

Background: Histone chaperone Spt6 is required for CSR, but the mechanism remains unknown.
Results: Spt6 preserves H3K4me3 mark on the target chromatin, which is required for AID induced DNA cleavage of CSR and SHM.
Conclusion: Target-specific DNA breaks and AID expression are controlled by two distinct modes of histone epigenetic regulation by Spt6.
Significance: Chromatin plays a key role in controlling AID-induced genomic instability.
Keywords: Chromatin, DNA Damage, DNA Recombination, Gene Regulation, Genomic Instability, Histone Chaperone, Histone Modification, Immunology, Mutagenesis Mechanisms
Abstract
H3K4me3 plays a critical role in the activation-induced cytidine deaminase (AID)-induced DNA cleavage of switch (S) regions in the immunoglobulin heavy chain (IgH) locus during class-switch recombination (CSR). The histone chaperone complex facilitates chromatin transcription (FACT) is responsible for forming H3K4me3 at AID target loci. Here we show that the histone chaperone suppressor of Ty6 (Spt6) also participates in regulating H3K4me3 for CSR and for somatic hypermutation in AID target loci. We found that H3K4me3 loss was correlated with defects in AID-induced DNA breakage and reduced mutation frequencies in IgH loci in both S and variable regions and in non-IgH loci such as metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) and small nucleolar RNA host gene 3 (SNHG3). Global gene expression analysis revealed that Spt6 can act as both a positive and negative transcriptional regulator in B cells, affecting ∼5% of the genes that includes suppressor of Ty4 (Spt4) and AID. Interestingly, Spt6 regulates CSR and AID expression through two distinct histone modification pathways, H3K4me3 and H3K36me3, respectively. Tandem SH2 domain of Spt6 plays a critical role in CSR and H3K4me3 regulation involving Set1 histone methyltransferase. We conclude that Spt6 is a unique histone chaperone capable of regulating the histone epigenetic state of both AID targets and the AID locus.
Introduction
The classical view that genetic recombination relies on a DNA motif recognized by a specific DNA-cleaving enzyme is changing rapidly; a growing body of research points to the involvement of chromatin modifications in regulating recombination. Recent studies of histone epigenetic regulation have brought new perspectives to various aspects of recombination from the meiotic crossover breakpoints in germ cells to the antigen receptor gene rearrangement in T and B cells (1–6). Recombination breakpoints appear to be epigenetically marked by H3K4me3 in three distinct programmed recombinations: Spo11-induced meiotic recombination, RAG-induced V(D)J recombination, and AID3-induced class-switch recombination (CSR) (3, 6, 7).
Although depleting H3K4me3, either globally or at specific sites, decreases all three of these recombinations, H3K4me3 involvement in the target recognition process is not fully understood (6, 8). The RAG2 PHD domain recognizes H3K4me3, which explains how H3K4me3 can tether the RAG complex to transcriptionally active loci. H3K4me3 also has a stimulatory effect that suggests it is involved in both the recruiting and the enzymatic activity of V(D)J recombinase (9). In the case of meiotic recombination, the hotspot-specific H3K4me3 methylase Prdm9, found in mammalian germ cells, is critical for positioning the recombination sites (1, 2). It has recently been demonstrated that hotspot-specific H3K4me3 marks can be distinguished from transcription-associated H3K4me3 marks (7). However, meiotic recombination-specific and transcription-specific H3K4me3 are both mediated by the single histone methyltransferase Set1 in budding yeast, and disrupting Set1 leads to defective meiotic recombination (8).
CSR requires not only AID but also germ line transcription of the recombining switch (S) regions (10–12). The functional role of germline transcript (GLT) in CSR has long been debated ever since it became clear that transcripts per se do not encode protein. The transcripts may serve to form a heteroduplex with transcribed DNA, thus forming an R-loop (13). An obvious consequence of transcription is the introduction of aberrant supercoiling, positive to the front and negative to the rear of the transcription machinery. This accumulation of aberrant supercoiling can enhance non-B DNA structure formation, especially in highly repetitive and palindromic sequences such as S regions (14–17). Non-B structure formation coupled with transcription has been extensively discussed with respect to triplet repeats and other repetitive sequences that suffer from genome instability (17, 18). Our recent studies on the FACT complex show that transcription has a function associated with the chromatin epigenetic regulation in CSR. In CH12F3–2A cells, which efficiently switch to IgA, knocking down FACT components, either structure-specific recognition protein 1 (Ssrp1) or suppressor of Ty16 homolog (Spt16), drastically reduces CSR by specifically reducing the histone posttranslational modification (PTM) H3K4me3 marks. This reduction in H3K4me3, which is not linked to S-region histone loss or transcriptional status, suppressed DNA cleavage of the target S regions (6). The requirement of H3K4me3 as a DNA cleavage mark in CSR led us to speculate that the same molecular mechanism is shared between CSR and meiotic and V(D)J recombinations (6).
Clearly, the FACT complex is not the only chromatin regulator. We recently reported that AID interacts with Spt6, another histone chaperone associated with transcription elongation (19). Unexpectedly, knocking down Spt6 reduced CSR but not SHM in a GFP reporter construct. Moreover, an extensive protein-protein interaction analysis between Spt6 and various AID mutants revealed that interaction of Spt6 with AID is unlikely to contribute in CSR (19). Besides chaperoning histones in transcription, Spt6 acts to transport and splice mRNA by forming a complex with Iws1 and Pol II phosphorylated at the C-terminal domain on serine 2 (Ser2P-CTD-Pol II) (20). Iws1 interacts directly with the mRNA export factor REF1/Aly (21), which in turn recruits the nuclear exosome component Rrp6 in the elongation complex to form messenger ribonucleoprotein particle (mRNP) particles (22). Thus, Spt6 involvement in processing mRNA is not direct but depends on the Iws1 and exosome complexes. Moreover, nine exosome subunits are copurified with Drosophila Spt6 (23); if this is the case in higher eukaryotes as well, exosome-associated components may influence Spt6 function in CSR.
In this study we screened for new chromatin modulators in CSR regulation and identified only FACT and Spt6. The role of Spt6 in CSR has never been studied from a histone chaperone point of view; therefore, we investigated the molecular mechanism by which Spt6 regulates CSR. Depleting Spt6 in CH12F3–2A cells effectively blocked AID-induced DNA breaks and concomitantly reduced H3K4me3 in the Sμ and Sα regions. We also found that Spt6 knockdown abolished SHM and H3K4me3 marks in the IgH variable (VH) region and in newly identified AID target genes but not in the GFP transgene. Spt6 knockdown in the human Burkitt's lymphoma cell line BL2 produced similar effects. Therefore, we conclude that Spt6 regulates both CSR and SHM through H3K4me3 marks on AID target loci.
EXPERIMENTAL PROCEDURES
Cell Lines and RNAi Oligonucleotide Transfection
The mouse B-cell lymphoma line CH12F3–2A and a human Burkitt's lymphoma line BL2 used in this study have been described previously (6, 24). The BL2 cells used express Jp8BdelER, a hypermutating form of AID fused with the hormone binding domain of the estrogen receptor. Jp8BdelER was activated using 4-hydroxytamoxifen.
Chemically modified RNAi oligonucleotides (Stealth RNAi siRNA) from Invitrogen were transfected into 1 × 106 BL2 or CH12F3–2A cells. Three kinds of stealth siRNA were examined per target gene, and the siRNA with the highest knockdown efficiency was selected. In CSR rescue experiments, FLAG-Spt6 constructs and siRNA against Spt6 were co-transfected into cells using a Nucleofector 96-well electroporation system (Amaxa). The CM150 and DS150 programs were used for CH12F3–2A and BL2 cells, respectively. After electroporation, the cells were cultured for a 24-h period, stimulated by CD40L, IL4, and TGFβ (CIT) or 4-hydroxytamoxifen, and cultured for another 24 h before harvesting and analysis.
CSR and SHM Assays
CH12F3–2A cells were cultured and stimulated to induce class switching, as previously described (25). The cells were analyzed by FACS after 24 h of CIT stimulation. Surface IgM and IgA were stained with FITC-conjugated anti-IgM and phosphatidylethanolamine-conjugated anti-IgA antibodies, respectively. Dead cells were excluded by propidium iodide staining, and all the analyses were performed on a FACS Calibur (BD Biosciences).
To analyze SHM in BL2 cells, cells expressing Jp8BdelER (24) were treated with 4-hydroxytamoxifen (1 μm) for 24 h, and the genomic DNA was purified by phenol-chloroform extraction. PCR was performed using Pyrobest polymerase (TaKaRa) with the following amplification conditions: 95 °C for 5 min, 30 cycles at 95 °C for 30 s, 58 °C for 30 s, and 68 °C for 1 min. After purification, the PCR fragments were cloned into Zero Blunt vectors (Invitrogen). The mutation frequency was calculated as the number of mutations per total bases from independent experiments.
Chromatin Immunoprecipitation
ChIP assays were performed using the ChIP-IT Express Kit (Active Motif) according to the manufacturer's instructions. In brief, 5 × 106 cells were fixed in the presence of 1% formaldehyde for 5–8 min at room temperature. Glycine was added to a final concentration of 0.125 m to stop the reaction. Cell lysis and sonication yielded a soluble chromatin fraction containing fragmented DNA of 500–2000 bp. The lysate was immunoprecipitated by incubation with 3 μg of antibody. Protein-G magnetic bead-bound ChIPed DNA was eluted, and locus-specific enrichment was detected by real-time PCR (RT-PCR) normalized to the input DNA. To show the relative occupancy, one of the data points was set as 100%, as described earlier (6, 26). ChIP results were verified in independent experiments, and S.D. was calculated from three experiments.
RT-PCR Analysis
For all siRNA-related experiments, the total RNA was extracted from cells using TRIzol (Invitrogen). Oligo d(T)-primed cDNA was synthesized from 1 μg of RNA using SuperScript II. The SYBR Green Master Mix was used for RT-PCR with gene-specific primers. The Hprt and Gapdh genes were used as reference controls for CH12F3–2A and BL2 cells, respectively.
Labeling of DNA-break Ends with Biotin-dUTP
The DNA break assay was conducted as described previously (27).
siRNA-resistant Spt6 and Its Mutants
Full-length mouse Spt6 was amplified by RT-PCR of the RNA isolated from CH12F3–2A cells. The Spt6 cDNA was then cloned into the BamHI and SalI sites of the pCMVTag-2A vector (Sigma) to generate N-terminally FLAG-tagged wild type Spt6. The appropriate primers were designed to create serial deletions from either the N or C terminus; the resulting fragments were inserted in-frame with the N-terminal FLAG sequence in the vector. Internal deletions were generated separately by overlapping PCR amplification, and the resulting fragments were joined and fused in-frame with the FLAG epitope. Mutagenesis was performed using the QuikChange Multi-site-directed mutagenesis kit (Stratagene) to alter the siRNA (Spt6, MSS209821 Invitrogen) target sequence (gagaccttctacattggaaagctta/3475–3499 bp of Ref seq. BC072657) by preserving the coded amino acid sequence (gaAacGttTtaTatCggGaaActCa). Each construct's expression was examined by lysing 1 × 106 cells in RIPA buffer supplemented with protease inhibitor. Expression of FLAG tagged constructs was confirmed by immunoblot analysis using cell lysates or proteins pulled down by FLAG beads.
Gene Expression Array Analysis
RNA samples were prepared from CIT-stimulated CH12F3–2A cells pretreated with either control or Spt6 siRNA. For microarray analysis, we used a 3D-Gene Mouse Oligo chip 24k (Toray Industries Inc., Tokyo) with 23,522 distinct genes. Total RNA was labeled with Cy5 using the Amino Allyl MessageAMP II aRNA Amplification kit (Applied Biosystems, CA). The Cy5-labeled aRNA was hybridized for 16 h using the supplier's protocol (3d-gene website). Hybridization signals were scanned with a ScanArray Express Scanner (PerkinElmer Life Sciences) and were processed by GenePixPro Version 5.0 (Molecular Devices). Subtracting the mean intensity of the background signal normalized the raw data of each spot (determined by the signal intensities of all blank spots with 95% confidence intervals). Intensities greater than 2 S.D. of the background signal intensity were considered valid. The signals detected for each gene were subjected to global normalization (the median of the detected signal intensity was adjusted to 30). Primers, antibodies, and siRNA oligonucleotides used are listed in the supplemental reagent list.”
RESULTS
siRNA Screening to Identify Histone Chaperones Critical for CSR
In this screen we focused on histone chaperones and chromatin remodelers that showed distinct histone selectivity or interaction during transcription, replication, or repair processes (28, 29). We first tested the effect of knocking down Caf1 or Asf1, which are replication-associated histone chaperones, on CSR. Three siRNAs against Caf1 showed different degrees of knockdown efficiency that correlated well with reductions in CSR and cell viability. In fact, when the siRNA with the highest knockdown efficiency was introduced, no class-switched cells were detected (Fig. 1A). On the other hand, depleting Asf1 did not inhibit CSR or affect cell viability even though Asf1 acts as a histone donor (H3-H4 and H3.1-H4) for Caf1 during replication-coupled chromatin assembly. Asf1 also transfers H3.3-H4 to the Hira histone chaperone in replication-independent chromatin assembly (30), but Hira knockdown did not cause any observable CSR defect. We thus concluded that the effect of Caf1 knockdown on CSR was primarily due to cell death.
FIGURE 1.

Screen for chromatin modulators critical for CSR. A, shown is FACS analysis of CH12F3–2A cell IgA-switching efficiency after knockdown of the genes by siRNAs as indicated. Numbers in the FACS profile indicate the percentage of the IgA switched population. B, shown is estimation of individual gene knockdown efficiency by qRT-PCR of the extracted RNA. C, CSR efficiency data were compiled from three independent experiments.
We also examined the effects of knocking down Daxx, Dek, and Atrx, as these histone chaperones were recently reported to have locus-specific involvement in H3.3-H4 assembly (31), but there was no significant effect on CSR (supplemental Table 1 and Fig. 1). We next tested Nap1l1 and Nap1l3, nucleosome assembly protein family members that interact with H2A-H2B and with linker histones. Although Nap1l1 knockdown inhibited CSR to some degree, Nap1l3 knockdown did not change the CSR efficiency.
Knocking down the Snf2-related CREBBP-activator protein Srcap, a component of the H2AZ-associated histone chaperone complex, inhibited CSR by about 50%. A similar reduction in CSR was seen when a dominant-negative form of H2AZ was overexpressed in CH12F3–2A cells,4 suggesting that specific histone variants may contribute to CSR. Depleting DNA repair-associated chromatin remodelers such as the ATP-dependent chromatin assembly factor Acf1, the SWI/SNF-related chromatin regulator Smarcal1, and Ino80 minimally affected CSR. In contrast, knocking down any of the elongation-associated histone chaperones, Spt6, or the FACT components structure-specific recognition protein 1 (Ssrp1) and suppressor of Ty16 homolog (Spt16) strongly inhibited CSR (Fig. 1C). These results along with characteristic features of the histone chaperones and chromatin remodelers examined are summarized in supplemental Table 1.
Spt6 Depletion Inhibits S-region Breakage but Not Transcription
We next investigated how the histone chaperone activity of Spt6 affects CSR. We first confirmed that depleting Spt6 in activated CH12F3–2A cells did not inhibit μGLT or αGLT production from the Iμ and Iα promoters, respectively (19). Spt6 knockdown slightly enhanced μGLT transcription (Fig. 2, A and B, transcript b). On the other hand, knocking down Spt6 modestly decreased the αGLT and AID transcripts (Fig. 2A,B; transcripts d and f). Since aberrant transcription initiation was reported in yeast Spt6 mutants (32), we looked for possible cryptic transcription initiation from the Sμ, Sα, and AID loci after Spt6 depletion. As depicted in Fig. 2A, we designed primers within the S-region introns to detect type-c and type-e transcripts from Sμ and Sα, respectively. In the absence of Spt6 we found increased accumulations of type-c transcripts from Sμ (Fig. 2Bc, Sμ-internal) but no increase in type-e transcripts from Sα. A similar approach for the AID locus using primers located in intron1 and exon2 did not detect any such transcripts, suggesting that the aberrant intronic transcript initiation in the absence of Spt6 was specific for Sμ.
FIGURE 2.

Spt6 regulates transcription and DNA breakage. A, shown is a schematic representation of Sμ- and Sα-derived germline transcripts. The spliced-out region in the transcript is indicated by dotted lines. B, CH12F3–2A cells treated with the indicated siRNAs were left untreated (−) or were stimulated (+) by CIT for 24 h. The transcriptional status of Sμ, Sα, and AID was analyzed by RT-PCR of the extracted RNA. C, DNA double-strand breaks in Sμ and Sα, analyzed by γH2AX ChIP are shown. Sμ- and Sα-specific qPCR was performed with ChIPed DNA from the three different conditions indicated. Each sample was normalized against input DNA. D, quantitative representation of biotin-dUTP incorporation at DNA-break ends is shown; see “Experimental Procedures.” E and F, shown is a comparison of the 5′-Sμ mutation frequency in the genomic DNA of CH12F3–2A cells treated as indicated. A pie diagram shows the total number of mutated clones (center) and the mutation frequency (the number of mutations/clone, periphery).
Next we examined whether Spt6 knockdown inhibits DNA breakage in the S regions. To assess S-region DNA cleavage, we performed quantitative DNA-break assays using two methods, γH2AX ChIP and direct cleaved-end labeling (27, 33). Quantitative DNA-breakage data from the two independent assays (Fig. 2, C and D) showed that knocking down Spt6 strongly reduced DNA breakage signals in both the Sμ and Sα regions. To validate this observation further, we examined DNA cleavage-dependent mutation signatures at the 5′ flanking region of Sμ. The mutation frequency and distribution were analyzed by sequencing nearly 48,000 bp in DNA derived from control cells and cells treated with siRNA against Spt6. The mutation frequency in the Spt6-depleted sample was one-fourth that in the control sample (Fig. 2E). Consistent with this finding, we also observed significantly reduced numbers of mutated clones after depleting Spt6 (Fig. 2F). Thus, these three independent lines of evidence indicate that Spt6 depletion inhibits CSR due to reduced S-region DNA cleavage.
Spt6 Regulates S-region Chromatin Modification
Because the S-region DNA cleavage in CSR is regulated through histone modification by the FACT complex (6), we examined the effect of Spt6 knockdown on S-region chromatin by analyzing the histone occupancy and histone modifications associated with transcription elongation. A comparison of the S-region histone profiles of control and Spt6-knocked down CH12F3–2A cells indicated that there was a 50% average loss of core histones specifically from the Sμ region, but not from the Sα region, in Spt6 knocked down cells (Fig. 3B). However, the H3K4me3 and H3K36me3 histone-PTMs were reduced in both the Sμ and Sα regions (Fig. 3C), suggesting that Spt6 differentially regulates chromatin assembly and chromatin marks in the two S regions.
FIGURE 3.

Spt6 regulates IgH-locus chromatin. A, shown is a schematic diagram of the IgH locus showing the position of ChIP analysis primers in Sμ, Sγ1, and Sα. B, shown is the Spt6 knockdown effect on Sμ and Sα core histones; ChIP analysis of H2A, H2B, and H3 in CIT-stimulated CH12F3–2A cells treated with either control siRNA (black bars) or siRNA against Spt6 (gray bars). The relative enrichment (y axis) profile was determined by quantitative PCR of the ChIPed DNA normalized against input; target regions are indicated on the x axis. C, histone-PTM down-modulation after Spt6 knockdown is shown; ChIP analysis of H3K4me3 and H3K36me3 in Sμ and Sα. D, Spt6 and H3K27me3 occupancy were determined by ChIP analysis of Sμ, Sγ1, and Sα in CIT-stimulated and unstimulated CH12F3–2A cells.
Next, we examined whether the presence of Spt6 on chromatin correlated with the histone-PTM status of the locus by performing ChIP against endogenous Spt6. We conducted anti-Spt6 ChIP for the Iμ-Sμ, Iα-Sα, and Iγ1-Sγ1 regions, which represent transcriptionally constitutive, inducible, and silent loci, respectively. Spt6 was detected on both the constitutive (Iμ) and inducible (Iα) promoters and their respective S-regions. However, Spt6 was minimal in the Eμ enhancer region located upstream of Iμ and undetectable in the Iγ1-Sγ1 region (Fig. 3D, top). Consistent with its transcriptional silence, the Iγ1-Sγ1 region was enriched with the silent histone modification H3K27me3 (Fig. 3D, bottom). Thus, the presence of Spt6 was strongly correlated with H3K PTMs and a transcriptionally active state of the locus. These results suggest that Spt6 cooperates in determining DNA cleavage targets by modifying H3K, which agrees with our previous finding that the FACT complex is involved in the H3K4me3 formation at AID target loci (6).
Requirement of Spt6-mediated H3K4me3 for SHM
Because AID-induced mutations and DNA breaks at non-Ig loci can cause tumorigenic DNA lesions, including translocations, in various lymphomas (34–36), we examined whether Spt6 also regulates H3K4me3 modifications in non-Ig AID targets. We recently identified novel gene loci that are prone to AID-induced breakage and mutations in the SHM-proficient BL2 cell line (37). Spt6 knockdown in this cell line did not reduce SHM in an integrated GFP transgene (19). In this study, therefore, we analyzed the effect of Spt6 knockdown on AID-induced mutation rates in the VH region, a GFP transgene, and two novel AID-target genes, MALAT1 and SNHG3 (37). To our surprise, we found that knocking down Spt6 reduced mutations in SNHG3, the VH region, and the MALAT1 loci (Fig. 4A) but not in the GFP transgene. Curiously, Spt6 knockdown reduced H3K4me3 at all the loci subject to AID-induced mutations with the exception of the GFP transgene, and this H3K4me3 loss correlated well with the reductions in mutation frequency at these loci (Fig. 4B). At the GFP locus, however, H3K4me3 and its mutation rate were slightly augmented, in agreement with our previous observation that Spt6 knockdown did not affect SHM at the GFP transgene (19). Interestingly, the Cμ exon, where H3K4me3 enrichment was hardly detectable, was also completely devoid of AID-induced mutations. As expected, Spt6 depletion affected neither the steady-state mRNA expression nor the H3 occupancy in the loci tested (Fig. 4, C and D).
FIGURE 4.

Spt6-dependent regulation of SHM and H3K4me3 in IgH and non-IgH target loci. A, shown are mutation analysis of IgH-V and the breakpoint regions of SNHG3 and MALAT1 loci in BL2 cells containing Jp8BdelER (a hypermutating form of AID fused with estrogen receptor). Before AID activation, the cells were treated with either control siRNA (gray bars) or Spt6 siRNA (black bars). The GFP transgene mutation frequencies were taken from our previous work (19). B, H3K4me3 occupancy, determined by ChIP analysis, in the indicated genomic loci using the same cell line with and without Spt6 knockdown is shown. C, transcripts were analyzed by qRT-PCR of RNA extracted from siControl- and siSpt6-treated cells. D–F, ChIP analysis of H3, H3R2, and Spt6 at the indicated genomic loci is shown. Experimental conditions and cell treatment were as described for A–C.
As the RAG2 PHD domain recognizes both H3R2 and H3K4me3, and asymmetric R2 dimethylation on the H3K4me3 peptide increases the PHD domain's binding affinity, we also examined whether Spt6 knockdown affected H3 arginine (R2) methylation. Although the two modifications exist in close proximity, Spt6 depletion did not affect this modification at all (Fig. 4E), emphasizing the specificity of Spt6's regulation of histone-PTMs. Finally, we compared the relative occupancy of Spt6 in the VH region and the SNHG3 and MALAT1 breakpoints. Although the Spt6 level was highest in the VH region, Spt6 depletion naturally reduced the Spt6 occupancy at all the loci (Fig. 4F), which agreed well with the reduction of H3K4me3 and mutation frequencies at these loci. Taken together, these results suggest that Spt6-dependent H3K4me3 marks on a particular chromatin locus are also involved in regulating SHM targets.
Requirement of tandem SH2 domain of Spt6 for CSR
To examine the specificity of Spt6 knockdown and to determine the critical structural requirements of Spt6, we generated a siRNA-resistant, non-degradable Spt6 construct by mutating the siRNA target region in the cDNA. Transfection of this siRNA-resistant wild type Spt6 construct, along with the siRNA against Spt6, completely rescued CSR (Fig. 5, B and C, and supplemental Fig. 2, C and D). Anti-Spt6 siRNA decreased the endogenous Spt6, but the FLAG epitope-tagged Spt6 proteins proved resistant to degradation (Fig. 5C and supplemental Fig. 2B). We also confirmed that H3K4me3 loss in the Sμ region after Spt6 knockdown could be fully restored by expressing the siRNA-resistant Spt6 construct (supplemental Fig. 2E).
FIGURE 5.

Requirement of tandem SH2 domain of Spt6 for CSR. A, shown is a schematic representation of mouse wild type and tandem SH2 domain-deleted Spt6 constructs. Filled boxes represent various structural domains present in Spt6, and the scale above shows the amino acid numbers. B, shown is a FACS profile of the IgA-switching efficiency of CH12F3–2A cells expressing wild type and mutant Spt6 constructs resistant to siRNA-mediated degradation. Cells were co-transfected with siRNA (control or Spt6) and plasmid (an empty vector or siSpt6-resistant Spt6 construct) as indicated above each FACS profile. C, Western blots show the expression status of endogenous Spt6 and FLAG-tagged Spt6. The plot below shows the average CSR rescue efficiency by wild type and mutant Spt6 constructs; data were taken from three independent experiments. D, shown is ChIP analysis of H3K4me3 and H3K36me3 in Sμ and Sα before and after wild type and mutant Spt6-mediated CSR complementation. E, evaluation of S-region and AID transcriptional status by RT-qPCR.
Recently, the crystal structure of Spt6 revealed that Spt6 has an unusual type of tandem SH2 domain (SH2(N) and SH2(C)) in which SH2(C) is a noncanonical type (38–40). We became curious to know whether the tandem SH2 domain plays any special role in CSR regulation by Spt6. It revealed that loss of tandem SH2 domain, but not the individual SH2 domain, makes Spt6 defective in CSR rescue (Fig. 5, A and B, supplemental Fig. 2). Strikingly, this mutant also failed to rescue H3K4me3 but not H3K36me3 in the S regions (Fig. 5D), suggesting that the CSR defect was likely due to the loss of H3K4me3 from the locus. Interestingly, whereas wild type Spt6 effectively suppressed the Sμ internal transcription (cryptic transcripts), the mutant failed to suppress this excessive cryptic transcription (Fig. 5E). As expected, we did not observe any adverse effect of this mutant on S region and AID transcription (Fig. 5E), and consistently, Pol II distribution was intact (data not shown).
As CSR was strongly dependent on CxxC1 and Wdr82 (6), the Set1 histone methyl transferase-specific co-factors (41, 42), we verified the knockdown effect of Set1A and Set1B in CSR (Fig. 6, A and B). Depletion of either one showed a significant CSR inhibitory effect; therefore, we asked whether the depletion of Spt6 leads to Set1 recruitment in the S-region and whether Set1 can associate with Spt6. As native Spt6 IP clearly showed Set1A association (data not shown), we tested whether there is any difference in this interaction between wild type versus tandem SH2 domain-deleted mutant. It is evident that although IP efficiency by FLAG epitope is very good for both the Spt6 proteins, a clear association of Set1A can be seen with the wild type but not with the mutant Spt6 (Fig. 6C). We also found that Set1A recruitment in the S region was defective due to the deletion of tandem SH2 domain in Spt6. Consistently, a strong reduction of H3K4me3 in the S-region was observed by the knockdown of either Set1A or Set1B or both (Fig. 6D). Although Set1B knockdown showed the strongest effect, we are unable to examine Set1B association or distribution in the S-region due to the unavailability of good antibody. We did not observe any change in Pol II distribution, which was consistent with the unperturbed S region transcriptions.
FIGURE 6.

Interaction of Spt6 and Set1 histone methyltransferase mediates H3K4me3 formation in the S-region. A, shown is a FACS profile of CSR efficiency after Set1A and Set1B knockdown in CH12-F32A. B, shown is CSR inhibition and knockdown efficiency data from three independent experiments. C, representative co-immunoprecipitation (IP) data with anti-FLAG show that Set1A associates with wild type Spt6 but not with the mutant Spt6, in which both the SH2 domains were deleted. D, ChIP analysis of Set1A, H3K4me3, and Pol II in various conditions as indicated is shown. FSC, forward scatter; WCE, whole cell extract.
The CSR rescue activity of Spt6 with the concomitant recovery of H3K4me3 suggests that Spt6 is critical for regulating chromatin but not transcription in CSR. Taken together it can be postulated that Spt6, through its tandem SH2 domain, helps recruit the Set1 complex in Sμ and Sα, thereby regulating H3K4me3 formation for DNA cleavage and CSR.
We additionally tested several other Spt6 mutants with N-terminal and C-terminal truncations and with internal deletions specific for functional Spt6 domains (supplemental Fig. 2A). Mutant Spt6 in which the first 300 amino acids were deleted (ΔNmut4) or the C terminus was truncated at amino acid residue 1426 (ΔCmut4) showed decreased expression compared with that of wild type Spt6 (supplemental Fig. 2C), yet these mutants also rescued CSR. Two domain-specific mutants (ΔYgg1, ΔS1) with less efficient expression (not shown) were also complemented CSR defect (supplemental Fig. 2D). Our observations were somewhat unexpected, because similar deletion of either the N or C terminus of Spt6 has been reported to cause transcriptional defects (43), and the Spt6 C-terminal region has also been reported to interact with AID (19). Furthermore, a ΔSH2 mutant of Spt6 in which the C-terminal SH2(N) domain is deleted fails to interact with the elongating form of Pol II (Ser2P) (21). However, a mutant with deleted SH2(N) domain (ΔSH2) was able to rescue CSR and was expressed as strongly as the wild type Spt6 (supplemental Fig. 2, A–D).
RNA-processing Activity of Spt6 Is Not Involved in CSR
To examine whether Spt6 influences CSR through its mRNA export and exosome-associated activities (20, 21, 23), we tested the effect of knocking down Iws1 and specific exosome components such as Exosc3/Rrp40, which is associated with nuclease activity, and Dis3/Rrp44, which is not associated with nuclease activity (44). Quantitative RT-PCR showed that at least two independent siRNAs effectively depleted individual target genes but did not dramatically affect CSR (Fig. 7). Knocking down additional exosome components, such as Exosc1/Csl4, Exosc2/Rrp4, and Exosc10/Rrp6, had similar results. We thus failed to identify CSR involvement of components known to partner with Spt6 in RNA processing; this strengthens the idea that Spt6 is critically involved in AID-induced DNA breakage through its histone-chaperone and histone-PTM activities.
FIGURE 7.
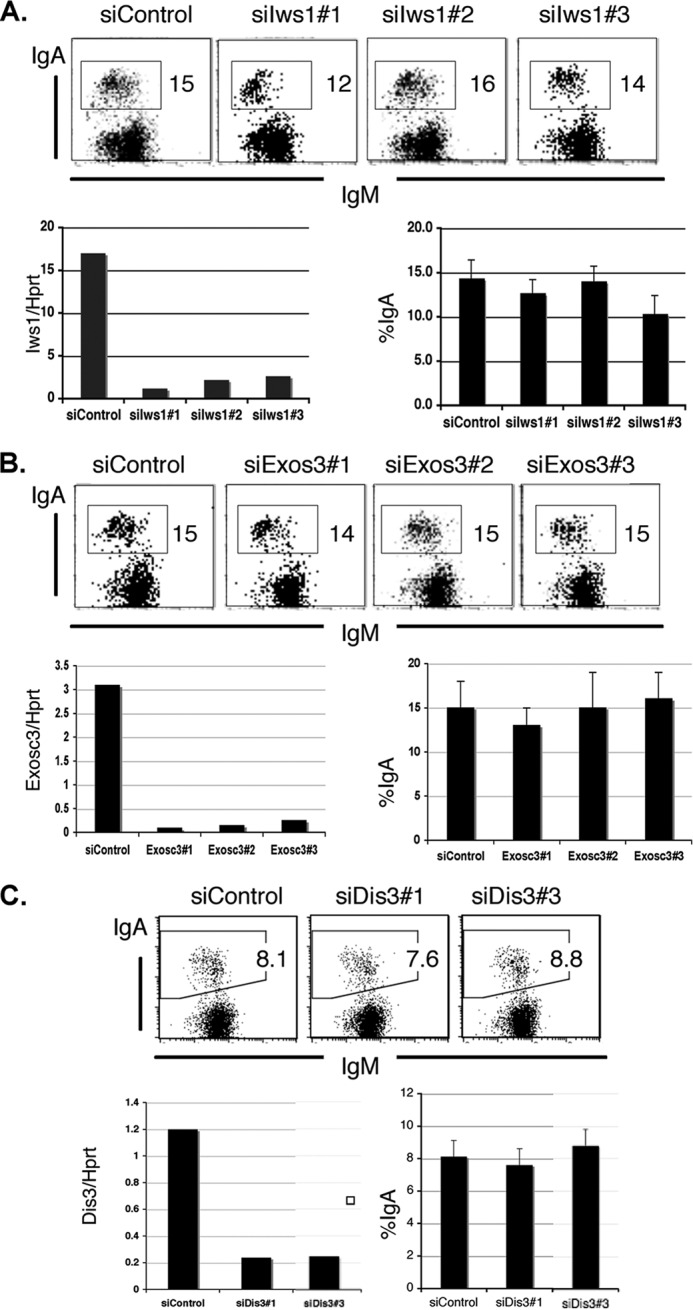
Spt6 regulation of CSR does not depend on Iws1 or exosome association. A, shown are FACS profiles of the IgA-switching efficiency of CH12F3–2A cells after Iws1 knockdown by three independent siRNAs, knockdown efficiency as determined by qRT-PCR of the extracted RNA, and a summary of CSR efficiency in three independent experiments. B, shown are FACS profiles of the IgA-switching efficiency after Exosc3 knockdown by three independent siRNAs, knockdown efficiency as determined by qRT-PCR of the extracted RNA, and a summary of CSR efficiency in three independent experiments. C, FACS profiles of IgA-switching efficiency after Dis3 knockdown by two independent siRNAs, knockdown efficiency as determined by qRT-PCR of extracted RNA, and a summary of CSR efficiency in three independent experiments.
Spt6 Modulates a Limited Set of Genes That Includes AID
Finally, we wanted to confirm that Spt6 does not influence CSR through the transcriptional control of other genes required for CSR. We compared gene expression profiles of activated CH12F3–2A cells with or without anti-Spt6 siRNA treatment. Of nearly 24,000 genes analyzed in the microarray, 95% were not affected by Spt6 depletion (Fig. 8A); of the remaining 5%, 3% were up-regulated, and only 2% were down-regulated. Of the genes most strongly up-regulated by Spt6 depletion (Fig. 8B), none were known to be involved in CSR except Spt4 (Supt4ht), which positively regulates CSR.5 In fact, the expression levels of many of these genes were very low (Fig. 8C, scores lower than 100, and Fig. 8D, lane 1), and some genes were specific to germ cells or testes (see “Supplementary Discussion”).
FIGURE 8.

Spt6 regulated transcriptional activation and suppression. A, a pie diagram shows the total of differentially regulated genes after Spt6 knockdown in CIT-activated CH12F3–2A cells. B, the top 11 up-regulated genes (>8-fold, red bars) and 14 other selected up-regulated genes (>2-fold, light red bars) are shown. The gene name symbol and -fold up-regulation are indicated just below the plot. C, normalized gene expression signal score in the microarray for all genes regulated up or downward and plotted in B is shown. Genes with expression scores above 100 (highlighted by black) are readily detectable by 25 cycles of PCR. D, a representative example of Spt6-up-regulated genes with low expression levels is shown. Expression of the three indicated genes was totally absent in CH12F3–2A cells but could be detected by 35 cycles of PCR after Spt6 knockdown and CIT stimulation. Spt6 also differentially regulated two Spata1 isoforms. E, shown are more examples of isoform-specific gene up-regulation by Spt6 (e.g. the Ms4a, Ly6, and histone families). F, the right side of the plot shows the top 12 down-regulated genes (black bars) and 24 selected down-regulated genes (gray bars), respectively, including the AID (Aicda) gene. The -fold down-regulation and gene name symbol are indicated just above the plot. Gene knockdowns that did not affect CSR are marked by black asterisks; the Hax1 gene is marked by a gray asterisk as CSR defects were reported in Hax1-KO mice (45). See supplemental Discussion.
The 12 genes down-regulated to the greatest degree by Spt6 depletion along with another 24 genes down-regulated at least 2-fold are shown in Fig. 8F. In this list the Ccl22 chemokine had the highest expression level, which was reduced 3.8-fold by Spt6 depletion. Ccl22, which can be induced by CD40L, interacts with T cells through the CCR4 receptor; thus, it does not have any direct effect in this CH12F3–2A-based in vitro CSR assay. Similarly, none of the 12 genes most down-regulated have been reported to regulate CSR. We then looked specifically at genes that were down-regulated 2-fold or more for those that might critically influence CSR by modulating chromatin. We then knocked down selected chromatin-regulating genes individually (marked by asterisks in Fig. 8F) to determine whether CSR would be altered. However, CSR was not significantly affected by the knockdown of any of these genes. We also previously showed down-regulation of AID due to Spt6 depletion is not the direct cause of CSR defect, because the defect cannot be complemented by exogenous AID expression (19). In addition, CSR was assessed for a short period in our study, during which the majority of genes was expressed at substantial levels. It is thus unlikely that genes regulated by Spt6 caused secondary CSR defects in the absence of Spt6.
Spt6-regulated H3K36me3 Affects AID Expression
As AID is the most critical gene for CSR and SHM, we wondered whether Spt6 remodels chromatin in the AID locus as in the IgH locus. We thus examined Spt6 distribution in the AID locus and monitored chromatin alterations upon Spt6 depletion. To obtain an overview of the entire AID locus, we performed histone ChIPs using sets of primers in the promoter and transcribed regions, as indicated in Fig. 9, top. H3K4me3 was significantly enriched from the promoter area to the middle of the first intron followed by a region with no H3K4me3. H3K36me3 marks increased from this point and remained at high levels throughout the coding region down to the 3′ end of the gene. Although the H3K4me3 and H3K36me3 distribution patterns were consistent with many transcriptionally active gene loci, remarkably, they were down-modulated from the AID locus in parallel by Spt6 depletion (Fig. 9, B and C). It is worth noting that H3 remained stable in the AID locus (Fig. 9D) despite Spt6 knockdown and drastically reduced H3K-PTMs, strengthening the idea that Spt6 can act as a histone chaperone, a histone-PTM regulator, or both in a locus-specific manner. Spt6 was also distributed throughout the AID locus (Fig. 9E), which is consistent with both the distribution of H3K4me3 and H3K36me3 and their regulation by Spt6.
FIGURE 9.

Spt6 preserves specific AID-locus chromatin marks in cooperation with H3K36-histone methyltransferases. A, shown is a schematic representation of the AID locus, with ChIP primer positions. B–D, ChIP analysis of H3K4me3, H3K36me3, and H3 using CH12F3–2A cells treated with either control siRNA or siRNA against Spt6 is shown. E, Spt6 distribution in the AID locus with and without CIT induction is shown. F, occupancy of Pol II (Ser5P) in the AID locus before and after Spt6 knockdown is shown. G and H, shown is the effect of Sted2 and Nsd1 knockdown on H3K36me3 status of the AID locus, AID expression, and IgA switching. I, Spt6 regulates CSR and AID through two different chromatin modifications. Non-stimulation and CIT-stimulated conditions are indicated by (−) and (+), respectively.
To gain further insight into the mechanism of transcriptional regulation of the AID gene by Spt 6, we examined the Pol II occupancy. Knocking down Spt6 did not drastically reduce either the total Pol II (not shown) or the Ser5P-CTD-Pol II (Fig. 9F). Thus, H3K4me3 loss in the AID locus cannot be fully explained by Ser5P-CTD-Pol II occupancy, which is known to associate with the H3K4me3 methylase complex (20, 46) to help recruit the complex in the promoter proximal area. In addition, H3K4me3 does not appear to have any direct role in transcribing the AID gene, because AID was unaffected even when H3K4me3 was globally reduced by Wdr5 or Ash2 knockdown (6). Interestingly, however, depletion of the major H3K36me3 methyltransferase Setd2 down-regulated AID by about 50%, with a concomitant loss of H3K36me3 from the locus (Fig. 9G). Spt6 loss has been reported to reduce H3K36me3 due to defective loading of the Setd2-Ser2P-CTD-Pol II complex (20, 47). However, we could not confirm the relative occupancy of Ser2P-CTD form of Pol II due to the poor efficiency of ChIP in CH12F3–2A cells with the antibodies available. It has been reported that Spt6 stimulates the catalytic activity of the yeast Setd2. Surprisingly, knocking down Setd2 did not cause any apparent CSR defect, although H3K36me3 was reduced in both Sμ and Sα (Fig. 9H). We examined another H3K36me3 methylase, Nsd1, which was down-regulated 3.3-fold by Spt6 knockdown (Fig. 8F). We observed a correlation between H3K36me3 and AID similar to that seen for Setd2 (Fig. 9, G and H). These results together may indicate that Spt6 employs independent mechanisms to regulate CSR and AID by H3K4me3 and H3K36me3, respectively.
DISCUSSION
FACT complex-mediated H3K4me3 modification is critical for DNA cleavage in CSR (6); in this study we screened several other histone chaperones and chromatin remodelers for their possible involvement in CSR. The screening of two dozen chromatin modulators identified Spt6 along with the FACT complex as the most critical components regulating CSR. We clearly demonstrated that Spt6 regulates H3K4me3 DNA-cleavage target marks in CSR. Knocking down Spt6 simultaneously reduced the H3K4me3 marks and DNA cleavage in the S region. CSR could be fully rescued after Spt6 knockdown by wild type Spt6 or by Spt6 mutants with C-terminal truncations, including SH2(N) domain-deleted mutant, which mediates Spt6 interaction with the phosphorylated form of Pol II (21, 39). Because AID interacts with the very C-terminal region of Spt6, these results suggest that interaction between Spt6 and AID is not directly important for Spt6 regulation of CSR (19).
Requirement of tandem SH2 domain of Spt6 for Set1interaction provides a logical explanation of H3K4me3 formation in the S-region by Spt6 and its relationship with CSR. We also screened a series of proteins that interact with Spt6 and are linked to mRNA processing and exosome-associated functions, such as Iws1, Exos3, and Dis3; CSR was not noticeably reduced by knocking down any of these proteins. However, there is a report showing that depletion of exosomes reduces CSR efficiency (48). The reason for such a discrepancy is not clear, although we frequently observed excessive proliferation of CH2F3–2A cells upon exosome knockdown, which may negatively affect CSR in CH2F3–2A. Alternatively, the difference in results could be due to the knockdown system applied, lentivirally-introduced stable shRNA expression versus transiently transfection of siRNA oligonucleotides. However, the same transient siRNA introduction system reproducibly worked well for many genes we tested, including Spt6 and FACT components.
We also tested whether other genes regulated by Spt6 might affect CSR indirectly. The analysis of cDNA microarrays revealed that Spt6 knockdown raised or lowered the expression of 962 (5%) of the genes examined. We individually knocked down several genes to determine their effects on CSR and did not find any genes other than AID that might link Spt6 knockdown with reduced CSR. However, our previous data showed that the CSR defects induced by Spt6 knockdown are not caused by AID reduction, as the CSR defect was not rescued by the expression of exogenous AID fused with estrogen receptor (19). Taken together, the results confirm that Spt6 regulates CSR through H3K4me3 modification of the target chromatin.
We also asked why Spt6 knockdown did not affect SHM of the GFP transgene in BL2 cells (19). Although H3K4me3 marks the GFP locus, Spt6 knockdown did not regulate the H3K4me3 mark in the GFP transgene. In contrast, Spt6 depletion affects the VH gene and the novel SHM-target genes SNHG3 and MALAT1 (37) in terms of AID-induced mutation frequency and H3K4me3 occupancy at the target site. These results clearly indicate that Spt6 regulates the H3K4me3 marking at both CSR and SHM targets, further supporting the conclusion that AID-induced cleavage depends on H3K4me3 marking.
Finally, we analyzed the histone modification profile of the AID locus. Knocking down Spt6 decreased H3K4me3 and H3K36me3, which are distributed mostly in the 5′ and 3′ regions of the AID locus, respectively. However, CSR was not affected when the histone methyltransferases Setd2 and Nsd1 specifically reduced H3K36me3. These results indicate that although Spt6 regulates both H3K4me3 and H3K36me3, H3K4me3 is important for DNA cleavage in CSR, whereas H3K36me3 is involved in AID gene transcription.
Traditionally, histone chaperones have been thought to interact with histones for their stability, transport, and exchange (29). Emerging research, including our previous and present work on FACT and Spt6, indicates that histone chaperones act as regulators at specific genomic loci by imprinting and erasing the histone marks (6, 49). The FACT complex is reported to be involved in H2B ubiquitination, leading to H3K4me3 modification in specific gene loci (50, 51). Yeast studies suggest that H3K4me3 and H3K36me3 histone methyltransferase complexes are sequentially loaded onto the Pol II complex during transcription elongation (52). Presumably, histone chaperones or elongation factors collaborate with site-specific histone modifiers or recruitment complexes. The identification of locus-specific histone methyltransferase complexes is important for understanding the mechanism of locus-specific histone epigenetic regulation by Spt6 and the FACT complex. It is possible that DNA cleavage or repair complexes travel with selective histone methyltransferase complexes and finally tether to H3K4me3 and other histone marks at the target locus. For instance, H3K36me3 can often be found in the intronic and coding regions and is proposed to play a role in repair and splicing through specific histone-PTM readers. It is also reported that a group of long non-coding gene loci (53, 54), including MALAT1, which is a target of chromosomal translocation, are highly enriched with H3K36me3. Although we do not know how many histone codes other than H3K4me3 are involved in CSR and SHM, we assume that a combinatorial histone code with site-specific readers would have tremendous potential to generate chromatin target specificity (55–57).
Because H3K4me3 is not only the marker of an actively transcribed locus but is also a common signature of DSB hotspots in CSR, V(D)J, and meiotic recombinations (4, 6, 7), understanding how H3K4me3 formation is regulated is of special interest. It remains to be elucidated exactly how histone chaperones like Spt6 and FACT regulate chromatin marks, collaborating with various histone modifiers and readers to promote beneficial DNA breaks while protecting the genome from aberrant DNA damage.
Supplementary Material
Acknowledgment
We thank Y. Shiraki for editorial assistance.
This research was supported by Grant-in-aid for Specially Promoted Research 17002015 and Grant-in-Aid for Scientific Research 24590352 from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.

This article contains supplemental Discussion, reagent list, GE dataset, Table 1, and Figs. 1 and 2.
A. Stanlie, N. A. Begum, and T. Honjo, unpublished information.
The DSIF subunits Spt4 and Spt5 have distinct roles at various phases of immunoglobulin class switch. Stanlie, A., Begum, N. A., Akiyama, H., and Honjo, T. (2012) PLoS Genet. e1002675.
- AID
- activation-induced cytidine deaminase
- CSR
- class-switch recombination
- GLT
- germ line transcription
- S
- switch
- PTM
- posttranslational modification
- SHM
- somatic hypermutation
- CIT
- CD40L, IL4, and TGFβ
- qRT-PCR
- real-time PCR
- Pol II
- polymerase II
- RT-PCR
- reverse transcription-PCR
- RAG
- recombination-activating gene
- FACT
- facilitates chromatin transcription.
REFERENCES
- 1. Parvanov E. D., Petkov P. M., Paigen K. (2010) Prdm9 controls activation of mammalian recombination hotspots. Science 327, 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baudat F., Buard J., Grey C., Fledel-Alon A., Ober C., Przeworski M., Coop G., de Massy B. (2010) PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327, 836–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Matthews A. G., Kuo A. J., Ramón-Maiques S., Han S., Champagne K. S., Ivanov D., Gallardo M., Carney D., Cheung P., Ciccone D. N., Walter K. L., Utz P. J., Shi Y., Kutateladze T. G., Yang W., Gozani O., Oettinger M. A. (2007) RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature 450, 1106–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu Y., Subrahmanyam R., Chakraborty T., Sen R., Desiderio S. (2007) A plant homeodomain in RAG-2 that binds hypermethylated lysine 4 of histone H3 is necessary for efficient antigen-receptor-gene rearrangement. Immunity 27, 561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ji Y., Resch W., Corbett E., Yamane A., Casellas R., Schatz D. G. (2010) The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell 141, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stanlie A., Aida M., Muramatsu M., Honjo T., Begum N. A. (2010) Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 107, 22190–22195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smagulova F., Gregoretti I. V., Brick K., Khil P., Camerini-Otero R. D., Petukhova G. V. (2011) Genome-wide analysis reveals novel molecular features of mouse recombination hotspots. Nature 472, 375–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borde V., Robine N., Lin W., Bonfils S., Géli V., Nicolas A. (2009) Histone H3 lysine 4 trimethylation marks meiotic recombination initiation sites. EMBO J. 28, 99–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shimazaki N., Tsai A. G., Lieber M. R. (2009) H3K4me3 stimulates the V(D)J RAG complex for both nicking and hairpinning in trans in addition to tethering in cis. Implications for translocations. Mol. Cell 34, 535–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Muramatsu M., Kinoshita K., Fagarasan S., Yamada S., Shinkai Y., Honjo T. (2000) Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102, 553–563 [DOI] [PubMed] [Google Scholar]
- 11. Jung S., Rajewsky K., Radbruch A. (1993) Shutdown of class switch recombination by deletion of a switch region control element. Science 259, 984–987 [DOI] [PubMed] [Google Scholar]
- 12. Stavnezer-Nordgren J., Sirlin S. (1986) Specificity of immunoglobulin heavy chain switch correlates with activity of germ line heavy chain genes prior to switching. EMBO J. 5, 95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yu K., Roy D., Bayramyan M., Haworth I. S., Lieber M. R. (2005) Fine-structure analysis of activation-induced deaminase accessibility to class switch region R-loops. Mol. Cell. Biol. 25, 1730–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dunnick W., Hertz G. Z., Scappino L., Gritzmacher C. (1993) DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res. 21, 365–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kataoka T., Miyata T., Honjo T. (1981) Repetitive sequences in class-switch recombination regions of immunoglobulin heavy chain genes. Cell 23, 357–368 [DOI] [PubMed] [Google Scholar]
- 16. Nikaido T., Nakai S., Honjo T. (1981) Switch region of immunoglobulin Cmu gene is composed of simple tandem repetitive sequences. Nature 292, 845–848 [DOI] [PubMed] [Google Scholar]
- 17. Zhao J., Bacolla A., Wang G., Vasquez K. M. (2010) Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 67, 43–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hubert L., Jr., Lin Y., Dion V., Wilson J. H. (2011) Topoisomerase 1 and single-strand break repair modulate transcription-induced CAG repeat contraction in human cells. Mol. Cell. Biol. 31, 3105–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Okazaki I. M., Okawa K., Kobayashi M., Yoshikawa K., Kawamoto S., Nagaoka H., Shinkura R., Kitawaki Y., Taniguchi H., Natsume T., Iemura S., Honjo T. (2011) Histone chaperone Spt6 is required for class switch recombination but not somatic hypermutation. Proc. Natl. Acad. Sci. U.S.A. 108, 7920–7925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoh S. M., Lucas J. S., Jones K. A. (2008) The Iws1·Spt6·CTD complex controls cotranscriptional mRNA biosynthesis and HYPB/Setd2-mediated histone H3K36 methylation. Genes Dev. 22, 3422–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoh S. M., Cho H., Pickle L., Evans R. M., Jones K. A. (2007) The Spt6 SH2 domain binds Ser2-P RNAPII to direct Iws1-dependent mRNA splicing and export. Genes Dev. 21, 160–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jensen T. H., Dower K., Libri D., Rosbash M. (2003) Early formation of mRNP. License for export or quality control? Mol. Cell 11, 1129–1138 [DOI] [PubMed] [Google Scholar]
- 23. Andrulis E. D., Werner J., Nazarian A., Erdjument-Bromage H., Tempst P., Lis J. T. (2002) The RNA processing exosome is linked to elongating RNA polymerase II in Drosophila. Nature 420, 837–841 [DOI] [PubMed] [Google Scholar]
- 24. Nagaoka H., Ito S., Muramatsu M., Nakata M., Honjo T. (2005) DNA cleavage in immunoglobulin somatic hypermutation depends on de novo protein synthesis but not on uracil DNA glycosylase. Proc. Natl. Acad. Sci. U.S.A. 102, 2022–2027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakamura M., Kondo S., Sugai M., Nazarea M., Imamura S., Honjo T. (1996) High frequency class switching of an IgM+ B lymphoma clone CH12F3 to IgA+ cells. Int. Immunol. 8, 193–201 [DOI] [PubMed] [Google Scholar]
- 26. Glover-Cutter K., Kim S., Espinosa J., Bentley D. L. (2008) RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat. Struct. Mol. Biol. 15, 71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Doi T., Kato L., Ito S., Shinkura R., Wei M., Nagaoka H., Wang J., Honjo T. (2009) The C-terminal region of activation-induced cytidine deaminase is responsible for a recombination function other than DNA cleavage in class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 106, 2758–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Das C., Tyler J. K., Churchill M. E. (2010) The histone shuffle. Histone chaperones in an energetic dance. Trends Biochem. Sci. 35, 476–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Koning L., Corpet A., Haber J. E., Almouzni G. (2007) Histone chaperones. An escort network regulating histone traffic. Nat. Struct. Mol. Biol. 14, 997–1007 [DOI] [PubMed] [Google Scholar]
- 30. Tagami H., Ray-Gallet D., Almouzni G., Nakatani Y. (2004) Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116, 51–61 [DOI] [PubMed] [Google Scholar]
- 31. Lewis P. W., Elsaesser S. J., Noh K. M., Stadler S. C., Allis C. D. (2010) Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. U.S.A. 107, 14075–14080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaplan C. D., Laprade L., Winston F. (2003) Transcription elongation factors repress transcription initiation from cryptic sites. Science 301, 1096–1099 [DOI] [PubMed] [Google Scholar]
- 33. Begum N. A., Kinoshita K., Kakazu N., Muramatsu M., Nagaoka H., Shinkura R., Biniszkiewicz D., Boyer L. A., Jaenisch R., Honjo T. (2004) Uracil DNA glycosylase activity is dispensable for immunoglobulin class switch. Science 305, 1160–1163 [DOI] [PubMed] [Google Scholar]
- 34. Okazaki I. M., Kotani A., Honjo T. (2007) Role of AID in tumorigenesis. Adv. Immunol. 94, 245–273 [DOI] [PubMed] [Google Scholar]
- 35. Ramiro A. R., Jankovic M., Eisenreich T., Difilippantonio S., Chen-Kiang S., Muramatsu M., Honjo T., Nussenzweig A., Nussenzweig M. C. (2004) AID is required for c-myc/IgH chromosome translocations in vivo. Cell 118, 431–438 [DOI] [PubMed] [Google Scholar]
- 36. Pasqualucci L., Bhagat G., Jankovic M., Compagno M., Smith P., Muramatsu M., Honjo T., Morse H. C., 3rd, Nussenzweig M. C., Dalla-Favera R. (2008) AID is required for germinal center-derived lymphomagenesis. Nat. Genet. 40, 108–112 [DOI] [PubMed] [Google Scholar]
- 37. Kato L., Begum N. A., Burroughs A. M., Doi T., Kawai J., Daub C. O., Kawaguchi T., Matsuda F., Hayashizaki Y., Honjo T. (2012) Nonimmunoglobulin target loci of activation-induced cytidine deaminase (AID) share unique features with immunoglobulin genes. Proc. Natl. Acad. Sci. U.S.A. 109, 2479–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Close D., Johnson S. J., Sdano M. A., McDonald S. M., Robinson H., Formosa T., Hill C. P. (2011) Crystal structures of the S. cerevisiae Spt6 core and C-terminal tandem SH2 domain. J. Mol. Biol. 408, 697–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu J., Zhang J., Gong Q., Xiong P., Huang H., Wu B., Lu G., Wu J., Shi Y. (2011) Solution structure of tandem SH2 domains from Spt6 protein and their binding to the phosphorylated RNA polymerase II C-terminal domain. J. Biol. Chem. 286, 29218–29226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Diebold M. L., Loeliger E., Koch M., Winston F., Cavarelli J., Romier C. (2010) Noncanonical tandem SH2 enables interaction of elongation factor Spt6 with RNA polymerase II. J. Biol. Chem. 285, 38389–38398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee J. H., Tate C. M., You J. S., Skalnik D. G. (2007) Identification and characterization of the human Set1B histone H3-Lys-4 methyltransferase complex. J. Biol. Chem. 282, 13419–13428 [DOI] [PubMed] [Google Scholar]
- 42. Wu M., Wang P. F., Lee J. S., Martin-Brown S., Florens L., Washburn M., Shilatifard A. (2008) Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol. Cell. Biol. 28, 7337–7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Endoh M., Zhu W., Hasegawa J., Watanabe H., Kim D. K., Aida M., Inukai N., Narita T., Yamada T., Furuya A., Sato H., Yamaguchi Y., Mandal S. S., Reinberg D., Wada T., Handa H. (2004) Human Spt6 stimulates transcription elongation by RNA polymerase II in vitro. Mol. Cell. Biol. 24, 3324–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Houseley J., LaCava J., Tollervey D. (2006) RNA-quality control by the exosome. Nat. Rev. Mol. Cell Biol. 7, 529–539 [DOI] [PubMed] [Google Scholar]
- 45. Peckl-Schmid D., Wolkerstorfer S., Königsberger S., Achatz-Straussberger G., Feichtner S., Schwaiger E., Zaborsky N., Huemer M., Gratz I. K., Schibli R., Lamers M., Crameri R., Moser K., Luger E. O., Achatz G. (2010) HAX1 deficiency. Impact on lymphopoiesis and B-cell development. Eur. J. Immunol. 40, 3161–3172 [DOI] [PubMed] [Google Scholar]
- 46. Selth L. A., Sigurdsson S., Svejstrup J. Q. (2010) Transcript elongation by RNA polymerase II. Annu. Rev. Biochem. 79, 271–293 [DOI] [PubMed] [Google Scholar]
- 47. Kizer K. O., Phatnani H. P., Shibata Y., Hall H., Greenleaf A. L., Strahl B. D. (2005) A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol. Cell. Biol. 25, 3305–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Basu U., Meng F. L., Keim C., Grinstein V., Pefanis E., Eccleston J., Zhang T., Myers D., Wasserman C. R., Wesemann D. R., Januszyk K., Gregory R. I., Deng H., Lima C. D., Alt F. W. (2011) The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell 144, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Avvakumov N., Nourani A., Côté J. (2011) Histone chaperones. Modulators of chromatin marks. Mol. Cell 41, 502–514 [DOI] [PubMed] [Google Scholar]
- 50. Pavri R., Zhu B., Li G., Trojer P., Mandal S., Shilatifard A., Reinberg D. (2006) Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell 125, 703–717 [DOI] [PubMed] [Google Scholar]
- 51. Fleming A. B., Kao C. F., Hillyer C., Pikaart M., Osley M. A. (2008) H2B ubiquitylation plays a role in nucleosome dynamics during transcription elongation. Mol. Cell 31, 57–66 [DOI] [PubMed] [Google Scholar]
- 52. Sims R. J., 3rd, Belotserkovskaya R., Reinberg D. (2004) Elongation by RNA polymerase II. The short and long of it. Genes Dev. 18, 2437–2468 [DOI] [PubMed] [Google Scholar]
- 53. Guttman M., Amit I., Garber M., French C., Lin M. F., Feldser D., Huarte M., Zuk O., Carey B. W., Cassady J. P., Cabili M. N., Jaenisch R., Mikkelsen T. S., Jacks T., Hacohen N., Bernstein B. E., Kellis M., Regev A., Rinn J. L., Lander E. S. (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Khalil A. M., Guttman M., Huarte M., Garber M., Raj A., Rivea Morales D., Thomas K., Presser A., Bernstein B. E., van Oudenaarden A., Regev A., Lander E. S., Rinn J. L. (2009) Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 106, 11667–11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Taverna S. D., Li H., Ruthenburg A. J., Allis C. D., Patel D. J. (2007) How chromatin-binding modules interpret histone modifications. Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14, 1025–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Suganuma T., Workman J. L. (2011) Signals and combinatorial functions of histone modifications. Annu. Rev. Biochem. 80, 473–499 [DOI] [PubMed] [Google Scholar]
- 57. Fuchs S. M., Krajewski K., Baker R. W., Miller V. L., Strahl B. D. (2011) Influence of combinatorial histone modifications on antibody and effector protein recognition. Curr. Biol. 21, 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.