby is essential for the stimulatory effects of aldosterone on ENaC. This large complex undoubtedly contains additional protein components, and a key next step is their identification and characterization both at the plasma membrane and in other intracellular compartments. In this latter regard, it will be of particular interest to identify the compartment(s) in which GILZ1, which is not present in the plasma membrane ERC, interacts with ERC components such as SGK1. Furthermore, understanding the precise mechanism of ERC function will require knowledge of the order and kinetics of its assembly and disassembly and ascertaining which of the protein-protein interactions are direct and which result indirectly from complex formation.
Supplementary Material
Acknowledgments
We thank Roland Bainton and Catherine Gleason for critical reading of the manuscript and helpful suggestions.
This work was supported, in whole or in part, by National Institutes of Health Grants K01-DK078679 (to R. S.), R01-DK56695 (to D. P.), and R01-DK85101 (to D. P.). This work was also supported by a Manning Foundation award (to D. P.).

This article contains supplemental Figs. 1 and 2.
- ENaC
- epithelial sodium channel
- GILZ1
- glucocorticoid-induced leucine zipper protein 1
- CCD
- cortical collecting duct
- SGK1
- serum- and glucocorticoid-induced kinase 1
- Nedd4-2
- neural precursor cell-expressed, developmentally down-regulated protein
- CNK3
- connector enhancer of kinase suppressor of Ras isoform 3
- PDZ
- PSD-95/DLG-1/ZO-1
- NHERF
- Na+/H+ exchanger-regulatory factor
- IP
- immunoprecipitation
- Ab
- antibody
- BN
- blue native
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- NHS
- N-hydroxysuccinimide.
REFERENCES
- 1. Soundararajan R., Pearce D., Hughey R. P., Kleyman T. R. (2010) Role of epithelial sodium channels and their regulators in hypertension. J. Biol. Chem. 285, 30363–30369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Butterworth M. B. (2010) Regulation of the epithelial sodium channel (ENaC) by membrane trafficking. Biochim. Biophys. Acta 1802, 1166–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Capuano P., Bacic D., Stange G., Hernando N., Kaissling B., Pal R., Kocher O., Biber J., Wagner C. A., Murer H. (2005) Expression and regulation of the renal Na/phosphate cotransporter NaPi-IIa in a mouse model deficient for the PDZ protein PDZK1. Pflugers Arch. 449, 392–402 [DOI] [PubMed] [Google Scholar]
- 4. Sandberg M. B., Riquier A. D., Pihakaski-Maunsbach K., McDonough A. A., Maunsbach A. B. (2007) ANG II provokes acute trafficking of distal tubule Na+-Cl− cotransporter to apical membrane. Am. J. Physiol. Renal Physiol. 293, F662–F669 [DOI] [PubMed] [Google Scholar]
- 5. Snyder P. M. (2005) Minireview: regulation of epithelial Na+ channel trafficking. Endocrinology 146, 5079–5085 [DOI] [PubMed] [Google Scholar]
- 6. Welker P., Böhlick A., Mutig K., Salanova M., Kahl T., Schlüter H., Blottner D., Ponce-Coria J., Gamba G., Bachmann S. (2008) Renal Na+-K+-Cl− cotransporter activity and vasopressin-induced trafficking are lipid raft-dependent. Am. J. Physiol. Renal Physiol. 295, F789–F802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Donowitz M., Mohan S., Zhu C. X., Chen T. E., Lin R., Cha B., Zachos N. C., Murtazina R., Sarker R., Li X. (2009) NHE3 regulatory complexes. J. Exp. Biol. 212, 1638–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ismailov I. I., Berdiev B. K., Bradford A. L., Awayda M. S., Fuller C. M., Benos D. J. (1996) Associated proteins and renal epithelial Na+ channel function. J. Membr. Biol. 149, 123–132 [DOI] [PubMed] [Google Scholar]
- 9. McDonald F. J., Welsh M. J. (1995) Binding of the proline-rich region of the epithelial Na+ channel to SH3 domains and its association with specific cellular proteins. Biochem. J. 312, 491–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dimke H. (2011) Exploring the intricate regulatory network controlling the thiazide-sensitive NaCl cotransporter (NCC). Pflugers Arch. 462, 767–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hernando N., Gisler S. M., Reining S. C., Déliot N., Capuano P., Biber J., Murer H. (2010) NaPi-IIa interacting proteins and regulation of renal reabsorption of phosphate. Urol. Res. 38, 271–276 [DOI] [PubMed] [Google Scholar]
- 12. Soundararajan R., Melters D., Shih I. C., Wang J., Pearce D. (2009) Epithelial sodium channel regulated by differential composition of a signaling complex. Proc. Natl. Acad. Sci. U.S.A. 106, 7804–7809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tanimura A., Yamada F., Saito A., Ito M., Kimura T., Anzai N., Horie D., Yamamoto H., Miyamoto K., Taketani Y., Takeda E. (2011) Analysis of different complexes of type IIa sodium-dependent phosphate transporter in rat renal cortex using blue-native polyacrylamide gel electrophoresis. J. Med. Invest. 58, 140–147 [DOI] [PubMed] [Google Scholar]
- 14. Staruschenko A., Medina J. L., Patel P., Shapiro M. S., Booth R. E., Stockand J. D. (2004) Fluorescence resonance energy transfer analysis of subunit stoichiometry of the epithelial Na+ channel. J. Biol. Chem. 279, 27729–27734 [DOI] [PubMed] [Google Scholar]
- 15. Benos D. J., Saccomani G., Brenner B. M., Sariban-Sohraby S. (1986) Purification and characterization of the amiloride-sensitive sodium channel from A6 cultured cells and bovine renal papilla. Proc. Natl. Acad. Sci. U.S.A. 83, 8525–8529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li X., Zhang H., Cheong A., Leu S., Chen Y., Elowsky C. G., Donowitz M. (2004) Carbachol regulation of rabbit ileal brush border Na+-H+ exchanger 3 (NHE3) occurs through changes in NHE3 trafficking and complex formation and is Src dependent. J. Physiol. 556, 791–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berger S., Bleich M., Schmid W., Cole T. J., Peters J., Watanabe H., Kriz W., Warth R., Greger R., Schütz G. (1998) Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc. Natl. Acad. Sci. U.S.A. 95, 9424–9429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Christensen B. M., Perrier R., Wang Q., Zuber A. M., Maillard M., Mordasini D., Malsure S., Ronzaud C., Stehle J. C., Rossier B. C., Hummler E. (2010) Sodium and potassium balance depends on αENaC expression in connecting tubule. J. Am. Soc. Nephrol. 21, 1942–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Soundararajan R., Wang J., Melters D., Pearce D. (2010) Glucocorticoid-induced Leucine zipper 1 stimulates the epithelial sodium channel by regulating serum- and glucocorticoid-induced kinase 1 stability and subcellular localization. J. Biol. Chem. 285, 39905–39913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Debonneville C., Flores S. Y., Kamynina E., Plant P. J., Tauxe C., Thomas M. A., Münster C., Chraïbi A., Pratt J. H., Horisberger J. D., Pearce D., Loffing J., Staub O. (2001) Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J. 20, 7052–7059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Snyder P. M., Olson D. R., Thomas B. C. (2002) Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J. Biol. Chem. 277, 5–8 [DOI] [PubMed] [Google Scholar]
- 22. Thomas S. V., Kathpalia P. P., Rajagopal M., Charlton C., Zhang J., Eaton D. C., Helms M. N., Pao A. C. (2011) Epithelial sodium channel regulation by cell surface-associated serum- and glucocorticoid-regulated kinase 1. J. Biol. Chem. 286, 32074–32085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou R., Patel S. V., Snyder P. M. (2007) Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J. Biol. Chem. 282, 20207–20212 [DOI] [PubMed] [Google Scholar]
- 24. Wang J., Barbry P., Maiyar A. C., Rozansky D. J., Bhargava A., Leong M., Firestone G. L., Pearce D. (2001) SGK integrates insulin and mineralocorticoid regulation of epithelial sodium transport. Am. J. Physiol. Renal Physiol. 280, F303–F313 [DOI] [PubMed] [Google Scholar]
- 25. Ziera T., Irlbacher H., Fromm A., Latouche C., Krug S. M., Fromm M., Jaisser F., Borden S. A. (2009) Cnksr3 is a direct mineralocorticoid receptor target gene and plays a key role in the regulation of the epithelial sodium channel. FASEB J. 23, 3936–3946 [DOI] [PubMed] [Google Scholar]
- 26. Fritz R. D., Radziwill G. (2005) The scaffold protein CNK1 interacts with the angiotensin II type 2 receptor. Biochem. Biophys. Res. Commun. 338, 1906–1912 [DOI] [PubMed] [Google Scholar]
- 27. Lim J., Zhou M., Veenstra T. D., Morrison D. K. (2010) The CNK1 scaffold binds cytohesins and promotes insulin pathway signaling. Genes Dev. 24, 1496–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bilder D. (2001) PDZ proteins and polarity: functions from the fly. Trends Genet. 17, 511–519 [DOI] [PubMed] [Google Scholar]
- 29. Hernando N., Gisler S. M., Pribanic S., Déliot N., Capuano P., Wagner C. A., Moe O. W., Biber J., Murer H. (2005) NaPi-IIa and interacting partners. J. Physiol. 567, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Biber J., Gisler S. M., Hernando N., Wagner C. A., Murer H. (2004) PDZ interactions and proximal tubular phosphate reabsorption. Am. J. Physiol. Renal Physiol. 287, F871–F875 [DOI] [PubMed] [Google Scholar]
- 31. Cunningham R., Biswas R., Steplock D., Shenolikar S., Weinman E. (2010) Role of NHERF and scaffolding proteins in proximal tubule transport. Urol. Res. 38, 257–262 [DOI] [PubMed] [Google Scholar]
- 32. Soundararajan R., Zhang T. T., Wang J., Vandewalle A., Pearce D. (2005) A novel role for glucocorticoid-induced leucine zipper protein in epithelial sodium channel-mediated sodium transport. J. Biol. Chem. 280, 39970–39981 [DOI] [PubMed] [Google Scholar]
- 33. Invitrogen (2006) ViraPower Lentiviral Expression Systems User Manual, Invitrogen, Carlsbad, CA [Google Scholar]
- 34. Soundararajan R., Wang J., Melters D., Pearce D. (2007) Differential activities of glucocorticoid-induced leucine zipper protein isoforms. J. Biol. Chem. 282, 36303–36313 [DOI] [PubMed] [Google Scholar]
- 35. Camacho-Carvajal M. M., Wollscheid B., Aebersold R., Steimle V., Schamel W. W. (2004) Two-dimensional blue native/SDS gel electrophoresis of multi-protein complexes from whole cellular lysates: a proteomics approach. Mol. Cell. Proteomics 3, 176–182 [DOI] [PubMed] [Google Scholar]
- 36. Swamy M., Siegers G. M., Minguet S., Wollscheid B., Schamel W. W. (2006) Blue native polyacrylamide gel electrophoresis (BN-PAGE) for the identification and analysis of multiprotein complexes. Sci. STKE 2006, pl4. [DOI] [PubMed] [Google Scholar]
- 37. Reisinger V., Eichacker L. A. (2008) Solubilization of membrane protein complexes for blue native PAGE. J. Proteomics 71, 277–283 [DOI] [PubMed] [Google Scholar]
- 38. Fiala G. J., Schamel W. W., Blumenthal B. (2011) Blue native polyacrylamide gel electrophoresis (BN-PAGE) for analysis of multiprotein complexes from cellular lysates. J. Vis. Exp. 2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Benos D. J., Saccomani G., Sariban-Sohraby S. (1987) The epithelial sodium channel. Subunit number and location of the amiloride binding site. J. Biol. Chem. 262, 10613–10618 [PubMed] [Google Scholar]
- 40. Xia H., von Zastrow M., Malenka R. C. (2002) A novel anterograde trafficking signal present in the N-terminal extracellular domain of ionotropic glutamate receptors. J. Biol. Chem. 277, 47765–47769 [DOI] [PubMed] [Google Scholar]
- 41. Prince L. S., Welsh M. J. (1998) Cell surface expression and biosynthesis of epithelial Na+ channels. Biochem. J. 336, 705–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pearce D. (2003) SGK1 regulation of epithelial sodium transport. Cell. Physiol. Biochem. 13, 13–20 [DOI] [PubMed] [Google Scholar]
- 43. Loffing J., Flores S. Y., Staub O. (2006) Sgk kinases and their role in epithelial transport. Annu. Rev. Physiol. 68, 461–490 [DOI] [PubMed] [Google Scholar]
- 44. Lang F., Artunc F., Vallon V. (2009) The physiological impact of the serum and glucocorticoid-inducible kinase SGK1. Curr. Opin. Nephrol. Hypertens. 18, 439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee I. H., Campbell C. R., Cook D. I., Dinudom A. (2008) Regulation of epithelial Na+ channels by aldosterone: role of Sgk1. Clin. Exp. Pharmacol. Physiol. 35, 235–241 [DOI] [PubMed] [Google Scholar]
- 46. Lang F., Böhmer C., Palmada M., Seebohm G., Strutz-Seebohm N., Vallon V. (2006) (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol. Rev. 86, 1151–1178 [DOI] [PubMed] [Google Scholar]
- 47. Wulff P., Vallon V., Huang D. Y., Völkl H., Yu F., Richter K., Jansen M., Schlünz M., Klingel K., Loffing J., Kauselmann G., Bösl M. R., Lang F., Kuhl D. (2002) Impaired renal Na+ retention in the sgk1-knockout mouse. J. Clin. Investig. 110, 1263–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fejes-Tóth G., Frindt G., Náray-Fejes-Tóth A., Palmer L. G. (2008) Epithelial Na+ channel activation and processing in mice lacking SGK1. Am. J. Physiol. Renal Physiol. 294, F1298–F1305 [DOI] [PubMed] [Google Scholar]
- 49. Flores S. Y., Loffing-Cueni D., Kamynina E., Daidié D., Gerbex C., Chabanel S., Dudler J., Loffing J., Staub O. (2005) Aldosterone-induced serum and glucocorticoid-induced kinase 1 expression is accompanied by Nedd4-2 phosphorylation and increased Na+ transport in cortical collecting duct cells. J. Am. Soc. Nephrol. 16, 2279–2287 [DOI] [PubMed] [Google Scholar]
- 50. Bhalla V., Daidié D., Li H., Pao A. C., LaGrange L. P., Wang J., Vandewalle A., Stockand J. D., Staub O., Pearce D. (2005) Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Mol. Endocrinol. 19, 3073–3084 [DOI] [PubMed] [Google Scholar]
- 51. Diakov A., Korbmacher C. (2004) A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel's α-subunit. J. Biol. Chem. 279, 38134–38142 [DOI] [PubMed] [Google Scholar]
- 52. Wiemuth D., Lott J. S., Ly K., Ke Y., Teesdale-Spittle P., Snyder P. M., McDonald F. J. (2010) Interaction of serum- and glucocorticoid regulated kinase 1 (SGK1) with the WW-domains of Nedd4-2 is required for epithelial sodium channel regulation. PLoS One 5, e12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Itani O. A., Stokes J. B., Thomas C. P. (2005) Nedd4-2 isoforms differentially associate with ENaC and regulate its activity. Am. J. Physiol. Renal Physiol. 289, F334–F346 [DOI] [PubMed] [Google Scholar]
- 54. Staub O., Dho S., Henry P., Correa J., Ishikawa T., McGlade J., Rotin D. (1996) WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle's syndrome. EMBO J. 15, 2371–2380 [PMC free article] [PubMed] [Google Scholar]
- 55. Henry P. C., Kanelis V., O'Brien M. C., Kim B., Gautschi I., Forman-Kay J., Schild L., Rotin D. (2003) Affinity and specificity of interactions between Nedd4 isoforms and the epithelial Na+ channel. J. Biol. Chem. 278, 20019–20028 [DOI] [PubMed] [Google Scholar]
- 56. Bens M., Vallet V., Cluzeaud F., Pascual-Letallec L., Kahn A., Rafestin-Oblin M. E., Rossier B. C., Vandewalle A. (1999) Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J. Am. Soc. Nephrol. 10, 923–934 [DOI] [PubMed] [Google Scholar]
- 57. Soundararajan R., Pearce D., Ziera T. (2012) The role of the ENaC-regulatory complex in aldosterone-mediated sodium transport. Mol. Cell. Endocrinol. 350, 242–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jaffe A. B., Aspenström P., Hall A. (2004) Human CNK1 acts as a scaffold protein, linking Rho and Ras signal transduction pathways. Mol. Cell. Biol. 24, 1736–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shaw A. S., Filbert E. L. (2009) Scaffold proteins and immune-cell signalling. Nat. Rev. Immunol. 9, 47–56 [DOI] [PubMed] [Google Scholar]
- 60. Yun C. C., Chen Y., Lang F. (2002) Glucocorticoid activation of Na+/H+ exchanger isoform 3 revisited. The roles of SGK1 and NHERF2. J. Biol. Chem. 277, 7676–7683 [DOI] [PubMed] [Google Scholar]
- 61. Ardura J. A., Wang B., Watkins S. C., Vilardaga J. P., Friedman P. A. (2011) Dynamic Na+-H+ exchanger regulatory factor-1 association and dissociation regulate parathyroid hormone receptor trafficking at membrane microdomains. J. Biol. Chem. 286, 35020–35029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bacic D., Wagner C. A., Hernando N., Kaissling B., Biber J., Murer H. (2004) Novel aspects in regulated expression of the renal type IIa Na/Pi-cotransporter. Kidney Int. Suppl. S5–S12 [DOI] [PubMed] [Google Scholar]
- 63. Ilatovskaya D. V., Pavlov T. S., Levchenko V., Negulyaev Y. A., Staruschenko A. (2011) Cortical actin binding protein cortactin mediates ENaC activity via Arp2/3 complex. FASEB J. 25, 2688–2699 [DOI] [PubMed] [Google Scholar]
- 64. Zuckerman J. B., Chen X., Jacobs J. D., Hu B., Kleyman T. R., Smith P. R. (1999) Association of the epithelial sodium channel with Apx and α-spectrin in A6 renal epithelial cells. J. Biol. Chem. 274, 23286–23295 [DOI] [PubMed] [Google Scholar]
- 65. Blazer-Yost B., Cox M. (1985) Aldosterone-induced proteins: characterization using lectin-affinity chromatography. Am. J. Physiol. Cell Physiol. 249, C215–C225 [DOI] [PubMed] [Google Scholar]
- 66. Blazer-Yost B., Geheb M., Preston A., Handler J., Cox M. (1982) Aldosterone-induced proteins in renal epithelia. Biochim. Biophys. Acta 719, 158–161 [DOI] [PubMed] [Google Scholar]
- 67. Ausiello D. A., Stow J. L., Cantiello H. F., de Almeida J. B., Benos D. J. (1992) Purified epithelial Na+ channel complex contains the pertussis toxin-sensitive Gαi-3 protein. J. Biol. Chem. 267, 4759–4765 [PubMed] [Google Scholar]
- 68. Chen S. Y., Bhargava A., Mastroberardino L., Meijer O. C., Wang J., Buse P., Firestone G. L., Verrey F., Pearce D. (1999) Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc. Natl. Acad. Sci. U.S.A. 96, 2514–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Robert-Nicoud M., Flahaut M., Elalouf J. M., Nicod M., Salinas M., Bens M., Doucet A., Wincker P., Artiguenave F., Horisberger J. D., Vandewalle A., Rossier B. C., Firsov D. (2001) Transcriptome of a mouse kidney cortical collecting duct cell line: effects of aldosterone and vasopressin. Proc. Natl. Acad. Sci. U.S.A. 98, 2712–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Spindler B., Mastroberardino L., Custer M., Verrey F. (1997) Characterization of early aldosterone-induced RNAs identified in A6 kidney epithelia. Pflugers Arch. 434, 323–331 [DOI] [PubMed] [Google Scholar]
- 71. Stockand J. D., Spier B. J., Worrell R. T., Yue G., Al-Baldawi N., Eaton D. C. (1999) Regulation of Na+ reabsorption by the aldosterone-induced small G protein K-Ras2A. J. Biol. Chem. 274, 35449–35454 [DOI] [PubMed] [Google Scholar]
- 72. Alvarez de la Rosa D., Zhang P., Náray-Fejes-Tóth A., Fejes-Tóth G., Canessa C. M. (1999) The serum and glucocorticoid kinase sgk increases the abundance of epithelial sodium channels in the plasma membrane of Xenopus oocytes. J. Biol. Chem. 274, 37834–37839 [DOI] [PubMed] [Google Scholar]
- 73. Shi H., Asher C., Chigaev A., Yung Y., Reuveny E., Seger R., Garty H. (2002) Interactions of β and γ ENaC with Nedd4 can be facilitated by an ERK-mediated phosphorylation. J. Biol. Chem. 277, 13539–13547 [DOI] [PubMed] [Google Scholar]
- 74. Booth R. E., Stockand J. D. (2003) Targeted degradation of ENaC in response to PKC activation of the ERK1/2 cascade. Am. J. Physiol. Renal Physiol. 284, F938–F947 [DOI] [PubMed] [Google Scholar]
- 75. Bhalla V., Hallows K. R. (2008) Mechanisms of ENaC regulation and clinical implications. J. Am. Soc. Nephrol. 19, 1845–1854 [DOI] [PubMed] [Google Scholar]
- 76. Bhargava A., Wang J., Pearce D. (2004) Regulation of epithelial ion transport by aldosterone through changes in gene expression. Mol. Cell. Endocrinol. 217, 189–196 [DOI] [PubMed] [Google Scholar]
- 77. Kamynina E., Staub O. (2002) Concerted action of ENaC, Nedd4-2, and Sgk1 in transepithelial Na+ transport. Am. J. Physiol. Renal Physiol. 283, F377–F387 [DOI] [PubMed] [Google Scholar]
- 78. Brône B., Eggermont J. (2005) PDZ proteins retain and regulate membrane transporters in polarized epithelial cell membranes. Am. J. Physiol. Cell Physiol. 288, C20–C29 [DOI] [PubMed] [Google Scholar]
- 79. Hernando N., Wagner C. A., Gisler S. M., Biber J., Murer H. (2004) PDZ proteins and proximal ion transport. Curr. Opin. Nephrol. Hypertens. 13, 569–574 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.