

Over the last thirty years, substantial advances in stereoselective synthesis have enabled the preparation of exquisitely complex and therapeutically valuable polyketide-derived natural products.[1] While impressive, these accomplishments are typically composed of multistep sequences that are required to manage the reactivity of synthetic intermediates in the course of iterative chain growth (i.e. protecting/functional group manipulations and carbonyl redox chemistry). This is particularly evident in synthesis strategies for deoxypropionate-containing natural products (Figure 1). Reduced polypropionate architecture is found in natural products that possess a broad range of biological properties and, as such, has been the target of numerous studies in stereoselective synthesis.[2] A variety of iterative synthetic strategies to deoxypropionates have been described that can be characterized in one of two ways: (1) Those that forge single C–C bonds in the iterative chain elongation process (i.e. methods based on sequential enolate alkylation,[3] SN2’ chemistry,[4] and carbonyl olefination/asymmetric hydrogenation),[5] and (2) those that proceed by constructing 2 C–C bonds in the course of chain elongation (i.e. methods based on asymmetric carbometalation/oxidation/carbonyl olefination,[6] and stereoselective 1,4-addition/carbonyl olefination[7]).[8, 9] While these iterative synthesis strategies to deoxypropionates are powerful and stereochemically flexible, they uniformly suffer from the requirement of long sequences of chemical transformations to construct a complex deoxypropionate chain and, as such, suffer from poor step-economy (3–4 steps required for 2-carbon chain elongation).[10]
Figure 1.

Representative deoxypropionate-containing natural products.
Accepting the value of convergent strategies in chemical synthesis, and the impact that such processes have on synthesis efficiency, it is surprising that the general approaches to deoxypropionate synthesis remain largely dependent on synthetic methods that target iterative chain growth.[11] Here, we describe a chemical solution for the convergent synthesis of deoxypropionates that is enabled by metallacycle-mediated allylic alcohol–alkyne cross-coupling and stereoselective hydrogenation (Figure 2). Based on the stereoselective course of metallacycle-mediated coupling, and the established selectivity of hydroxyl-directed hydrogenation, a range of stereoisomeric deoxypropionate tetrads can be accessed with high step economy and stereoselectivity – albeit, only a subset of the stereoisomers possible can be prepared with this strategy. In addition to establishing the convergent method, we demonstrate the utility of this process in natural product synthesis by achieving an efficient synthesis of the C1–C11 subunit of borrelidin and completing a concise total synthesis of (–)-vittatalactone.
Figure 2.

A convergent strategy for deoxypropionate synthesis.
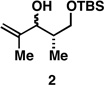
Recently, we described a stereoselective metallacycle-mediated reductive cross-coupling reaction between allylic alcohols and alkynes that proceeds with regio- and stereochemical control.[12, 13] With our sights set on securing the convergent approach to deoxypropionates illustrated in Figure 2, we initiated efforts to accomplish the allylic alcohol–alkyne coupling reaction depicted in Figure 3. While initial attempts with the homopropargylic alcohol substrate 1 were met with failure, when the free hydroxy group was masked as its corresponding methyl ether (4), Ti-mediated reductive cross-coupling with allylic alcohol 2 (1.5 equiv) delivered the stereodefined 1,4-diene product 5 in 73% yield as a single regio- and stereoisomer (ds ≥ 20:1).[14] Regio-and stereochemical control for this process is consistent with our previous studies and the empirical model depicted. In short, regioselective alkoxide directed carbometalation proceeds proximal to the Me-group and distal to the branched substitution of the Ti–alkyne complex, and stereochemical control derives from minimization of A-1,2 strain in a boat-like transition state. Subsequent syn-elimination of the fleeting stereodefined oxametallacyclobutane intermediate then delivers the (Z,E)-1,4-diene product 5. In this manner, stereoselective reductive cross-coupling ensues independent of the relative stereochemistry of the allylic alcohol (as depicted in the bottom portion of Figure 3).
Figure 3.

Reductive cross-coupling process.
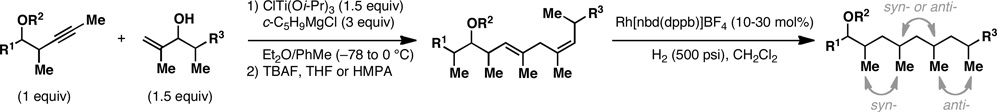
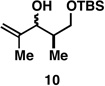
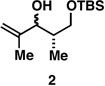
With what appeared to be a robust coupling reaction en route to polyunsaturated substrates bearing the appropriate Me-substitution pattern for the synthetic goal in mind, effort was focused on exploring the substrate scope of this process. Specifically, attention was focused on exploring substrates that contained varying stereochemistry and substitution patterns of relevance in natural product synthesis. As depicted in entries 1–3 of Table 1, substrates derived from propargylation (6 and 9)[15] and Grignard addition to a readily available chiral aldehyde (3-benzyl- or 3-silyloxy-2-methylpropanal; 2 and 10) were competent reaction partners for reductive cross-coupling. In each case the crude reaction mixture was advanced by subsequent deprotection (TBAF, HMPA, 35 °C) to deliver the stereodefined diols 7, 11, and 13 with similarly high levels of stereoselection (yields reported are for the combined two-step process).
Table 1.
Deoxypropionates by metallacycle-mediated alkyne–allylic alcohol cross-coupling and directed hydrogenation.
 | ||||||
|---|---|---|---|---|---|---|
| entry | alkyne | allylic alcohol |
combined yield for coupling/deprotection (%) |
1,4-diene | yield for conversion to deoxypropionate (%) |
deoxypropionate |
| 1 | 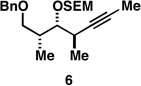 |
 |
50a | 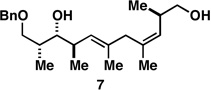 |
80c | 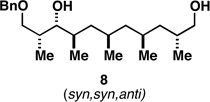 |
| 2 | 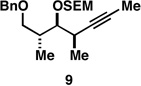 |
 |
60a | 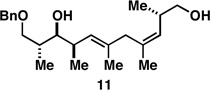 |
82c | 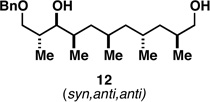 |
| 3 | 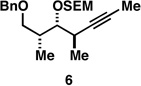 |
 |
50a | 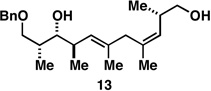 |
82c | 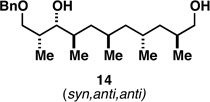 |
| 4 | 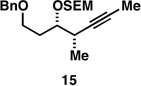 |
 |
56a | 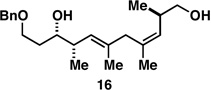 |
80c | 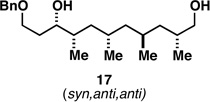 |
| 5 | 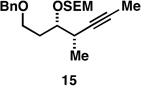 |
 |
62a | 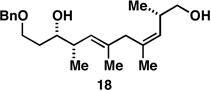 |
79c | 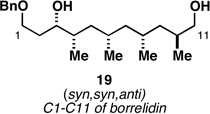 |
| 6 | 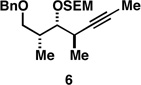 |
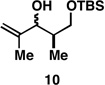 |
72b | 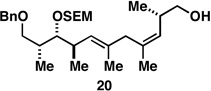 |
87c | 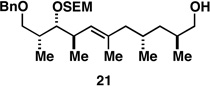 |
Reaction conditions:
For Ti-mediated reductive cross-coupling: ClTi(Oi-Pr)3, c-C5H9MgCl, PhMe, Et2O; For global desilylation: TBAF, HMPA, 35 °C.
Mono desilylation: TBAF, THF.
Rh[nbd(dppb)]BF4, CH2Cl2, H2 (500 psi) – reactions were typically run using 30 mol% of the Rh-catalyst; similar yields were observed when catalyst loading was reduced to 10 mol%. See Supporting Information for experimental details.
Moving on to stereoselective functionalization of the 1,4-diene products, we directed our attention to the use of hydroxyl-directed Rh-catalyzed hydrogenation.[5a, 16] As anticipated from the early reports of Evans, these hydroxyl-directed hydrogenation reactions [Rh[nbd(dppb)] BF4, CH2Cl2, H2 (500–700 psi] were uniformly successful and resulted in the formation of the stereodefined deoxypropionates 8, 12, and 14.[17]
As illustrated in entries 4 and 5, internal alkyne substrates bearing less branching were also viable substrates in this coupling process. Here the 1,4-diene-containing diol products 16 and 18 were produced in 56 and 62% yield over the two-step process, and subsequent hydroxyl-directed hydrogenation delivered the stereodefined deoxypropionates 17 and 19 in 80 and 79% yield, respectively. In the case of entry 5, this three-step process, consisting of Ti-mediated coupling, desilylation and directed hydrogenation, delivers the C1–C11 deoxypropionate core of the natural product borrelidin (19).[11a, 18]
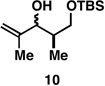
Finally, selective deprotection of 1,4-diene products derived from the reductive cross-coupling reaction leads to intermediates that can be functionalized in a site-selective manner. As depicted in entry 6, coupling of alkyne 6 with allylic alcohol 10, followed by selective removal of the primary TBS ether delivers the stereodefined 1,4-diene 20 in 72% overall yield. Such substrates are ideal for chemoselective hydroxyl-directed hydrogenation, in this case delivering the unsaturated deoxypropionate product 21 in 87% yield. While not investigated in the context of this study, the remaining alkene of 21 can be functionalized by a variety of well-established reactions to deliver more highly oxygenated products than those depicted in entries 1–5.
While our stereochemical assignments of the deoxypropionate products depicted in Table 1 were supported by application of Breit’s empirical model,[17] we selected to secure an alternative stereochemical proof by applying this process in natural product synthesis. Vittatalactone is a deoxypropionate natural product that was isolated in a search for sex pheromones specific to the cucumber beetle (Acylymma vittatum) – an insect that causes major damage to the cucurbit crops in North America (Figure 4A). Despite its relative structural simplicity, reported laboratory syntheses of this natural product proceed in up to 27 linear steps – a fact that supports the great difficulty/inefficiency associated with the synthesis of deoxypropionate targets.[19] While our convergent coupling chemistry is ideally suited for convergent union of larger/more complex coupling partners than would be required to prepare vittatalactone, our focus on this target was solely based on our desire to confirm the stereochemical assignments made previously in Table 1.
Figure 4.

Structure and laboratory synthesis of vittatalactone.
Reaction conditions: (a) ClTi(Oi-Pr)3, c-C5H9MgCl, PhMe, Et2O (−78 to 0 °C); (b) TBAF, HMPA; (c) Rh[nbd(dppb)]BF4, H2 (500 psi), CH2Cl2; (d) TsCl, pyridine, CH2Cl2; (e) LiEt3BH, THF; (f) Pd(OH)2, EtOH; (g) 4-methoxy-TEMPO, NaClO, CH2Cl2/H2O, then NaClO2, t-BuOH/H2O; (h) TsCl, pyridine.
As depicted in Figure 4B, our synthesis begins with titanium-mediated reductive cross-coupling of the stereodefined homopropargylic ether 9 (available from propargylation of 3-benzyloxy-2-methylpropanal) with allylic alcohol 2. Desilylation, and directed hydrogenation then delivers the deoxypropionate product 22 in 55% yield (over three steps). Site-selective tosylation and reduction furnishes 23 in 84% yield, and benzyl deprotection, site-selective oxidation to the acid, and lactonization delivers (–)-vittatalactone (57% yield over three steps).
In summary, a convergent route to deoxypropionates is described that proceeds by metallacycle-mediated cross-coupling and stereoselective hydrogenation. Overall, this approach has been demonstrated to be useful for a considerable subset of deoxypropionate stereoisomers, providing a solution to the assembly of such targets that is no longer limited to iterative C–C bond formation. While the stereochemical scope of the current study is constrained by the use of hydroxyl-directed hydrogenation, we anticipate that the identification of other methods for substrate- or catalyst-controlled hydrogenation of the complex stereodefined 1,4-diene intermediates will further increase the utility of this approach. From a broader perspective, the chemical pathway described lays a foundation to polyketide synthesis where alkene–alkyne coupling chemistry plays a central role.[20, 21] In addition to reporting the general aspects of this synthesis pathway, convergent and stereoselective syntheses of (–)-vittatalactone and the C1–C11 subunit of borrelidin are described. We look forward to future developments that emerge from these initial findings.
Supplementary Material
Acknowledgments
The authors acknowledge financial support by the National Institutes of Health –NIGMS (GM80266 and GM80266-04S1). The authors thank the Periana laboratory for assistance with all hydrogenation reactions, and Professor Kevin Burgess for helpful discussions in the pursuit of a catalyst-controlled stereoselective hydrogenation.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For recent reviews on the stereoselective synthesis of polyketides, see: Dalby SM, Paterson I. Curr. Opin. Drug. Disc. 2010;13:777–794. Paterson I. In: Asymmetric Synthesis (2nd edition) Christmann M, Braese S, editors. Weinheim, Germany: Wiley-VCH; 2008. pp. 293–298. Wang Y, Xing Y, Zhang Q, O’Doherty GA. Chem. Commun. 2011;47:8493–8505. doi: 10.1039/c1cc11791b. Morris JC, Phillips AJ. Nat. Prod. Rep. 2010;27:1186–1203. doi: 10.1039/b919366a. Schetter B, Mahrwald R. Angew. Chem. Int. Ed. 2006;45:7506–7525. doi: 10.1002/anie.200602780. Paterson I, Cowden CJ, Wallace DJ. In: Modern Carbonyl Chemistry. Otera J, editor. Weinheim, Germany: Wiley-VCH; 2001. pp. 249–297. Chemler SR, Roush WR. In: Modern Carbonyl Chemistry. Otera J, editor. Weinheim, Germany: Wiley-VCH; 2001. pp. 403–490.
- 2.For reviews on the stereoselective synthesis of deoxypropionates, see: Hanessian S, Giroux S, Mascitti, V V. Synthesis. 2006:1057–1076. ter Horst B, Feringa BL, Minnaard AJ. Chem. Commun. 2010;46:2535–2547. doi: 10.1039/b926265b.
- 3.a) Myers AG, Yang BH, Chen H, Kopecky DJ. Synlett. 1997:457–459. [Google Scholar]; b) Brand GJ, Studte C, Breit B. Org. Lett. 2009;11:4668–4670. doi: 10.1021/ol901944b. [DOI] [PubMed] [Google Scholar]
- 4.a) Breit B, Herber C. Angew. Chem. Int. Ed. 2004;43:3790–3792. doi: 10.1002/anie.200453990. [DOI] [PubMed] [Google Scholar]; b) Herber C, Breit B. Angew. Chem. Int. Ed. 2005;44:5267–5269. doi: 10.1002/anie.200501612. [DOI] [PubMed] [Google Scholar]; c) Herber C, Breit B. Chem. Eur. J. 2006;12:6684–6691. doi: 10.1002/chem.200600343. [DOI] [PubMed] [Google Scholar]; d) Herber C, Breit B. Eur. J. Org. Chem. 2007:3512–3519. [Google Scholar]; e) Schmidt Y, Lehr K, Breuninger U, Brand G, Reiss T, Breit B. J. Org. Chem. 2010;75:4424–4433. doi: 10.1021/jo100383u. [DOI] [PubMed] [Google Scholar]
- 5. Evans DA, Morrissey MM, Dow RL. Tetrahedron Lett. 1985;26:6005–6008. Attempts to accomplish catalyst-controlled asymmetric hydrogenation of the 1,4-diene products have been unsuccessful. For recent examples of related hydrogenations, see: Zhou J, Burgess K. Angew. Chem. Int. Ed. 2007;46:1129–1131. doi: 10.1002/anie.200603635. Zhou J, Ogle JW, Fan Y, Banphavichit(Bee) V, Zhu Y, Burgess K. Chem. Eur. J. 2007;13:7162–7170. doi: 10.1002/chem.200700390. Zhou J, Zhu Y, Burgess K. Org. Lett. 2007;9:1391–1393. doi: 10.1021/ol070298z.
- 6.a) Negishi E, Tan Z, Liang B, Novak T. Proc. Natl. Acad. Sci, U.S.A. 2004;101:5782–5787. doi: 10.1073/pnas.0307514101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tan Z, Negishi E. Angew. Chem. Int. Ed. 2004;43:2911–2914. doi: 10.1002/anie.200353429. [DOI] [PubMed] [Google Scholar]; c) Zhu G, Negishi E. Org. Lett. 2007;9:2771–2774. doi: 10.1021/ol0707259. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhu G, Liang B, Negishi E. Org. Lett. 2008;10:1099–1101. doi: 10.1021/ol703056u. [DOI] [PubMed] [Google Scholar]
- 7.a) Oppolzer W, Moretti R, Bernardinelli G. Tetrahedron Lett. 1986;27:4713–4716. [Google Scholar]; b) Hanessian S, Yang Y, Giroux S, Mascitti V, Ma J, Raeppel F. J. Am. Chem. Soc. 2003;125:13784–13792. doi: 10.1021/ja030139k. [DOI] [PubMed] [Google Scholar]; c) Hanessian S, Mascitti V, Giroux S. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11996–12001. doi: 10.1073/pnas.0401637101. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Des Mazery R, Pullez M, López F, Harutyunyan SR, Minnaard AJ, Feringa BL. J. Am. Chem. Soc. 2005;127:9966–9967. doi: 10.1021/ja053020f. [DOI] [PubMed] [Google Scholar]; e) Hanessian S, Chahal N, Giroux S. J. Org. Chem. 2006;71:7403–7411. doi: 10.1021/jo061098o. [DOI] [PubMed] [Google Scholar]; f) ter Horst B, Feringa BL, Minnaard AJ. Chem. Commun. 2007:489–491. doi: 10.1039/b612593j. [DOI] [PubMed] [Google Scholar]; g) ter Horst B, Feringa BL, Minnaard AJ. Org. Lett. 2007;9:3013–3015. doi: 10.1021/ol071078o. [DOI] [PubMed] [Google Scholar]; h) Lum T-K, Wang S-Y, Loh T-P. Org. Lett. 2008;10:761–764. doi: 10.1021/ol702715q. [DOI] [PubMed] [Google Scholar]; i) der Hartog T, van Dijken DJ, Minnard AJ, Feringa BL. Tetrahedron: Asym. 2010;21:1574–1584. [Google Scholar]; j) Wang S-Y, Loh T-P. Chem. Commun. 2010;46:8694–8703. doi: 10.1039/c0cc03211e. [DOI] [PubMed] [Google Scholar]
- 8.For other routes to deoxypropionates, see: Hanessian S, Thavonekham B, DeHoff B. J. Org. Chem. 1989;54:5831–5833. Marshall JA, Blough BE. J. Org. Chem. 1990;55:1540–1547. Hanessian S, Cooke NG, DeHoff B, Sakito Y. J. Am. Chem. Soc. 1990;112:5276–5290. Gambacorta A, Tofani D, Lupattelli P, Tafi A. Tetrahedron Lett. 2002;43:2195–2198. Lautens M, Colucci JT, Hiebert S, Smith ND, Bouchain G. Org. Lett. 2002;4:1879–1882. doi: 10.1021/ol025872f. Sugimura T, Sato Y, Im CY, Okuyama T. Org. Lett. 2004;6:4439–4442. doi: 10.1021/ol0483596. Cook MJ, Rovis T. J. Am. Chem. Soc. 2007;129:9302–9303. doi: 10.1021/ja073269s. Sugimura T, Im CY, Sato Y, Okuyama T. Tetrahedron. 2007;63:4027–4038. Cook MJ, Rovis T. J. Am. Chem. Soc. 2007;129:9302–9303. doi: 10.1021/ja073269s.
- 9.For a non-iterative approach to deoxypropionates of 7-carbon length based on stereoselective [3,3]-rearrangement, asymmetric hydrogenation, and diastereoselective alkylation, see ref. 19d.
- 10.Wender PA, Verma VA, Paxton TJ, Pillow TH. Acc. Chem. Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 11.For other solutions to convergent deoxypropionate synthesis in natural product synthesis, see: Duffey MO, LeTiran A, Morken JP. J. Am. Chem. Soc. 2003;125:1458–1459. doi: 10.1021/ja028941u. Shimp HL, Micalizio GC. Tetrahedron. 2009;65:5908–5915.
- 12. Kolundzic F, Micalizio GC. J. Am. Chem. Soc. 2007;129:15112–15113. doi: 10.1021/ja075678u. For a related reaction for the (Z)-selective coupling of 1,1-disubstituted allylic alcohols with vinylsilanes, see: Belardi JK, Micalizio GC. J. Am. Chem. Soc. 2008;130:16870–16872. doi: 10.1021/ja8074242. See also, Lysenko IL, Kim K, Lee HG, Cha JK. J. Am. Chem. Soc. 2008;130:15997–16002. doi: 10.1021/ja806440m.
- 13.a) Jeso V, Micalizio GC. J. Am. Chem. Soc. 2010;132:11422–11424. doi: 10.1021/ja104782u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Diez PS, Micalizio GC. J. Am Chem. Soc. 2010;132:9576–9578. doi: 10.1021/ja103836h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.1H NMR analysis of the purified product did not show evidence of a minor isomer. Due to the well-established difficulties associated with separation of simple alkene isomers by column chromatography, we are confident that this coupling reaction proceeds with high levels of stereocontrol (≥ 20:1). Related coupling reactions of 1,1-disubstituted alkenes have previously been shown to proceed with high levels of (Z)-selectivity (ref 12).
- 15. Bahadoor AB, Micalizio GC. J. Am. Chem. Soc. 2005;127:3694–3695. doi: 10.1021/ja050039+. For a recent review, see: Marshall JA. J. Org. Chem. 2007;72:8153–8166. doi: 10.1021/jo070787c.
- 16.Stork G, Kahne DE. J. Am. Chem. Soc. 1983;105:1072–1073. [Google Scholar]
- 17.1H and 13C spectra of the purified products from this sequence are provided in the Supporting Information. Hydroxyl-directed hydrogenation reactions of homoallylic alcohols of the nature explored here are well known to proceed with exquisite levels of stereoselection (see ref 5a). Also, the relative stereochemistry of these products was assigned by use of a recently established empirical model, and is consistent with that expected from hydroxyl-directed hydrogenation of isolated homoallylic alcohols of this nature: Schmidt Y, Breit B. Org. Lett. 2010;12:2218–2221. doi: 10.1021/ol1005399.
- 18.For routes to this subunit of borrelidin, and completed total syntheses, see: Hanessian S, Yang Y, Giroux S, Mascitti V, Ma J, Raeppel F. J. Am. Chem. Soc. 2003;125:13784–13792. doi: 10.1021/ja030139k. Vong BG, Abraham S, Xiang AX, Theodorakis EA. Org. Lett. 2003;5:1617–1620. doi: 10.1021/ol034243i. Vong BG, Kim SH, Abraham S, Theodorakis EA. Angew. Chem. Int. Ed. 2004;43:3947–3951. doi: 10.1002/anie.200460203. Nagamitsu T, Takano D, Fukuda T, Otoguro K, Kuwajima I, Harigaya Y, Omura S. Org. Lett. 2004;6:1865–1867. doi: 10.1021/ol049356w. e) Ref 4c. Yadav JS, Bezawada P, Chenna V. Tetrahedron Lett. 2009;50:3772–3775.
- 19.For total syntheses and lead references regarding vittatalactone, see: Schmidt Y, Breit B. Org. Lett. 2009;11:4767–4769. doi: 10.1021/ol901591t. b) Ref 4e. Yadav JS, Yadav NN, Rao TS, Reddy BVS, Al Ghamdi AAK. Eur. J. Org. Chem. 2011:4603–4608. Weise CF, Pischl M, Pfaltz A, Schneider C. Chem. Commun. 2011;47:3248–3250. doi: 10.1039/c0cc05215a.
- 20.For strategies to polyketide targets based on alkene–alkyne coupling (specifically, Alder-Ene chemistry), see: Trost BM, Amans D, Seganish WM, Chung CK. J. Am. Chem. Soc. 2009;131:17087–17089. doi: 10.1021/ja907924j. Trost BM, Gunzner JL. J. Am. Chem. Soc. 2001;123:9449–9450. doi: 10.1021/ja011424b. Trost BM, Probst GD, Schoop A. J. Am. Chem. Soc. 1998;120:9228–9236.
- 21.For a route to polyketides through terpene functionalization, see: Winter P, Vaxelaire C, Heinz C, Christmann C. Chem. Commun. 2011;47:394–396. doi: 10.1039/c0cc02332a.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.