

Abstract
The role of epigenetic mechanisms in control of gene expression during mammalian development is well established. Associations between specific DNA or histone modifications and numerous neurodevelopmental and neurodegenerative disorders implies significant consequences of epigenetic dysregulation in both the developing and mature brain, the latter of which is the general focus of this review. Accumulating evidence suggests that epigenetic changes are involved in normal cognitive processes in addition to neurological and psychiatric disorders. Recent investigations into the regulation of epigenetic modifications in the adult brain have revealed novel and surprisingly dynamic mechanisms for controlling learning and memory-related behaviors as well as long-term synaptic plasticity. DNA methylation and histone acetylation have also been implicated in the modulation of basal synaptic transmission and the balance between excitation and inhibition in various brain regions. Studies have begun to uncover some of the alterations in gene expression that appear to mediate many of these effects, but an understanding of the precise mechanisms involved is still lacking. Nevertheless, the fundamental importance of epigenetic processes in influencing neuronal activity is becoming increasingly evident.
Keywords: HDAC, DNA methylation, MeCP2, Neurotransmission, Synaptic Plasticity
The classic definition of epigenetics is the study of potentially heritable changes in gene expression without alteration of the actual DNA sequence. During development, epigenetic mechanisms are important for determining the functional identity of each cell type in an organism. Many of these mechanisms were first discovered as processes by which differentiated cells pass on their unique gene expression patterns onto daughter cells (Goldberg, Allis, & Bernstein, 2007). Recent data over the past decade has called into question the canonical definition of epigenetics, through findings that include observations of epigenetic changes occurring within non-dividing cells to bring about both stable and dynamic functional alterations. One particular area of interest where these epigenetic alterations have a strong impact is in the brain, where epigenetic modifications are not static, but dynamically change in response to external stimuli including synaptic activity (Crepaldi & Riccio, 2009).
The expression of genes is thought to be primarily regulated by the status of chromatin, the complex of DNA and histone proteins that makes up chromosomes. Heterochromatin is genetically inactive due to a tight association between DNA and histones, while euchromatin is the more loosely associated form that allows for active gene transcription. Changes in chromatin structure can be affected by epigenetic modifications, of which the two most extensively investigated mechanisms are histone modifications and DNA methylation. Histone modifications encompass a number of post-translational additions to specific amino acid residues found on histone tails, including acetylation, phosphorylation, methylation, ubiquitination, and SUMOylation. DNA methylation is the covalent addition of methyl groups to the 5’-position of cytosine residues within CpG dinucleotides. While in most instances these mechanisms are studied in isolation with respect to cellular and molecular outcomes, it has been well demonstrated that many function together to bring about changes in gene expression. The abundance of chromatin modifications and the patterns in which they associate with one another are extensively reviewed elsewhere (Berger, 2007; Kouzarides, 2007).
The etiologies of many neurological diseases appear to involve epigenetic processes. The neurodevelopmental disorder, Rett syndrome, is caused by mutations in the methyl-CpG-binding protein 2 (Mecp2) gene, which codes for a protein important for interpreting both DNA methylation and histone acetylation (Amir et al., 1999). Another neurodevelopmental disorder, Rubinstein-Taybi syndrome, results from mutations in a known gene involved in controlling histone acetylation, CREB-binding protein (Cbp) (Petrij et al., 1995). Fragile X syndrome arises from a hypermethylated trinucleotide repeat stretch of DNA on the X chromosome (Sutcliffe et al., 1992). A number of neurodegenerative disorders have also been linked to changes in DNA methylation and/or histone modifications, including Alzheimer’s and Huntington’s diseases (Graff & Mansuy, 2009). Furthermore, chromatin modifications may also be involved in the pathogenesis of psychiatric disorders such as depression, anxiety, and addiction (Renthal & Nestler, 2009). For instance, hypermethylation of the reelin (Reln) promoter and subsequent suppression of reelin expression have been implicated in schizophrenia (Grayson et al., 2005). Given these numerous examples in which epigenetic dysregulation dictates neurological phenotypes, it is becoming increasingly apparent that epigenetic mechanisms play a critical role in regulating neuronal function, cognition, and behavior both during development and in the adult brain.
A number of recent reviews have addressed the role of epigenetics in nervous system development (Fagiolini, Jensen, & Champagne, 2009; MacDonald & Roskams, 2009), so here we will focus on the functional consequences of alterations in histone modifications and DNA methylation in the mature brain. This review will first describe how epigenetics plays an important role in learning and memory-related behaviors and then focus on how the epigenome influences synaptic transmission as well as how synaptic activity can drive changes in epigenetic modifications in return. Complex behavior is dependent upon communication between neural networks in the different areas of the brain. Current research on the involvement of chromatin modifications in the control of long-term synaptic plasticity, basal synaptic transmission, and the balance between excitation and inhibition in brain regions known to be involved in learning and memory behaviors will be discussed. Given that alterations in cognitive function and behavior are ultimately brought about by changes in synaptic activity, it is important to have a detailed understanding of how epigenetic modifications influence synaptic transmission and vice versa.
Epigenetic mechanisms in cognition and behavior
Just as epigenetic mechanisms are utilized during mammalian development by differentiating cells in order to pass along cellular ‘memory’, we are beginning to discover how these same mechanisms can be employed by the adult nervous system to regulate behaviors related to memory formation (Barrett & Wood, 2008; Levenson & Sweatt, 2005, 2006). It has been known for quite some time that the formation of long-term memories is largely dependent upon gene expression, so the notion that neurons can exploit epigenetic modifications in this process is not that surprising. However, the role of chromatin modifications in cognition and behavior is turning out to be an expanding field of study, in that many different histone modifications, DNA methylation, and a number of chromatin-binding proteins have been shown to be dynamically regulated in the adult brain. Furthermore, these discoveries are helping to uncover the means by which these modifications work together to control gene expression, particularly in neurons.
The most extensively studied histone modification in relation to learning and memory is histone acetylation. Histone acetylation occurs on the amino side chain of specific lysine residues found within histone tails, thereby neutralizing their positive charge and interrupting their association with negatively-charged DNA, allowing for the activation of transcription. Histone acetyltransferases (HATs) are the enzymes responsible for utilizing the cofactor acetyl-CoA in this acetylation reaction (Varga-Weisz & Becker, 1998). Histone deacetylases (HDACs) repress transcription through removal of the acetyl group, which then strengthens the histone-DNA interaction and blocks access of the transcriptional machinery to the DNA template (Fischle, Wang, & Allis, 2003). HDACs are separated into classes based on structure and functional similarities, expression patterns, and subcellular localization. The ‘classical’ HDACs, demonstrating confirmed histone deacetylase activity, include those in classes I and II. Class I HDACs (1–3, and 8) are widely expressed and function in protein complexes within the nucleus, while class II HDACs (4–7, 9, and 10) have more specific expression patterns and can be shuttled between the cytoplasm and nucleus in response to phosphorylation or dephosphorylation (Fischle, Kiermer, Dequiedt, & Verdin, 2001).
Studies investigating the effects of histone acetylation on learning and memory have utilized inhibitors of these enzymes, as well as transgenic mice that have altered expression levels of individual HATs and HDACs. Some of the first indications for the involvement of histone acetylation came from studying animal models of Rubenstein-Taybi syndrome. This human neurodevelopmental disorder can result from mutations in the CREB-binding protein gene, Cbp, which codes for a protein with known histone acetyltransferase activity. Several Cbp mutant mice were discovered to display impairments in long-term, but not short-term, memory in a number of different learning and memory tests, including passive avoidance, fear conditioning, Morris water maze, and object recognition (Alarcon et al., 2004; Bourtchouladze et al., 2003; Korzus, Rosenfeld, & Mayford, 2004; Oike et al., 1999; M. A. Wood et al., 2005). A recent study of Cbp conditional knockout (KO) mice found similar deficits in long-term memory behaviors along with impaired short-term associative and object-recognition memory (Chen, Zou, Watanabe, van Deursen, & Shen, 2010). Administration of the Class I HDAC inhibitors, trichostatin A (TSA) or suberoylanilide hydroxamic acid (SAHA), to Cbp mutant mice reversed their deficits in associative and declarative memory (Alarcon et al., 2004; Korzus et al., 2004). When TSA was administered systemically to the conditional KO mice, which completely lack expression of CBP in excitatory neurons of the forebrain, no rescue effect on associative memory was observed (Chen et al., 2010). The authors of this study speculate that the inability of the HDAC inhibitor to rescue memory deficits in the conditional Cbp KO mice was due to the complete deletion of CBP from excitatory neurons, compared to the previous effects of HDAC inhibition on heterozygous or dominant negative Cbp mice in which some CBP HAT activity likely remained. This difference in remaining HAT activity may also be why the conditional KO mice displayed additional short-term memory deficits compared to other Cbp mutant mice. Together, the data strongly suggest that CBP can promote memory formation through its transcriptional activation of genes important for learning and memory (Figure 1).
Figure 1. Epigenetic control of behavior and synapse function in mature hippocampal neurons.
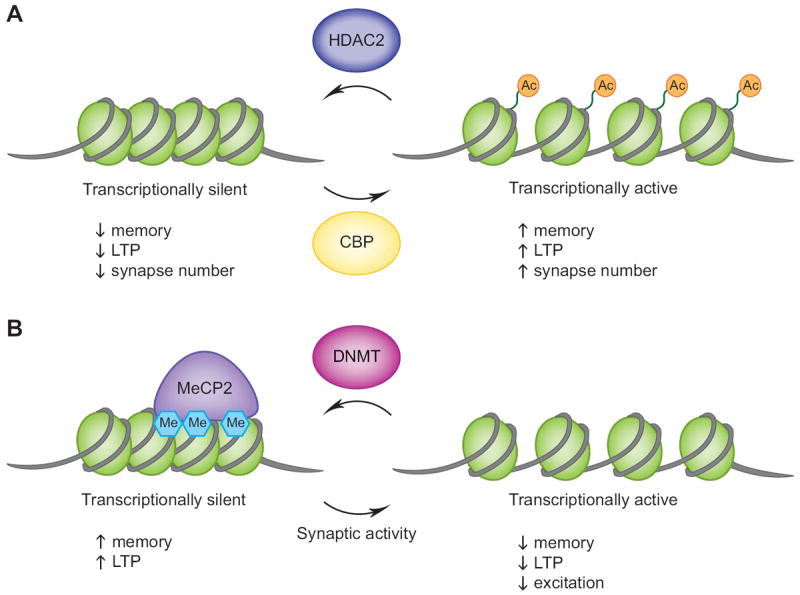
(A) Studies utilizing transgenic mice and pharmacological inhibitors indicate that histone acetylation, likely via CBP’s HAT activity, leads to enhanced memory behaviors and long-term potentiation (LTP) and an increase in the number of synapses. Conversely, histone deacetylation by HDAC2 results in memory and LTP deficits and fewer synapses. (B) DNA methylation in the adult hippocampus can be catalyzed by the DNA methyltransferase enzymes DNMT1 and/or DNMT3a, which leads to repressed gene expression and enhanced memory and LTP. MeCP2 is an example of a methyl-binding protein suggested to be involved in mediating some of these effects. DNA demethylation has been shown to occur at specific gene promoters in response to synaptic activity or with the loss of DNMT activity, which results in impairments in LTP and memory formation as well as indications of decreased excitatory activity.
Histone acetyltransferase activity has been directly measured and found to increase in the amygdala following cued fear conditioning, a measure of amygdala- and hippocampal-dependent associative learning (Yeh, Lin, & Gean, 2004). This study went on to show that TSA injection directly into the amygdala was able to enhance long-term memory seen with fear-potentiated startle. Memory consolidation and extinction during hippocampal-dependent contextual fear conditioning have been shown to increase as a result of intrahippocampal infusions of HDAC inhibitors (Lattal, Barrett, & Wood, 2007; Vecsey et al., 2007). Similar effects were also seen after intraperitoneal injections of the HDAC inhibitors valproic acid (VPA) and sodium butyrate (NaB) (Bredy & Barad, 2008; Bredy et al., 2007; Lattal et al., 2007). The time course of alterations in histone acetylation during learning and memory processes appears to be rapid and short-lived. An increase in histone H3 acetylation in area CA1 of the hippocampus was seen 1 hour after fear conditioning, but had returned to baseline levels after 24 hours (Levenson et al., 2004). These dynamic changes in response to memory formation are supported by the previously mentioned studies using acute exposures to HDAC inhibitors. Chronic administration into the amygdala has been shown to result in increased acetylation of histones H3 and H4 accompanied by a deficit in cue-dependent fear conditioning (Adachi, Autry, Covington, & Monteggia, 2009). A recent study helped delineate which specific HDAC enzymes are important for bringing about many of the memory enhancements seen with non-specific HDAC inhibitors. It was discovered that mice with HDAC2 overexpression, but not HDAC1, have impaired learning and memory, while mice lacking neuronal expression of HDAC2 display enhanced hippocampal-dependent memory formation, suggesting that HDAC2 is a negative regulator of memory formation (J. S. Guan et al., 2009). All together, these findings highlight the importance of histone acetylation and deacetylation in controlling an animal’s ability to form long-term memories (Figure 1).
Alterations in histone phosphorylation have been shown to be closely correlated with learning and memory induced changes in acetylation. Phosphorylation occurs on serine/threonine residues of histones H3, H2A, and H2B and is generally associated with transcriptional activation (Berger, 2007). In addition to inducing increases in histone H3 acetylation, contextual fear conditioning also appears to increase H3 phosphorylation and phosphoacetylation in the hippocampus (Chwang, O’Riordan, Levenson, & Sweatt, 2006; Lubin & Sweatt, 2007). Histone methylation is another modification that has been recently implicated in learning and memory. Histones can be mono-, di-, or trimethylated, and each level of modification can have different effects on gene expression. A recent study found deficits in long-term memory in mice deficient in the histone H3 lysine (K) 4-specific methyltransferase, Mll (Gupta et al., 2010). In normal mice, contextual fear conditioning resulted in immediate increases in H3K4 trimethylation and H3K9 dimethylation in the hippocampus. H3K4 trimethylation is believed to be a marker of transcriptional activation, however, recent evidence suggests that alterations in histone methylation may have differing effects on gene expression depending on the presence of other histone modifications (Berger, 2007). Despite this caveat, given what is currently understood about histone modifications and transcriptional control, it appears that memory-related behaviors are positively associated with histone modifications, particularly on histone H3, which favor increased gene expression – acetylation, phosphorylation, and K4 trimethylation.
Besides histone modifications, epigenetic control of gene expression can occur via DNA methylation. In mammals, DNA methylation occurs predominantly on the cytosine residues of CpG dinucleotides, which can sometimes be found in repetitive lengths referred to as CpG islands. These methyl groups are covalently added to DNA by the enzymes DNA methyltransferases (DNMTs). There are three main DNMTs expressed in mammals: DNMT1, 3a and 3b. DNMT3a and DNMT3b are de novo methyltransferases that establish methylation patterns at specific sites within the genome (Okano, Bell, Haber, & Li, 1999). DNMT1 is responsible for the maintenance of these methylation patterns during DNA replication (Hermann, Goyal, & Jeltsch, 2004). Currently, the initiation signals for DNA methylation and how DNMTs are targeted to specific gene promoters remain unclear.
While DNA methylation has long been recognized as a marker for transcriptional repression, researchers have recently uncovered many new findings regarding this epigenetic marker, particularly pertaining to dynamic changes in DNA methylation in the adult brain. Just as alterations in histone modifications have been observed during learning and memory tasks in rodents, somewhat surprisingly, so have changes in DNA methylation. Contextual fear conditioning has been found to both increase and decrease methylation patterns on different promoters of the brain-derived neurotrophic factor (Bdnf) gene, which is known to be involved in many behaviors including learning and memory (Lubin, Roth, & Sweatt, 2008). Contextual fear conditioning also increased expression of both DNMT3a and DNMT3b in the CA1 region of the hippocampus (Miller & Sweatt, 2007). Intrahippocampal infusions of inhibitors of DNMT activity, 5-aza-deoxycytidine (5-aza) and zebularine (Zeb) resulted in deficits in fear memory consolidation (Lubin et al., 2008; Miller & Sweatt, 2007). Accordingly, mice lacking expression of both DNMT1 and DNMT3a in forebrain postmitotic neurons showed impaired memory consolidation during fear conditioning as well as impaired spatial learning and memory in the Morris water maze (Feng et al., 2010). The question of why hippocampal-dependent associative memory appears to be associated with DNA methylation changes that favor transcriptional repression while the opposite is typically seen for histone modifications remains to be answered (Figure 1). The simplest explanation would be that distinct sets of target genes are regulated by histone modification when compared to DNA methylation in the rodent brain, resulting in opposing effects on behavior and synaptic transmission, but this has yet to be verified.
Often grouped into epigenetics with histone modifications and DNA methylation are proteins that are known to bind to these markers and help translate them into functional alterations in gene expression, typically by recruiting additional proteins and transcription factors to the promoters of specific genes. Once such protein is methyl-CpG-binding protein 2 (MeCP2), which contains a methyl-binding domain (MBD) important for interacting with methylated cytosines and a transcriptional repression domain that helps recruit co-repressor proteins such as HDACs (Nan et al., 1998). In humans, mutations in the Mecp2 gene are associated with the neurodevelopmental disorder Rett syndrome, a predominantly female occurring disorder that manifests itself with autonomic and cognitive deficits related to mental retardation and autism (Van den Veyver & Zoghbi, 2001). Mecp2 mutant mice have impairments in many behaviors, including motor coordination, social interaction, anxiety, and spatial and associative learning and memory (Adachi et al., 2009; Gemelli et al., 2006; McGill et al., 2006; Moretti et al., 2006; Shahbazian et al., 2002). Conversely, MeCP2 overexpressing mice show enhanced motor and contextual learning, however, having too much MeCP2 also appears to be detrimental in that many of these mice go on to develop seizures and have shortened life spans (Collins et al., 2004). Interestingly, mice lacking expression of another methyl-binding domain-containing protein, MBD1, also display deficits in a number of behavioral tests, including those for social interaction, anxiety- and depression-related behaviors, and spatial and associative learning (Allan et al., 2008; Zhao et al., 2003). This suggests that DNA methylation, and the proteins important for interpreting methylation patterns, play very important roles in the adult brain, especially with respect to learning and memory.
Epigenetic control of long-term synaptic plasticity
It has been known for quite some time now that changes in gene expression coupled with alterations in neuronal activity can lead to persistent long-term potentiation (LTP) of synaptic strength (Castellucci et al., 1986), the widely accepted cellular mechanism behind long-term memory. LTP is characterized as a long-lasting enhancement in synapse-specific neurotransmission in response to repetitive, high frequency stimulation. Long-term facilitation of excitatory synaptic transmission has been observed in numerous areas of the mammalian brain, including the hippocampus, amygdala, striatum, and cortex, however the molecular changes that modulate LTP in different brain regions can vary significantly (Malenka & Bear, 2004). Enhancements and impairments in spatial memory are often seen in animals that display increases or decreases in hippocampal LTP, respectively, with similar correlations observed in amygdala-dependent fear memory and LTP. Many of the studies looking at epigenetics in the control of learning and memory have also explored the importance of these modifications in the induction of LTP.
Histone acetylation was first linked with changes in long-term synaptic plasticity at the sensory motor synapse of the large sea slug, Aplysia. It was observed that serotonin-induced long-term facilitation resulted in increases in histone H3 and H4 acetylation, and that treatment with the HDAC inhibitor TSA was sufficient to turn short-term facilitation into long-term facilitation (Z. Guan et al., 2002). Furthermore, this study found opposing effects on histone acetylation associated with induction of long-term depression (LTD), an activity-dependent long-lasting weakening of synaptic efficacy, which could be blocked by treatment with TSA. HDAC inhibitors have since been shown to promote long-term potentiation in mice. TSA and NaB treatment of hippocampal slices enhanced induction of LTP at Schaffer-collateral synapses (Levenson et al., 2004; Vecsey et al., 2007). Treatment of amygdala-containing slices with TSA also resulted in enhancement of forskolin-induced LTP (Yeh et al., 2004). Heterozygous Cbp mutant mice were discovered to have impaired hippocampal late-phase LTP (L-LTP) in response to typical theta-burst stimulation; however this deficit was no longer seen using a stronger stimulation protocol (Alarcon et al., 2004). HDAC2 overexpressing mice also displayed deficits in hippocampal LTP, while LTP in Hdac2 forebrain-specific KO mice was enhanced (J. S. Guan et al., 2009). Treatment of hippocampal slices with SAHA enhanced L-LTP in wild-type mice, ameliorated the deficit seen in Cbp+/- mice, and had no effect on the enhancement of LTP seen in Hdac2 KO mice (Alarcon et al., 2004; J. S. Guan et al., 2009). Collectively, these data emphasize the importance of histone acetylation in the regulation of long-term synaptic plasticity, with CBP and HDAC2 possibly acting as the predominant enzymes responsible for mediating such effects (Figure 1).
DNA methylation and DNA methyl-binding proteins have also been shown to play roles in the induction of long-term synaptic plasticity. Treatment of hippocampal slices with the DNMT inhibitors, Zeb and 5-aza, was found to reduce the magnitude of theta-burst stimulated LTP (Levenson et al., 2006). These effects appear to be modulated by both the maintenance methyltransferase, DNMT1, and the de novo methyltransferase, DNMT3a. Hippocampal neurons lacking only one of these enzymes display normal LTP, whereas loss of both enzymes results in significant L-LTP deficits (Feng et al., 2010). DNMT1 and DNMT3a also appear to function redundantly in the induction of LTD, all of which is somewhat surprising given the current understanding of their distinct functionalities. Mecp2 mutant mice have also been found to have impairments in hippocampal LTP and LTD, as well as cortical LTP (Asaka, Jugloff, Zhang, Eubanks, & Fitzsimonds, 2006; Moretti et al., 2006), while MeCP2 overexpressing mice show enhanced LTP in the hippocampus (Collins et al., 2004). The LTP deficits appear to be age dependent, in that younger (<4 weeks old) presymptomatic mice display normal LTP while older (≥4 weeks old) symptomatic mice show reductions in LTP magnitude and maintenance (Asaka et al., 2006; Dani & Nelson, 2009; Moretti et al., 2006). Furthermore, LTD that is dependent on presynaptic mechanisms was impaired in mice expressing a truncated form of Mecp2 (Mecp2308/y), while LTD that relies on postsynaptic mechanisms was normal (Moretti et al., 2006). Mice lacking expression of MBD1 were found to display impairments in LTP of the dentate gyrus (Zhao et al., 2003), an area in the hippocampus where neurogenesis occurs. This deficit was determined to likely be due to a reduction in the birth of new neurons in these mice. In light of all of these studies, it is becoming clear that DNA methylation is involved in controlling long-term synaptic plasticity in many areas of the brain. Research on mice lacking methyl-binding proteins indicates that DNA methylation’s effects on synaptic plasticity may be a result of alterations in presynaptic function or neurogenesis, mediated through proteins such as MeCP2 or MBD1.
Epigenetic control of basal synaptic transmission
Long-term synaptic plasticity is just one aspect of communication between CNS neurons, which relies on proper functioning of each component of synaptic activity, including presynaptic neurotransmitter release and postsynaptic neurotransmitter receptor responsiveness. Alterations in LTP and LTD could result in changes in basal synaptic transmission, while fundamental alterations in presynaptic and postsynaptic function could also impact the ability to induce long-term plasticity. For the sake of this review, the discussion of basal synaptic transmission will encompass assessments of everything from spontaneous activity to short-term plasticity. While LTP has a well-established behavioral correlate in learning and memory, observed alterations in basal neurotransmission are associated with defined synaptic properties. Short-term changes in synaptic strength, on time scales of milliseconds to seconds, are believed to have an inverse relationship to changes in the probability of neurotransmitter release from presynaptic terminals (Zucker & Regehr, 2002). Spontaneous, non-action potential-driven release has been implicated in influencing firing rates, stabilizing synaptic function and structural integrity, and activating distinct postsynaptic signaling pathways (Sutton & Schuman, 2009). Changes in miniature events also have functional correlates in that frequency is linked to presynaptic neurotransmitter release or synapse number and amplitude is linked to postsynaptic receptor number or responsiveness.
Many of the studies identifying LTP and LTD deficits associated with epigenetic modifications in mice also evaluated a number of measures of basal synaptic transmission. Hippocampal slices from heterozygous Cbp mutant mice revealed no alterations in input-output relationships, a measure of synaptic connectivity, or in short-term synaptic plasticity, assayed via paired pulse facilitation (Alarcon et al., 2004). These measurements were also unchanged following acute treatment of hippocampal slices with various HDAC inhibitors – SAHA, TSA, or NaB (Alarcon et al., 2004; Levenson et al., 2004) – or in mice overexpressing HDAC2 in the brain (J. S. Guan et al., 2009). These data suggest that synaptic connections and neurotransmitter release are unaffected by histone acetylation, despite observed robust changes in long-term synaptic plasticity. In the case of the HDAC2 overexpressing mice, the hippocampal LTP deficits have been attributed to a decrease in synapse number, seen as a decrease in dendritic spines and synaptophysin staining in CA1 pyramidal neurons, while Hdac2 KO mice showed enhanced LTP and increased synapse number (J. S. Guan et al., 2009). Alterations in synapse number have also been observed as a result of altering HDAC activity in dissociated hippocampal neurons in culture. In very young cultures (5 days in vitro), treatment with the Class I HDAC inhibitor, TSA, caused a significant increase in excitatory, but not inhibitory, synapse number (Akhtar et al., 2009). This change was correlated with a large increase in the frequency of miniature excitatory postsynaptic currents (mEPSCs). Double deletion, but not single deletion, of floxed Hdac1 and Hdac2 using a lentiviral Cre-recombinase system in young hippocampal neurons also caused a significant increase in mEPSC frequency, suggesting that both HDAC1 and HDAC2 are important for controlling synapse number in immature neurons. However, in older (16 days in vitro) hippocampal cultures, treatment with TSA or deletion of Hdac2 alone resulted in a decrease in the frequency of mEPSCs with no changes in excitatory synapse number (Akhtar et al., 2009; Nelson, Kavalali, & Monteggia, 2006). Accordingly, HDAC2 overexpression caused an increase in mEPSC frequency (Akhtar et al., 2009). Altogether, these data suggest that histone acetylation can regulate the formation of synapses in hippocampal neurons, with HDAC1 and HDAC2 playing both redundant and distinct roles during the time course of neuronal maturation. Some aspects of basal synaptic transmission, such as subtle changes in presynaptic neurotransmitter release, may also be controlled by histone acetylation.
Evidence now supports the idea that DNA methylation is another epigenetic modification that plays a role in the induction of long-term synaptic plasticity; therefore its role in basal synaptic function has also been evaluated. Similar to what was observed with HDAC inhibitors, acute treatment of hippocampal slices with inhibitors of DNMT activity, Zeb and 5-aza, did not reveal any major deficits in input-output curves, paired pulse facilitation, or basal synaptic efficacy, despite the fact that LTP was significantly impaired (Levenson et al., 2006). Comparable results were seen in hippocampal slices from Dnmt1 and Dnmt3a conditional KO mice (Feng et al., 2010). Very subtle alterations in NMDA receptor function and synaptic connectivity were actually observed in these studies, findings that were somewhat supported by experiments done in hippocampal cultures. Treatment of mature dissociated hippocampal neurons with a DNMT inhibitor resulted in a decrease in mEPSC frequency with no change in amplitudes or miniature inhibitory postsynaptic currents (mIPSCs) (Nelson et al., 2006; Nelson, Kavalali, & Monteggia, 2008). Spontaneous synaptic vesicle fusion and the magnitude of miniature NMDA receptor currents were also significantly reduced. However, no changes were seen in paired pulse ratios or short-term synaptic depression in response to 10 Hz train stimulation (Nelson et al., 2008), both measures of activity-driven presynaptic neurotransmitter release. Whether these minor changes in synaptic efficacy are responsible for bringing about the significant deficits in LTP seen with impaired DNMT activity is unclear.
The role of MeCP2 in controlling synaptic transmission has been reviewed elsewhere (Monteggia & Kavalali, 2009), therefore the topic will only be briefly discussed here. Field recordings of acute hippocampal slices from Mecp2 mutant mice have revealed enhanced synaptic connectivity, decreased inhibitory rhythmic activity, hyperexcitability in response to high frequency stimulation, and reduced paired pulse facilitation – interpreted as an enhancement in the probability of neurotransmitter release (Asaka et al., 2006; Moretti et al., 2006; Zhang, He, Jugloff, & Eubanks, 2008). Hippocampal neurons from MeCP2 overexpressing mice display enhanced paired pulse facilitation, suggesting reduced presynaptic release probability. Together, these alterations suggest that MeCP2 is involved in basal synaptic function and neuronal circuitry in the hippocampus. Dissociated hippocampal neurons from MeCP2-deficient mice display reduced paired pulse ratios, faster response depression to a brief 10 Hz train stimulation, decreased mEPSC frequency, reduced glutamatergic response amplitudes, and a decrease in excitatory synapse number suggesting impairments in excitatory synapse function (Chao, Zoghbi, & Rosenmund, 2007; Nelson et al., 2006). Similar deficits have been observed in cortical slices lacking MeCP2, including reductions in excitatory input, spontaneous EPSC frequency and amplitudes, and circuit connectivity (Dani et al., 2005; Tropea et al., 2009; L. Wood, Gray, Zhou, Greenberg, & Shepherd, 2009; L. Wood & Shepherd, 2010). Spontaneous inhibitory postsynaptic current (IPSC) amplitudes were also found to be increased in the cortex (Dani et al., 2005). Altogether, these findings implicate MeCP2 in controlling the balance between excitation and inhibition in the mature brain, with cortical and hippocampal regions of MeCP2 mutant mice having a propensity to favor inhibition over excitation. In accordance with this premise, an imbalance between excitatory and inhibitory synaptic inputs has been proposed as the cellular basis for a number of neurodevelopmental disorders, including Rett syndrome (Rubenstein & Merzenich, 2003).
The basal synaptic transmission deficits in Mecp2 mutant mice insinuate that more general epigenetic mechanisms may also be important for controlling the balance between excitation and inhibition in the mature brain. Some observations that support this hypothesis include: the regulation of excitatory synapse formation by HDACs1 and 2 (Akhtar et al., 2009; J. S. Guan et al., 2009), the decrease in mEPSC frequency but not mIPSC properties following treatment with DNMT inhibitors (Nelson et al., 2008), the presence of handling-induced seizures in HDAC4 mutant mice (Rajan et al., 2009), and an increase in neuronal excitability genes and decrease in inhibition genes following treatment of rat cortical neurons with an HDAC inhibitor (Fukuchi et al., 2009).
Gene-specific epigenetic modifications related to behavior and synaptic transmission
Global changes in epigenetic markers in response to fear conditioning have been demonstrated (Chwang et al., 2006; Levenson et al., 2004; Lubin & Sweatt, 2007). Elucidating the specificity of these modifications with respect to gene targets is essential to understanding how the resulting changes in gene expression may be regulating synaptic activity and learning and memory behaviors. Using candidate gene approaches, a number of promising gene-specific epigenetic modifications have been identified. Epigenetic markers on the promoter regions of the Bdnf gene have been the most extensively studied, including alterations in histone acetylation, phosphorylation, methylation, and DNA methylation associated with memory behavior (Bredy et al., 2007; Gupta et al., 2010; Lubin et al., 2008), as well as activity-dependent changes in DNA methylation (Martinowich et al., 2003; Nelson et al., 2008). Additional genes known to be involved in the regulation of long-term memory and synaptic plasticity, such as protein phosphatase 1 (Pp1) and Reln, have been shown to be modified by DNA methylation during these processes, and treatment with DNMT inhibitors resulted in increased expression of these two genes (Levenson et al., 2006; Miller & Sweatt, 2007). However, Pp1 and Reln expression levels were unchanged in the hippocampus of Dnmt1 and Dnmt3a double knockout mice that display altered memory behavior and synaptic transmission (Feng et al., 2010). Additional works suggests that PP1 itself may actually be responsible for regulating histone modifications at the promoters of the other target genes, Creb and NF-kB, during memory formation (Koshibu et al., 2009).
Most of the gene identification studies performed up to this point have assayed for only one type of epigenetic marker, leaving the question as to whether additional modifications are taking place to ultimately affect gene expression. While some of these studies also went on to monitor expression levels of these genes, the candidate gene approach fails to take into consideration alternate gene possibilities. This problem can be somewhat remedied using more global, gene chip expression and modification assays (Feng et al., 2010). But, again, the issues of not evaluating the entire array of epigenetic modifications at specific gene promoters, along with the fact that gene chips fail to cover a large portion of the genome, means current technology has not yet reached the levels needed to fully assess the gene expression changes responsible for mediating many of the epigenetically associated phenotypes in the adult rodent brain.
Conclusion
We are now only beginning to unravel the role of epigenetics in controlling neuronal functions that may ultimately underlie behavioral adaptations. At this time, it is difficult to draw firm conclusions about how the epigenetic changes observed during basal synaptic functioning may be related to those modifications regulated in conjunction with behavior and long-term synaptic plasticity. Perhaps the enhancements in LTP and memory seen with HDAC inhibitors and the loss of HDAC2 are due in part to an increase in excitatory synapse number, which was observed in both Hdac2 KO mice and young hippocampal cultures lacking HDAC1 and HDAC2 (Akhtar et al., 2009; J. S. Guan et al., 2009). With respect to those results seen with reduced DNMT function or expression, it is conceivable that the deficits in LTP and memory could be attributable to minor alterations in NMDA receptor function or synaptic connectivity that were observed in a few individual studies (Feng et al., 2010; Levenson et al., 2006; Nelson et al., 2008). And it is likely that the methyl-binding proteins, MeCP2 or MDB1, are important for interpreting these changes in DNA methylation into alterations in gene expression. Additional research is needed to clarify these observations and support such hypotheses. The use of genetic mice to explore the role of individual HDACs, HATs, DNMTs, and other genes involved in epigenetic mechanisms in the brain will be important to further delineate the role of these factors in behavior and synaptic transmission. These animal models will also be important in examining whether these enzymes have specific downstream targets or whether there is a convergence on particular targets. Addressing these challenges will help expand our knowledge on the role of epigenetics in the brain and may lead to clinical applications for a variety of neurological and psychiatric illnesses in the future.
Research Highlights.
Epigenetic mechanisms in cognition and behavior
Epigenetics and long-term synaptic plasticity
Epigenetic mechanisms and basal synaptic transmission
Excitation – Inhibition balance in the brain
Gene-specific epigenetic modifications related to behavior and synaptic transmission
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi M, Autry AE, Covington HE, 3rd, Monteggia LM. Mecp2-mediated transcription repression in the basolateral amygdala may underlie heightened anxiety in a mouse model of rett syndrome. Journal of Neuroscience. 2009;29:4218–4227. doi: 10.1523/JNEUROSCI.4225-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar MW, Raingo J, Nelson ED, Montgomery RL, Olson EN, Kavalali ET, et al. Histone deacetylases 1 and 2 form a developmental switch that controls excitatory synapse maturation and function. Journal of Neuroscience. 2009;29:8288–8297. doi: 10.1523/JNEUROSCI.0097-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, et al. Chromatin acetylation, memory, and ltp are impaired in cbp+/- mice: A model for the cognitive deficit in rubinstein-taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Allan AM, Liang X, Luo Y, Pak C, Li X, Szulwach KE, et al. The loss of methyl-cpg binding protein 1 leads to autism-like behavioral deficits. Human Molecular Genetics. 2008;17:2047–2057. doi: 10.1093/hmg/ddn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in x-linked mecp2, encoding methyl-cpg-binding protein 2. Nature Genetics. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the mecp2-null mouse model of rett syndrome. Neurobiology of Disease. 2006;21:217–227. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Barrett RM, Wood MA. Beyond transcription factors: The role of chromatin modifying enzymes in regulating transcription required for memory. Learning & Memory. 2008;15:460–467. doi: 10.1101/lm.917508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Bourtchouladze R, Lidge R, Catapano R, Stanley J, Gossweiler S, Romashko D, et al. A mouse model of rubinstein-taybi syndrome: Defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proceedings of the National Academy of Science U S A. 2003;100:10518–10522. doi: 10.1073/pnas.1834280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredy TW, Barad M. The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learning & Memory. 2008;15:39–45. doi: 10.1101/lm.801108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual bdnf gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learning & Memory. 2007;14:268–276. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellucci VF, Frost WN, Goelet P, Montarolo PG, Schacher S, Morgan JA, et al. Cell and molecular analysis of long-term sensitization in aplysia. Journal of Physiology (Paris) 1986;81:349–357. [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. Mecp2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Zou X, Watanabe H, van Deursen JM, Shen J. Creb binding protein is required for both short-term and long-term memory formation. Journal of Neuroscience. 2010;30:13066–13077. doi: 10.1523/JNEUROSCI.2378-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwang WB, O’Riordan KJ, Levenson JM, Sweatt JD. Erk/mapk regulates hippocampal histone phosphorylation following contextual fear conditioning. Learning & Memory. 2006;13:322–328. doi: 10.1101/lm.152906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, et al. Mild overexpression of mecp2 causes a progressive neurological disorder in mice. Human Molecular Genetics. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- Crepaldi L, Riccio A. Chromatin learns to behave. Epigenetics. 2009;4:23–26. doi: 10.4161/epi.4.1.7604. [DOI] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of rett syndrome. Proceedings of the National Academy of Science U S A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Nelson SB. Intact long-term potentiation but reduced connectivity between neocortical layer 5 pyramidal neurons in a mouse model of rett syndrome. Journal of Neuroscience. 2009;29:11263–11270. doi: 10.1523/JNEUROSCI.1019-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiolini M, Jensen CL, Champagne FA. Epigenetic influences on brain development and plasticity. Current Opinion in Neurobiology. 2009;19:207–212. doi: 10.1016/j.conb.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, et al. Dnmt1 and dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nature Neuroscience. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Kiermer V, Dequiedt F, Verdin E. The emerging role of class ii histone deacetylases. Biochemistry and Cell Biology. 2001;79:337–348. [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Current Opinion in Cell Biology. 2003;15:172–183. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- Fukuchi M, Nii T, Ishimaru N, Minamino A, Hara D, Takasaki I, et al. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neuroscience Research. 2009;65:35–43. doi: 10.1016/j.neures.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM. Postnatal loss of methyl-cpg binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of rett syndrome in mice. Biological Psychiatry. 2006;59:468–476. doi: 10.1016/j.biopsych.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: A landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Graff J, Mansuy IM. Epigenetic dysregulation in cognitive disorders. European Journal of Neuroscience. 2009;30:1–8. doi: 10.1111/j.1460-9568.2009.06787.x. [DOI] [PubMed] [Google Scholar]
- Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, et al. Reelin promoter hypermethylation in schizophrenia. Proceedings of the National Academy of Science U S A. 2005;102:9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, et al. Hdac2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Giustetto M, Lomvardas S, Kim JH, Miniaci MC, Schwartz JH, et al. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, et al. Histone methylation regulates memory formation. Journal of Neuroscience. 2010;30:3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann A, Goyal R, Jeltsch A. The dnmt1 DNA-(cytosine-c5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. Journal of Biological Chemistry. 2004;279:48350–48359. doi: 10.1074/jbc.M403427200. [DOI] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. Cbp histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshibu K, Graff J, Beullens M, Heitz FD, Berchtold D, Russig H, et al. Protein phosphatase 1 regulates the histone code for long-term memory. Journal of Neuroscience. 2009;29:13079–13089. doi: 10.1523/JNEUROSCI.3610-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lattal KM, Barrett RM, Wood MA. Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behavioral Neuroscience. 2007;121:1125–1131. doi: 10.1037/0735-7044.121.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, O’Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. Journal of Biological Chemistry. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, et al. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. Journal of Biological Chemistry. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nature Review Neuroscience. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms: A common theme in vertebrate and invertebrate memory formation. Cell and Molecular Life Sciences. 2006;63:1009–1016. doi: 10.1007/s00018-006-6026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of bdnf gene transcription in the consolidation of fear memory. Journal of Neuroscience. 2008;28:10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD, Sweatt JD. The ikappab kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron. 2007;55:942–957. doi: 10.1016/j.neuron.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JL, Roskams AJ. Epigenetic regulation of nervous system development by DNA methylation and histone deacetylation. Progress in Neurobiology. 2009;88:170–183. doi: 10.1016/j.pneurobio.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. Ltp and ltd: An embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, et al. DNA methylation-related chromatin remodeling in activity-dependent bdnf gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- McGill BE, Bundle SF, Yaylaoglu MB, Carson JP, Thaller C, Zoghbi HY. Enhanced anxiety and stress-induced corticosterone release are associated with increased crh expression in a mouse model of rett syndrome. Proceedings of the National Academy of Science U S A. 2006;103:18267–18272. doi: 10.1073/pnas.0608702103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Kavalali ET. Rett syndrome and the impact of mecp2 associated transcriptional mechanisms on neurotransmission. Biological Psychiatry. 2009;65:204–210. doi: 10.1016/j.biopsych.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, et al. Learning and memory and synaptic plasticity are impaired in a mouse model of rett syndrome. Journal of Neuroscience. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, et al. Transcriptional repression by the methyl-cpg-binding protein mecp2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. Mecp2-dependent transcriptional repression regulates excitatory neurotransmission. Current Biology. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. Journal of Neuroscience. 2008;28:395–406. doi: 10.1523/JNEUROSCI.3796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oike Y, Hata A, Mamiya T, Kaname T, Noda Y, Suzuki M, et al. Truncated cbp protein leads to classical rubinstein-taybi syndrome phenotypes in mice: Implications for a dominant-negative mechanism. Human Molecular Genetics. 1999;8:387–396. doi: 10.1093/hmg/8.3.387. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases dnmt3a and dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, et al. Rubinstein-taybi syndrome caused by mutations in the transcriptional co-activator cbp. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Rajan I, Savelieva KV, Ye GL, Wang CY, Malbari MM, Friddle C, et al. Loss of the putative catalytic domain of hdac4 leads to reduced thermal nociception and seizures while allowing normal bone development. Public Library of Science One. 2009;4:e6612. doi: 10.1371/journal.pone.0006612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Nestler EJ. Chromatin regulation in drug addiction and depression. Dialogues in Clinical Neurosciences. 2009;11:257–268. doi: 10.31887/DCNS.2009.11.3/wrenthal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes, Brain and Behavior. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, et al. Mice with truncated mecp2 recapitulate many rett syndrome features and display hyperacetylation of histone h3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, et al. DNA methylation represses fmr-1 transcription in fragile x syndrome. Human Molecular Genetics. 1992;1:397–400. doi: 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. Partitioning the synaptic landscape: Distinct microdomains for spontaneous and spike-triggered neurotransmission. Science Signaling. 2009;2:pe19. doi: 10.1126/scisignal.265pe19. [DOI] [PubMed] [Google Scholar]
- Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, et al. Partial reversal of rett syndrome-like symptoms in mecp2 mutant mice. Proceedings of the National Academy of Science U S A. 2009;106:2029–2034. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Veyver IB, Zoghbi HY. Mutations in the gene encoding methyl-cpg-binding protein 2 cause rett syndrome. Brain & Development. 2001;23(Suppl 1):S147–151. doi: 10.1016/s0387-7604(01)00376-x. [DOI] [PubMed] [Google Scholar]
- Varga-Weisz PD, Becker PB. Chromatin-remodeling factors: Machines that regulate? Current Opinion in Cell Biology. 1998;10:346–353. doi: 10.1016/s0955-0674(98)80010-0. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via creb:Cbp-dependent transcriptional activation. Journal of Neuroscience. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood L, Gray NW, Zhou Z, Greenberg ME, Shepherd GM. Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in an rna interference model of methyl-cpg-binding protein 2 deficiency. Journal of Neuroscience. 2009;29:12440–12448. doi: 10.1523/JNEUROSCI.3321-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood L, Shepherd GM. Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in a mutant mouse model of rett syndrome. Neurobiology of Disease. 2010 doi: 10.1016/j.nbd.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AM, Lombardi TL, et al. Transgenic mice expressing a truncated form of creb-binding protein (cbp) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learning & Memory. 2005;12:111–119. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh SH, Lin CH, Gean PW. Acetylation of nuclear factor-kappab in rat amygdala improves long-term but not short-term retention of fear memory. Molecular Pharmacology. 2004;65:1286–1292. doi: 10.1124/mol.65.5.1286. [DOI] [PubMed] [Google Scholar]
- Zhang L, He J, Jugloff DG, Eubanks JH. The mecp2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus. 2008;18:294–309. doi: 10.1002/hipo.20389. [DOI] [PubMed] [Google Scholar]
- Zhao X, Ueba T, Christie BR, Barkho B, McConnell MJ, Nakashima K, et al. Mice lacking methyl-cpg binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proceedings of the National Academy of Science U S A. 2003;100:6777–6782. doi: 10.1073/pnas.1131928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annual Review of Physiology. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]