

Abstract
In contrast to the F-type ATPases, which use a proton gradient to generate ATP, the V-type enzymes use ATP to actively transport protons into organelles and extracellular compartments. We describe here the structure of the H-subunit (also called Vma13p) of the yeast enzyme. This is the first structure of any component of a V-type ATPase. The H-subunit is not required for assembly but plays an essential regulatory role. Despite the lack of any apparent sequence homology the structure contains five motifs similar to the so-called HEAT or armadillo repeats seen in the importins. A groove, which is occupied in the importins by the peptide that targets proteins for import into the nucleus, is occupied here by the 10 amino-terminal residues of subunit H itself. The structural similarity suggests how subunit H may interact with the ATPase itself or with other proteins. A cleft between the amino- and carboxyl-terminal domains also suggests another possible site of interaction with other factors.
The vacuolar proton-translocating ATPases play an important role in the acidification of extracelluar compartments and organelles and are found in most eukaryotic cells. In contrast to the F-type ATPases, which generate ATP from a proton gradient across a membrane, the V-type enzymes use ATP to acidify compartments for receptor-mediated endocytosis, intracellular trafficking, and protein degradation (1, 2). The enzyme is composed of two functionally distinct complexes V1 and V0 (Fig. 1). V0 is integral to the membrane and is thought to consist of at least five different subunits with a total molecular mass of about 260 kDa. This complex is involved in translocation of protons across the membrane. The V1 complex is more hydrophilic and is composed of at least eight different subunits totaling a molecular mass of about 570 kDa. Because of the relatively high sequence identity between the catalytic subunits α and β of the F-type ATPases and subunits B and A of the V-type ATPases, the V1 complex is thought to use ATP to drive the proton translocation across the membrane. The rotary catalytic mechanism of ATP hydrolysis and proton transportation (3, 4) is thought to be very similar among the two classes of enzymes, but the different subunit stoichiometry and architectural appearance in electron micrographs suggests that the V-type ATPases are more complex (5, 6).
Figure 1.

Schematic overview of the V-type ATPases [adapted from Wilkens et al. (6)]. Subunit H, also named Vma13p, is highlighted in green and only one subunit is shown.
Because the V-type enzymes are involved in key biochemical processes, such as the regulation of pH, and also because they consume rather than generate energy, their function is likely to be under tight control. Previous studies have revealed that most components of the V-type ATPase are essential for assembly of the enzyme complex (1). Characterization of subunit H, also known as Vma13p, however, indicates that this polypeptide is essential for the activity of the enzyme but not for targeting or assembly (7).
The regulatory function of subunit H recently was demonstrated by Parra et al. (8). Binding of the subunit to isolated, cytosolic V1 particles inhibits CaATP hydrolysis. The amino acid sequence of subunit H consists of 478 residues and, other than subunits of other V-type ATPases, does not appear to be related to any other known protein. It also does not appear to contain membrane-spanning regions or repetitive sequence motifs. There is no known counterpart to subunit H in the F-type ATPases.
Materials and Methods
X-Ray Data Collection and Structure Determination.
Subunit H was purified from Escherichia coli cultures expressing the recombinant protein. The purified protein was found to fully reconstitute V-ATPase activity when added back to vacuolar membranes isolated from vma13Δ yeast mutants (unpublished work). Crystals were obtained as described (9) in space group P3221 with one molecule per asymmetric unit.
For heavy-atom labeling the crystals were soaked for 1–21 days in mother liquor consisting of 50 mM Tris (pH 7.0), 100 mM NaCl, 10 mM DTT, and 1 mM heavy atom.
Selenomethionine-derivatized protein was prepared as described (10). Because of the high solvent content of the crystals flash-freezing proved difficult. The best procedure was to take a crystal in the above mother liquor, briefly submerge it in paratone N, and then mount and flash-freeze it. Using this procedure, native, multiple isomorphous replacement, and multiple wavelength anomalous dispersion (MAD) data sets were collected at Stanford Synchrotron Research Laboratory beamlines 9–2 and 1–5. Data integration and reduction were performed with mosflm and scala (11). The intensities of the reflections were strong to about 6.0 Å and fell off rapidly at higher angles, explaining the relatively high Rsym (Table 1). The difficulty in flash-freezing, and the weakness in the higher-resolution diffraction data, can be attributed to the high solvent content of about 73% (VM = 4.4 Å3/Da) (12) and to solvent channels, 80 Å in diameter, extending through the crystal.
Table 1.
X-ray data collection, phase determination, and refinement statistics
| Single-wavelength data
|
MAD data [NAT(Se)]
|
|||||
|---|---|---|---|---|---|---|
| NAT1 | NAT2(Se) | Eur1(Se) | λ1 | λ2 | λ3 | |
| Source/beamline | SSRL/9-2 | SSRL/1-5 | SSRL/1-5 | SSRL/9-2 | SSRL/9-2 | SSRL/9-2 |
| Wavelength (Å) | 0.979 | 0.9796 | 0.9796 | 0.97938 | 0.97919 | 0.9640 |
| Resolution (Å) | 2.95 | 4.5 | 3.8 | 4.0 | 4.0 | 4.0 |
| Reflections | ||||||
| Total | 87,426 | 16,965 | 44,602 | 57,998 | 57,476 | 57,777 |
| Unique | 20,090 | 4,826 | 8,672 | 8,434 | 8,422 | 8,396 |
| Completeness (%) | 99.8 | 83.5 | 99.8 | 99.7 | 99.7 | 99.6 |
| I/σ(I) | 7.4 | 4.2 | 3.5 | 4.0 | 3.0 | 3.7 |
| Rsym (%) | 7.4 (45.4) | 13.7 (35.7) | 16.7 (39.8) | 14.2 (34.1) | 13.1 (37.5) | 14.7 (37.2) |
Rsym gives the agreement between independent measurements of equivalent reflections with the values for the outermost shell of data in parentheses. For single-wavelength data, unique reflections are to 3.8 Å. For MAD data, both total and unique reflections are up to 3.5 Å.
The best heavy-atom data set [Eur1(Se)] was obtained from a selenomethionine protein crystal soaked in 1 mM europium for 2 weeks. This, together with the isomorphous selenomethionine data set (NAT2), allowed the position of a single europium binding site to be immediately identified by visual inspection of the difference Patterson map. A second, minor, site was identified by calculating cross-Fourier-difference maps and was further confirmed by additional runs of shelxs (13). Scaling and initial phasing calculations were performed with scaleit and mlphare (14). The refined positions and occupancies of the two europium atoms were subsequently input into the program sharp (15), and the resulting phases were modified by solvent leveling using dm or solomon (14, 16). The resultant single isomorphous replacement electron density map based on the europium isomorphous replacement differences plus single-wavelength anomalous scattering data and calculated to 4.2-Å resolution revealed much of the overall fold of the molecule, although at this stage individual amino acids could not be identified.
By using the same europium-based single isomorphous replacement phases, four of the five expected selenium sites could be located by difference Fourier calculations. [Because of the poor quality of the diffraction data (Table 1), prior attempts to locate the selenium atoms by inspection of isomorphous or anomalous Patterson maps had failed, as did direct methods as well as the automated Patterson-search methods solve (17), snb, and cns (18).] The europium-based single isomorphous replacement phases, together with MAD phasing based on the selenium atoms, then was used to aid in the refinement of the selenium sites (15). The overall figure of merit was 0.48. The resultant electron density map clearly showed the arrangement of the α-helices and the topology of the structure (Fig. 2A).
Figure 2.

(A) Stereo ribbon diagram showing the overall structure of subunit H. The amino-terminal domain is highlighted in yellow and the C-terminal domain in green. The amino-terminal peptide including residues 2–10 is colored in red. This and other related figures were prepared with bobscript (41), molscript (42), and raster3d (43). (B) Stereo diagram showing the electron density map with experimental phases in the region between helices α7, α9, and α12. The resolution is 3.5 Å, and the map is contoured at 1.1 σ. (C) Electron density for residues 2–10. The coefficients are (2 Fo−Fc), phases are from the refined structure, the resolution is 2.9 Å, and the map is contoured at 1.2 σ. For clarity, only parts of the contacting helices α1, α8, and α11 are shown.
Using a higher-resolution native data set (NAT1, Table 1) and the best phases from the europium derivative and selenium MAD data, additional runs of phase extension, solvent flattening, and histogram matching from 5.0 Å to 3.5 Å were performed (14). The resulting map to 2.95-Å resolution (Fig. 2B) confirmed the correct hand by the appearance of the α-helices and allowed the identification of many of the large residues. During model building some maps were calculated with an artificial overall temperature factor of B = −70 Å2 to provide additional details.
Two possible binding sites for calcium ions can be inferred from the positions of the europium atoms used in the phasing process. The first is located close to Glu-34 and Asp-88, and the second site is formed by residue Glu-35 and by residue Glu-352 from a symmetry-related molecule (Fig. 2A). The first binding site would be expected to be retained in solution, although not necessarily the second.
Refinement.
Model building was performed by using o (19) and whatif (20). An initial polyalanine model (Rcryst = 45.0%) was subjected to several runs of torsion-angle molecular dynamics in cns (21, 22) in combination with bulk solvent correction, initial anisotropic B-value correction, and phase combination with the best experimental phase set. Refinement and map calculations were performed with weighted coefficients as defined by Read (23). The final refinement runs were performed with tnt (24, 25). Throughout the refinement an invariant set of reflections (7% of the total) was set aside to calculate Rfree.
The positions of the selenium atoms, the relative positions of proline and glycine residues, and the identification of the larger residues, especially within helices α14, α15, and α21, guided the building of the polypeptide chain. The positions of the two loops, coil1 (residues 52–71) and coil2 (residues 223–237), could not be identified in the initial maps but became partially visible after several rounds of torsion angle molecular dynamics of a preliminary model that included about 75% of the residues. Statistics for the current refined structure are given in Table 2. The high overall Wilson B-value for the native data (65 Å2) contributes to the high overall B-values of the model.
Table 2.
Phase determination and refinement statistics
| Phase determination statistics | |
| Resolution (Å) | 4.2 |
| Rcullis (%) | 46(Eur), 58(Se2) |
| Phasing power | 2.3(Eur), 1.4(Se2) |
| Figure of merit | 0.39(Se1), 0.41(Se2), 0.16(Eur) |
| Overall FOM | 0.48 |
| FOM after solvent flattening and phase extension | 0.72 |
| Refinement statistics | |
| Resolution (Å) | 2.95 |
| Rcryst/Rfree | 22.1/31.2 |
| Deviation from ideal geometry | |
| Bonds (Å) | 0.015 |
| Angles (°) | 2.0 |
FOM is the figure of merit; Rcryst and Rfree give the agreement between the observed structure factors and those calculated from the refined model. As verified by using the program procheck (45), 98.6% of the Ramachandran angles have “allowed” values and the remaining 1.4% are “generously allowed.”
Results
Overall Structure.
Subunit H has an elongated structure consisting of two distinct domains (Fig. 2A). The larger amino-terminal domain contains amino acids 2–352 and is primarily α-helical. Its most striking feature is a repetitive arrangement of α-helices.
Between residues Ala-52 and Leu-353 there are 17 consecutive α-helices that are arranged in a shallow, right-handed spiral or superhelix of six turns (Fig. 2A). Each turn of the superhelix is made up of three α-helices. In successive turns, individual α-helices are stacked, one on top of the other, at angles that vary from about 18° to 33°. The mode in which adjacent, stacked helices pack together is, in some respects, similar to that seen in leucine zippers. This also is reflected in the fact that the most abundant amino acids in the protein as a whole are leucine and isoleucine, with respective frequencies of 14.6% and 8.4%. Analysis of the amino acid sequence fails to detect any apparent repetition between residues within successive turns of the superhelix.
The individual α-helices that constitute the superhelical spiral vary considerably in length, as do the loops that connect them. Toward the beginning of the spiral, between helices α3 and α4, there is a region extending from residues 52 to 71 where the electron density is very weak, suggesting disorder or the presence of multiple conformations.
The superhelical structure results in a shallow groove that extends from one end of the amino-terminal domain to the other. The center of this groove is occupied by the extreme amino-terminal segment of the polypeptide chain including the first 10 residues, which adopt an extended conformation (Fig. 2 A and C). This amino-terminal peptide appears to be anchored in the groove predominantly by hydrophobic contacts with residues Ile-6 and Leu-7 (Fig. 2C). Following the amino-terminal peptide there is an α-helix (α1) plus an extended strand that connects to the start of the superhelix.
The second domain of subunit H consists of about 120 aa (residues 353–478) and includes eight α-helices (Fig. 2A). These helices form two turns of a right-handed superhelix, which is similar in overall topology to that seen in the amino-terminal domain although the arrangement of the individual helices is less regular.
The two domains of the protein, which are connected by a four-residue loop, do not seem to be tightly packed against each other, suggesting structural flexibility.
Structural Comparisons with Other Proteins.
A search for structurally similar proteins was performed with dali (26). Despite the apparent lack of any sequence homology, the algorithm identified structural similarity with the importins. These proteins, also called karyopherins, recognize proteins that bear a nuclear localization signal (NLS) and facilitate their transport from the cytosol into the nucleus (27, 28). Structurally, the importins are built up from multiple copies of so-called armadillo or HEAT motifs each of which is about 42 aa in length and consists of three α-helices. Together these α-helical motifs produce a superhelical structure with a shallow groove on one site of the protein that has been shown to be the binding site for the NLS signal peptide. In the present case, five turns of the superhelix within the amino-terminal domain of subunit H (residues 77–349) superimpose on the first five armadillo repeats of karyopherin α (residues 87–329) with a rms deviation of about 3 Å (Fig. 3) (27). The same superposition also shows that the NLS peptide of the karyopherin structure (PDB code 1EE4) occupies a position very similar to the extended amino-terminal residue of subunit H. Moreover a recent study indicates that the extended amino terminus of mammalian importin α (residues 44–54) occupies the same position in the groove thereby autoinhibiting the protein from binding to other peptides (28). This structure provides a regulatory switch that allows importin α to bind peptides with either high or low affinity.
Figure 3.
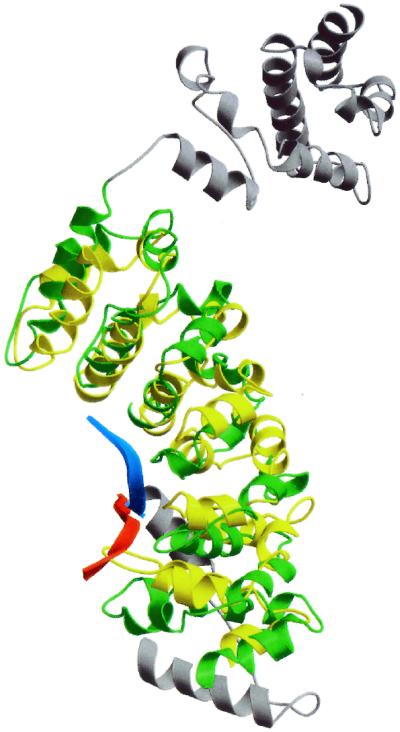
Superposition of subunit H (green) and residues 87–329 of karyopherin α (PDB code 1EE4) (yellow) using the overlay matrix determined by dali. The parts of subunit H that were not included in the superposition are colored gray. The amino-terminal peptide of subunit H (residues 2–10) is highlighted in red. The NLS peptide bound to karyopherin α is shown in blue.
Given the structural similarity with this class of proteins it suggests that subunit H may use a similar mechanism to bind other proteins. The crystal structure suggests that residues Ala-3, Ile-6, and Leu-7 of the amino-terminal peptide interact with the hydrophobic residues Leu-104, Phe-144, Val-185, and Leu-189 of helices α8 and α11 (Fig. 2c). Comparison with the sequences of the known mammalian homologs of subunit H (Fig. 4) reveals that Leu-104 is conserved, but not Phe-144, Val-185, or Leu-189.
Figure 4.

Hidden Markov sequence alignment of subunit H from yeast with related sequences from Homo sapiens, Drosophila melanogaster, Arabidopsis thaliana, and Caenorhabditis elegans. Sequence identification numbers from the National Center for Biotechnology Information are gi/6325293, gi/6563196, gi/7801655, gi/10728827, and gi/1086810. The secondary structure of yeast subunit H is shown below the alignment. The regions labeled coil1 and coil2 are disordered and could not be reliably determined. The sequences that form structural repeats are underlined in orange and are labeled repeat 1 through repeat 5. Residues in the alignment that are identical are shown in red boxes; those that are similar are shown in yellow boxes. The sequence highlighted in the pink box corresponds to the alternatively spliced sequence found in mammalian subunit H sequences. The figure was prepared with the program alscript (44).
Interactions of Subunit H with Other Subunits of the ATPase.
Subunit H of the bovine ATPase has been shown by crosslinking experiments to interact with subunits E and F (compare Fig. 1) (29). The corresponding subunits in the yeast complex (respectively Vma4p and Vma7p) are essential for assembly (30–32). None of these subunits is present in the F-type ATPases. Modification of Cys-254 of subunit A of the bovine ATPase by cystine causes subunit H to dissociate from the complex (29). It remains to be determined whether this is the result of steric interference at the interface between subunits A and H or a transmitted structural change. Landolt-Marticorena et al. (33) also demonstrated that subunit H forms a direct interaction with subunit a of the V0 domain in vitro and in vivo.
The fact that several different subunits can interact with subunit H suggests that there may be a number of binding sites on the surface of the protein. The analysis of the structure suggests two candidates. The first of these is the hydrophobic groove that binds the amino-terminal peptide. The second possible site is between the two domains. In this context, the experimentally and model-phased maps both show weak electron density that could correspond to a peptide up to 10 aa in length in the crevice between the amino-terminal and carboxyl-terminal domains. On the other hand, refinement of a model of this peptide resulted in low occupancy, and the coordinates therefore were not included in the final model.
Sequence Homology to Subunit H in Other Eukaryotic ATPases.
The identity between the amino acid sequence of yeast subunit H and the corresponding subunit in the mammalian ATPases is weak, ranging from about 16% (bovine subunit H) to 14% (human NBP-1). The sequence identity appears to be more pronounced for the carboxyl-terminal 280 residues (34) (Fig. 4).
Studies on the bovine subunit have shown that it is expressed in two different, alternatively spliced, versions named SFD-α and SFD-β. SFD-α contains an 18-aa insertion at position 175 (35). Similar differentially spliced versions of subunit H also have been identified for the porcine enzyme (36). Because the sequence correspondence within the first 200 aa is so poor, the location of the alternatively spliced insert region of the bovine and porcine sequences cannot be positioned unambiguously in the yeast structure. The alignment of Zhou et al. (35) placed the deletion at a position that corresponds to helix α11 of the yeast enzyme. This position, however, seemed inconsistent with the apparent importance of this helix in helping to maintain the overall fold. An attempt therefore was made to improve the accuracy of the alignment by using a hidden-Markov model (37) to align all possibly related ATPase subunits. This multiple-sequence alignment, a subset of which is shown in Fig. 4, positions the deletion at a site that corresponds to a loop between helices α10 and α11. In the bovine enzyme this loop presumably can be removed without interfering with the integrity of the protein. A second insertion of 9 aa was found at position 404, in agreement with the previous alignments (35). This region corresponds to the last turn of an α-helix plus six residues in a surface loop.
The highest degree of sequence conservation within the amino-terminal domain was observed for residues 283–291 of helix α17. These residues are exposed at the C-terminal end of the shallow groove, creating a positively charged patch on the surface of the groove (Fig. 5).
Figure 5.
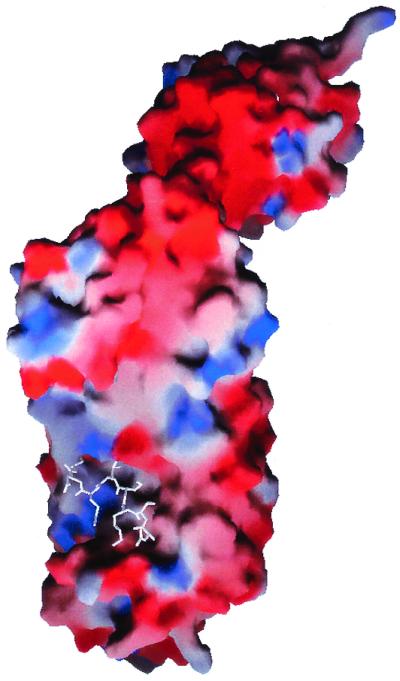
grasp representation (38) of the molecular surface of subunit H showing its electrostatic potential. The amino-terminal residues 2–10 were omitted from the calculation and are displayed in white. Negative surface charge is shown in red, positive in blue. An accumulation of negative charge occurs between the two domains of the protein.
Within the C-terminal domain the highest degree of sequence conservation is between residues 353 and 383. The aromatic side chains of residues Phe-356, Tyr-359, Trp-370, Phe-379, and Trp-380 form the hydrophobic core of the domain and are likely to be conserved for structural reasons. The main chain between residues 365 and 388 follows the overall superhelical topology of an armadillo repeat unit but differs substantially in detail. In particular, it does not appear to be stabilized by helix–helix packing interactions. Rather, the interactions of the aromatic residues noted above, together with a conserved salt bridge between residues His-374 and Asp-426 appear to be most important.
Discussion
The observation that recombinantly expressed subunit H can reactivate subunit H-depleted ATPase demonstrates the regulatory role of this subunit. The crystal structure of the subunit suggests that it may have multiple binding capabilities. The long elongated shape of the protein, the groove, the two-domain structure, and the cleft between the domains provide a variety of interfaces to interact with other subunits of the ATPase.
The most pronounced sequence similarity between yeast subunit H and the other V-type ATPase subunits is within the C-terminal half of the protein (34). This, together with the observation that an amino-terminal, 180-aa deletion of subunit H does not destroy ATPase activity (7) (unpublished work), suggests that the binding sites to other ATPase subunits may be located in the carboxyl-terminal half of the protein. This region includes a part of the amino-terminal domain and the C-terminal domain of the protein plus the cleft between them (Fig. 2A). An electrostatic potential surface representation (38) of this area shows an accumulation of negative charges on the surface of the cleft (Fig. 5). As mentioned, the two domains are only connected by a short 4-aa linker. The linker region could allow for structural flexibility between the two domains and could possibly function as an interface for subunit interactions to the ATPase via electrostatic interactions. The biochemical evidence, however, also suggests this area interacts with various other non-ATPase factors.
The similarity of subunit H to the importin family of proteins may provide a further structural basis for its regulatory function. Because the importins are target proteins that carry a NLS sequence, it raises the question of related function for subunit H. In the importin (or karyopherin) case the binding and dissociation of imported proteins is triggered by association with GTP-binding proteins (39). Two similar regulatory mechanisms seem plausible with subunit H. First, because the activity but not the assembly of the yeast V-ATPase depends on the presence of subunit H, the regulation of binding of this subunit to the ATPase could be triggered by a yet-to-be-identified cofactor. The regulation of the binding affinity to the ATPase could occur before or even while bound (40). Second, the amino-terminal peptide of subunit H could play an autoinhibitory role similar to the amino terminus in karyopherin α. Such an autoinhibitory function of subunit H would imply that the on and off states of the subunit are controlled by the amino-terminal peptide. Dissociation of the peptide would lead to an increase in binding affinity and vice versa. It therefore seems possible that the amino-terminal peptide also could bind to another subunit. It also might help form dimers of subunit H, which, in turn, could lead to an inhibited or activated state. At concentrations above 4 mg/ml the protein precipitates and forms oligomers at high concentrations of DTT (data not shown), supporting this hypothesis. On the other hand, in contrast to the NLS peptides, the amino-terminal peptide of subunit H does not appear to be conserved among the eukaryotic ATPases. Given this low sequence similarity of the yeast and mammalian subunits binding of the peptide within the groove may not be critical in regulating the ATPase activity in these organisms. It remains to be determined whether the shallow groove of the amino-terminal domain is also a binding site for other factors as the structural similarity to the karyopherin family suggests.
Interactions with Other Proteins.
Several non-ATPase proteins have been shown to interact specifically with subunit H. A human protein termed Nef-binding protein-1 (NBP-1) that shares 19% sequence identity to yeast subunit H coimmunoprecipitates with the Nef protein of the HIV-1 virus (34). The interaction has been confirmed by two-hybrid interaction assays. Expression of this 55-kDa protein can complement the loss of subunit H in yeast, suggesting that NBP-1 functions as subunit H of the human V-type ATPase.
Ynd1p is a yeast ectoapyrase that also has recently been identified to bind in vitro and in vivo to subunit H (40). The inhibition of the association of the two peptides in subunit H-depleted cells results in a dramatic increase of the apyrase activity. The studies suggest that subunit H is actively involved in the regulation of glycosylation reactions in the Golgi compartments. The interaction of subunit H with other non-ATPase proteins may suggest the involvement of proton pumping in other biochemical processes.
Acknowledgments
We thank Leslie Gay, Laurie Graham, and Andrew Flannery for expert technical assistance, Dr. Andy Hausrath for helpful discussions, and the staff at the Stanford Synchrotron Research Laboratory for invaluable help with data collection. This work was supported in part by National Institutes of Health Grants GM20066 (to B.W.M.) and GM38006 (to T.H.S.).
Abbreviations
- MAD
multiple wavelength anomalous dispersion
- NLS
nuclear localization signal
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID 1HO8).
References
- 1.Stevens T H, Forgac M. Annu Rev Cell Dev Biol. 1997;13:779–808. doi: 10.1146/annurev.cellbio.13.1.779. [DOI] [PubMed] [Google Scholar]
- 2.Forgac M. J Biol Chem. 1999;274:12951–12954. doi: 10.1074/jbc.274.19.12951. [DOI] [PubMed] [Google Scholar]
- 3.Abrahams J P, Leslie A G W, Lutter R, Walker J E. Nature (London) 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 4.Boyer P D. Annu Rev Biochem. 1997;66:717–749. doi: 10.1146/annurev.biochem.66.1.717. [DOI] [PubMed] [Google Scholar]
- 5.Boekema E J, van Breemen J F L, Brisson A, Ubbink-Kok T, Konings W N, Lolkema J S. Nature (London) 1999;401:37–38. doi: 10.1038/43369. [DOI] [PubMed] [Google Scholar]
- 6.Wilkens S, Vasilyeva E, Forgac M. J Biol Chem. 1999;273:31804–31810. doi: 10.1074/jbc.274.45.31804. [DOI] [PubMed] [Google Scholar]
- 7.Ho M N, Hirata R, Umemoto N, Ohya Y, Takatsuki A, Stevens T H, Anraku Y. J Biol Chem. 1993;268:18286–18292. [PubMed] [Google Scholar]
- 8.Parra K J, Keenan K L, Kane P M. J Biol Chem. 2000;275:21761–21767. doi: 10.1074/jbc.M002305200. [DOI] [PubMed] [Google Scholar]
- 9.Sagermann, M. & Matthews, B. W. (2000) Acta Crystallogr. D 56475–56477. [DOI] [PubMed]
- 10.Van Duyne G D, Standaert R F, Karplus P A, Schreiber S L, Clardy J. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 11.Leslie A G W. In: Crystallographic Computing 5, From Chemistry to Biology. Moras D, Podjarny A D, Thierry J C, editors. Oxford: Oxford Univ. Press; 1991. pp. 50–61. [Google Scholar]
- 12.Matthews B W. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 13.Sheldrick, G. M., Dauter, Z., Wilson, K. S., Hope, H. & Sieker, L. C. (1993) Acta Crystallogr. D 4918–4923. [DOI] [PubMed]
- 14.CCP4, Collaborative Computational Project Number 4 (1994) Acta Crystallogr. D 50760–50763.
- 15.de La Fortelle E D, Bricogne G. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 16.Abrahams, J. P. & Leslie, A. G. W. (1996) Acta Crystallogr. D 5230–5242.
- 17.Terwilliger, S. C. & Berendzen, J. (1999) Acta Crystallogr. D 55849–55861.
- 18.Grosse-Kunstleve, R. W. & Brunger, A. T. (1999) Acta Crystallogr. D 551568–551577.
- 19.Jones T A. Methods Enzymol. 1997;277:173–208. doi: 10.1016/s0076-6879(97)77012-5. [DOI] [PubMed] [Google Scholar]
- 20.Vriend G. J Mol Graphics. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 21.Adams P D, Pannu N S, Read R J, Brunger A T. Proc Natl Acad Sci USA. 1997;94:5018–5023. doi: 10.1073/pnas.94.10.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brunger A T, Rice L M. Methods Enzymol. 1997;277:243–269. doi: 10.1016/s0076-6879(97)77015-0. [DOI] [PubMed] [Google Scholar]
- 23.Read R J. Methods Enzymol. 1997;277:110–128. doi: 10.1016/s0076-6879(97)77009-5. [DOI] [PubMed] [Google Scholar]
- 24.Tronrud, D. E., Ten Eyck, L. F. & Matthews, B. W. (1987) Acta Crystallogr. A 43489–43503.
- 25.Tronrud D E. J Appl Crystallogr. 1996;29:100–104. [Google Scholar]
- 26.Holm L, Sander C. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 27.Peifer M, Berg S, Reynolds A B. Cell. 1994;76:789–790. doi: 10.1016/0092-8674(94)90353-0. [DOI] [PubMed] [Google Scholar]
- 28.Kobe B. Nat Struct Biol. 1999;6:388–397. doi: 10.1038/7625. [DOI] [PubMed] [Google Scholar]
- 29.Xu T, Vaselyeva E, Forgac M. J Biol Chem. 1999;274:28909–28915. doi: 10.1074/jbc.274.41.28909. [DOI] [PubMed] [Google Scholar]
- 30.Graham L A, Hill K J, Stevens T H. J Biol Chem. 1994;269:25974–25977. [PubMed] [Google Scholar]
- 31.Ho M N, Hill K J, Lindorfer M A, Stevens T H. J Biol Chem. 1992;268:221–227. [PubMed] [Google Scholar]
- 32.Graham L A, Stevens T H. J Bioenergetics Biomemb. 1999;31:39–47. doi: 10.1023/a:1005455411918. [DOI] [PubMed] [Google Scholar]
- 33.Landolt-Marticorena C, Williams K M, Correa J, Chen W, Manolson M F. J Biol Chem. 2000;275:15449–15457. doi: 10.1074/jbc.M000207200. [DOI] [PubMed] [Google Scholar]
- 34.Lu X, Yu H, Liu S H, Brodsky F M, Peterlin B M. Immunity. 1998;8:647–656. doi: 10.1016/s1074-7613(00)80569-5. [DOI] [PubMed] [Google Scholar]
- 35.Zhou Z, Peng S-B, Crider B P, Slaughter C, Xie X-S, Stone D K. J Biol Chem. 1998;273:5878–5884. doi: 10.1074/jbc.273.10.5878. [DOI] [PubMed] [Google Scholar]
- 36.Hui D, Deppe A, Wen G, Leeb T, Masabanda J, Robic A, Baumgartner B G, Fries R, Brenig B. Mamm Genome. 1999;10:266–270. doi: 10.1007/s003359900984. [DOI] [PubMed] [Google Scholar]
- 37.Karplus K, Barret C, Hughey R. Bioinformatics. 1998;14:846–856. doi: 10.1093/bioinformatics/14.10.846. [DOI] [PubMed] [Google Scholar]
- 38.Nicholls A, Sharp K A, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 39.Cingolani G, Petosa C, Weis K, Müller C W. Nature (London) 1999;399:221–229. doi: 10.1038/20367. [DOI] [PubMed] [Google Scholar]
- 40.Zhong X, Malhotra R, Guidotti G. J Biol Chem. 2000;275:35592–35599. doi: 10.1074/jbc.M006932200. [DOI] [PubMed] [Google Scholar]
- 41.Esnouf R M. J Mol Graphics. 1997;15:133–138. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 42.Kraulis P. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 43.Merrit, E. & Murphy, M. E. P. (1994) Acta Crystallogr. D 50869–50873.
- 44.Barton G J. Protein Eng. 1993;6:37–40. doi: 10.1093/protein/6.1.37. [DOI] [PubMed] [Google Scholar]
- 45.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]