

Abstract
This article reviews the current knowledge and experimental research about the mechanisms by which fatty acids and their derivatives control specific gene expression involved during carcinogenesis. Changes in dietary fatty acids, specifically the polyunsaturated fatty acids (PUFAs) of the ω-3 and ω-6 families and some derived eicosanoids from lipoxygenases (LOXs), cyclooxygenases (COXs), and cytochrome P-450 (CYP-450), seem to control the activity of transcription factor families involved in cancer cell proliferation or cell death. Their regulation may be carried out either through direct binding to DNA as peroxisome proliferator–activated receptors (PPARs) or via modulation in an indirect manner of signaling pathway molecules (e.g., protein kinase C [PKC]) and other transcription factors (nuclear factor kappa B [NFκB] and sterol regulatory element binding protein [SREBP]). Knowledge of the mechanisms by which fatty acids control specific gene expression may identify important risk factors for cancer, and provide insight into the development of new therapeutic strategies for a better management of whole-body lipid metabolism.
Keywords: Fatty acids, signaling, gene expression, cancer
Introduction
1. Basic concepts of fatty acid signaling and its bioactive derivatives
The molecular basis of the activities regulated by fatty acids (FAs) and their derivatives, such as eicosanoids, include changes in receptor signaling, the composition of rafts, cell metabolism, and membrane structures that lead to changes in the function of transcription factors and their target genes, a key step essential for the function of FA-activated signaling. Here, we review the basis of FA signalling and its associated gene expression changes that play a role during carcinogenesis.
a) Lipid metabolism: Biosynthesis of fatty acids
Lipid metabolism involves exogenous lipids obtained through the diet and endogenous lipids synthesized de novo in the body. Fatty acids are composed of a hydrocarbon chain with a carbonyl group at one end and a methyl group at the other, and are fundamental components of the different lipids. They form ester links with an alcohol group, finally forming the triacylglycerols, phospholipids, and cholesterol esters, among other lipids.
In the cell, endogenous or exogenous FAs are activated first to the acyl-coenzyme A (CoA) thiol-ester and then incorporated into membrane phospholipids. Exogenous FAs require that their transport among and into tissues occurs through specific mechanisms that allow their rapid and controlled distribution, so that their uptake can be regulated according to metabolic needs and to avoid the possible harmful effects of excess FA accumulation. The uptake of FAs can be by simple diffusion or principally by a selective expression and/or regulation of specific sets of membrane proteins. The membrane proteins implicated in FA uptake in mammalian cells include FAT/CD36 (fatty acid translocase), FATP (fatty acid transport protein) and FABPpm (fatty acid–binding protein-plasma membrane) [1,2]. Further evidence suggests that not only proteins but also lipid microdomains play an important part in the uptake process. The increased raft association of FAT/CD36 leads to an increased FA uptake, and dynamic association of FAT/CD36 with lipid rafts may regulate the process [3].
De novo synthesis of FAs begins with acetyl CoA originated either from FA oxidation or carbohydrate and protein metabolism. The synthesis of new FAs is carried out by the successive addition of 2-carbon fragments (acetyl CoA) at the carboxyl end of the growing acyl chain. Polyunsaturated fatty acids (PUFAs) have one or more double bonds in the carbon chain, whereas saturated fats have no double bonds [4]. For instance, the synthesis of 16:0 PA (palmitic acid) from saturated FAs begins in the cytosol and is catalyzed by FA synthase (FAS). Next, chain elongation and desaturation occur in the membrane of the smooth endoplasmic reticulum. In this way, 16:0 is elongated to form 18:0 SA (stearic acid). Also, the insertion of double bonds between carbons 9 and 10 of the acyl chain by Δ9-desaturase transforms 16:0 into 16:1, ω-7, PA (palmitoleic acid) and 18:0 into 18:1, ω-9 OA (oleic acid) (Figure 1).
Fig. 1.
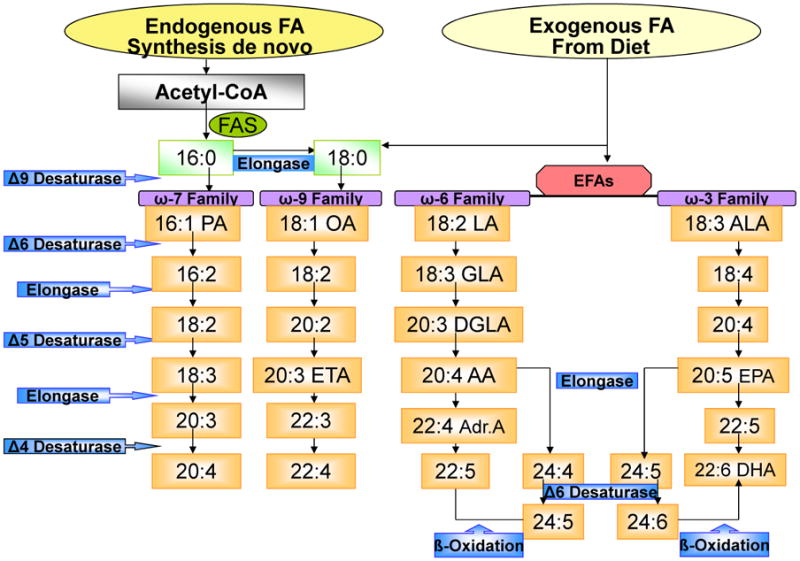
Fatty Acids Biosynthesis. The figure shows the exogenous (incorporated by diet) as well as the endogenous (de novo) saturated and unsaturated fatty acids (FAs) biosynthesis. The endogenous FAs require acetyl-CoA and fatty acyl-synthetase being the 16:0, the first produced FA and elongated to form 18:0. Exogenous FA incorporation through diet includes: 16:0, 18:0 and the essential fatty acids (EFAs) which cannot be synthesized de novo as ω-6 and ω-3 families. The implicated enzymes in these pathways are: Δ9, Δ6, Δ5 and Δ 4 desaturases and elongases.
There are at least four families of PUFAs, depending on the parent FA from which they are synthesized: the ω-3 series derived from α-linolenic acid (ALA: 18:3, ω-3), the ω-6 series derived from linoleic acid (LA: 18:2, ω-6), the ω-9 series derived from OA, and the ω-7 series derived from PA [5]. Plants can insert double bonds between carbons 12 and 13 via conversion of OA to LA by Δ12-desaturase; further desaturation can occur via the insertion by Δ15-desaturase of a double bond between carbons 15 and 16 to form ALA. However, mammals lack Δ12- and Δ15-desaturase; therefore, they cannot synthesize de novo LA and ALA. These two FAs are required for mammalian survival and are thus called essential fatty acids (EFAs). Both must be incorporated through the diet before the synthesis of their series PUFAs and derivatives can begin. Mammalian cells metabolize EFAs by successive elongation and desaturation reactions. Implicated in these pathways are the same set of enzymes, elongases, and the Δ5- and Δ6-desaturases that metabolized the different families of fatty acids (ω-3, ω-6, ω-9 and ω-7) (Figure 1).
The PUFA precursors compete with each other for the same enzymes, with hierarchical preference given for ω-3 over ω-6, and ω-6 over ω-9 and ω-7. This preference for the higher degree of desaturation in 20–carbon chain PUFA precursors has major implications for dietary habits. Hence, under normal physiological conditions, ω-9 derivatives are formed in small amounts, and a significant increase in 20:3, ω-9 (mead acid), a metabolite of OA, suggests a deficiency of ω-3 and ω-6 essential fatty acids [6].
Controversy exists about the mechanisms involved in the last part of FA synthesis, in particular about the roles played by desaturation and elongation of arachidonic acid (AA: 20:4, ω-6) and eicosapentaenoic acid (EPA: 20:5, ω-3) to form other compounds. It was long accepted that AA and EPA were first elongated and then, via Δ4-desaturation, transformed into 22:5, ω-6 and docosahexaenoic acid (DHA: 22:6 ω-3), respectively. However, an alternative pathway involving the peroxisome has been proposed in mammals—the so-called “Sprecher pathway” [7]. This pathway involves sequential elongations of EPA or AA to C24 substrates followed by a second Δ6-desaturation step to 24:6, ω3 and 24:5, ω6, which would finally undergo ß-oxidation in the peroxisome to produce DHA 22:6 ω-3 and 22:5 ω-6. The current literature suggests that both the direct mechanism involving elongation, followed by Δ4 desaturation and the Sprecher pathway that assumes a role for peroxisomes, are implicated in the biosynthesis of the long-chain PUFAs [8-9].
The activities of desaturases and elongases involved in the metabolism of PUFAs can be influenced by different factors, including saturated fats, cholesterol, transfatty acids formed by vegetable oil processing, alcohol, adrenaline, and glucocorticoids, which inhibit Δ6- and Δ5-desaturases. Pyridoxine, zinc, and magnesium are also necessary for normal desaturase activity [10].
In summary, FA families differ in their structure, including the quantities of carbons in the acyl chain and the presence of double bonds. The two families of PUFAs that cannot be synthesized de novo—ω-6 and ω-3—are EFAs and must be obtained via dietary sources to ensure survival. The following section discusses the key roles of FAs in cells, not only as part of the phospholipid membrane but also as eicosanoids and other biolipids, and as regulators of signaling in gene expression.
b) Major functions of FAs in cells
In addition to serving as energy sources in cells, FAs have many other functions. They serve as membrane constituents, playing a vital role in the maintenance of membrane fluidity. They determine and influence the behavior of membrane proteins and membrane-bound enzymes and receptors. They also may act as precursors for the synthesis of eicosanoids and other biolipid derivatives of PUFAs, which in turn influence intracellular signaling processes affecting transcription factor activities, such as the regulation of gene expression.
As membrane components, endogenous or exogenous FAs incorporated into membrane phospholipids play important roles in membrane structure, potentially influencing membrane functions and transmembrane and intracellular protein activity via a number of mechanisms. Changes in lipid composition alter the physical properties of the membrane bilayer, a process which, in turn, regulates a wide range of membrane protein function in a seemingly nonspecific manner. [11,12].
Many functional proteins require a specific lipid to provide a proper microenvironment around hydrophobic regions [4, 13-15]. A number of membrane-bound enzymes, transporters, and receptors have been shown to be particularly sensitive to their FA environments. These include adenylate cyclase, 5′-nucleotidase, the Na+/K+ ATPase, and the insulin receptors [16]. Saturated FAs such as myristic: 14:0 and PA are the predominant FAs found attached to proteins in eukaryotic cells [17]. Both FAs are attached to distinct classes of proteins, some of which are involved in signal transduction, such as Src family kinases, G-proteins, growth factor receptors, mitogenic-activated protein kinase (MAPK), protein kinase C (PKC), and a number of oncogene products [18]. These proteins are targeted to specific regions of the plasma membrane known as “rafts,” which act as signal platforms for various signaling components [19]. Lipid rafts are discrete patches of plasma membrane with distinct lipid composition that differ from other regions of the membrane. These microdomains are rich in sphingolipids and cholesterol, and the FA chains of lipids are more saturated and tend to be tightly packed. Lipid rafts seem to be involved directly or indirectly in lipid-mediated cell regulation for generation of intracellular signals in response to extracellular stimuli.
It is known that the reactivity of PUFAs with molecular oxygen depends on the number and position of their double bonds. This can happen nonenzymatically, contributing to oxidative stress, or through the actions of different oxygenases. The enzymatic oxygenation of 20-carbon PUFAs, particularly the EFAs, generates a series of bioactive metabolites broadly termed eicosanoids [22].
Arachidonic acid, a relatively minor PUFA found at the n-2 position of cell membrane glycerophospholipids, is released either by the action of phospholipase A2 (PLA2) or from membrane phosphatidylinositol-4,5-biphosphate by the actions of phospholipase C (PLC) and diacylglycerol lipase (DAGL) [23]; it also may be directly supplied to tissues from dietary sources or formed from the EFA linoleic acid (LA). In Western-fed populations, AA is the main precursor of eicosanoids, and these bioactive lipids have more potent biological functions than those released from dihomo-γ-linolenic acid (DGLA) or eicosapentaenoic acid (EPA). In contrast to other, more abundant unsaturated FAs such as oleic, linoleic, or linolenic acid, levels of unesterified AA are strictly controlled within mammalian cells, and pathways of AA uptake, incorporation, and remodelling in glycerolipids are well documented [24]. As shown in Figure 2, eicosanoids belong to a family of biolipid mediators that are oxygenated derivatives of the 20-carbon PUFAs such as prostaglandins (PGs), thromboxanes (TXs) formed by the enzymatic activity of cyclooxygenases (COXs) or leukotrienes (LTs), lipoxins (LXs), hepoxilins (HXs), dihydroxyeicosatrienoic acid (DiHETEs), and hydroxyeicosatetraenoic acids (HETEs derived from enzymatic activity of lipoxygenases (LOXs). Eicosanoids are also formed by the cytochrome P450 enzymes that metabolize AA in PGs, epoxyeicosatrienoic acid (EET), and HETEs, among others [25-27]. LA is the precursor of hydroxyoctadecadienoic acids (HODEs). The DGLA: 20:3, n-6 is the precursor of the 1 series of PGs and TXs (Figure 2).
Fig. 2.
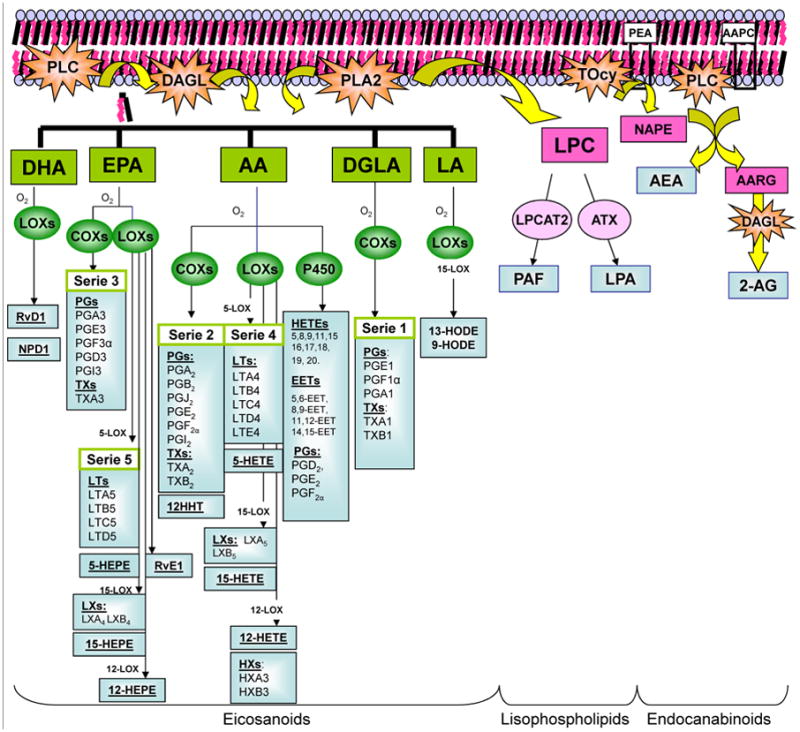
Biosynthesis of Lipids Derivatives. The figure shows the biosynthesis of some lipid derivatives as eicosanoids, lisophospholipids and endocanabinoids. After phospholipase C (PLC) activation diacylglycerol (DGA) is liberated from membrane phospholipids. DGA lipase and phospholipase A2 (PLA2) activities produced fatty acids (FA) liberation from membrane phospholipids as well DHA: docosahexaenoic acid; EPA: eicosapentaenoic acid, AA: arachidonic acid, DGLA: dihomo-gamma-linoleic acid and LA: linoleic acid. These unsaturated fatty acids can be function as substrate of lipoxigenases (LOXs), cyclooxygenases (COXs) and cytochrome P-450 (CYP-450) giving rise to resolvins (Rvs), neuroprotectins (NPs), prostaglandins (PGs), leukotrienes (LTs), thromboxanes (TXs), hydroxyeicosatetraenoic acids (HETEs), hydroxyeicosapentaenoic acids (HEPEs), lipoxins (LXs) and hepoxilins (HXs). By another way, PLA2 is implicated in lisophospholipid production. The activity of this enzyme yields to the 1-O-alkyl-lysophosphatidyl coline (LPC). This molecule is metabolized by LPC acetyl-transferase-2 giving the platelet-activating factor (PAF) and by autoxine enzyme (ATX) it is obtained lisophosphatidic acid (LPA). Indeed, the endocannabinoids synthesis takes place when a transacylase (TRCY) catalyses the phosphatidylethanolamine (PEA) in N-acyl-phosphatidyl etanolamine (NAPE). These molecules are substrate for PLC and for PLD, resulting in the liberation of arachidonylethanolamide (anandamide: AEA) and 2-arachidonylglicerol (2-AG), considered the two main endogenous endocannabinoid compounds.
By another pathway, AA is the precursor of the 2 series of PGs, TXs, and 12- HHT, and the 4 series of LTs, LXs, HXs, DiHETEs, HETEs, and EETs [28] (Figure 2). EPA forms the 3 series of PGs and TXs and the 5 series of LTs, LXs, and hydroxyeicosapentaenoic acids (HEPEs), as well as the E series of new compounds known as resolvins [12, 13, 29]. Docosahexaenoic acid (DHA) is also a precursor of resolvins of the D series. Other bioactive members from DHA with conjugated triene structures are docosatrienes (DTs), termed neuroprotectins (NPDs) [30] (Figure 2). Indeed, when PLA2 catalyzes the hydrolysis of FA ester linkage at the 2 position of glycerol, a variety of free FAs (usually unsaturated) and a variety of lysophospholipids are formed. The first recognized lysophospholipid-type lipid derivative was platelet-activating factor (PAF). Its biosynthesis involves production of 1-O-alkyl-lysophosphatidil choline (LPC) by PLA2, followed by its acetylation by LPC acyltranferase-2 (LPCAT-2). The other derivate is lysophosphatidic acid (LPA), obtained from LPC by autotoxin enzyme activity [26] (Figure 2).
Interestingly, other eicosanoids resulting from the oxidative metabolism of related lipids are those of the endocannabinoid system (ECBS). These endogenous lipids activate cannabinoid receptors CB1 and CB2 and regulate numerous cellular process. [31]. Although various new molecules with ECBS activity have been identified, arachidonoyl-ethanolamide (Anandamide: AEA) and 2-arachidonoylglycerol (2-AG) are considered to be the two main endogenous cannabinoid compounds. ECBS are released from cells upon activation of G-protein–coupled receptors or increase of calcium levels. Increasing calcium activates trans-acylase, which catalyzes the first step of ECBS biosynthesis converting the phospholipid phosphatidylethanolamine into N-acyl-phosphatidylethanolamine (NAPE). Phospholipase C (PLC) and phospholipase D (PLD) can cleave NAPE to yield AEA [32]. However, in vivo experiment using the PLD knockout mice demonstrate that NAPE cleavage was minimally impaired, with low resultant calcium concentrations, thus providing evidence of the involvement of additional pathways in this process. The other endocannabinoid, 2-AG, is produced by action of PLC on the sn-1, 2-diarachidonoyl-phosphatidylcholine (AAPC); the product formed by sn-1, acyl-2-arachidonyl glycerol (AARG) is degraded by the diacylglycerol lipase (DAGLAN) to 2-AG. Dietary uptake of various PUFAs may modify ECBS production, potentially deeply affecting ECBS activities [33,34]. It is interesting to consider that cells can use either free FAs taken up from the extracellular pool or etherified FAs released from cell membrane phospholipids by PLA2 for eicosanoid production [22]. For example, studies of the relative utilization of exogenous versus endogenous etherified AA studied in collagen-stimulated platelets under physiological conditions (in plasma) have shown that endogenous EFA is used for thromboxane production, but that exogenous plasma lipid is preferentially used for 12-HETE production by 12-LOX [35,36]. This difference may be due to the efficiency of coupling of exogenous versus endogenous AA to downstream enzymes, depending on their subcellular localization [37] (Figure 2). Substantial epidemiological and experimental evidence shows that FAs such as PUFAs and their derivatives modulate many metabolic processes. Indeed, PUFAs and their LOXs, COXs, and CYP-450 products activate cellular signaling pathways to produce specific changes in gene expression affecting proliferation, cell death, migration, and extracellular matrix production. When the lipids are modified by dietary manipulation, this can give rise to pathological processes such as carcinogenesis, in part by altering the activity of transcription factors regulating gene expression implicated in carcinogenesis.
2. Role of transcription factors in FA signaling–regulated gene expression
a) Direct effects of FAs on transcription factors
To modify the function of a transcription factor, one of the common approaches is to affect the direct binding between the ligand and the protein. Indeed, several FAs have been identified to interact with transcription factors and mediate the process of transcription. In this section, we will summarize a classic lipid-activated transcription factor, the peroxisome proliferator-activated receptor (PPAR), as an example of the direct effects of FAs.
Peroxisome proliferator-activated receptors
The PPARs are a group of nuclear receptor proteins belonging to the sterol/thyroid superfamily, and function as transcription factors regulating the expression of genes. Three subtypes of PPAR (α, β/δ, and γ) have been described, each with organ-specific expression. PPAR contains a DNA-binding domain and a ligand-binding domain (LBD) that interacts with various ligands. The first identification of the PPAR activator was done by Green's group in the early 90s [38]. Green's group transfected cultured cells with reporter construct containing the PPAR ligand-binding domain. By treating the cells with fibrate hypolipidemic agents, they were able to stimulate reporter gene expression, suggesting that fibrate serves as a PPAR activator [38]. Following this finding, more natural compounds, including several FAs, have been found to potentiate PPAR activation [39-42]. The LBD of PPAR contains a large Y-shaped cavity with a total volume of 1300(X001FA) to 1400(X001FA). The cavity branches off two extended arms from the surface of the protein and forms a hydrophobic entrance and a polar pocket. In this pocket, Thr289, His323, His449, and Tyr473 of PPARβ/δ (respectively Ser280, Tyr314, His440, and Tyr464 of PPAR-α, and Ser289, His323, His449, and Tyr473 of PPARγ) construct a hydrogen network that interacts with the carboxyl group of FAs or eicosanoic acids [43]. Nielsen et al proposed a “mouse trap” model to explain the regulation of ligand-stimulated PPAR activation [44]. Based on this model, structural analysis has been conducted on the retinoid X receptor (RXR) and its activating function-2 domain (AF-2). The PPAR-RXR heterodimers bind specifically to PPAR response elements (PPREs), which are direct repeats (DRs) of 5′-AGGTCA separated by one or, in a few cases, two nucleotide(s) [44]. In the heterodimer, PPAR occupies the 5′ repeat, whereas RXR occupies the 3′ repeat [45]. The activation of transcription by the PPARs relies on two activation domains, i.e., activation function one (AF-1) located in the N terminus and activation function two (AF-2) located in the C-terminal LBD [46]. The activity of AF-2 is regulated by the binding of ligands, such as fatty acids and fatty acid derivatives. This type of binding leads to a conformational change of the C-terminal domain, the AF-2 helix in particular favoring the interactions with transcriptional coactivators [47]. Because PPARs function as sensors for FAs and their derivatives, this family mostly controls the gene expression involved in lipid metabolism and energy homeostasis [48].
Substantial evidence suggests that PPAR plays a role in cancer development. Activation of PPAR-β/δ is involved in the development of breast, prostate, and liver cancers. However, considerable controversy remains as to whether PPAR-β/δ stimulates or inhibits cancer cell proliferation [49-51].
PUFAs such as LA, ALA, gammalinoleic acid (GLA), AA, EPA, and DHA are natural ligands and activators of PPAR-α and PPAR-δ. Several CLA isomers have also been shown to bind and activate PPAR-α [52]. As described before, the binding capacity of different PUFAs to PPARs may prevent cancer development. In contrast, saturated FAs do not bind or activate these transcription factors.
PPAR-α is required for the induction of genes involved in mitochondrial and peroxisomal β-oxidation [53]. In rats and mice, these PPAR ligands have been associated with rapid peroxisome proliferation and liver enlargement, and their long-term administration led to liver carcinogenesis, perhaps as a result of the enhancement of lipid peroxidation products [54]. However, the antineoplastic effects of PPARs have been shown in several types of cancer cells. These effects are thought to be related to programmed cell death because, apart from its well-established metabolic actions, PPAR-γ induces apoptosis through an up-regulation of certain proapoptotic proteins by the release of cytochrome C and subsequent activation of several effector caspases [55].
Imbalance of peroxisome proliferator levels results in the sustained activity of PPAR-α, which causes DNA damage and hepatocellular proliferation by deregulating responsive genes [56,57], thereby leading to the development of liver cancer [58].
Although a variety of compounds have been shown to activate PPARs, identification of physiologically relevant ligands has been difficult [59]. The PPARs' regulation is directly done by FAs interacting with PPAR ligand-binding domain and potentiates the transcription activity. The behavior of PPAR in cancer may be modulated by PUFAs and their derivated biomolecules [60] (Figure 3).
Fig. 3.
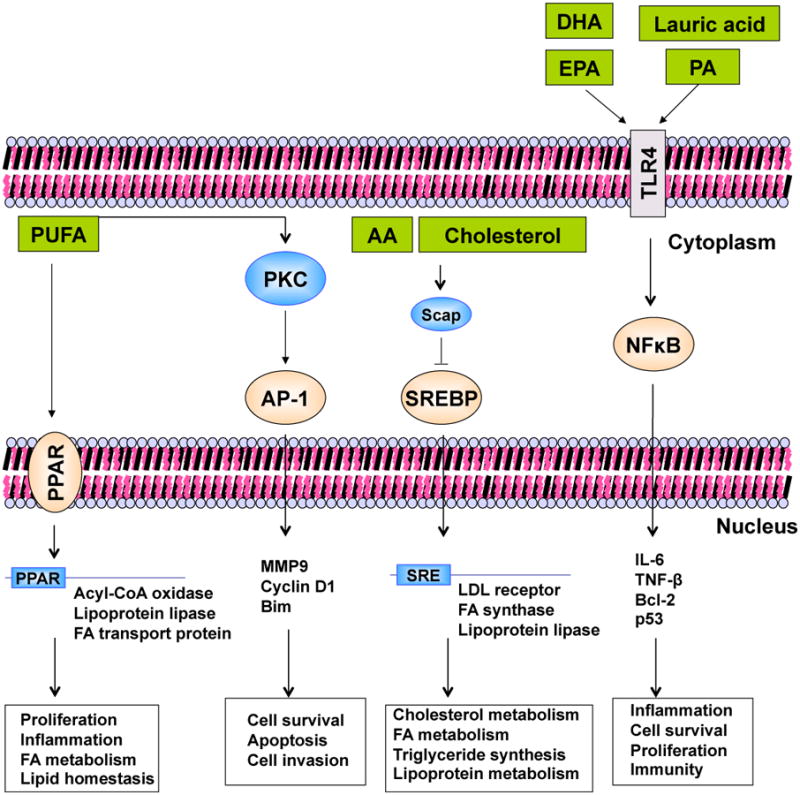
Interaction of Fatty Acids and Transcription Factors. The figure summarizes examples of the effect FAs have on transcription factors. In peroxisome proliferator-activated receptor (PPAR) regulation, FAs directly interact with PPAR ligand-binding domain and potentiate the transcription activity. Active PPAR associates with PPAR response element (PPRE) in the target gene promoter and recruits transcription-coactivator complex to initiate gene expression. AP-1, sterol regulatory element-binding protein (SREBP), and NFκB represent the indirect effect of FAs to transcription factors. FAs and cholesterol modulate the upstream molecules (e.g. PKC, Scap, TLR4) and cause activation and nuclear translocation of the downstream transcription factors, and as a result change the target gene expression.
b) Indirect effects of FAs on transcription factors
In addition to directly affecting transcription factors, as with PPAR (described in the previous section), FAs can also regulate transcription factors by interacting with their regulatory pathways. Other signaling molecules, such as PKC, also have indirect effects on FAs and transcription factors. We will discuss the indirect effects of FAs, using nuclear factor kappa B (NFκB) and sterol regulatory element—binding proteins (SREBPs) as examples.
PKC signaling
PKC is a family of phospholipid-dependent serine/threonine kinases that play important roles in signal transduction associated with a variety of cellular responses, including cell growth and differentiation, gene expression, hormone secretion, apoptosis, and membrane function. PKC consists of at least 11 isoforms that show diversity in structure, cellular distribution, and biological functions and have been divided into three groups based on their structures and cofactor requirements: conventional, novel, and atypical. The conventional PKC isoforms α, βI, βII, and γ require phosphatidylserine and Ca2+ for activity. The novel PKC isoforms δ, ε, η, and θ do not require Ca2+ as a cofactor, but bind to phosphatidylserine when activated. Both conventional and novel PKCs are activated by phorbol 12-myristate 13-acetate (PMA) or 1,2-Diacyl-sn-Glycerol (DAG) [61]. The interaction of DAG with PKC has implications for many PKC-dependent cellular functions because PKC is a substrate of neuromodulin, myristoylated alanine-rich C kinase, G-protein coupled receptors, growth factor receptors, metabolic enzymes, signaling enzymes such as mitogen-activated protein kinase, cytoskeletal proteins, nuclear proteins, and proto-oncogenes such as pp60c-src [62]. PUFAs of the ω-3 and ω-6 families, such as LA, AA, GLA, EPA, and DHA, compete to activate PKC-α. Once activated, PKC-α induces mitogenic signals by activating Fos, Jun, and AP-1 transcription factors. In this regard, Fields and Murray showed that a diet high in ω-6 fatty acids increased cancer colon cell proliferation [61]. This was associated with an increment in cell membrane PKCβII. In contrast, ω-3 FAs inhibited colon carcinogenesis in vivo, by direct inhibition of PKCβII activity, which was mediated by COX-2 protein expression and transforming growth factor receptor-β II (TGF-β RII) expression [61] (Figure 3).
Nuclear factor kappa B (NFκB)
NFκB is a transcription factor that controls genes involved in apoptosis, inflammation, cell adhesion, proliferation, the adaptive immune response, the stress response, and tissue remodeling. Disruption of NFκB signaling results in inflammatory diseases and cancer. In the inactive state, NFκB forms a heterodimer complex with the inhibitory factor, IκBα, in the cytoplasm. But, when the IκB is phosphorylated by kinase, IκK unlocks the inhibitory effect of NFκB and the active form migrates into the nucleus where it regulates gene expression. Studies of the modulation of NFκB activity by ω-3 PUFAs have shown that impeding phosphorylation of IκB selectively decreased NFκB activation, preventing the nuclear translocation of NFκB [63,64]. In addition to the effect on the IκB molecule, FAs have a more complex effect on the Toll-like receptor (TLR) family and affect downstream NFκB activity. TLRs are membrane-bound receptors that recognize the pathogen-associated molecular patterns (PAMPs) of invading microorganisms. Upon recognition of a specific ligand, the responding TLR can trigger various downstream signaling by conjugating to different mediators, including MyD88, MyD88 adaptor-like (Mal), TIR domain–containing adaptor protein (TIRAP), TIR-domain–containing adaptor including interferon-beta (TRIF), and TRIF-related adaptor molecule (TRAM) [65]. Among the representative agonists of TLRs, the TLR4 agonist lipopolysaccharide (LPS) and TLR2 agonist lipopeptides contain a saturated FA moiety (lauric acid, myristic acid, and palmitic acid) [66]. Lee and colleagues [67] identified these saturated FAs (lauric acid and palmitic acid) as inductors of NFκB expression through TLR4 signaling. Interestingly, PUFAs such as DHA and EPA saturated FA activation [68]. These studies suggest that saturated FAs and unsaturated FAs regulate TLR in opposite ways. Even though the detailed mechanism is currently unclear, several lines of evidence indicate that FAs play a role in TLR trafficking to lipid rafts [69, 70]. Consistent with these observations, a recent finding shows that lauric acid, a saturated FA, induced dimerization and recruitment of TLR4 in lipid rafts [71]. In this report, the authors found that lauric acid and LPS enhanced the association between TLR4 and its downstream adaptors, TRIF and MyD88, by gathering them in lipid rafts. Treatment with the ω-3 DHA inhibited this dimerization and recruitment. More research is needed to further elucidate the role of PUFAs in lipid raft regulation of TLR4.
In summary, NF-kB is constitutively active in most tumor cells (hematopoietic, prostate, breast cancers). Suppression of NFkB activity in tumors inhibits proliferation, causes cell cycle arrest, and leads to apoptosis, indicating the crucial role of NFkB in cell proliferation and survival. Findings show that PUFAs can selectively affect the TLR family and influence downstream NFκB activity. These interactions form a crucial interface with other signaling pathways in the cell. In fact, the regulatory ability of PUFAs to behave as either agonists or antagonists provides a plausible explanation for the cell-type– and stimulus-specific effects of NFκB, such as the ability to either inhibit or facilitate apoptosis. These pathways also provide important routes through which FAs or their derivatives can modulate NFκB activity. (Figure 3).
Sterol regulatory response element–binding proteins
SREBPs are another group of transcription factors that mediate the effects of dietary FAs on gene expression. The 3 members of the SREBP family (SREBP-1a, SREBP-1c, and SREBP-c) are membrane-bound transcription factors that function as transcription activators for genes involved in the biosynthesis and intake of cholesterol, FAs, triglycerides, and phospholipids [72]. Members of the SREBP family are encoded by two unique genes, SREBP1 [73-76] and SREBP2. All of them share three similar features on the N-terminal basic-helix-loop-helix-leucine zipper (bHLH-LZ) domain— two hydrophobic transmembrane regions and a C-terminal regulatory domain. Spiegelman's group first implicated a regulatory role for SREBPs in FA synthesis [77]. By screening the rat cDNA of adipose tissue for proteins that contain the bHLH domain to recognize E box, they isolated ADD-1, a rat homologue of human and mouse SREBP-1c, and showed that overexpression of ADD-1 stimulated transcription through a polymerized set of Eboxes located on the 5′-flanking FAS promoter [78]. Subsequently, it was shown that SREBPs directly stimulated the FAS promoter by coordinating with the consensus sequence adjacent to E boxes, a sterol regulatory element (SRE)-like sequence.
Interestingly, while SREBP plays an important role in the synthesis of FAs and cholesterol, the activation of SREBPs is also a cholesterol-regulated process [79,80]. When the cells are replete with cholesterol, SREBPs anchor on the ER membrane by two transmembrane segments, forming a hairpin structure with the N-terminal bHLH-LZ domain and the C-terminal regulatory domain extending into the cytoplasm [81]. The C-terminus binds to SREBP cleavage–activating protein (Scap), a variety of protein that associates with cholesterol, and promotes the conjugation between the SREBP-Scap complex and the ER-resident insulin-induced gene (Insig) [82]. This process retains SREBP on the ER membrane, preventing interaction with Golgi-localized proteases [83]. When the cells are lacking in cholesterol, decreased levels of sterols are released, allowing binding between SREBP-Scap and Insig and the exit of SREBP-Scap to the Golgi by COPII-coated vesicles through interaction with Sec23/24 [84,85]. In the Golgi, SREBPs are proteolytically cleaved by site 1 protease and site 2 protease, releasing the N-terminal transcription factor domain into the nucleus [86]. Nuclear SREBPs include ∼480 amino acids on the protein N-terminus that form an acidic domain, followed by an activation domain, bHLH-LZ. This segment endows SREBPs with the ability to bind to SRE and activate transcription. As would be expected given the function of the bHLH-LZ domain, SREBPs in which this domain is deleted retain their DNA-binding ability but have an attenuated transcriptional activity, turning them from transactivators into inhibitors [87]. Furthermore, the shorter acidic domain in SREBP-1c makes it a weaker transactivator compared with the other two family members [88], suggesting that the length of the acidic head also affects the activity of SREBPs. To function as a transcription factor, nuclear SREBPs first recognize SRE in the promoter and bind to the DNA elements as dimers. After binding occurs, the activation domains of the nuclear SREBPs recruit coactivator CBP/p300 and SWI-SNF, a nucleosome remodeling complex, to modulate target gene expression [89].
In this context, AA is almost the unique PUFA that significantly affected the SREBP-1 transcription factor, which controls genes for the low-density lipoprotein receptor (LDL) and 3-hydroxyl-3-methylglutaryl coenzyme A (HMG-CoA) synthase. Indeed, it has been shown that AA had inhibitory effects on SREBP-1a and SREBP-1c in cultured human cells [5]. Moreover, alterations of SREBP and their downstream-regulated genes such as FAS were involved in the development and progression of cancer. Indeed, SREBP-1 overexpression is highly linked to some cancer tissues.
In summary, the cholesterol-dependent ER localization and transcriptional activity of SREBPs, in which AA and other FAs may competitively inhibit activation of the endogenous SREBP-1 gene, suggest that intracellular levels of FAs and cholesterol should regulate preneoplastic changes. The dependence of SREBP-ER localization on cholesterol could explain the intracellular accumulation of cholesterol observed in some types of cancers. Consequently, disrupting the mechanisms that increase sterol levels has become an important checkpoint implicated in cancer development (Figure 3).
c) Lipid derivatives and cell signaling
LOX derivatives
Some AA-LOX metabolites, such as leukotriene B4, 8-HETE, 15-HETE, 9 HODE, and 13-HODE, activate signal transduction pathways that lead to cell proliferation or elicit a positive modulation of cell behavior and phenotype that include: 1) cell surface G-protein activation; 2) nuclear PPAR activation; 3) Ras signaling pathway through activation of regulation of Ras-GAP; 4) activation of immediate early genes such as c-fos and Egr-1; 5) PKC activation; 6) activation of the Raf-1/Mek/Erk pathway; 7) ROS-induced NFκB activation; 8) activation of P21-activated kinase; 9) phosphoinositol 3 kinase (PI3K) activation; and 10) interaction with steroid receptor coactivator-1 [90-91]. It is generally acknowledged that abnormal expression of specific PKC isoforms is closely associated with cellular transformation and cancer progression. Also, 12-HETE may act as a second messenger activating PKC [92].
A different mechanism was proposed by Zeng et al [93], who reported that 5-HETE and 12-HETE stimulate endothelial cell growth via Jak-2/STAT and phosphatidylinositol-3-kinase/AKT signaling, inducing the expression of fibroblast growth factor-2. Furthermore, 12 (HETE) activated ERK ½ and p38 MAPk pathways in 3T6 fibroblast cultures [94], whereas 20-HETE also induced the Ras/MAPk pathway in vascular smooth muscle cells [95].
COX derivatives
COX-derived products, such as PGE2, PGF2α, and the arachidonate product of thromboxane synthase (TXA2), may stimulate cell growth and proliferation by enhancing the self production of PGs. TXA2 has a number of potent and diverse bioactivities, including platelet aggregative activity and smooth muscle constrictive activity, that are important both to the maintenance of health and the development of disease and are mediated by TXA2 receptors (TPs) [60]. Two isoforms of TP, TPα and TPβ, have been identified. TPs can also activate mitogenic pathways including epidermal growth factor receptor (EGFR), extracellular signal-regulated kinase (ERK), Akt/protein kinase B (PKB), and glycogen synthase kinase (GSK) [96]. It has been shown that TXA2-TP signaling axis may regulate cell migration through Rho factor activation and cytoskeleton reorganization in prostate carcinoma cells [97].
Indeed, it was demonstrated that PGE2 increased the activity of the a2b1 integrin via EP1, PLC, PKC, c-Src, and NFkB-dependent pathways and enhanced migration of human chondrosarcoma cells [98]. Also, PGE2 is known to play an important role as a physiological stimulator of cAMP synthesis in the activation of mechanogrowth factor (MGF), a second messenger implicated in the synthesis of myoblasts and differentiated myotubes in murine and human cells [99]. In HCA-7 human colon cancer cells, PGE2 enhanced the upregulation of vascular endothelial growth factor (VEGF) receptor-1 (VEGFR-1) expression. The PGE2-stimulated increase in VEGFR-1 expression was accompanied by an increase in the cellular migration of HCA-7 cells. This PGE2 activity included a signaling pathway involving a receptor-mediated activation of phosphatidylinositol 3-kinase (PI3K) and other extracellular signal-regulated kinases [100]. Yang et al [101] showed that PGE2 treatment of oral cancer cells produced cell migration and ICAM-1 expression-linked to an upregulation of PKCδ and c-Src and activation of the AP-1 signaling pathway.
The human gastric carcinoma cell line MGC803 exhibits high expression levels of PPAR-γ, an agonist of 15-deoxy-Delta12,14-prostaglandin J2 (15d-PGJ2) that has been shown to inhibit growth and induce apoptosis and G(1)/G(0) cell cycle arrest in these cells in a concentration-dependent and time-dependent manner. The inhibition of PPAR-γ function by COX derivatives may be a potentially important and novel modality for the treatment and prevention of gastric carcinoma [102].
P450 derivatives
P450-derived eicosanoids have activities closely linked to inflammation, angiogenesis, and cardiovascular function rather than to cancer pathways [103]. Nevertheless, among AA p450 derivatives, 20-HETE is the major metabolite of the ω-hydroxylation by cytochrome-P4A (CYP4A) and the CYP4F pathway. This metabolite is a proinflammatory mediator that markedly stimulates the production of inflammatory cytokines/chemokines in endothelial cells, including IL-8, IL-13, IL-4, and PGE2. 20-HETE activates vascular endothelial cells by its participation in the cellular mechanisms that trigger activation of the signaling pathways NFκB and MAPK/ERK [104]. Also, 20-HETE induces angiogenic responses in vivo and in vitro, and CYP ω-hydroxylase promotes tumor angiogenesis and metastasis by up-regulation of VEGF and MMP-9 via PI3 K and ERK1/2 signaling in human non-small cell lung cancer cells [105].
The above-discussed roles played by PUFAs and their derivatives in molecular mechanisms could explain the in vitro, in vivo, and clinical effects observed after diverse PUFA treatments. In animals, FAs are key molecules that participate in various biological processes. The structural properties of FAs, such as their chain length, the degree of desaturation, and the position of the double bonds, largely determine the nature of these processes [4].
Summing up, eicosanoids derived from LOXs, COXs, and CYP450 enzyme activity can interfere with different transcription factors activating or inhibiting the expression of gene products involved in carcinogenesis.
3. PUFAs: a context-dependent oncogene-tumor suppressor pathway
Perturbation of lipid metabolism is an early event in carcinogenesis and a central hallmark of many cancers. However, the precise molecular lipid profile in tumor cells remains poorly characterized because variability is a common feature of cancer cells. Nonetheless, in the following section we attempt to characterize the major lipid profile characteristics of a tumor cell.
a) Lipid profile of tumor cells
Cancer cells synthesize de novo large amounts of FAs and cholesterol, irrespective of the circulating lipid levels, and benefit from these increased levels in terms of growth advantage, self-survival, and drug resistance. Key lipogenic alterations that are common in cancer cells include overexpression of the FAS enzyme and deregulation of the 5-AMP–activated protein kinase (AMPK) [106]. It is well known that many tumors and cancer cell lines are deficient in Δ5, Δ6 desaturase and/or contain low levels of Δ5, Δ6 desaturated EFAs because loss of Δ-5 and Δ-6 desaturating ability is relevant to the process of malignant transformation [107,108]. Remodeling the FA composition of membrane lipids can alter cell metabolism as well as affect the complex interacting array of signaling cascades that govern a range of physiological responses. Fatty acids can function as signaling molecules that not only trigger FA immune response but also elicit the general “cancer” signaling network [109]. Recent studies on the lipid profile of cancer membrane cells carried out in gastric cancer cell lines have shown that a lipid kinase (Sphingsine Kinase -1 [Sphk1] is overexpressed in this line and is linked to tumor growth, cell transformation, and poor prognosis [110]. The determination of the global lipid profile in 267 human breast cancer cells was correlated to protein expression. It was observed that the increased products of de novo FA synthesis incorporated into membrane phospholipids were mainly palmitate-containing phosphatidylcholine, which renders membranes less fluid and more rigid. The increased concentration of these lipids was associated with cancer progression and a higher expression of lipid metabolism–related genes. It was also shown that the expression of many genes in mammals is positively or negatively modulated by FAs through changes in the rate of transcriptional or post-transcriptional modifications that could be dependent on the viscosity of the plasma membrane [111,112].
Exogenous and endogenous PUFAs and their metabolites can significantly alter protein expression and metabolism to influence the cellular tissue microenvironment and cell-cellinteractions. However, it was also proposed that ω-6 FAs enhance tumor cell growth, whereas ω-3 Fas are beneficial because they arrest cell growth. The cytotoxic effects of ω-3 PUFAs on various cancer cells depends on the type and concentration of FAs used and of the cancer cell being tested.
We will next discuss experimental approaches in vivo as well as clinical research on the effects of treatment with different PUFA families on cancer development.
b) Targeting cancer cells with omega-6 PUFAs: LA, AA, and γ-linoleic acid (GLA)
In vitro studies
It is interesting to note that different cell lines showed diverse sensitivity to LA or AA depending on the cell line, as well as the dose of FAs. Lu et al [113] showed that low concentrations of LA (≤ 200 μM) promote colorectal cancer cell growth, whereas high levels (≥ 200 μM) induce apoptosis in vitro in these cancer cells by inducing differential formation of lipid peroxides. After exposure to increasing concentrations of AA, hepatomoma cancer cell lines (HepG2) showed decreased cell growth and increased apoptosis. These effects were linked to down-regulation of the gene expression of lipogenic enzymes such as FAS and Hydroxy-Methyl-glutaryl CoA-Reductase (HMG-CoAR) [114].
Jiang et al [115] showed that GLA reduced adhesion of colon and breast cancer cells to the endothelium by inhibiting the tyrosine phosphorylation of connnexin-43, a protein that formed the gap junction communication in these cells with the endothelium. GLA was shown to increase E-cadherin expression in a human squamous cell carcinoma line. This cadherin is a cell adhesion molecule highly concentrated at the zonulae occludens; its expression is decreased in highly malignant carcinoma. For these reasons, it is considered an accurate parameter of tumor differentiation. Interestingly, 20:3, ω-9 is increased in the EFA deficiency selectively and blocks the beneficial prodifferentiation effects of GLA. [116].
Using cultures of human tumor cells, our group examined the effects of exposure to GLA and EPA, ω-6 and ω-3 EFAs, respectively, on E-cadherin expression and 15-HETE and 13-HODE liberation. In bladder (T-24), colon (HRT-18), and mammary gland (MCF-7) cancer cell lines, we found significant levels of 15-HETE and 13-HODE synthesis following GLA or EPA treatment. In addition, the predominant metabolites produced by the three types of cancer cells via the LOX pathway were 15-HETE and 13-HODE [117]. The high levels of 15-HETE and 13-HODE in cancer cells may be explained by the preferential activity of Δ5 desaturase on exogenous and endogenous EPAs and the partial suppression of Δ6 desaturase in these cancer lines. We observed that levels of both LOX products were well correlated to e-cadherin expression.
Another group showed that AA is a potent stimulator of prostate cancer cell (PC-3) invasion by inducing bone marrow adipocyte formation. PC-3 cells treated by AA induced destruction of the adipocyte and subsequent formation of a bone metastasis [118]. Other studies with fresh surgical explants of tumors from 22 patients with five varieties of malignancy exposed to different concentrations of GLA and ALA using an in vitro analysis of chemosensitivity testing system and fluorescent cytoprint assay demonstrated antitumor activity by both PUFAs. The antitumor activity mechanism observed for PUFAs was the selective perturbation of the biophysical properties of the malignant plasma membrane [119].
Other in vitro experiments showed that GLA induced apoptosis of tumor cells without harming normal cells. GLA increased free radicals and lipid peroxides, suppressed the expression of the oncogenes ras and Bcl-2, and enhanced the activity of p53. GLA also seemed to produce mitochondrial depolarization, lipid accumulation, and overexpression of c-Myc and p53 to bring about their tumoricidal action [120].
Exogenous AA and inhibitors of AA metabolism that lead to the accumulation of unesterified AA are cytotoxic to the colon cancer cell line, HCT-116. Additionally, exogenous AA and triasin C, an inhibitor of AA acylation, induced apoptosis by caspase-3 activity in a transcriptionally dependent manner [121].
Animal studies
Our group studies with experimental animals showed the effects of the manipulation of dietary lipids on the generation of eicosanoids linked to metastatic growth in a murine lung alveolar carcinoma model. Feeding the mice a diet containing corn oil rich in n-6 PUFAs modulated the synthesis of eicosanoids (increasing 12-HETE and decreasing 12 HHT). These findings were positively correlated with an increment in apoptosis, diminution of mitosis and, concomitantly, with a decrease in tumoral growth [122]. Experimental studies on implanted C6 glioma cells into rat brains locally infused with 200 microM-2 mM of GLA have been shown the most active local concentration of GLA for antitumor activity was 2 mM, infused at 1 microl/h over 7 days. This concentration of GLA-induced tumor regression, increased apoptosis, and decreased proliferation in rats' tumors with a small the effect detected on normal neuronal tissue [123]. Also, these findings were recently confirmed in a similar model of glioma in rats (orthotopic C6 glioma model) [124].
Clinical studies
Clinical studies were carried out in more than 30 patients with intractable glioma tumor who had different grades of advanced disease (grade III or IV glioma). All were intratumorally injected with GLA, given at increased rates (0.5-1 mg/day for 7 - 10 days). Inducing GLA produced a significant reduction in tumor size, with significant increases in survival without any acute adverse clinical effects [120,125]. In an extension of this study, the authors, using intracerebral injection of GLA on the brains of patients with malignant gliomas, showed that this PUFA can induce regression of cancer cells without having cytotoxic effects on normal cells, suggesting that GLA is a safe antitumor agent and recommend its use in the management of human gliomas [126].
c) Tumor cell treatment with omega-3: ALA and EPA
In vitro studies
The molecular basis for the health benefits of omega-3 fatty acids is poorly understood. The most immediate effect is an increased incorporation of EPA and DHA into the sn-2 position of plasma membrane phospholipids, where AA is usually esterified. Thus, EPA and DHA replace LA and AA, the main substrates for the production of eicosanoids by the COX, LOX, and cytochrome P-450 pathways [127]. Numerous subsequent studies confirm that AA and EPA compete at all steps of prostanoid biosynthesis, resulting in specific shifting in metabolite production and action because respective ω-3 or ω-6 metabolites are synthesized [128].
Similarly, the competition between AA and EPA for the production of leukotrienes provides an interesting explanation for the anti-inflammatory effect of dietary ω-3 PUFAs. Arachidonic acid is metabolized by the 5-lipoxygenase to leukotriene B4 (LTB4) that induces inflammation and acts as a powerful chemoattractant of neutrophils. In contrast, the same pathway yields LTB5 from EPA, a metabolite at least 30 times less potent than LTB4 [129]. However, recent studies reveal that the anti-inflammatory effects of EPA and DHA are due to an exchange of the classical AA-derived proinflammatory eicosanoids for their less potent ω-3 counterparts. EPA and DHA are the precursors of novel lipid mediators, termed resolvins and protectins, which have potent anti-inflammatory and pro-resolution properties and may play a major role in the protection against various inflammatory diseases [130].
In other in vitro studies using BT-474 and SKBr-3 breast cancer cells, which naturally exhibit amplification of the HER2 oncogene, Menéndez et al [131] showed that ALA suppresses overexpression of the HER2 oncogene at the transcriptional level and thereby interacts synergistically with anti-HER2 immunotherapy. It has been suggested that ALA may be a potential dietary alternative or adjunct to currently used drugs in the management of HER2-positive breast carcinomas. Moreover, a low ω-6/ω-3 PUFA ratio and elevated levels of monounsaturated fats (e.g., ω-9), the two prominent fat features of the Mediterranean diet, should be extremely efficient at blocking HER2 expression in breast cancer cells [131].
Other investigators sought to determine whether EPA or DHA was more responsible for the increased apoptosis in cancer cells that was observed with the higher consumption of fish oils. They measured a number of endpoints, including phosphatidylserine staining with Annexin-V, Bcl-2 expression, and Bid, caspase 3, 8, and 9 expression, as well as PARP cleavage, in LT97 human colon adenoma and HT29 human colon cancer cells treated with EPA, DHA or LA. They observed that DHA was more effective at inducing apoptosis than EPA, and that LT97 cells were more prone to DHA- and EPA-induced apoptosis than HT29 cells. These authors concluded that cancer cells are highly susceptible to ω-3 PUFA–induced apoptosis, and that longer chains and higher numbers of double bonds produced greater anticancer effects [132].
Siddiqui et al [133] induced Jurkat leukemic programmed cell death by DHA treatment, suggesting that one mechanism through which DHA may control cancer cell growth is through apoptosis involving PP1/PP2B protein phosphatase (PP) activities. Activation of these PPs increases the susceptibility of apoptotic cell death. Indeed, introduction of DHA and EPA into the plasma membrane led to disruption of the lipid raft domains, perhaps causing an increasing fluidity of the bilayer, thus displacing signaling metastatic proteins such as CXCR4 in MDA-MB-231 breast cancer cell lines [134]. Human mammary cancer cells were treated with ω-3 FAs to assess the potential of these FAs as an adjuvant therapy against the transcription factor Sp1 [135]. The transcription factor Sp1 governs genes involved in cell cycle regulation, apoptosis, and lipogenesis. Sp1 is a target of acetylation, and is associated with loss of DNA binding at promoters related to cell cycle arrest and cell death in a colon cell line. The findings of this study suggest that the use of ω-3 FAs as adjuvant therapy prevents recurrence and metastasis of breast cancer.
Animal studies
Changes in dietary lipids may modify the eicosanoid production. In this way, experimental data from our laboratory with murine models of mammary cancer fed with Chia oil (Salvia Hispanica) rich in ω-3 showed that this diet modifies tumor cells, enriching membranes with ω-3 PUFAs with a concomitant decrease in the AA- derivative eicosanoids 12-HETE and 12-HHT—both associated with cancer cell proliferation and lymphocyte migration. Increased apoptosis and infiltration of T-lymphocytes and inhibition of tumor growth and metastasis formation were also observed in association to minor production of these eicosanoids [136].
Studies by other investigators in Lewis lung carcinoma-bearing C57BL/6 mice have shown that a diet containing 5% fish oil (rich in ω-3 PUFAS) resulted in the substantial slower growth of primary tumors, a lower mortality rate, and decreased metastatic spread, compared with tumor-bearing mice on a diet containing 5% soybean oil rich in ω-6 PUFAS [137].
Clinical Studies
In patients with prostate cancer, consumption of fish rich in ω-3 PUFAs was inversely related to cancer development [138]. In contrast, low levels of ALA 18:3, ω-3, a precursor of EPA and DHA in mammary adipose tissue, were associated with an increased risk of breast cancer in women [139]. Liang et al [140] examined the effects of omega-3 FA parenteral supplementation postoperatively on clinical outcomes and immunomodulation in colorectal cancer patients. They evaluated routine blood tests, biochemistry, systemic levels of IL-6 and TNF-α, and the percentage of CD3+, CD4+, and CD8+ lymphocytes preoperatively and on postoperative days 1 and 8. Patient outcome was evaluated considering mortality during the hospital stay, the length of the postoperative hospital stay, and the occurrence of infectious complications. They concluded that postoperative supplementation with omega-3 FAs may have a favorable effect on outcomes in colorectal cancer patients undergoing radical resection by lowering the magnitude of inflammatory responses and modulating the immune response.
d) Tumor cell treatment with omega-9
In vitro studies
Our laboratory examined the effects of the administration of abnormal PUFA: ETA (20:3, n-9)-affected parameters linked to tumor progression and metastases in several human cancer cells lines. In the T-24 bladder cancer cell line, cell peroxidation and expression of the cell-cell adhesion molecule e-cadherin were decreased. In the MCF-7 breast cancer cell line, e-cadherin expression was increased, lipid peroxidation was enhanced, and cell proliferation was reduced. In the HRT-18 colon cancer cell line, ETA produced an increment in e-cadherin expression and lipid peroxidation but also increased proliferation. We concluded that this 18:1 derivative produced some procarcinogenic effects on three human cancer cell lines [141]. Other experiments in which ETA was added at a low concentration resulted in a reduction in the expression of e-cadherin, and to a lesser degree, of desmoglein, along with increased invasion of human squamous cell carcinoma (SCC) cells in vitro. At higher concentrations, ETA stimulated the growth of SCC cells [141, 142]. The promoting effects of OA and its metabolites on cancer cells were recently confirmed. Soto-Guzman et al [143] demonstrated that stimulation of MDA-MB-231 breast cancer cells with 200 μM of OA (18:1, n-9) induced an increase in MMP-9 secretion through a PKC-, Src-, and EGFR-dependent pathway (as revealed by gelatin zymography assays), increasing the invasiveness of breast cancer cells. In contrast, studies using MDA-MB-231 human breast cancer cells showed that treatment OA augments focal adhesion kinase (FAK) activation and promotes migration [144].
The procarcinogenic action of OA was demonstrated by experimental research studying the activity of stearoyl CoA desaturase (SCD), the enzyme that produces monounsaturated fatty acids, in tumoral cells. This enzyme impairs lung cancer cell proliferation, survival, and invasiveness, and dramatically reduces tumor formation in mice. In this way, Hess et al [145] have demonstrated that exogenous OA treatment in human lung cancer cells restored the decreased membrane lipid levels resulting from inhibition of SCD activity, a key enzyme involved in the biosynthesis of unsaturated fatty acids. These data suggest that active lipid synthesis is required for the FA-mediated restoration of proliferation in SCD1-inhibited cancer cells [145]. Although some evidence exists for the antiproliferative potential of OA in vitro, most studies have shown OA and its derivatives to have procarcinogenic effects.
Animal studies
Recent studies in our laboratories using an experimental murine model of mammary gland adenocarcinoma showed that diets rich in ω-9 FAs reduce tumor growth, metastasis, and tumor leukocyte infiltration by inhibiting LOX activity, thereby reducing the formation of protumorigenic eicosanoids such as 12 (S)-HETE and 15 (S)-HETE; increasing the synthesis of apoptosis-inducing 12 (S)-HHT; and decreasing the production of proinflammatory PGE2 [146]. Our group also conducted studies examining the modulating effects of diets rich in ω-9, ω-6, and ω-3 on the development of murine mammary gland adenocarcinoma. Different tumor parameters were analyzed. Our findings showed that a diet rich in OA induced an essential FA deficiency and increased the incidence and the number of metastases when compared with the controls [147].
Conclusion
The understanding the mechanisms of FA-regulated signaling and its molecular bioactive derivatives is a critical issue to determine their involvement in cell proliferation and apoptosis impacting of the promotion, progression, or regression of the carcinogenic process. The intervention on dietary FAs induces changes in membrane lipids modifying their bioactive lipids derivated from specific biosynthetic pathways in response to extracellular stimuli. These molecules, in turn, modulate transcription factors involved in many physiological processes, and their deregulations have been linked to diseases such as inflammation, metabolic syndrome, and cancer. Knowledge of the mechanisms by which FAs control specific gene expressions may provide insight into the development of new therapeutic strategies for better management of whole body lipid metabolism, which, as discussed in this review, are important risk factors for cancer. Finally, to better understand the complex roles played by PUFAs in tumorigenesis, further research will be needed, using a wide variety of experimental models and cell tumor lines as well as different FA concentrations.
Acknowledgments
We are indebted to Dr. Kimberly K. McGhee (PhD) for technical English revision. This work was supported by Grants from Consejo Nacional de Investigaciones Cientìficas y Técnicas (CONICET- Argentina), Ministerio de Ciencias y tecnología de Córdoba (MINCYT-CBA) y Secretaria de Ciencia Y Tecnología de la Universidad Nacional de Còrdoba (SECYT-UNC).
References
- 1.Calderón RO, Eynard AR. Fatty acids specifically related to the anisotropic properties of plasma membrane from rat urothelium. Biochimica et. Biophysica Acta. 2000;1483(1):174–184. doi: 10.1016/s1388-1981(99)00173-0. [DOI] [PubMed] [Google Scholar]
- 2.Abumrad N, Coburn C, Ibrahimi A. Membrane proteins implicated in long-chain fatty acid uptake by mammalian cells: CD36, FATP and FABPm. Biochimica et Biophys ica Acta. 1999;1441(1):4–13. doi: 10.1016/s1388-1981(99)00137-7. [DOI] [PubMed] [Google Scholar]
- 3.Ehehalt R, Sparla R, Hasan Kulaksiz H, Herrmann T, Füllekrug J J, Stremmel W. Uptake of long chain fatty acids is regulated by dynamic interaction of FAT/CD36 with cholesterol/sphingolipid enriched microdomains. (lipid rafts) BMC Cell Biology. 2008;9:45. doi: 10.1186/1471-2121-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calder PC, Budge GC. Fatty acids. In: Nicolaou A, Kokotos G, editors. Bioactive Lipids. England: The Oil Press; 2004. pp. 1–36. [Google Scholar]
- 5.Das UN. Essential fatty acids: biochemistry, physiology and pathology. Biotechnology Journal. 2006;4:420–439. doi: 10.1002/biot.200600012. [DOI] [PubMed] [Google Scholar]
- 6.Eynard AR. Role of dietary polyunsaturated fatty acids (PUFA) on tumorigenesis. The cancer journal. 1996;9(3):142–144. [Google Scholar]
- 7.Qiu X. Biosynthesis of docosahexaenoic acid (DHA, 22:6-4, 7,10,13,16,19): two distinct pathways. Prostaglandins Leukot Essent Fatty Acids. 2003;68(2):181–186. doi: 10.1016/s0952-3278(02)00268-5. [DOI] [PubMed] [Google Scholar]
- 8.Martinez M, Ichaso N, Setien F, Durany N, Qiu X, Roesler W. The Δ4-desaturation pathway for DHA biosynthesis is operative in the human species: differences between normal controls and children with the Zellweger syndrome. Lipids in Health and Disease. 2010;9:98. doi: 10.1186/1476-511X-9-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Monroig O, Zhang L, Wang S, Zheng X, Dick JR, You C, Tocher DR. Vertebrate fatty acyl desaturase with Δ4 activity. Proceedings of the National Academy of Sciences of Unites State of America. 2010;107(39):16840–16845. doi: 10.1073/pnas.1008429107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das UN. Can essential fatty acids reduce the burden of disease(s)? Lipids in Health and Disease. 2008;7:19. doi: 10.1186/1476-511X-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lundbæk JA, Collingwood SA, Ingolfsson HI, Kapoor R, Andersen OS. Lipid bilayer regulation of membrane protein function: gramicidin channels as molecular force probes. Journal of the Royal Society, Interface. 2010;7(44):373–395. doi: 10.1098/rsif.2009.0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das UN. Essential Fatty acids: Bichemistry and Physiology. In: DAs UN, editor. Metabolic Syndrome Pathophysiology: The role of Essential Fatty acids. Wiley-Blackwell; California, USA: 2010. pp. 181–200. [Google Scholar]
- 13.Calder PC. Polyunsaturated Fatty acids and inflammation. Prostaglandins Leukotrienes and Essential Fatty Acids. 2006;75(3):197–202. doi: 10.1016/j.plefa.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 14.Arnold C, Konkel A, Fisher R, Schunck WH. Cytochrome p450-dependent metabolism of ω-6 and ω-3 long-chain polyunsaturated fatty acids. Pharmacological Reports. 2010;62(3):536–547. doi: 10.1016/s1734-1140(10)70311-x. [DOI] [PubMed] [Google Scholar]
- 15.Weaver KL, Ivester P, Seeds M, Case LD, Arm JP, Chilton FH. Effect of dietary fatty acids on inflammatory gene expression in healthy humans. Journal of Biological Chemistry. 2009;284(23):15400–15407. doi: 10.1074/jbc.M109.004861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphy MG. Dietary fatty acids and membrane protein function. Journal of Nutritional Biochemistry. 1990;1(2):68–79. doi: 10.1016/0955-2863(90)90052-m. [DOI] [PubMed] [Google Scholar]
- 17.Gurr MI, Harwood JL, Frayn KN. Lipid Biochemistry: an introduction. Blackwell Science; Oxford: 2002. [Google Scholar]
- 18.Calder PC, Yaqoob P. Lipid rafts Composition, Characterization and Controversies. Journal of Nutrition. 2007;137(3):545–547. doi: 10.1093/jn/137.3.545. [DOI] [PubMed] [Google Scholar]
- 19.Pike LJ. Lipid rafts: bringing order to chaos. Journal of Lipid Research. 2003;44(4):655–667. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Jahn KA, Su Y, Braet F. Multifaceted nature of membrane microdomains in colorectal cancer. World Journal of Gastroenterology,1. 2011;7(6):681–690. doi: 10.3748/wjg.v17.i6.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris R, Cox H, Mombelli E, Quinn PJ. Rafts, little caves and large potholes: how lipid structure interacts with membrane proteins to create functionally diverse membrane environments. Sub-cellular Biochemistry. 2004;37:35–118. doi: 10.1007/978-1-4757-5806-1_2. [DOI] [PubMed] [Google Scholar]
- 22.Brash AR. Arachidonic acid as bioactive molecule. The Journal of Clinical Investigation. 2001;107(11):1339–1345. doi: 10.1172/JCI13210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lands WE. Stories about acyl chains. Biochimica et Biohysica Acta. 2000;1483(1):1–14. doi: 10.1016/s1388-1981(99)00177-8. [DOI] [PubMed] [Google Scholar]
- 24.Chilton FH, Fontech AN, Surette ME, Triggiani M, Winkler JD. Control of arachidonic levels within inflammatory cells. Biochim Biophys Acta. 1996;1299(1):1–15. doi: 10.1016/0005-2760(95)00169-7. [DOI] [PubMed] [Google Scholar]
- 25.Nebert DW, Russell DW. Clinical importance of the cytochromes P450. Lancet. 2002;360(9340):1155–1162. doi: 10.1016/S0140-6736(02)11203-7. [DOI] [PubMed] [Google Scholar]
- 26.Murakami M. Mediators in Life Sciences. Experimental Animals . 2011;60:7–20. doi: 10.1538/expanim.60.7. [DOI] [PubMed] [Google Scholar]
- 27.Wang D, Du Bois RN. Eicosanoids and cancer. Nat Rev Cancer . 2010;2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panigrahy D, Kaipainen A, Greene ER, Huang S. Cytochrome P450-derived eicosanoids: the neglected pathway in cancer. Cancer Metastasis Reviews. 2010;29(4):723–735. doi: 10.1007/s10555-010-9264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation:dual anti-inflammatory and pro-resolution lipid mediators. Nature Reviews Immunology. 2008;8(5):349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serhan CN, Gotlinger K, Hong S, Arita M. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their aspirin-triggered endogenous epimers: an overview of their protective roles in catabasis. Prostaglandins & Other Lipid Mediators. 2004;73(3-4):155–172. doi: 10.1016/j.prostaglandins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 31.Di Marzo V. The endocannabinoid System: its general strategy of action, tools fo(r its pharmacological manipulation and potential therapeutic explotation. Pharmacological Research. 2009;60(2):77–84. doi: 10.1016/j.phrs.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, et al. A biosynthetic pathway for anandamide. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(36):13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasqualini ME, Berra MA, Yurawecz MP, Repossi G, Eynard AR. Dietary manipulation of precursor PUFAs modulates eicosanoid and endocanabinoid synthesis: a potential tool to control tumor development. Current Nutrition and Food Science. 2008;4:161–175. [Google Scholar]
- 34.Das UN, Repossi G, Dain A, Eynard AR. Is insulin resistance a disorder of the brain? Frontiers in Biosciences.: a journal and virtual library, 1. 2011;16:1–12. doi: 10.2741/3671. [DOI] [PubMed] [Google Scholar]
- 35.Hwang DH. Characteristics of the formation of the platelet lipoxygenase product from endogenous arachidonic acid. Lipids. 1982;17(12):845–847. doi: 10.1007/BF02534576. [DOI] [PubMed] [Google Scholar]
- 36.Eynard AR, Galli G, Tremoli E, Maderna P, Magni F, Paoletti R. Aspirin inhibits platelet 12-hydroxy-eicosatetraenoic acid formation. The Journal of Laboratory and Clinical Medicine. 1986;107(1):73–78. [PubMed] [Google Scholar]
- 37.Brash AR, Ingram CD. Lipoxygenase metabolism of endogenous arachidonate in leukocytes: GC-MS analyses of incubations in H2180 Buffers. Prostaglandins Leukotrienes and Medicine. 1986;23(2-3):149–154. doi: 10.1016/0262-1746(86)90178-2. [DOI] [PubMed] [Google Scholar]
- 38.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347(6294):645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 39.Gottlicher M, Widmark E, Li Q, Gustafsson JA. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(10):4653–4657. doi: 10.1073/pnas.89.10.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keller H, Dreyer C, Medin J, Mahfoudi A, Ozato K, Wahli W. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(6):2160–2164. doi: 10.1073/pnas.90.6.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisely GB, Koble CS, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(9):4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Molecular cell. 1999;3(3):397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- 43.Zoete V, Grosdidier A, Michielin O. Peroxisome proliferator-activated receptor structures: ligand specificity, molecular switch and interactions with regulators. Biochimica et Biophysica Acta. 2007;1771(8):915–925. doi: 10.1016/j.bbalip.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 44.Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, Denissov S, Borgesen M, Francoijs KJ, Mandrup S, Stunnenberg HG. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev . 2008;22:2953–2967. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.IJpenberg A, Jeannin E, Wahli W, Desvergne B. Polarity and specific sequence requirements of peroxisome proliferator-activated receptor (PPAR)/retinoid X receptor heterodimer binding to DNA. A functional analysis of the malic enzyme gene PPAR response element. J Biol Chem . 1997;272:20108–20117. doi: 10.1074/jbc.272.32.20108. [DOI] [PubMed] [Google Scholar]
- 46.Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 47.Nagy L, Schwabe JW. Mechanism of the nuclear receptor molecular switch. Trends Biochem Sci. 2004;29:317–324. doi: 10.1016/j.tibs.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 48.Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nature Medicine. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- 49.Yin Y, Russell RG, Dettin LE, Bai R, Wei ZL, Kozikowski AP, et al. Peroxisome proliferator-activated receptor delta and gamma agonists differentially alter tumor differentiation and progression during mammary carcinogenesis. Cancer Research. 2005;65:3950–3957. doi: 10.1158/0008-5472.CAN-04-3990. [DOI] [PubMed] [Google Scholar]
- 50.Stephen RL, Gustafsson MC, Jarvis M, Tatoud R, Marshall BR, Knight D, et al. Activation of peroxisome proliferator-activated receptor delta stimulates the proliferation of human breast and prostate cancer cell lines. Cancer Research. 2004;64:3162–3170. doi: 10.1158/0008-5472.can-03-2760. [DOI] [PubMed] [Google Scholar]
- 51.Glinghammar B, Skogsberg J, Hamsten A, Ehrenborg E. PPARdelta activation induces COX-2 gene expression and cell proliferation in human hepatocellular carcinoma cells. Biochemica et Biophysica Research Communication. 2003;308:361–368. doi: 10.1016/s0006-291x(03)01384-6. [DOI] [PubMed] [Google Scholar]
- 52.Zhai JJ, Liu ZL, Li JM, Chen JP, Jiang L, Wang DM, Yuan J, Shen JG, Yang DP, Chen JQ. Different mechanisms of cis-9,trans-11- and trans-10,cis-12- conjugated linoleic acid affecting lipid metabolism in 3T3-L1 cells. The Journal of Nutrition Biochemistry. 2010;21(11):1099–1105. doi: 10.1016/j.jnutbio.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 53.Le May C, Pineau T, Bigot K, Kohl C, Girard J, Pegorier JP. Reduced hepatic fatty acid oxidation in fasting PPAR alpha-null mice is due to impaired mitochondrial hydroxymethylglutaryl-CoA synthase gene expression. FEBS Letters. 2002;475(3):163–166. doi: 10.1016/s0014-5793(00)01648-3. [DOI] [PubMed] [Google Scholar]
- 54.Ide T, Kobayashi H, Ashakumary L, et al. Comparative effects of.perilla and fish oils on the activity and gene expression of fatty acid oxidation enzymes in rat liver. Biochimia et Biophysica Acta. 2000;1485(1):23–35. doi: 10.1016/s1388-1981(00)00026-3. [DOI] [PubMed] [Google Scholar]
- 55.Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator-activated receptor gamma agonists. Lancet Oncology. 2004;5(7):119–129. doi: 10.1016/S1470-2045(04)01509-8. [DOI] [PubMed] [Google Scholar]
- 56.Chu R, Lin Y, Rao MS, Reddy JK. Cloning and identification of rat deoxyuridine triphosphatase as an inhibitor of peroxisome proliferator-activated receptor alpha. Journal of Biolical Chemistry. 1996;271:27670–27676. doi: 10.1074/jbc.271.44.27670. [DOI] [PubMed] [Google Scholar]
- 57.Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annual Review of Nutrition. 2001;21:193–230. doi: 10.1146/annurev.nutr.21.1.193. [DOI] [PubMed] [Google Scholar]
- 58.Peters JM, Cheung C, Gonzalez FJ. Peroxisome proliferator-activated receptor-alpha and liver cancer: where do we stand? Journal of Molecular Medicine. 2005;83:774–785. doi: 10.1007/s00109-005-0678-9. [DOI] [PubMed] [Google Scholar]
- 59.Narala VR, Adapala RK, Suresh MV, Brock TG, Peters-Golden M, Reddy RC. Leukotriene B4 is physiologically relevant endogenous peroxisome proliferators activated receptor-alpha.agonist. The Journal of Biological Chemistry. 2010;285(29):22067–22074. doi: 10.1074/jbc.M109.085118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eynard AR, Tremoli E, Caruso D, Magni F, Sirtori CR, Galli G. Platelet formation of 12-hydroxyeicosatetraenoic acid and thromboxane B2 is increased in type IIA hypercholesterolemic subjects. Atherosclerosis. 1986;60(1):61–66. doi: 10.1016/0021-9150(86)90088-2. [DOI] [PubMed] [Google Scholar]
- 61.Fields AP, Murray NR. Protein kinase C isozymes as therapeuthic targets for treatment of human cancers. Advance in Enzyme Regulation. 2008;48:166–178. doi: 10.1016/j.advenzreg.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rozengurt E. Protein kinase D signaling: multiple biological functions in health and disease. Physiology (Bethesda) 2011;26(1):23–33. doi: 10.1152/physiol.00037.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao Y, Joshi-Barve S, Barve S, Chen LH. Eicosapentaenoic acid prevents LPS-induced TNF-alpha expression by preventing NF-kappaB activation. Journal of the American College of Nutrition. 2004;23:71–78. doi: 10.1080/07315724.2004.10719345. [DOI] [PubMed] [Google Scholar]
- 64.Novak TE, Babcock TA, Jho DH, Helton WS, Espat NJ. NF-kappa B inhibition by omega -3 fatty acids modulates LPS-stimulated macrophage TNF-alpha transcription. America Journal of Physiology. Lung Cellular and Molecular Physiology. 2003;284:L84–89. doi: 10.1152/ajplung.00077.2002. [DOI] [PubMed] [Google Scholar]
- 65.Lee JY, Hwang DH. The modulation of inflammatory gene expression by lipids: mediation through Toll-like receptors. Molecular Cells. 2006;21:174–185. [PubMed] [Google Scholar]
- 66.Munford RS, Hall CL. Detoxification of bacterial lipopolysaccharides (endotoxins) by a human neutrophil enzyme. Science. 1986;234:203–205. doi: 10.1126/science.3529396. [DOI] [PubMed] [Google Scholar]
- 67.Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. Journal of Biolical Chemistry. 2001;276:16683–166689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- 68.Lee JY, Plakidas A, Lee WH, Heikkinen A, Chanmugam P, Bray G, et al. Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. Journal of Lipid Research. 2003;44:479–486. doi: 10.1194/jlr.M200361-JLR200. [DOI] [PubMed] [Google Scholar]
- 69.Lee HK, Dunzendorfer S, Soldau K, Tobias PS. Double-stranded RNA-mediated TLR3 activation is enhanced by CD14. Immunity. 24:153–163. doi: 10.1016/j.immuni.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 70.Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. The Journal of Experimental Medicine. 2006;203:2377–2389. doi: 10.1084/jem.20060845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wong SW, Kwon MJ, Choi AM, Kim HP, Nakahira K, Hwang DH. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. The Journal of Biological Chemistry. 2009;284:27384–27392. doi: 10.1074/jbc.M109.044065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Procedings Nationational Academy of Sciences of United State of America. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hua X, Wu J, Goldstein JL, Brown MS, Hobbs HH. Structure of the human gene encoding sterol regulatory element binding protein-1 (SREBF1) and localization of SREBF1 and SREBF2 to chromosomes 17p11.2 and 22q13. Genomics. 1995;25:667–673. doi: 10.1016/0888-7543(95)80009-b. [DOI] [PubMed] [Google Scholar]
- 74.Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X, et al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell. 1993;75:187–197. [PubMed] [Google Scholar]
- 75.Shimomura I, Bashmakov Y, Shimano H, Horton JD, Goldstein JL, Brown MS. Cholesterol feeding reduces nuclear forms of sterol regulatory element binding proteins in hamster liver. P rocedings Nationational Academy of Sciences of United State of America. 1997;94:12354–12359. doi: 10.1073/pnas.94.23.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. The Journal of Clinical Investigation. 1997;99:838–845. doi: 10.1172/JCI119247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tontonoz P, Kim JB, Graves RA, Spiegelman BM. ADD1: a novel helix-loop-helix transcription factor associated with adipocyte determination and differentiation. Molecular Cell Biology. 1993;13:4753–4759. doi: 10.1128/mcb.13.8.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Magana MM, Osborne TF. Two tandem binding sites for sterol regulatory element binding proteins are required for sterol regulation of fatty-acid synthase promoter. The Journal of Biological Chemistry. 1996;271:32689–32694. doi: 10.1074/jbc.271.51.32689. [DOI] [PubMed] [Google Scholar]
- 79.Rawson RB, DeBose-Boyd R, Goldstein JL, Brown MS. Failure to cleave sterol regulatory element-binding proteins (SREBPs) causes cholesterol auxotrophy in Chinese hamster ovary cells with genetic absence of SREBP cleavage-activating protein. The Journal of Biological Chemistry . 1999;274:28549–28556. doi: 10.1074/jbc.274.40.28549. [DOI] [PubMed] [Google Scholar]
- 80.Matsuda M, Korn BS, Hammer RE, Moon YA, Komuro R, Horton JD, et al. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes & Development. 2001;15:1206–1216. doi: 10.1101/gad.891301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Espenshade PJ, Cheng D, Goldstein JL, Brown MS. Autocatalytic processing of site-1 protease removes propeptide and permits cleavage of sterol regulatory element-binding proteins. The Journal of Biological Chemistry. 1999;274:22795–22804. doi: 10.1074/jbc.274.32.22795. [DOI] [PubMed] [Google Scholar]
- 82.Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 83.Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, Goldstein JL. Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Molecular Cell. 2004;15:259–268. doi: 10.1016/j.molcel.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 84.Gong Y, Lee JN, Lee PC, Goldstein JL, Brown MS, Ye J. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metabolism. 2006;3:15–24. doi: 10.1016/j.cmet.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 85.Gurkan C, Stagg SM, Lapointe P, Balch WE. The COPII cage: unifying principles of vesicle coat assembly. Nature Review. Molecular Cell Biology. 2006;7:727–738. doi: 10.1038/nrm2025. [DOI] [PubMed] [Google Scholar]
- 86.Nohturfft A, Yabe D, Goldstein JL, Brown MS, Espenshade PJ. Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell. 2000;102:315–323. doi: 10.1016/s0092-8674(00)00037-4. [DOI] [PubMed] [Google Scholar]
- 87.Sato R, Yang J, Wang X, Evans MJ, Ho YK, Goldstein JL et al. Assignment of the membrane attachment, DNA binding, and transcriptional activation domains of sterol regulatory element-binding protein-1 (SREBP-1) The Journal of Biological Chemistry. 1004;269:17267–17273. [PubMed] [Google Scholar]
- 88.Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. The Journal of Clinical Investestigation. 1997;99:846–854. doi: 10.1172/JCI119248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee YS, Sohn DH, Han D, Lee HW, Seong RH, Kim JB. Chromatin remodeling complex interacts with ADD1/SREBP1c to mediate insulin-dependent regulation of gene expression. Molecular Cell Biology. 2007;27:438–452. doi: 10.1128/MCB.00490-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moreno JJ. New aspecs of the role of hydroxyeicosatertraenoic acids in cell growth and cancer development. Biochemical Pharmacological. 2009;77(1):1–10. doi: 10.1016/j.bcp.2008.07.033. [DOI] [PubMed] [Google Scholar]
- 91.Tang DG, La E, Kern J, Kehrer JP. Fatty Acid Oxidation and signaling in apoptosis. Biological Chemistry. 2002;383(3-4):425–442. doi: 10.1515/BC.2002.046. [DOI] [PubMed] [Google Scholar]
- 92.Szekeres CK, Trikha M, Honn KV. 12(S)-HETE, pleiotropic functions, multiple signaling pathways. Advances in Experimental Medicine and Biology. 2002;507:509–515. doi: 10.1007/978-1-4615-0193-0_78. [DOI] [PubMed] [Google Scholar]
- 93.Zeng ZZ, Yellaturu CR, Neeli I, Rao GN. 5 (S)-Hydroxyeicosatetraenoic acid stimulates DNA synthesis in human microvascular endothelial cells via activation of Jak/STAT and phophatidilylinositol 3-kinase/AKT signaling, leading to induction of expression of basic fibroblast growth factor-2. The Journal of Biological Chemistry. 2002;277(43):41213–41219. doi: 10.1074/jbc.M204508200. [DOI] [PubMed] [Google Scholar]
- 94.Nieves D, Moreno JJ. Enantioselective effect of 12 (S)- Hydroxyeicosatetraenoic acid on 3T6 fibroblast growth throughERK ½ and p38 MAPK pathways and cyclin D1 activation. Biochemical Pharmacology. 2008;76(5):654–661. doi: 10.1016/j.bcp.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 95.Muthalif MM, Benter IF, Karzoun N, et al. 20-Hydroxyeicosatetraenoic acid mediates calcium/calmodulin -dependent protein kinase II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. Proceedings of National Academy of Sciences of Unied States of America. 1998;95(21):12701–12706. doi: 10.1073/pnas.95.21.12701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wei J, Yan W, Li X, Chang WC, Tai HH. Activation of thromboxane receptor-α induces expression of cyclooxygenase-2 through multiple signaling pathways in A549 human lung adenocarcinoma cells. Biochemical Pharmacology. 2007;74(5):787–800. doi: 10.1016/j.bcp.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nie D, Guo Y, Yang D, Tang Y, et al. Thromboxane A2 receptors in prostate carcinoma: expression and its role in regulating cell motility via small GTPase Rho. Cancer Research. 2008;68(1):115–121. doi: 10.1158/0008-5472.CAN-07-1018. [DOI] [PubMed] [Google Scholar]
- 98.Liu JF, Fong YC, Chang CS, Huang CY, Chen HT, Yang WH, Hsu CJ, Jeng LB, Chen CY, Tang CH. Cyclooxygenase-2 enhances alpha2beta1 integrin expression and cell migration via EP1 dependent signaling in human condrosarcoma cells. Mol Cancer . 2010;9:43. doi: 10.1186/1476-4598-9-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kravchenko IV, Furalyov VA, Lisitsina ES, Popov VO. Stimulation of mechano-growth factor expression by second messengers. Archives of Biochemistry and Biophysics, 507 (2), 323-331)pathway in human chondrosarcoma cells. Molecular Cancer. 2011;9:43–57. doi: 10.1016/j.abb.2010.12.028. [DOI] [PubMed] [Google Scholar]
- 100.Fujino H, Toyomura K, Chen XB, Regan JW, Murayama T. Prostaglandin E2 regulates cellular migration via induction of vascular endothelial growth factor receptor-1 in HCA-7 human colon cancer cells. Biochemical Pharmacological. 2011;81(3):379–387. doi: 10.1016/j.bcp.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 101.Yang SF, Chen MK, Hsieh YS, et al. Prostaglandin E2/EP1 signaling pathway enhances intercellular adhesion molecule 1 (ICAM-1) expression and cell motility in oral cancer cells. The Journal of Biological Chemistry. 2010;285(39):29808–29816. doi: 10.1074/jbc.M110.108183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ma XM, Yu H, Huai N. Peroxisome proliferator-activated receptor-gamma is essential in the pathogenesis of gastric carcinoma. World Journal of Gastroenterology. 2009;15(31):3874–3883. doi: 10.3748/wjg.15.3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Konkel A, Schunck WH. Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acid. Biochimic et Biophysica Acta. 2011;1814(1):210–222. doi: 10.1016/j.bbapap.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 104.Ishizuka T, Cheng J, Singh H, Vitto MD, Manthati VL, et al. 20-Hydroxyeicosatetraenoic acid stimulates nuclearfactor-kappaB activation and the production of inflammatory cytokines in human endothelial cells. The Journal of Pharmacology and Experimental Therapeutics. 2008;324(1):103–110. doi: 10.1124/jpet.107.130336. [DOI] [PubMed] [Google Scholar]
- 105.Yu W, Chen L, Yang YQ, et al. Cytochrome P450 ω-hydroxylase promotes angiogenesis and metastasis by up-regulation of VEGF and MMP-9 in non-small cell lung cancer. Cancer Chemotherapy Pharmacology. 2010 Dec 1; doi: 10.1007/s00280-010-1521-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Flavin R, Zadra G, Loda M. Metabolic alterations and target therapies in prostate cancer. The Journal of Pathology. 2011;223(2):283–294. doi: 10.1002/path.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Grammatikos S, Harvey M, Subbaiah PV, Victor T, Miller W. Loos of fatty acid Δ-6 desaturating ability in human mammary epithelial cells that express an activated c-Ha-ras-oncogen. International Journal of Oncology. 1995;6(5):1039–1046. doi: 10.3892/ijo.6.5.1039. [DOI] [PubMed] [Google Scholar]
- 108.Sinclair HM. And History of essential fatty acids. In: Horrobin DF, editor. Omega-6 Essential Fatty acids: Pathophysiology and roles in Clinical Medicine. Alan R Liss, Inc; 1990. pp. 1–20. [Google Scholar]
- 109.Hilvo M, Denkert C, Lehtinen L, Muller B, et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Research March. 2011 Mar 17; doi: 10.1158/0008-5472.CAN-10-3894. [DOI] [PubMed] [Google Scholar]
- 110.Fuereder T, Hoeflmayer D, Jaeger-Lansky A, Rasin-Streden D et al. Sphingosine kinase 1 is a relevant molecular target in gastric cancer. Anticancer Drugs. 2011;22(3):245–252. doi: 10.1097/cad.0b013e328340bd95. [DOI] [PubMed] [Google Scholar]
- 111.Duplus E, Glorian M, Forest C. Fatty acid regulation of gene transcription. The journal of Biological Chemistry, 275 (40), 30749-30752);(Huang, ZH., Gu, D., Mazzone, T. (2004) Oleic acid modulates the post-translational glycosylation of macrophage ApoE to increase its secretion. The Journal of Biological Chemistry. 2000;279(28):29195–29201. doi: 10.1074/jbc.M402631200. [DOI] [PubMed] [Google Scholar]
- 112.Pegorier JP, Le May C, Girard J. Control of gene expression by fatty acids. The Journal of Nutrition. 2004;1348(9):24445S–24495S. doi: 10.1093/jn/134.9.2444S. [DOI] [PubMed] [Google Scholar]
- 113.Lu X, Yu H, Ma Q, Shen S, Das UN. Linoleic acid suppresses colorectal cancer cell growth by inducing oxidant stress and mitochondrial dysfunction. Lipids in Health and Disease. 2010;9:106–117. doi: 10.1186/1476-511X-9-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Notarnicola M, Messa C, Refolo MG, Tutito V, et al. Polyunsaturated fatty acids reduce fatty acid synthase and hydroxy-methyl-glutaryl CoA-reductase gene expression and promote apoptosis in HepG2 cell line. Lipids in Health and Disease. 2011;10:10. doi: 10.1186/1476-511X-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jiang WG, Bryce RP, Mansel RE. Gamma linoleic acid regulates gap junction communication in endothelial cells and their interation with tumor cells. Prostaglandins Leukotrienes and Essential Fatty Acids. 1997;56(4):307–316. doi: 10.1016/s0952-3278(97)90575-5. [DOI] [PubMed] [Google Scholar]
- 116.Eynard AR, Jiang WG, Mansel RE. Eicosatrienoic acid (20:3 n-9) inhibits the expression of E-cadherin and desmoglein in human squamous cell carcinoma in vitro. Prostaglandins Leukotrienes and Essential Fatty Acids. 1998;59(6):371–377. doi: 10.1016/s0952-3278(98)90098-9. [DOI] [PubMed] [Google Scholar]
- 117.Pasqualini ME, Heyd VL, Manzo P, Eynard AR. Association between E-cadherin expression by human colon, bladder and breast cancer cells and the 13-HODE: 15-HETE ratio. A possible role of their metastatic potential. Prostaglandins Leukotrienes and Essential Fatty Acids 2003. 2003;68(1):9–16. doi: 10.1016/s0952-3278(02)00230-2. [DOI] [PubMed] [Google Scholar]
- 118.Brown MD, Hart C, Gazi E, Gardner P, et al. Influence of omega-6 PUFA arachidonic acid and bone marrow adipocytes on metastatic spread from prostate cancer. British Journal of Cancer. 2009;102(2):403–413. doi: 10.1038/sj.bjc.6605481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Scheim DE. Cytotoxicity of unsaturated fatty acids in fresh human tumor explants: concentration thresholds and implications for clinical efficacy. Lipids in Health and Disease. 2009;8:54–65. doi: 10.1186/1476-511X-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Das UN. γ-linoleic acid therapy of human glioma- a review of in vitro, in vivo and clinical studies. Medical Science Monitor. 2007;13(7):119–131. [PubMed] [Google Scholar]
- 121.Monjazeb AM, High KP, Connoy A, Hart LS, Koumenis C, Chilton FH. Arachidonic acid-induced gene expression in colon cancer cells. Carcinogenesis. 2006;27(10):1950–1960. doi: 10.1093/carcin/bgl023. [DOI] [PubMed] [Google Scholar]
- 122.Pasqualini ME, Berra MA, Calderón RO, et al. Dietary lipids modulate eicosanoid release and apoptosis of cells of a murine lung alveolar carcinoma. Prostaglandins Leukotrienes and Essential Fatty Acids. 2005;72(4):235–240. doi: 10.1016/j.plefa.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 123.Leaver HA, Wharton SB, Bell HS, Leaver-Yap IM, Whittle IR. Highly unsaturated fatty acid induced tumour regression in glioma pharmacodynamics and bioavailability of gamma linolenic acid in an implantation glioma model: effects on tumour biomass, apoptosis and neuronal tissue histology. Prostaglandins Leukotrienes and Essential Fatty Acids. 2002;67(5):283–292. doi: 10.1054/plef.2002.0431. [DOI] [PubMed] [Google Scholar]
- 124.Miyake JA, Benadiba M, Colquhoun A. Gamma-linolenic acid inhibits both tumour cell cycle progression and angiogenesis in the orthotopic C6 glioma model through changes in VEGF, Flt1, ERK1/2, MMP2, cyclin D1, pRb, p53 and p27 protein expression. Lipids Health Dis. 2009;17:8–8. doi: 10.1186/1476-511X-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Naidu MR, Das UN, Kishan A. Intratumoral gamma-linolenic acid therapy of human gliomas. Prostaglandins Leukotrienes and Essential Fatty Acids. 1992;45(3):181–184. doi: 10.1016/0952-3278(92)90110-5. [DOI] [PubMed] [Google Scholar]
- 126.Das UN, Prasad VV, Reddy DR. Local application of gamma-linolenic acid in the treatment of human gliomas. Cancer Letters. 1995;94(2):147–55. doi: 10.1016/0304-3835(95)03844-m. [DOI] [PubMed] [Google Scholar]
- 127.Smith WL, Murphy RC. In: The eicosanoids: cyclooxygenase, lipoxygenase, and epoxygenase pathways, in Biochemistry of Lipids, Lipoproteins and Membranes. Vance DE, Vance JE, editors. Elsevier; New York.: Smith and Murphy; 2002. pp. 341–372. [Google Scholar]
- 128.Arnold C, Markovic M, Blossey K, Wallukat G, et al. Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of {omega}-3 fatty acids. The Journal of Biological Chemistry. 2010;285(43):32720–32733. doi: 10.1074/jbc.M110.118406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Terano T, Salmon JA, Moncada S. Biosynthesis and biological activity of leukotriene B5. Prostaglandins. 1984;27(2):217–232. doi: 10.1016/0090-6980(84)90075-3. [DOI] [PubMed] [Google Scholar]
- 130.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation:dual anti-inflammatory and pro-resolution lipid mediators. Nature Reviews. Immunology, 2008. 2008;8(5):349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Menéndez JA, Vázquez-Martín A, Ropero S, Colomer R, Lupu R. HER2 (erbB-2)-targeted effects of the omega-3 polyunsaturated fatty acid, alpha-linolenic acid (ALA; 18:3n-3), in breast cancer cells: the “fat features” of the “Mediterranean diet” as an “anti-HER2 cocktail”. Clinical and Translocation Oncology. 2006;8(11):812–820. doi: 10.1007/s12094-006-0137-2. [DOI] [PubMed] [Google Scholar]
- 132.Habermann N, Schön A, Lund EK, Glei M. Fish fatty acids alter markers of apoptosis in colorectal adenoma and adenocarcinoma cell lines but fish consumption has no impact on apoptosis-induction ex vivo. Apoptosis. 2010;5(5):621–630. doi: 10.1007/s10495-010-0459-y. [DOI] [PubMed] [Google Scholar]
- 133.Siddiqui RA, Jenski LJ, Neff K, et al. Docosahexaenoic acid induces apoptosis in Jurkat cells by protein phosphatase mediated process. Biochimica et Biophysica Acta. 2001;1499(3):265–275. doi: 10.1016/s0167-4889(00)00128-2. [DOI] [PubMed] [Google Scholar]
- 134.Altenburg JD, Siddiqui RA. Omega -3 Polyunsaturated fatty acids down –modulate CXCR4 expression and function in MDA-MB-231 breast cancer cells. Molecular Cancer Research. 2008;7(17):1013–1020. doi: 10.1158/1541-7786.MCR-08-0385. [DOI] [PubMed] [Google Scholar]
- 135.Xia SH, Wang J, Kang JX. Decreased n-6/n-3 fatty acid ratio reduces the invasive potential of human lung cancer cells by downregulation of cell adhesion/invasion-related genes. Carcinogenesis. 2005;26(4):779 –784. doi: 10.1093/carcin/bgi019. [DOI] [PubMed] [Google Scholar]
- 136.Espada CE, Berra MA, Martinez MJ, Eynard AR, Pasqualini ME. Effect of chia oil (Salvia hispanica) rich in ω-3 fatty acids on the eicosanoid release, apoptosis and t-lymphocyte tumor infiltration in a murine mammary gland adenocarcinoma. Prostaglandins Leukotrienes and Essential Fatty Acids. 2007;77(1):21–28. doi: 10.1016/j.plefa.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 137.Yam D, Peled A, Husza M, Shinitzky M. Dietary fish oil suppresses tumor growth and metastasis of Lewis lung carcinoma in mice. Nutrition Biochemistry. 1997;8:619–622. [Google Scholar]
- 138.Larsson SC, Kumlin M, Ingelman-Sunderg M, Wolk A. Dietary long-chain n-3 fatty acids for prevention of cancer: a review of potential mechanisms. The American Journal of Clinical Nutrition. 2004;79(6):935–945. doi: 10.1093/ajcn/79.6.935. [DOI] [PubMed] [Google Scholar]
- 139.Bagga D, Anders HJ, Glaspy JA. Long-chain n-3-to-n-6 polyunsaturated fatty acid ratios in breast adipose tissue from women with and without breast cancer. Nutrition and Cancer. 2002;42(2):180–185. doi: 10.1207/S15327914NC422_5. [DOI] [PubMed] [Google Scholar]
- 140.Liang B, Wang S, Ye YJ, Yang XD, et al. Impact of postoperative omega-3 fatty acid-supplemented parenteral nutrition on clinical outcomes and immunomodulations in colorectal cancer patients. World Jo urnal of Gastroenterology: WJG. 2008;14(15):2434–2439. doi: 10.3748/wjg.14.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Heyd VL, Eynard AR. Effects of eicosatrienoic acid (20:3 n-9, Mead's acid) on some promalignant-related properties of three human cancer cell lines. Prostaglandins & Other Lipids Mediators. 2003;71(3-4):177–88. doi: 10.1016/s1098-8823(03)00037-6. [DOI] [PubMed] [Google Scholar]
- 142.Eynard AR, Jiang WG, Mansel RE. Eicosatrienoic acid (20:3 n-9) inhibits the expression of E-cadherin and desmoglein in human squamous cell carcinoma in vitro. Prostaglandins Leukotrienes and Essential Fatty Acids. 1998;59(6):371–377. doi: 10.1016/s0952-3278(98)90098-9. [DOI] [PubMed] [Google Scholar]
- 143.Soto-Guzman A, Navarro-Tito N, Castro-Sanchez L, et al. Oleic acid promotes MMP-9 secretion and invasion in breast cancer cells. Clinical & Experimental Metastasis. 2010;27(7):505–515. doi: 10.1007/s10585-010-9340-1. [DOI] [PubMed] [Google Scholar]
- 144.Navarro-Tito N, Soto-Guzman A, Castro-Sanchez L, et al. Oleic acid promotes migration on MDA-MB-231 breast cancer cells through an arachidonic acid-dependent pathway. The International Journal of Biochemistry & Cell Biology. 2010;42(2):306–317. doi: 10.1016/j.biocel.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 145.Hess D, Chisholm JW, Igal RA. Inhibition of stearoylCoA desaturase activity blocks cell cycle progression and induces programmed cell death in lung cancer cells. PLoS One. 2010;5(6):e11394. doi: 10.1371/journal.pone.0011394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Comba A, Maestri DM, Berra MA, et al. Effect of ω-3 and ω-9 fatty acid rich oils on lipoxygenases and cyclooxygenases enzymes and on the growth of a mammary adenocarcinoma model. Lipids in Health and Disease. 2010;9:112 –119. doi: 10.1186/1476-511X-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Muñoz SE, Piegari M, Guzmán CA, Eynard AR. Differential effects of dietary Oenothera, Zizyphus mistol, and corn oils, and essential fatty acid deficiency on the progression of a murine mammary gland adenocarcinoma. Nutrition. 1999;15(3):208–212. doi: 10.1016/s0899-9007(98)00181-6. [DOI] [PubMed] [Google Scholar]