

Abstract
Dihydropteroate synthase (DHPS) is the validated drug target for sulfonamide antimicrobial therapy. However, due to widespread drug resistance and poor tolerance, the use of the sulfonamide antibiotics is now limited. The pterin binding pocket in DHPS has a high degree of conservation and is distinct from the sulfonamide binding site, and therefore represents an attractive alternative target for the design of novel antibacterial agents. Previously, we have structurally characterized a known pyridazine inhibitor in the Bacillus anthracis DHPS pterin site and identified a number of unfavorable interactions that appear to compromise the binding. Using this structural information, a series of 4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazines was designed to improve binding affinity. Most importantly, the N-methyl ring substitution was removed to improve binding within the pterin pocket and the length of the side chain carboxylic acid was optimized to fully engage the pyrophosphate binding site. These inhibitors were synthesized and evaluated by an enzyme activity assay, X-ray crystallography, isothermal calorimetry, and surface plasmon resonance to obtain a comprehensive understanding of the binding interactions from structural, kinetic and thermodynamic perspectives. This study clearly demonstrates that compounds lacking the N-methyl substitution exhibit increased inhibition of DHPS, but the beneficial effects of optimizing the side chain length were less apparent.
Keywords: Heterocycles, Dihydropteroate synthase, Antimicrobial agents, Structure Based Drug design
Introduction
Since their discovery in the 1930’s, the sulfonamide class of drugs (sulfa drugs) has been widely used for the treatment of a broad spectrum of infectious diseases. [1, 2] These drugs target the essential folate pathway in micro-organisms, and combinations with dihydrofolate reductase (DHFR) inhibitors such as trimethoprim have proven to be highly effective in treating E. coli urinary infections, Pneumocystis carinii infections in immune-compromised patients and community acquired methicillin resistant Staphylococcus aureus (MRSA). [3, 4] Sulfa drugs target dihydropteroate synthase (DHPS), an enzyme encoded by the folP gene that acts at a key convergent point in folate biosynthesis [5], and mutations in the folP gene are associated with sulfa drug resistance. Resistance mutations have now been reported and characterized in many organisms, including drug resistant forms of Staphylococcus aureus and Pneumocystis carinii, and this phenomenon together with frequent severe side effects associated with allergenicity [6, 7] have become critical issues for the continued use of sulfa drugs as antimicrobials. Thus, to continue to take advantage of this valuable drug target, there is an urgent need for the development of alternatives to sulfa drugs that avoid resistance and overcome their poor tolerability.
To date, crystal structures of DHPS enzymes have been resolved from many microbial sources including complexes with substrate and product analogs.[8–15] The sulfa drugs mimic the substrate pABA and bind at the pABA-binding site that is comprised of two flexible loop regions where mutations that confer sulfa drug resistance can readily be accommodated.[11, 16] In contrast, the second substrate DHPP (Figure 1) binds in the pterin binding pocket, deep within the DHPS β-barrel where there is a high degree of conservation and in which sulfa drug resistance mutations have never been observed. The pterin binding site is therefore a very attractive alternative target for the design and development of novel antimicrobial agents, and we have been pursuing this goal.[17, 18]
Figure 1.

Structure of pyridazine inhibitor 1 and substrate DHPP.
One of the more potent pterin pocket targeted inhibitors that we have encountered is a pyridazine compound that was originally discovered by researchers at Burroughs Wellcome in the late 1970s.[19, 20] However, subsequent improvement was not possible due to the lack of structural information on the complex. We subsequently determined the crystal structure of Bacillus anthracis DHPS (BaDHPS) in complex with this molecule 1 (Figure 1) and confirmed that it does engage the pterin pocket.[21] The pyridazine pterin-like scaffold binds within the actual pocket as predicted and the carboxyl side chain partially occupies the anion pocket that normally accommodates the pyrophosphate moiety of the substrate.
The crystal structure revealed that the A ring of 1 engages the pterin pocket in a similar fashion to the actual pterin substrate DHPP with key interactions with conserved residues Asp184 and Asn120 (Figure 2a). [9, 21] Although the second B ring is more structurally divergent from the pterin substrate, the 5- carbonyl group of 1 mimics the hydrogen bond accepting N5-nitrogen of DHPP to form a strong hydrogen bond with the conserved Lys220. Elsewhere, the interactions are less ideal with two notable examples; the extra-cyclic carboxyl group attached to the pyridazine 6-position forms an apparently strained salt bridge with Arg254, and the methyl group at the 8-position prevents the N8-nitrogen from making a favorable hydrogen-bonding interaction with the conserved Asp101 as observed in the PtPP and DHPP complex structures.[9, 16] Using a ligand docking based approach to design and explore further analogs, we demonstrated that the removal of the N8-methyl and optimization of the C6 carboxylate side chain should facilitate a more optimal mode of binding that more accurately mimics the binding of DHPP.[13, 21] In the current study, we report the design and synthesis of new 2-aminopyrimido[4,5-c]pyridazines, and test whether they do indeed maximize these interactions and result in improved DHPS inhibitors.
Figure 2.

Crystal structures of complexes between BaDHPS and pyridazine-derived inhibitors. a) Details of the interactions of compound 1 with key residues within the pterin binding site of BaDHPS. [21] b–f) Similar representations for compounds 19 (b), 23 (c), 25 (d)*, 17 (e), and 24 (f). *25 (d) binds in two conformations within the BaDHPS dimer and these are superimposed in the figure.
Chemistry
In the first compound series the exo-cyclic C6 carboxylic acid side chain was systematically varied, which was chemically accessible via simple modifications of existing synthetic schemes.
The first molecule targeted was molecule 5 in which the carboxylate was placed directly adjacent to the ring system. Direct cyclization of 2-amino-6-chloropyrimidin-4(3H)-one 6 with diethyl 2-oxomalonate did not give the desired 4,5-dioxo pyridazine but instead produced the 3,5-dioxo analog. [20] Thus, compound 5 was obtained via an alternate route in which 2,4-diamino-6-chloropyrimidine 2 was treated with methylhydrazine in boiling water to give compound 3, which was condensed with diethyl 2-oxomalonate to give bicyclic compound 4. Simultaneous C4 deamination and saponification of 4 in hot aqueous sodium hydroxide followed by acidification afforded the target compound 5 (Scheme 1).
Scheme 1.

Reagent and conditions: a) methylhydrozine, water, reflux, 5 h, 27%; b) diethyl 2-oxomalonate, anhydrous methanol, reflux, 3 d, 44%; c) 1. 1 N NaOH, rt, overnight; 2, HCl, 47%.
In the next set of side chain variants, the carboxylate group was progressively extended away from the ring system by the addition of methyl branched or unbranched methylene spacers. These compounds 16–18 were obtained from 2-amino-6-chloropyrimidin-4(3H)-one 6 with the treatment of methylhydrazine in boiling water followed by condensation with diethyl appropriate α-keto ester to give 9–11, respectively. Saponification of 9–11 in aqueous sodium hydroxide followed by acidification afforded 16–18 (Scheme 2).[20]
Scheme 2.

Reagent and conditions: a) methylhydrozine, water, reflux, 5 h; or benzylhydrazine dihydrochloride, Et3N, water, reflux, overnight; b) water or methanol, reflux; c) 1. 1 N NaOH, rt, overnight; 2. HCl.
The synthesis of the N8 demethyl analogs proved to be more demanding. Initial attempts to directly demethylate the N8-position of 16 and 18 using β-(trimethylsilyl)ethyl chloroformate were unsuccessful.[22] We therefore adopted a benzyl protecting group strategy for N8. Benzylated 7-amino-1-benzylpyrimido[4,5-c]pyridazine-4,5(1H,6H)-diones 12–15 were synthesized from 6 with benzylhydrazine using a similar approach as described for 9–11.
Direct debenzylation and de-esterification of 13 was achieved by reaction with aluminum trichloride in boiling benzene (Scheme 3) to afford 19 in a moderate yield. However, attempts to extent this methodology to synthesize the other targeted analogs failed due to poor reaction yields and difficulty in separating product from aluminum salt byproducts.
Scheme 3.

Reagent and condition: AlCl3, toluene, 100 °C, 3 h
This led us to apply selective hydrogenolysis as an alternative strategy to perform debenzylation (Scheme 4).[23] Intermediates 12, 14, 15 were subject to debenzylation using formic acid and 10% palladium on carbon to give demethylated esters 20–22, [23] and these esters were subsequently saponified with aqueous sodium hydroxide to afford the target compounds 23–25, respectively.
Scheme 4.

Reagent and condition: a), HCOOH, 10% Pd/C; b) 1. 1 N NaOH, rt, overnight; 2. HCl
Enzyme inhibition studies
Compounds 5, 16–19, 22–25 were all tested for the inhibition of BaDHPS using a previously developed endpoint radiometric product detection assay (Table 1).[21] Compounds 16–18 and 5 directly probe the side chain moiety compared to the parent compound 1. The removal of the branched methyl side chain from 1 resulted in compound 16 with an approximately 50% poorer IC50 value, which mirrored the effect of methyl branch substitution in the N8 demethylated pair 19/23. Compounds 17/18 and 24/25 have an extra methylene sidechain spacer compared to parent compounds 1 and 19 respectively, which is seen to give rise to decreased inhibitory activity. In both the 17/18 and 24/25 pairs the inhibitors which were methyl branched (18 and 25) were weaker than the unbranched compounds 17 and 24, which is in contrast to the effects observed for the shorter chain branching pair of 1 and 16. The inhibitory activity of compound 5 in which the carboxylic acid is directly connected to the bicyclic ring dropped dramatically compared to the initial compound 1 and showed no detectable IC50 value. Thus, from this section of the study, the side chain substitution of 1 appears optimal.
Table 1.
Ba DHPS inhibitory activities of pyridazines
| # | Structure | Enzyme Inhibition Activity% @250μM | IC50 (μM) |
|---|---|---|---|
| 1 |
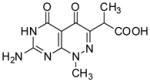
|
97.0 | 22 |
| 16 |
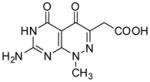
|
96.0 | 32 |
| 17 |
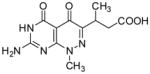
|
52.5 | 145 |
| 18 |
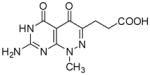
|
65.0 | ND |
| 5 |
|
32.4 | ND |
| 19 |
|
95.3 | 11 |
| 23 |
|
91.0 | 18 |
| 24 |
|
75.8 | 57 |
| 25 |
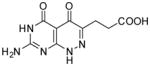
|
91.8 | 36 |
| 22 |
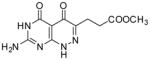
|
33.0 | 260 |
Compounds 19 and 23–25 probe the removal of the N8-methyl compared to 1 and 16–18, respectively. All of the demethylated compounds showed improved inhibition compared to their corresponding methylated analogs, and compounds 19 and 23 showed greater inhibition than the parent compound 1. Finally, to expand the SAR of the pyridazine analogs, a number of synthetic intermediates were also tested for inhibitory activity. The saponification products of N8-benzylated analogs 12–15 lacked significant enzyme inhibition (data not shown), consistent with the binding pocket at the N8-position being unable to accommodate a large substitution such as a benzyl group. The preference for a free terminal carboxylic acid was also confirmed by comparing the inhibitory activity of ester 22 with its free acid 25.
Thermodynamic and Kinetic Analyses
The binding affinities of the most potent derivative compounds compared to 1 were further examined with respect to thermodynamics and binding kinetics using isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR). Both techniques showed that all six compounds bind to BaDHPS at a 1:1 molar ratio with respect to the DHPS monomer (Table 2, see supplemental data figures S1–S3 for the specific ITC binding isotherms and SPR kinetic binding profiles). Comparing the N-methylated and demethylated pairs 1/19, both the ITC and SPR data showed an approximately two-fold increase in affinity upon demethylation. ITC reveals that the increase in free energy of binding of 19 over 1 results from an increased entropic contribution which over compensates for some loss to the enthalpy of binding,[24] With regard to the 16/23 pair, it was not possible to determine binding by ITC, but SPR shows a similar twofold level of improved binding upon removal of the N8-methyl group. ITC and SPR both show that increasing the side chain length in compounds 24 and 25 substantially reduces binding affinity, and SPR reveals that there is a pronounced and undesirable increase in the disassociation rate. A comparison between methyl substituted 24 and straight chain 25 analogs showed that the methyl substitution was overall disfavored but was overall more entropically favored but significantly enthapically penalized. Overall, the data from these experiments agree well with the results obtained from the enzyme assays (Table 1).
Table 2.
Summary of ITC and SPR binding data of the most potent inhibitors to BaDHPS
| # | Structure | ITC data | SPR data | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| KD (μM) | Na | ΔH | −TΔS | ΔG | ka (M−1 s−1) | kd (s−1) | KD (nM) | KD(eq) (nM) | ||
| (kcal/mol) | ||||||||||
| 1 |
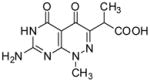
|
0.124 | 0.8 | −9.8 | 0.39 | −9.41 | 2.84 (1)× 105 | 7.95(4) × 10−2 | 280(2) | 267(2) |
| 16 |
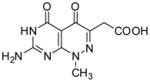
|
NDb | ND | ND | ND | ND | 3.42(3) × 105 | 6.94(6) × 10−2 | 203(2) | 190(2) |
| 19 |
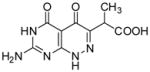
|
0.076 | 0.9 | −8.7 | −0.95 | −9.65 | 3.66(2) × 105 | 4.39(3) × 10−2 | 120(1) | 123(1) |
| 23 |
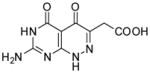
|
ND | ND | ND | ND | ND | 4.19(3) × 105 | 4.35(3) × 10−2 | 104(1) | 110(1) |
| 24 |
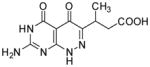
|
0.51 | 0.8 | −7.5 | −1.1 | −8.6 | 6.43(2) × 105 | 3.39(1) × 10−1 | 528(3) | 535(2) |
| 25 |
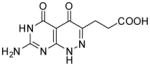
|
0.273 | 0.7 | −10.4 | 1.46 | −8.94 | 4.42(6) × 105 | 3.14(1) × 10−1 | 710(10) | 713(6) |
Stoichiometry of the interaction, experimental error is ±0.01; Number in parentheses is the standard error of last reported decimal place;
ND not determined
Structural Analyses
In the published crystal structure of the 1:DHPS complex, a network of H-bonding interactions between Asn120, Asp184, a structured water molecule (W1) and Lys220 anchors ring A to the pterin binding pocket in a similar fashion to PtPP and DHPP (Figure 2a; see supplementary data figure S5 for the complex between DHPP analogue—PtPP and BaDHPS).[9, 16] The B ring is bound via H-bonding interactions with Lys220 and a second structured water molecule (W2), a salt bridge between the acetate group of the small molecule and Arg254, van der Waals interactions between the methyl group of the linker and Phe189, and the methyl group at position 8 with the β carbon of Asp101 (Figure 2a).
We first structurally characterized the complex with 19 to directly observe the effect of removing the N8-methyl group (Figure 2b). The A ring is bound in the pterin pocket via an identical constellation of H-bonding interactions except that the H-bond between the 4-oxo group and Lys220 is missing. As predicted from the substrate analog complex,[21] the effect of removing the N8-methyl group from the B-ring is to allow the resulting NH group to now form a H-bond with the side chain of Asp101 which rotates to optimize this interaction. As regards the carboxylate side chain, its interactions with Arg254 within the anion binding pocket and with Phe189 via the branching methyl group are largely unchanged. A small but significant effect of removing the N8-methyl group is that the pterin-like ring structure rotates and moves to accommodate the new interaction with Asp101. Superimposing the 1 and 19 complexes with respect to the protein backbone shows this subtle movement and the rotation of Asp101 (Fig, 3a).
Figure 3.

Superimposed crystal structures of BaDHPS bound to compounds a) 1 (yellow) and 19(grey); b) 17 (yellow) and 19 (grey).
Compared to 19, compound 23 only lacks the methyl group in the carboxylate linker, and the crystal structures are unsurprisingly very similar (Figure 2c). The van der Waals interaction with Phe189 is missing and the 4-oxo group no longer interacts with the W1 structured water or Lys220. Compared to 23, compound 25 has one extra methylene group in the side chain linker, and the additional flexibility of the propionate group results in two conformations for this moiety in the two active sites of the crystallographic dimer (Figure 2d). The two conformations appear to be the result of alternate but equally accessible salt bridge interactions with Arg254 and Lys220. Note that each conformation does not affect interactions within the pterin pocket but does slightly impact the position of the pterin-like scaffold (Fig. 2d).
Compound 17 is identical to 1 but contains an extra methylene group in the side chain. The complex structures are almost identical except that the carboxylate group more intimately engages the anion pocket and Arg254 (Fig. 2e). Likewise, compound 24 is identical to 19 apart from the additional methylene group, and the structures are again very similar apart from the improved binding within the anion pocket (Fig. 2f). This effect of adding the extra methylene group was anticipated in the design of these compounds.[21] Note that in both complexes, although the carboxylate moiety interacts more favorably with the anion pocket, alternate hydrogen bonds to Ser218 and Lys220 are lost.
Discussion
Compound 1 was identified as a quite potent inhibitor of DHPS some 30 years ago in a study from Burroughs Wellcome designed to generate pterin-like antibacterial agents as alternatives to the pABA-like sulfa drugs.[19–21] Although the study generated impressive SAR information, it ultimately suffered from a lack of structural data. We addressed this problem some years ago and structurally characterized 1 in the pterin-binding pocket of DHPS from B. anthracis.[21] The structure suggested a number of possible ways in which 1 could potentially be further optimized, and we have reported here the results of this optimization. Specifically, we have designed and synthesized a series of 4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazines, and characterized their DHPS inhibitory properties using a direct enzyme assay, ITC and SPR measurements, and crystallography.
Two apparently non-ideal features of 1 were systematically analyzed, the length and branched nature of the carboxylate side chain at the 6 position of the two-ring scaffold, and the presence/absence of the methyl group at the N8 position. In the crystal structure of the compound 1 complex, the carboxylate group does not optimally fit into the anion pocket and the N8 methyl group prevents a hydrogen bonding interaction with Asp101. In addition, the methyl group on the carboxylate side chain of 1 was considered favorable due to a van der Waals interaction with the conserved Phe189 that creates part of the pterin-binding pocket. Based on our initial SAR analysis,[21] we would have expected that 24 would display the highest potency, but this is not the case. Although the crystal structure fully supports our prediction, the assay and the physical measurements consistently rank 24 below 1 in potency. Although this is a disappointing and unexpected result, a deeper analysis of the data does reveal possible explanations that will prove invaluable as we move forward with our goal of developing new DHPS-based lead compound.
Considering only derivatives with one methylene group in the carboxylate side chain, our SAR predictions are fully supported by the data. Thus, removing the N8 methyl group in going from 1 to 19 and from 16 to 23 results in a better inhibitor by all three metrics. Likewise, removing the side chain methyl group in going from 1 to 16 and 19 to 23 generates less potency. Consistent with these data, 16 that contains the N8 methyl and lacks the side chain methyl is the least potent inhibitor of the quartet. Inconsistencies appear when the carboxylate side chain is extended by one methylene group. Although the crystal structures clearly show that the extension allows more optimal docking within the anion pocket, this is not reflected by the potency values. Thus, in the pairings 1/17, 16/18, 19/24 and 23/25, the additional methylene group always reduces potency. In the context of the longer side chain, the beneficial effects of the branching methyl group are equivocal; in the 24/25 pairing, SPR suggests tighter binding whereas ITC and the assay suggests lower potency. However, it should be noted that the methyl group introduces chirality into the side chain, and measurements with these molecules involve both enantiomers whereas the crystal structures confirm that only one enantiomer (R) binds. Thus, the potency of the chiral compounds is likely to be higher than is apparent from the measurements. Another factor to consider in the loss of the methyl group from the extended side chain is the increased flexibility and the greater entropic penalty associated with its binding and consequent effects on structured water molecules near the site. Fig. 2d shows that 25 can indeed adopt two side chain conformations and the ITC data reveal that the binding of this compound is associated with the highest entropic penalty.
It should be noted that the side chain and the terminal carboxylate are clearly beneficial as can be seen by comparing 16 with 5 and 25 with 22, respectively. During the course of our studies, we also analyzed compound 21 which is the ester analog of compound 24. The crystal structure of the 21 complex (See supplementary data, Fig. S4) shows that the ester prevents interaction with Arg254 in the anion pocket, and this is reflected by significant decrease in inhibitory potency (21, IC50 376 μM).
The crystal structures reveal that removal of the N8 methyl group does allow the interaction with Asp101 as predicted, but the now smaller pyridazine scaffold moves to optimize this interaction. The precise positioning of the pyridazine scaffold and its interactions within the pterin pocket also appear to be modulated by the presence/absence of the side chain methyl group and the manner in which the terminal carboxylate engages the anion pocket. Although the moderate resolution of our complex structures (between 2.2 Å and 2.5 Å) precludes a detailed analysis, this movement and the subtle changes in the hydrogen bonding patterns are clearly apparent. These small changes are likely to have significant effects on binding affinity. The described molecules are essentially two linked fragments or pharmacophores that engage adjacent pterin and pyrophosphate binding pockets in DHPS, and the difficulty in connecting these units and capturing the maximum affinity gain is well documented because of subtle but important steric and conformational problems.[25]
Results from this study suggest that the demethylated pyridazine core is an optimized pterin mimic DHPS inhibitor, but the carboxylate side chain remains an area to be further optimized to generate higher affinity inhibitors. Our recent structural studies have shown that correct occupancy of the pyrophosphate pocket is required for stabilization of the outerloop structure of DHPS[16] and hence further effort is now ongoing to generate inhibitors that more fully contact this area.
Experimental section
Starting materials were purchased from commercial sources except dimethyl 3-methyl-2-oxopentanedioate and were used without further purification. Dimethyl 3-methyl-2-oxopentanedioate was synthesized as previous reported.[26] The reactions were monitored by thin layer chromatography (TLC) on pre-coated Merck 60 F254 silica gel plates and visualized by UV detection. The purity of final compounds was determined by UPLC/UV/ELSD/MS (see supplementary data for UPLC/UV/ELSD/MS method). All final compounds’ average UV and ELSD purity is > 95%.[27] Melting points were obtained on a Thomas Scientific Uni-melt capillary m.p. apparatus (Swedesboro, NJ) and were uncorrected. All 1H spectra were recorded on a Bruker ULTRASHIELD™ 400 plus. The chemical shift values were expressed in ppm (parts per million) relative to tetramethylsilane as internal standard; s = singlet, d = double, t = triplet, q = quartet, m = multiplet, bs = broad singlet. Coupling constants (J) are reported in hertz (Hz).
6-(1-Methylhydrazinyl)pyrimidine-2,4-diamine (3)
A stirred mixture of 4-chloro-2,6-diaminopyrimidine (0.694 g, 4.8 mmol) and methylhydrazine (0.553 g, 12 mmol) in methanol (30 mL) was heated under reflux under nitrogen for 18 h. After cooled overnight, the white solid was collected by filtration and dried to give 3 as a white solid (0.198 g, 27%): mp 215–217 °C; 1H NMR (400 MHz, DMSO-d6) δ 3.06 (s, 3 H), 4.33 (bs, 2 H), 5.37 (bs, 2 H), 5.41 (s, 1 H), 5.58 (bs, 2 H).
Methyl 5,7-diamino-1-methyl-4-oxo-1,4-dihydropyrimido[4,5-c]pyridazine-3-carboxylate (4)
A mixture of 3 (0.1 g, 0.649 mmol) and diethyl 2-oxomalonate (0.148 mL, 0.908 mmol) in anhydrous methanol (10 mL) was heated at reflux for 72 h under nitrogen. The solid was filtered out from the hot reaction mixture and washed with filtration to give 4 as a greenish solid (0.072 g, 44%): mp ~274 °C dec.; 1H NMR (400 MHz, DMSO-d6) δ3.78 (s, 3 H), 3.81 (s, 3 H), 7.12 (bd, 2 H), 7.98 (bs, 1 H), 8.82 (bs, 1 H).
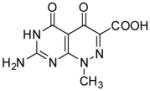
7-Amino-1-methyl-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazine-3-carboxylic acid (5)
A mixture of methyl 4 (0.25 g, 0.999 mmol) and 4N sodium hydroxide solution (12.5 mL) was stirred at reflux overnight. The white solid was filtered out and then dissolved in hot water. The resulted solution was acidified with diluted HCl to pH 5–6 and the precipitate was collected by filtration and dried to give 5 as a white solid (0.112 g, 47%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 3.89 (s, 3 H), 7.22 (bs, 1 H), 8.37 (bs, 1 H); HRMS m/z [M+H]+ calcd for C8H8N5O4: 238.0576, found: 238.0553.
2-Amino-6-(1-methylhydrazinyl)pyrimidin-4(3H)-one (7)
A stirred mixture of 2-amino-6-chloropyrimidin-4(3H)-one (1.75 g, 12mmol) and methylhydrazine (2.76 g, 60 mmol) in water (90 mL) was heated at reflux for 3 h and the resulting solution was allowed to stand at room temperature for 5 h before being cooled in freezer overnight. The precipitate was collected by filtration and dried under vacuum at 50 oC to give 7 as a white-off solid (1.37 g, 74%): mp ~275 °C dec.; 1H NMR (400 MHz, DMSO-d6) δ 3.11 (s, 3 H), 4.45 (bs, 2 H), 4.99 (s, 1 H), 6.17 (bs, 2 H), 9.84 (bs, 1 H).
2-Amino-6-(1-benzylhydrazinyl)pyrimidin-4(3H)-one (8)
A stirred mixture of 2-amino-6-chloropyrimidin-4(3H)-one (0.4 g, 2.75 mmol) and benzylhydrazine dihydrochloride (1.072 g, 5.50 mmol) along with triethylamine (2.857 mL, 20.61 mmol) in water (15 mL) was heated at reflux overnight. The reaction mixture was cooled to room temperature and the solid was filtered out and dried over P2O5 to give 8 as a off white solid (220 mg, 35%): mp ~280 °C dec.; 1H NMR (400 MHz, DMSO-d6) δ 4.36 (bs, 2 H), 4.86 (s, 2 H), 5.05 (s, 1 H), 6.22 (bs, 2 H), 7.19–7.33 (m, 5 H), 9.79 (bs, 1 H).
General method for the synthesis of 9–15
A mixture of 7 (or 8) and appropriate keto-ester in solvent (distiluted water expect 9 and 12 which were obtained from anhydrous methanol) was heated under reflux for 1.5–24 h. The resulting precipitate was collected by filtration from the hot mixture, washed with reaction solvent and dried under vacuum over P2O5 to give target compouns 9–11 (or 12–15).
Ethyl 2-(7-amino-1-methyl-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)acetate (9)
Compound 9 was obtained from 7 (0.50 g, 3.22 mmol) and diethyl 2-oxosuccinate (1.03 g, 0.904 ml, 5.48 mmol) following the general method described above after 3 h as a white solid (0.355 g, 39%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.18 (t, J = 7.2 Hz, 3 H), 3.47 (s, 2 H), 3.73 (s, 3 H), 4.06 (q, J = 7.2 Hz,2 H), 10.89 (bs, 1 H).
Methyl 3-(7-amino-1-methyl-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)butanoate (10)
Compound 10 was obtained from 7 (0.08 g, 0.43 mol) and dimethyl 3-methyl-2-oxopentanedioate (0.07 g, 0.43 mmol) following the general method described above after 3h as a light pink solid (0.03 g, 27%): mp >300 °C; 1NMR (400 MHz, DMSO-d6) δ 1.09 (d, J = 6.9 Hz, 3 H), 2.44 (dd, J = 15.9, 7.3 Hz, 1 H), 2.70 (dd, J = 15.9, 7.5 Hz, 1 H), 3.48 – 3.55 (m, 1 H), 3.56 (s, 3 H), 3.69 (s, 3 H), 10.80 (s, 1 H).
Methyl 3-(7-amino-1-methyl-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)propanoate (11)
Compound 11 was obtained from 7 (0.8 g, 5.16 mmol) and dimethyl 2-oxoglutarate (0.896 mL, 6.19 mmol) following the general method described above after 1.5 h as a yellow solid (0.71 g, 49%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.59 (t, J =7.6 Hz, 2 H), 2.57 (t, J = 7.6 Hz, 2 H), 3.59 (s, 3 H), 3.69 (s, 3 H), 10.81 (bs, 1 H).
Ethyl 2-(7-amino-1-benzyl-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)acetate (12)
Compound 12 was obtained from 8 (0.3 g, 1.297 mmol) and diethyl 2-oxosuccinate (0.642 mL, 3.89 mmol) following the general method described above after 2 d as a light pink solid (0.189 g, 41%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.15 (t, J = 7.2 Hz, 3 H), 3.49 (s, 2 H), 4.04 (q, J = 7.2 Hz, 2 H), 5.39 (s, 2 H), 7.26–7.35 (m, 5 H), 10.94 (bs, 1 H).
Ethyl 2-(7-amino-1-benzyl-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)propanoate (13)
Compound 13 was obtained from 8 (0.150 g, 0.649 mmol) and diethyl 2-methyl-3-oxosuccinate (0.240 mL, 1.297 mmol) following the general method described above after 3h as as a off white solid (0.185 g, 77%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.09 (t, J = 7.2 Hz, 3 H), 1.28 (d, J = 7.2 Hz, 3 H), 3.85 (t, J = 7.2 Hz, 2 H), 3.97–4.03 (m, 2 H), 5.38 (q, J = 14.8 Hz, 2 H), 7.26–7.36 (m, 5 H), 10.92 (bs, 1 H).
Methyl 3-(7-amino-1-benzyl-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)butanoate (14)
Compound 14 was obtained from 8 (0.29 g, 1.26 mol) and dimethyl 3-methyl-2-oxopentanedioate (0.26 g, 1.38 mmol) following the general method described above after 3h as a white solid (0.18 g, 39%): mp >300 °C; 1NMR (400 MHz, DMSO-d6) δ 1.08 (d, J = 6.9 Hz, 3 H), 2.34 (dd, J = 16.1, 7.0 Hz, 1 H), 2.42 – 2.48 (m, 1 H), 2.69 (m, 1 H), 3.45 (s, 3 H), 5.16 – 5.50 (m, 2 H), 7.19 – 7.42 (m, 5 H), 10.84 (s, 1 H), 12.02 (s, 1 H).
Methyl 3-(7-amino-1-benzyl-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)propanoate (15)
Compound 15 was obtained from 8 (0.3 g, 1.297 mmol) and dimethyl 2-oxopentanedioate (0.469 mL, 3.24 mmol) following the general method described above after 3 h as a off white solid (0.394 g, 85%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.60 (t, J = 7.2 Hz, 2 H), 2.78 (t, J = 7.2 Hz, 2 H), 3.49 (s, 3 H), 5.32 (s, 2 H), 7.26–7.35 (m, 5 H), 10.87 (bs, 1 H).
General method for the synthesis of 16–18
The suspension solution of compounds 9–11 in in THF (10 mL) and 1N sodium hydroxide solution (6 mL) was stirred at room temperature overnight. Solvent was removed by evaporation under vacuum to a small volue. The solution was neutralized with diluted HCl to pH 5–6. Fluffy solid was filtered out, dried over P2O5 to give target compounds 16–18.
2-(7-Amino-1-methyl-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)acetic acid (16)
Compound 16 was obtained from 9 (0.1 g, 0.358 mmol) following the general method described above as a white-off solid (0.065 g, 72%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 3.41 (s, 2 H), 3.73 (s, 3 H), 10.89 (s, 1 H), 12.36 (s, 1 H); HRMS m/z [M+H]+ calcd for C9H10N5O4: 252.0733, found: 252.0701.
3-(7-Amino-1-methyl-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)butanoic acid (17)
Compound 17 was obtained from 11 (0.035 g, 0.12 mmol) following the general method described above as a off-white solid (0.021g, 63%): mp ~295°C (dec.); 1H NMR (400 MHz, DMSO-d6) δ 1.08 (d, J = 6.9 Hz, 3 H), 2.33 (dd, J = 16.0, 7.4 Hz, 1 H), 2.64 (dd, J = 16.1, 7.4 Hz, 1 H), 3.50 (h, J = 7.1 Hz, 1 H), 3.70 (s, 3 H), 10.79 (s, 1 H), 12.01 (s, 1 H); HRMS m/z [M+H]+ calcd for C11H14N5O4: 280.1046, found: 280.1041.
3-(7-Amino-1-methyl-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)propanoic acid (18)
Compound 18 was obtained from 10 (0.3 g, 1.02 mmol) following the general method described above as a yellow solid (0.258 g, 95%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.52 (t, J = 7.6 Hz, 2 H), 2.78 (t, J = 7.6 Hz, 2 H), 3.49 (s, 3 H), 7.30 (bs, 2 H), 10.99 (bs, 1 H), 12.10 (bs, 1 H); HRMS m/z [M+H]+ calcd for C10H12N5O4: 266.0889, found: 266.0877.
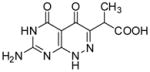
2-(7-Amino-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)propanoic acid (19)
A mixture of 13 (0.2 g, 0.541 mmol) and aluminum trichloride (0.289 g, 2.17 mmol) in anhydrous toluene (30 mL) was heated at reflux for 3 h. The solvent was decanted and the residue was dissolved in DMSO and purified through Waters Prep LC System with water (0.1% formic acid) and methanol (0.1% formic acid) as eluent to give 19 as a light yellow foam (0.035 g, 27%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.28 (d, J = 7.2 Hz, 3 H), 3.80 (q, J = 7.2Hz, 1 H), 7.08 (bs, 2 H), 10.76 (bs, 1 H), 12.22 (bs, 1 H),12.62 (bs, 1 H). HRMS m/z [M−H]− calcd for C9H8N5O4: 250.0576, found: 250.0595.
General method for the synthesis of 20–22
A solution of 12 (or 14–15) and 10% Pd/C 1:1 w/w) in formic acid (10 mL) was stirred at room temperature under nitrogen for 24 h. The reaction solution was filtered through celite pad and the filter cake was washed with warm formic acid (10 mL). The solvent was removed under reduced pressure to the minimum volume and the residue was purified through Waters PrepLC System with water (0.1% formic acid) and methanol (0.1% formic acid) as eluent to give compound 20 (or 21–22).
Ethyl 2-(7-amino-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)acetate (20)
Compound 20 was obtained from 12 (0.587 g, 1.652 mmol) following the general method described above as a white solid (0.07 g, 16%): mp >300°C; 1H NMR (400 MHz, DMSO-d6) δ 1.17 (t, J = 7.2 Hz, 3 H), 3.47 (s, 2 H), 4.06 (q, J = 7.2 Hz, 2 H), 7.15 (bs, 2 H), 10.89 (bs, 1 H), 12.70 (bs, 1 H).
Methyl 3-(7-amino-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)butanoate (21)
Compound 21 was obtained from 14 (0.11 g, 0.30 mmol) following the general method described above as a white solid (0.04 g, 48%): mp >300°C; 1H NMR (400 MHz, DMSO-d6) δ 1.08 (d, J = 6.9 Hz, 3 H), 2.44 (dd, J = 15.9, 7.1 Hz, 1 H), 2.71 (dd, J = 15.9, 7.7 Hz, 1 H), 3.54 (m, 1 H), 3.55 (s, 3 H), 7.02 (bs, 2 H), 10.70 (s, 1 H), 12.51 (s, 1 H).
Methyl 3-(7-amino-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)propanoate (22)
Compound 22 was obtained from 15 (0.3 g, 0.84 mmol)following the general method described above as a white solid (0.025 g, 11%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.61 (t, J = 6.8 Hz, 2 H), 2.76 (t, J = 6.8 Hz, 2 H), 3.58 (s, 3 H), 7.11 (bs, 2 H), 10.92 (bs, 1 H), 12.52 (bs, 1 H).
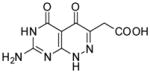
2-(7-Amino-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)acetic acid (23)
Compound 23 was obtained from 20 (0.025 g, 0.094 mmol) folowing the general method described for the synthesis of 16–18 as a white solid (0.02 g, 89%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6) 3.40 (s, 2 H), 7.15 (bs, 2 H), 10.74 (s, 1 H), 12.31 (bs, 1 H), 12.62 (s, 1 H); HRMS m/z [M+H]+ calcd for C8H8N5O4: 238.0576, found: 238.0553.
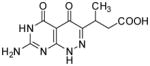
3-(7-Amino-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)butanoic acid (24)
Compound 24 was obtained from 21 (0.013 mg, 0.05 mmol) folowing the general method described for the synthesis of 16–18 as a white solid (0.005 mg, 41%): mp >300°C; 1H NMR (400 MHz, DMSO-d6) δ 1.07 (d, J = 6.9 Hz, 3 H), 2.33 (dd, J = 15.6, 7.1 Hz, 1 H), 2.63 (dd, J = 16.7, 7.7 Hz, 1 H), 3.50 (q, J = 7.0 Hz, 1 H), 10.65 (s, 1 H), 11.97 (s, 1 H), 12.49 (s, 1 H); HRMS m/z [M+H]+ calcd for C10H12N5O4: 266.0889, found: 266.0877.
3-(7-Amino-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)propanoic acid (25)
Compound 25 was obtained from 23 (0.02 g, 0.075 mmol) folowing the general method described for the synthesis of 16–18 as a white solid (0.012 g, 61%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6) 2.52 (t, J = 7.2 Hz, 2 H), 2.72 (t, J = 7.2 Hz, 2 H), 6.99 (bs, 2 H), 10.69 (s, 1 H), 12.06 (s, 1 H), 12.53 (s, 1 H); HRMS m/z [M+H]+ calcd for C9H11N5O4: 252.0733, found: 252.0701.
Enzyme assay
DHPS activity was measured in a 30 μL reaction containing 5 μM 14C pABA, 10 μM 6-hydroxymethyl-7,8-dihydropterin diphosphate, 10 mM magnesium chloride, 2% DMSO, 50 mM HEPES pH 7.6, and 10 ng DHPS.[28, 29] After 30 min incubation at 37 °C, the reactions were stopped by addition of 1 μL of 50 % acetic acid in an ice bath. The labeled product of the reaction, 14C dihydropteroate, was separated from 14C pABA by thin layer chromatography. Aliquots (15 μL) of the reaction mixture were spotted onto Polygram TLC plates (CEL 300 PEI) purchased from Macherey-Nagel and developed with ascending chromatography in 100 mM phosphate buffer pH 7.0. The plates were scanned using a Typhoon (GE Healthcare) and analyzed with ImageQuant TL. Inhibitor compounds were dissolved in DMSO, and inhibition was tested at 500 or 250 μM depending on solubility. The final concentration of DMSO in the reaction mixture was 2%. To determine the 50% inhibitory concentration (IC50) values, DHPS activities were measured in the presence of various concentrations of the compounds using the conditions described above but with 5 ng.
Crystallography
Compounds were dissolved in the crystallization mother liquor (1.45 M Li2SO4, 0.1 M Bis-Tris propane, pH 9.0) until saturated. Co-crystal structures of B. anthracis DHPS with compounds 17, 19, 21, 23, 24, and 25 were obtained by soaking the small molecules into pre-grown crystals, which were obtained as previously described.[9] After a 12-hour soaking period, the crystals were cryoprotected by a brief immersion in a mixture of 50% paratone-N: 50% mineral oil and flash frozen in liquid nitrogen. Diffraction data were collected at the SER-CAT 22-ID and 22-BM beam lines of the Advanced Photon Source and processed using HKL2000.[30] Structures were refined using refmac5, [31] CNS, [32] and Phenix, [33] and model building was performed using the programs Coot [34] and O. [35] Figures 2, 3 and 4 were rendered using PyMOL. [36] The atomic coordinates for the DHPS inhibitor bound structures have been deposited into the PDB data bank with ID codes: 23, 4DAI; 19, 4DAF; 17, 4D9P; 24, 4D8Z; 21, 4D8A; 25, 4DB7.
Isothermal Titration Calorimetry
The purified B. anthracis DHPS protein was dialyzed against 50 mM HEPES, 5mM MgCl2, pH 7.6. ITC titrations were performed in 5% DMSO, 40 mM HEPES, 4 mM MgCl2 at pH 7.6 and 25 °C. Nineteen injections of 2 μL each of 200 μM ligand solution were added to 203 μL of 20 μM protein solution. ITC titrations were performed on an Auto-iTC200 Isothermal Titration Calorimetry (MicroCal), and data was analyzed using MicroCal Origin 7.0 software using a one-site binding model.
SPR Methods
Binding studies were performed at 25 °C using a BIACORE T100 (GE Healthcare) surface plasmon resonance (SPR) instrument. 10xHis-Ba DHPS was immobilized on a nitrilotriacetic acid-derivatized carboxymethyldextran-coated gold surface (NTA Chip; GE Healthcare) to a level of ~5360 RU using the manufacturer’s protocol.
The kinetics of association and dissociation were monitored at a flow rate of 100 μl/min. Compounds were prepared in 20 mM Tris (pH 7.7), 150 mM NaCl, 5 mM MgCl2, 1 mM TCEP, 0.005% Tween20 and 5% DMSO as 3-fold serial dilutions decreasing from 3 μM to 37 nM for 1, 9 μM to 111 nM for 19, and 1 μM to 12.3 nM for 22. Each compound was injected in triplicate at each concentration. The data were processed, double-referenced, solvent corrected, and analyzed by kinetic and equilibrium affinity methods using the software package Scrubber2 (version 2.0b, BioLogic Software).
Supplementary Material
Table 3.
Crystallographic statistics of refinement*
| Parameter(unit) | Compound 17 | Compound 19 | Compound 21 | Compound 23 | Compound 24 | Compound 25 |
|---|---|---|---|---|---|---|
| Resolution range included (Å) | 87.0-2.2 | 30.0-2.5 | 35.6-2.2 | 30.0-2.5 | 29.0-2.2 | 30.0-2.4 |
| Rwork | 0.233 | 0.257 | 0.219 | 0.253 | 0.216 | 0.257 |
| Rfree | 0.270 | 0.286 | 0.259 | 0.282 | 0.259 | 0.283 |
see supplementary data table S2 and S3 for other crystallographic statistics of data collection and refinement.
Acknowledgments
Funding for this research was provided by National Institutes of Health grant AI070721, Cancer Center core grant CA21765, and the American Lebanese Syrian Associated Charities (ALSAC). We thank Matthew Frank from the Department of Infectious Diseases at St Jude Children’s Research Hospital for help with the radiometric enzyme inhibition assay. We also thank Jerrod Scarborough, Drs. Lei Yang and Bing Yan from Department of Chemical Biology and Therapeutics at St Jude Children’s Research Hospital for their help in analyzing and purification of final compounds, and Brett Waddell for assistance with the SPR experiments. Crystallography data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID and 22-BM beamlines at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
References
- 1.Miller AK. Proc Natl Acad Sci USA. 1944;57:151–153. [Google Scholar]
- 2.Woods DD. Br J Exp Path. 1940;21:74–90. [Google Scholar]
- 3.Skold O. Drug Resist Updates. 2000;3(3):155–160. doi: 10.1054/drup.2000.0146. [DOI] [PubMed] [Google Scholar]
- 4.Wiholm BE, Emanuelsson S. Eur J Haematol. 1996;57(S60):42–46. doi: 10.1111/j.1600-0609.1996.tb01644.x. [DOI] [PubMed] [Google Scholar]
- 5.Bermingham A, Derrick JP. BioEssays. 2002;24(7):637–648. doi: 10.1002/bies.10114. [DOI] [PubMed] [Google Scholar]
- 6.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Clin Infect Dis. 2009;48(1):1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 7.Dibbern DA, Jr, Montanaro A. Ann Allergy Asthma Immunol. 2008;100(2):91–101. doi: 10.1016/S1081-1206(10)60415-2. [DOI] [PubMed] [Google Scholar]
- 8.Achari A, Somers D, Champness JN, Bryant PK, Rosemond J, Stammers DK. Nat Struct Mol Biol. 1997;4(6):490–497. doi: 10.1038/nsb0697-490. [DOI] [PubMed] [Google Scholar]
- 9.Babaoglu K, Qi J, Lee RE, White SW. Structure. 2004;12(9):1705–1717. doi: 10.1016/j.str.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 10.Baca AM, Sirawaraporn R, Turley S, Sirawaraporn W, Hol WGJ. J Mol Biol. 2000;302(5):1193–1212. doi: 10.1006/jmbi.2000.4094. [DOI] [PubMed] [Google Scholar]
- 11.Haasum Y, Strom K, Wehelie R, Luna V, Roberts MC, Maskell JP, Hall LMC, Swedberg G. Antimicrob Agents Chemother. 2001;45(3):805–809. doi: 10.1128/AAC.45.3.805-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hampele IC, D’Arcy A, Dale GE, Kostrewa D, Nielsen J, Oefner C, Page MGP, Schonfeld HJ, Stuber D, Then RL. J Mol Biol. 1997;268(1):21–30. doi: 10.1006/jmbi.1997.0944. [DOI] [PubMed] [Google Scholar]
- 13.Hevener KE, Zhao W, Ball DM, Babaoglu K, Qi J, White SW, Lee RE. J Chem Inf Model. 2009;49(2):444–460. doi: 10.1021/ci800293n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawrence MC, Iliades P, Fernley RT, Berglez J, Pilling PA, Macreadie IG. J Mol Biol. 2005;348(3):655–670. doi: 10.1016/j.jmb.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 15.Levy C, Minnis D, Derrick JP. Biochem J. 2008;412(2):379–388. doi: 10.1042/BJ20071598. [DOI] [PubMed] [Google Scholar]
- 16.Yun M-K, Wu Y, Li Z, Zhao Y, Waddell MB, Ferreira AM, Lee RE, Bashford D, White SW. Science. 2012 doi: 10.1126/science.1214641. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Y, Hammoudeh D, Lin W, Das S, Yun MK, Li Z, Griffith E, Chen T, White SW, Lee RE. Bioconjugate Chem. 2011;22(10):2110–2117. doi: 10.1021/bc200346e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qi J, Virga KG, Das S, Zhao Y, Yun MK, White SW, Lee RE. Bioorg Med Chem. 2011;19(3):1298–1305. doi: 10.1016/j.bmc.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrison RW, Jr, Mallory WR, Styles VL. EP 0000383 (A1) Pyrimido[4,5-c]pyridazines, their use inpharmaceutical compositions, process and intermediates for their preparation. 1979
- 20.Morrison RW, Mallory WR, Styles VL. J Org Chem. 1978;43(25):4844–4849. [Google Scholar]
- 21.Hevener KE, Yun MK, Qi JJ, Kerr ID, Babaoglu K, Hurdle JG, Balakrishna K, White SW, Lee RE. J Med Chem. 2010;53(1):166–177. doi: 10.1021/jm900861d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell AL, Pilipauskas DR, Khanna IK, Rhodes RA. Tetrahedron Lett. 1987;28(21):2331–2334. [Google Scholar]
- 23.ElAmin B, Anantharamaiah GM, Royer GP, Means GE. J Org Chem. 1979;44(19):3442–3444. [Google Scholar]
- 24.Ladbury JE, Klebe G, Freire E. Nat Rev Drug Discov. 2010;9(1):23–27. doi: 10.1038/nrd3054. [DOI] [PubMed] [Google Scholar]
- 25.Hajduk PJ, Greer J. Nat Rev Drug Discov. 2007;6(3):211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 26.Milne C, Powell A, Jim J, Al Nakeeb M, Smith CP, Micklefield J. J Am Chem Soc. 2006;128(34):11250–11259. doi: 10.1021/ja062960c. [DOI] [PubMed] [Google Scholar]
- 27.Lemoff A, Yan B. J Comb Chem. 2008;10(5):746–751. doi: 10.1021/cc800100g. [DOI] [PubMed] [Google Scholar]
- 28.Aspinall TV, Joynson DHM, Guy E, Hyde JE, Sims PFG. J Infect Dis. 2002;185(11):1637–1643. doi: 10.1086/340577. [DOI] [PubMed] [Google Scholar]
- 29.Vinnicombe HG, Derrick JP. Biochem Biophys Res Commun. 1999;258(3):752–757. doi: 10.1006/bbrc.1999.0695. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W, Carter Charles W., Jr . Methods Enzymol. Vol. 276. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 31.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D Biol Crystallogr. 1997;53(3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 32.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 33.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr D Biol Crystallogr. 2010;66(2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60(12-1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Kleywegt GJ, Alwyn Jones T, Carter Charles WRMS., Jr . Methods Enzymol. Vol. 277. Academic Press; 1997. pp. 208–230. [DOI] [PubMed] [Google Scholar]
- 36.DeLano WL. In: The PyMOL molecular graphics system. Scientific D, editor. San Carlos, CA USA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.