

Abstract
Deletions of cryptococcal PIK1, RUB1, and ENA1 genes independently rendered defects in yeast survival in human CSF and within macrophages. We evaluated virulence potential of these genes by comparing wild-type Cryptococcus neoformans strain H99 with deletant and complement strains in a BALB/c mouse model of pulmonary infection. Survival of infected mice; pulmonary cryptococcal growth and pathology; immunological parameters; dissemination kinetics; and CNS pathology were examined. Deletion of each PIK1, RUB1, and ENA1 differentially reduced pulmonary growth and dissemination rates of C. neoformans and extended mice survival. Furthermore, pik1Δ induced similar pathologies to H99, however, with significantly delayed onset; rub1Δ was more efficiently contained within pulmonary macrophages and was further delayed in causing CNS dissemination/pathology; whereas ena1Δ was progressively eliminated from the lungs and did not induce pathological lesions or disseminate into the CNS. The diminished virulence of mutant strains was associated with differential modulation of pulmonary immune responses, including changes in leukocyte subsets, cytokine responses, and macrophage activation status. Compared to H99 infection, mutants induced more hallmarks of a protective Th1 immune response, rather than Th2, and more classical, rather than alternative, macrophage activation. The magnitude of immunological effects precisely corresponded to the level of virulence displayed by each strain. Thus, cryptococcal PIK1, RUB1, and ENA1 differentially contribute to cryptococcal virulence, in correlation with their differential capacity to modulate immune responses.
Cryptococcal infections are a major cause of meningoencephalitis-related deaths in immunocompromised hosts, but are also increasingly found in immunocompetent hosts. The successful clearance of Cryptococcus neoformans in the lungs and the prevention of systemic dissemination depend on the effector function of pulmonary CD4+ and CD8+ T cells and protective Th1 immune polarization, whereas the development of Th2 polarization is nonprotective.1–5 Our studies demonstrate that the overall balance between multiple Th cytokine responses in the C. neoformans–infected lungs translates into different spectra of macrophage activation.6–9 The M1-type macrophage activation, also referred to as classical activation, is driven by Th1 signals and is characterized by up-regulation of inducible nitric oxide synthase (iNOS)—an inducer of the fungicidal nitric oxide.6,9–12 The M2-type macrophage activation, also referred to as alternative activation, is driven by Th2 signals and is characterized by strong up-regulation of arginase (ARG1), which metabolizes arginine without yielding fungicidal nitric oxide.6,7,9–11 Thus, M1 macrophages serve as fungicidal effector cells, whereas M2 macrophages are prone to become parasitized by C. neoformans and harbor live/proliferating fungus.6,7,9–11 Apart from the distinct M1 and M2 phenotypes, intermediately activated macrophages that concurrently up-regulate Arg1 and iNOS were reported in the context of chronic cryptococcal infection, in which yeasts are contained, but not cleared, from lungs.6,7,9–11 Collectively, these studies underscore the role of macrophage activation status as a crucial determinant of clearance, persistence, or progression of C. neoformans infection.
Apart from the effects of host immune status, quantitative differences in the expression of multiple virulence factors define the ability of C. neoformans to persist in the infected host and to cause central nervous system (CNS) dissemination.13,14 Some of these factors have been shown to promote crucial steps in the pathogenesis of the yeast such as ability to grow in the lungs, disseminate from the lungs into other organs and tissues, and/or survive within the CNS.15–20 Although mechanisms of virulence for some factors have been at least partially clarified, and dozens of novel virulence factor candidate genes in C. neoformans have been identified, little is known about their role in the pathogenesis of cryptococcosis.13,16,17,21–23
To establish the role and/or mechanism of each potential virulence factor and facilitate the process of virulence evaluation, a variety of high-throughput methods has been introduced.14,24–27 These assays are important initial screening devices; however, it is not clear whether their outcomes translate into the global virulence in the infected host. On one hand, a correlation between the outcome of C. neoformans screening in macrophage co-culture assay and survival times of mice infected with different strains was reported.28–30 On the other hand, cryptococcal virulence attributes are also linked to their ability to inhibit T-cell responses and promote a nonprotective Th bias4,15,16,31,32 and/or to display high CNS tropism.16,19,33 Some of the latter mechanisms are likely to be unrelated to cryptococcal fitness in the macrophage co-culture and/or other simplified screening assays.
A recent global mutant screen study identified many cryptococcal genes with a possible role in cryptococcal growth and virulence.24 Three mutants with independent deletions of three cryptococcal genes encoding a cation ATPase transporter (ENA1), a ubiquitin-like protein (RUB1), and a phosphatidylinositol 4-kinase (PIK1) were selected for further studies; these mutants showed dramatically impaired survival in human cerebral spinal fluid (CSF) but retained the ability to grow in minimal media,24 suggesting virulence-specific defects, rather than simply impaired general viability. The mutants also demonstrated differentially decreased virulence in a rabbit model of meningitis and an invertebrate model of infection, and varied in fitness in a macrophage killing assay in vitro. Interestingly, the level of attenuation found for each mutant varied from assay to assay and/or at different time points. Thus, the role of these genes in pathogenesis, including primary lung infection, extrapulmonary dissemination, and eventual CNS colonization, needed to be evaluated in a relevant animal model.
The objective of the present study was to evaluate the effect of these mutations in the natural course of infection in a mouse model. We report that deletion of each of these cryptococcal genes differentially affected the course of pulmonary infection, extrapulmonary dissemination, and lung and brain pathology. These effects frequently deviated from the outcomes of simplified screening assays performed with these strains, but matched well with the changes in immunological processes we observed in the infected mice.
Materials and Methods
C. neoformans Strains
Deletant and complement strains were generated in J.R.P.'s laboratory from the wild-type C. neoformans H99 strain (ATCC 208821) as described previously.14,24–27 The null mutants had deleted: pik1Δ, a 1-phosphatidylinositol 4-kinase; rub1Δ, a ubiquitin-like protein; and ena1Δ, a P-type ATPase cation transporter. Reconstituted strains pik1Δ::PIK1, rub1Δ::RUB1, and ena1Δ::ENA1 represent each null mutant strain with an appropriate wild-type copy of PIK1, RUB1, or ENA1 reintroduced. Reconstitution was performed similarly to the previously described generation of the reconstituted ena1::ENA1 strain14,24–27 and confirmed by PCR, Southern hybridization, and growth in CSF. For infections, yeast were grown to stationary phase in Sabouraud Dextrose Broth (Difco, Detroit, MI) on a shaker for 72 hours at 37°C, washed twice with saline, counted on a hemocytometer, and then adjusted to the inoculum concentration.
Mice
All experimental procedures were approved by the VA and Duke University institutional animal care and use committees. Female wild-type BALB/c mice were obtained from Jackson Laboratories (Bar Harbor, ME). Mice were raised in specific-pathogen–free conditions in cages covered with a filter top and were fed sterile food/water ad libitum. Six- to 10-week-old mice were inoculated intranasally with 2 × 104 C. neoformans cells in a volume of 30 μL. Mice were humanely euthanized by CO2 inhalation.
Colony-Forming Unit Assay
Lung, brain, and spleen tissues were homogenized in 2 mL of sterile water. Serial 10-fold dilutions of the samples were plated in duplicates of 10-μL aliquots on Sabouraud Dextrose Agar and incubated at room temperature for 3 days. C. neoformans colonies were counted and expressed as colony-forming units per organ.
Lung Leukocyte Isolation
The lungs from each mouse were excised, washed in RPMI, dispersed using enzymatic digest procedure, and cells isolated as described previously.4–7,10,12 Leukocyte pellets were suspended in 5 mL of complete RPMI medium and enumerated on a hemocytometer following dilution in trypan blue (Sigma, St. Louis, MO).
Antibody Staining and Flow Cytometric Analysis
All staining reactions were performed according to the manufacturers' protocols. Data were collected on a FACS LSR II flow cytometer using FACSDiva software (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (Tree Star, San Carlos, CA). A minimum of 30,000 cells were analyzed per sample. Initial gates were set based on light-scatter characteristics and CD45 staining. Lymphocytes in the preselected CD45+ cell population were gated on forward and side scatter plots; small cells were selected by using anti-CD3 (for T cells), and subsets were identified using anti-CD8 and anti-CD4 antibodies.4,5 All antibody reagents were purchased from BioLegend (San Diego, CA).
Visual Identification of Leukocyte Populations
Macrophages, neutrophils, eosinophils, monocytes, and lymphocytes were counted in Wright-Giemsa–stained samples of lung cell suspensions cytospun onto glass slides. Samples were fixed, stained, and then counted under a microscope as described previously.4,5,7,10 The percentages of leukocyte subsets were multiplied by the total number of leukocytes to calculate absolute numbers in the sample.
Isolation of RNA from Pulmonary Leukocytes or Adherence-Enriched Pulmonary Macrophages
Isolated pulmonary leukocytes (10 × 106 cells/mL) were seeded in six-well plates and cultured at 37°C, 5% CO2 for 1.5 hours. Plates were washed twice using PBS to remove all nonadherent and loosely adherent cells. Total RNA was collected from fresh pulmonary leukocytes or adherent cells, and used for real-time reverse transcription and quantitative PCR analyses.7
Real-Time PCR
Total RNA was prepared using RNeasyPlus Mini Kit (Qiagen, Valencia, CA), and first-strand cDNA was synthesized using QuantiTect Reverse Transcription Kit (Qiagen) according to the manufacturer's instructions. Cytokine mRNA was quantified with SYBR Green–based detection using an MX 3000P system (Stratagene, La Jolla, CA) according to the manufacturer's protocols. Forty cycles of PCR (94°C for 15 seconds followed by 60°C for 30 seconds and 72°C for 30 seconds) were performed on a cDNA template. The primer sets used were previously published sequences.9,11,34–37 The mRNA levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels and relative expression shown as % of GAPDH.
Histological Analysis
Lungs were fixed by inflation with 1 mL of 10% neutral buffered formalin, excised, and then immersed in neutral buffered formalin. After paraffin embedding, 5-μm sections were cut and stained with mucicarmine with H&E counterstain. Sections were analyzed with light microscopy, and microphotographs were taken using Digital Microphotography system DFX1200 with ACT-1 software (Nikon, Tokyo, Japan).
Calculations and Statistics
All values are reported as means ± standard errors (SEM). Statistical significance was calculated using one-way analysis of variance whenever multiple groups were compared. For individual comparisons of multiple groups, Student-Newman-Keuls post hoc test was used to calculate P values. Survival study comparisons were performed using Kaplan-Meier analysis. Means with P values of <0.05 were considered significantly different. All statistical calculations were performed using Primer of Biostatistics.38
Results
Cryptococcal Genes PIK1, RUB1, and ENA1 Differentially Promote C. neoformans Virulence in a Mouse Model of Pulmonary Infection
Deletion of cryptococcal genes PIK1, RUB1, and ENA1 leads independently to dramatically reduced survival of the deletant strains in human CSF fluid.14,24–27 To determine whether independent deletion of these genes would render these strains avirulent in a murine model of pulmonary infection, BALB/c mice were infected with one of the following strains: null mutants pik1Δ, rub1Δ, and ena1Δ; complemented strains pik1Δ::PIK1, rub1Δ::RUB1, ena1Δ::ENA1; or the wild-type H99 via intranasal instillation of 2 × 104 cells. Survival of mice was assessed daily for up to 60 days postinfection (dpi).
Deletion of PIK1, RUB1, and ENA1 genes resulted in a significantly reduced virulence of the fungus and improved survival time of the infected mice (Figure 1). Compared to H99 infection with the median survival time of 22 dpi and 100% mouse mortality by 25 dpi, the pik1Δ mutant showed the least attenuated virulence (median survival time of 48 dpi and 90% mortality by 60 dpi, Figure 1A), rub1Δ showed intermediate attenuation (median survival time of 55.5 dpi and 60% mortality, Figure 1B), and ena1Δ showed the greatest attenuation with 0% mortality over 60 dpi (Figure 1C). Complementation of the deleted genes in each of the mutant strains virtually restored the original virulence to the level of the wild-type strain H99. The median survival time of mice infected with pik1Δ::PIK1, rub1Δ::RUB1, or ena1Δ::ENA1 was 28, 27, and 28 days, respectively, indicating that the attenuated virulence observed in each of the deletant strains was associated specifically with the deletion of the designated gene. Thus, cryptococcal expression of PIK1, RUB1, and ENA1 genes independently contribute to cryptococcal disease.
Figure 1.

Effect of cryptococcal PIK1, RUB1, and ENA1 deletions on mice survival following pulmonary C. neoformans infection. BALB/c mice were infected intranasally with 2 × 104 cells of three mutant strains, pik1Δ, rub1Δ, and ena1Δ, matching complement strains pik1Δ::PIK1, rub1Δ::RUB1, and ena1Δ::ENA1, or H99 wild-type strain. Mice were observed daily, and moribund animals were humanely euthanized and survival data recorded. Note improvement of survival time in the infected mice with pik1Δ showing the least attenuated virulence (A); rub1Δ showing intermediate virulence (B); and ena1Δ showing the most profound effect with no mortality over 60 days (C). All of the survival curves of complement strains are significantly different from their corresponding deletant strains, demonstrating that observed attenuation was gene specific for each mutant. The plots show data pulled from at least two independent experiments expressed as percentages of surviving mice out of n = 10 mice subjected to infection with each gene-deleted strain and H99, and n = 5 or above for the complements. *Significant difference in survival of mice infected with gene deletion mutant versus the wild type or the complement strain; †Significant difference in survival of mice infected with mutant strains compared to ena1Δ-infected mice.
Cryptococcal Genes PIK1, RUB1, and ENA1 Have a Differential Impact on Pulmonary Control of C. neoformans
To determine whether the improved survival of the mice was linked to decreased pulmonary growth of C. neoformans mutants, fungal burdens were evaluated at 3, 7, 14, 21 dpi, and postmortem. Consistent with the survival data, all deletant strains showed decreased pulmonary growth rate compared to the wild-type strain H99 (Figure 2, A–C); furthermore, the extent of pulmonary growth reduction corresponded to attenuation level of each mutant defined by the survival study. Compared to H99, pik1Δ, rub1Δ, and ena1Δ strains all showed significantly lower lung burden, beginning from day 3 to 21, with: pik1Δ showing slower but progressive growth and lung burden comparable to that of H99-infected mice postmortem (Figure 2A); rub1Δ showing even slower and stepwise growth dynamics with lower burden in mice that survived 60 dpi and comparable fungal burdens in mice that succumbed to the infection (Figure 2B); ena1Δ showing the most significant growth inhibition and a trend toward complete clearance from the infected lungs (Figure 2C). Thus, the effects of cryptococcal PIK1, RUB1, and ENA1 deletions on pulmonary growth mirrored their effects on the overall yeast virulence measured by host survival.
Figure 2.

Effect of cryptococcal PIK1, RUB1, and ENA1 deletions on pulmonary growth of C. neoformans. Lungs were harvested at days 3, 7, 14, and 21 postinfection with C. neoformans H99 (black bars in A, B, and C) or pik1Δ (A), rub1Δ (B), or ena1Δ (C) mutants, respectively. Panels B and C additionally show fungal burden in surviving mice at the conclusion of the survival study (day 60). Note that all strains showed significantly decreased fungal burden compared to H99 at all time points. Fungal burden of pik1Δ and rub1Δ were significantly higher than ena1Δ at days 14 and 21. Data are expressed as the mean colony forming units (CFU) per lung ± SEM; n = 5 mice and above per time point per group. Mutant-infected groups significantly different from *H99-, †ena1Δ-, and ‡rub1Δ-infected groups at the matching time points.
Cryptococcal PIK1, RUB1, and ENA1 Factors Differentially Modulate Pathology in the Lungs of Infected Mice
To evaluate how each of the cryptococcal genes of interest contributed to the development of lung pathology, histological analyses were performed at 21 dpi for H99-infected mice and around 60 dpi for infections with the mutants. C. neoformans H99 induced severe lung pathology, with organisms growing in virtually every area of the lungs and displacing lung tissue (cryptococcomas) (Figure 3, A and B). Inflammatory infiltrates occupied the margin of the infected areas, whereas the majority of cryptococci resided within the alveolar space unaccompanied by host inflammatory cells, a hallmark of uncontrolled growth. The lungs infected with pik1Δ and H99 showed similar pathology; however, pathology took a longer time to develop in pik1Δ-infected lungs (Figure 3, C and D), indicating that like H99, pik1Δ can escape host defenses, though with a substantial delay. The lesions in lungs infected with rub1Δ were more densely consolidated with numerous enlarged macrophages infiltrating alveolar spaces (Figure 3, E and F). Cryptococci were either contained within macrophages or surrounded by dense inflammatory infiltrates, suggesting that the host immune system recognized and attempted to control the rub1Δ mutant. However, many macrophages appeared to harbor live C. neoformans and/or were heavily laden with chitinase-like protein (YM1/2) crystals, indicating that they were alternatively or intermediately activated. These pathological features of lung macrophages resembled previously described pathologies found in chronic infection with cryptococcal persistence in the lungs and a progressing, inefficient inflammatory reaction.5,7,9
Figure 3.

Effect of cryptococcal PIK1, RUB1, and ENA1 deletions on development of pulmonary pathology in C. neoformans–infected mice. Lungs were collected at day 21 from H99-infected mice (A and B) and at day 60 from pik1Δ-infected (C and D), rub1Δ-infected (E and F), and ena1Δ-infected (G and H) mice and processed for histology (H&E and mucicarmine stains). The photographs were taken at ×10 and ×40 objective power. Note that lungs infected with H99 and pik1Δ show similar pathological lesions (A versus C at ×10 power, and B versus D at ×40 power), including wide-spread C. neoformans organisms in the lungs (black arrows) and tissue displacement by its rapid growth, and improved containment of the yeast within the macrophages in lungs infected with rub1Δ (E and F), including robust leukocyte infiltration (yellow arrows) with large macrophages. Note that these macrophages are heavily laden with YM1/2 crystals (red arrows) containing strongly stained fungal cells, characteristics of C. neoformans harbored in alternatively activated macrophages. Note the absence of a severe pathology or fungus in lungs infected with ena1Δ (G and H) and focused mononuclear cell infiltrates consistent with a protective immune response to C. neoformans.
In contrast with the severe lung pathologies induced by pik1Δ and rub1Δ infections, ena1Δ infection produced histological findings consistent with a protective immune response. The majority of lungs were almost free of inflammation, with only occasional inflammatory foci represented by tight mononuclear cell infiltrates and no discernible yeasts, consistent with resolution of pneumonia (Figure 3, G and H). Collectively, the pathological analysis of infected lungs revealed that the type of lung pathology that developed in response to each of the mutant strains corresponded with the level of virulence as measured by yeast growth and survival.
Cryptococcal Genes PIK1, RUB1, and ENA1 Have a Differential Impact on Pulmonary Leukocyte Recruitment in C. neoformans–Infected Lungs
Next, we characterized the compositions of the inflammatory responses to determine whether they correlate with differences in the lung pathology and mutant persistence. Flow cytometry and differential microscopic counts were used to evaluate leukocyte populations in the lungs at 21 dpi.
Flow cytometric analysis revealed significantly greater numbers of leukocytes (CD45+ cells) in the lungs of H99-infected mice compared to lungs infected with the mutants (Figure 4A). However, all mice infected with H99 and mutant strains showed similar numbers of CD4+ T cells (Figure 4B). Furthermore, significantly greater numbers of CD8+ T cells accumulated in the lungs infected with the mutants compared to H99-infected lungs (Figure 4C). Thus, deletions of PIK1, RUB1, and ENA1 resulted in alleviated inflammatory responses without reduced T-cell numbers, essential for anti-cryptococcal protection.1,10,39,40 Improved control of the mutant strains compared to H99 could be further explained by greater CD8+ T-cell numbers; however, variations in pulmonary control of pik1Δ, rub1Δ, and ena1Δ were not due to the differential CD8+ T-cell recruitment.
Figure 4.
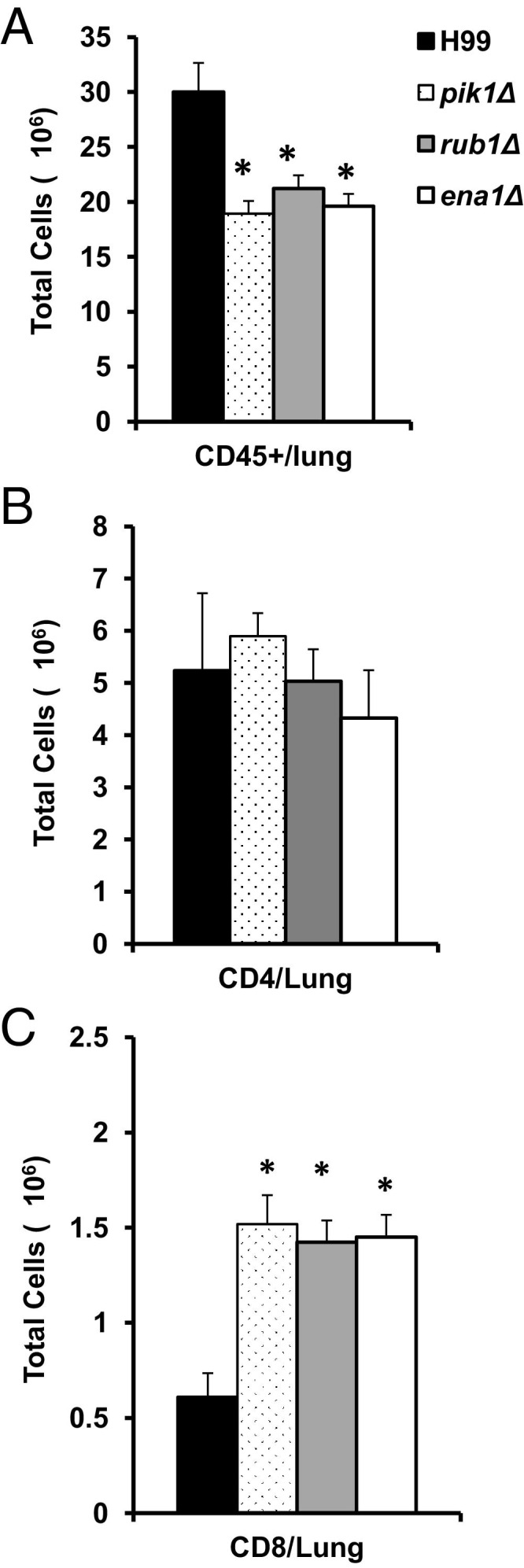
Effect of cryptococcal PIK1, RUB1, and ENA1 deletions on CD45+ cells and T lymphocyte recruitment in the lungs. Leukocytes were isolated from the lungs of H99-, pik1Δ-, rub1Δ-, or ena1Δ-infected mice at week 3 postinfection and then stained with specific antibodies. A: APC-labeled anti-CD45 was used for leukocytes staining. B and C: The lymphocytes in the preselected CD45+ cell population were gated on forward and side scatter plots, and the CD3+ T cells were selected (Percp-Cy5.5); CD4+ and CD8+ subsets were identified using fluorescein isothiocyanate–labeled anti-CD8 (for Tc cells) or anti-CD4 antibodies (for Th cells) and calculated to express a total number per lung. Note that pik1Δ-, rub1Δ-, or ena1Δ-infected mice had less inflammation in the lungs compared to the H99 group; however, they show increased CD8+ T-cell recruitment and equally robust CD4+ T-cell recruitment to the infected lungs at week 3. n = 4 or more mice per group. *Significantly different from H99-infected group.
Among myeloid subsets, the lowest percentage of lung mononuclear phagocytes and the highest percentages of eosinophils (a Th2 response marker) and neutrophils were observed in H99-infected mice compared to pik1Δ-, rub1Δ-, or ena1Δ-infected mice (Figure 5). Intermediate numbers of eosinophils and neutrophils were found in lungs infected with pik1Δ or rub1Δ, whereas ena1Δ-infected mice had the lowest numbers. Collectively, these data imply that anti-cryptococcal immune responses in the context of PIK1, RUB1, or ENA1 deletion were increasingly more protective, as illustrated by decreasing numbers of eosinophils and improved recruitment of CD8+ T cells.
Figure 5.

Effect of cryptococcal PIK1, RUB1, and ENA1 deletions on the recruitment of myeloid leukocyte subsets to C. neoformans–infected lungs. Leukocytes were isolated from the lungs of H99-, pik1Δ-, rub1Δ-, or ena1Δ-infected mice at week 3. Frequencies of each subset (left) were determined by microscopic enumeration of cytospun leukocyte isolates and calculated times the total CD45+ cells per lung to evaluate total number (right). Note that lungs infected with mutant strains had a significantly higher percentage of mononuclear phagocytes and lower percentages of neutrophils and eosinophils compared to H99-infected mice. All strains induced an equivalent recruitment of mononuclear cells but showed decreasing total numbers of neutrophils and eosinophils in order from highest to lowest virulence. n = 4 mice or more per group; mutant-infected groups significantly different from *H99-, †ena1Δ-, and ‡rub1Δ-infected groups. Mono & Mac, mononuclear cells and macrophages.
Cryptococcal Genes PIK1, RUB1, and ENA1 Have a Differential Impact on Cytokine Responses during Pulmonary Infection
We next evaluated the effect of cryptococcal PIK1, RUB1, and ENA1 deletions on pulmonary cytokine production. We assessed the cytokine mRNA expression patterns in leukocytes isolated from lungs infected with H99 or each of the mutant strains. In general, mice infected with deletant strains still showed relatively high cytokine levels, indicating an ongoing immune response in the infected lungs. Consistent with improved pulmonary control of C. neoformans, mutant strains elicited significantly lower expression of nonprotectiveTh2 cytokines, IL-4 and IL-13, compared to H99-infected mice (Figure 6). Expression of the Th1-driving IFN-γ tended to be elevated in leukocytes from lungs infected with the mutant strains compared to H99; however, this increase was significant in only the rub1Δ group. Since, the antigen levels differed among the groups, Th1/Th2 bias was normalized by calculating the IL-4/IFN-γ expression ratios for each infected group. Consistent with the least protective response in H99 infection, we observed a strong Th2 bias (greatest IL-4/IFN-γ ratio). The cytokine ratio in pik1Δ-infected lungs (the least attenuated) stayed close to a “neutral bias,” whereas lungs infected with rub1Δ and ena1Δ showed a robust shift toward a protective, Th1-biased cytokine profile (lower IL-4/IFN-γ ratio). In addition, lungs infected with mutant strains expressed significantly higher levels of protective, Th1-type cytokine TNF-α compared to H99-infected lungs, with most pronounced increase in the ena1Δ group, correlating with the most effective clearance of the fungus in these mice compared to the other groups.
Figure 6.

Effect of cryptococcal PIK1, RUB1, and ENA1 deletion on cytokine expression by pulmonary leukocytes from C. neoformans–infected mice. RNA was isolated from pulmonary leukocytes at week 3 postinfection, converted to cDNA, and analyzed by quantitative PCR for the expression of “Th polarizing” cytokines. Cytokine mRNA expression level was normalized using a housekeeping gene (GAPDH) and expressed as percent (relative expression). Note a highly Th2-biased phenotype in the H99-infected group, a Th1-biased cytokine profile in rub1Δ- and ena1Δ-infected groups, and pik1Δ showing mixed cytokine phenotype. n = 5 of infected mice for each of the strains; mutant-infected groups significantly different from *H99, †ena1Δ, and ‡rub1Δ groups.
The Th17 response has been shown to contribute to anticryptococcal host defenses; however, IL-17 expression levels did not vary between H99 infection and those infections with our mutant strains. Collectively, the cytokine analysis demonstrates that expression of each of the cryptococcal factors PIK1, RUB1, and ENA1 contributes to the development of a nonprotective Th2 response while opposing the protective Th1.
Cryptococcal Genes PIK1, RUB1, and ENA1 Have a Differential Impact on Pulmonary Macrophage Activation Status
Macrophage activation is a critical functional step in clearance of C. neoformans. To assess the M1/M2 macrophage polarization status, we quantified the mRNA expression of the M1 hallmark gene iNOS and the M2 hallmark Arg1 in adherence-enriched pulmonary macrophages at 21 dpi and calculated their expression ratio. Macrophages isolated from H99-infected lungs showed strong up-regulation of ARG1 and an ARG1/iNOS ratio above 8000, dramatically skewed macrophage activation toward M2 polarization (Figure 7). By contrast, macrophages from lungs infected with the mutant strains showed 100- to 200-fold lower ARG1 expression levels than H99-infected lungs (not shown). Furthermore, the ARG1/iNOS mRNA expression ratios showed a strong shift of macrophage activation in the rank order reflecting the level of protection, such that the most effectively cleared ena1Δ mutant induced the lowest ARG1/iNOS expression followed by the somewhat increased ARG1 to iNOS ratio found in the infections with the more virulent rub1Δ and pik1Δ strains, respectively (Figure 7). Thus, the shift in M1/M2 polarization in vivo that is associated with deletions of cryptococcal PIK1, RUB1, and ENA1 precisely reflects the level of virulence observed for each mutant.
Figure 7.
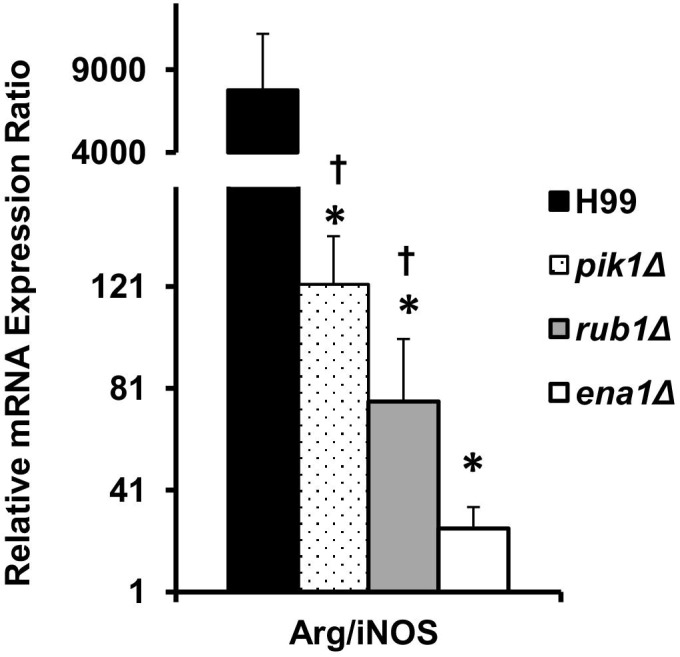
Effect of cryptococcal PIK1, RUB1, and ENA1 deletion on macrophage activation profile in C. neoformans–infected lungs. mRNA was isolated from adherence-enriched pulmonary macrophages at week 3 postinfection and converted to cDNA. Arginase (ARG1) and nitric oxide synthase (iNOS) expression levels were determined as in Figure 6, and the ratio between Arg1/iNOS is shown as readout of M2 versus M1 macrophage activation status. Note the profound M2 bias in macrophages from H99-infected lungs, shift of the balance toward M1 in pik1Δ-, rub1Δ-, and ena1Δ-infected groups, with the ena1Δ group showing the most significant shift toward he M1 phenotype. n = 5 or more infected mice for each of the strains; mutant-infected groups significantly different from *H99- and †ena1Δ-infected groups.
Cryptococcal Genes PIK1, RUB1, and ENA1 Have a Differential Impact on Cryptococcal Dissemination from the Infected Lungs
Our final goal in this study was to evaluate the impact of cryptococcal PIK1, RUB1, and ENA1 deletions on extrapulmonary dissemination and subsequent CNS invasion of the yeast. H99 disseminated rapidly to the CNS, showing the first positive brain cultures as early as 7 dpi (Table 1). The H99 burdens at 21 dpi approached tissue levels found in CNS postmortem, confirming that H99-infected mice were dying from disseminated cryptococcal disease. By contrast, the mutants showed nearly complete protection from both splenic and CNS dissemination during the initial 21 dpi. These outcomes changed at the onset of mortalities for both pik1Δ and rub1Δ infections, which both displayed a robust potential to cause disseminated extrapulmonary disease at later timepoints. The pik1Δ brain and spleen postmortem fungal burdens were 100% positive and were comparable to that of H99-infected mice at death (Table 1). Mice infected with rub1Δ demonstrated lower, but still considerable, fungal burdens in brain and spleen at the time of death. Furthermore, mice succumbing to H99, pik1Δ, or rub1Δ show similar brain pathology, with large cystoid lesions containing proliferating fungi consistent with the brain cultures (Figure 8, A–C). By contrast, ena1Δ showed no potential for causing extrapulmonary disease, evidenced by negative cultures and the absence of brain pathology. These results indicate that cryptococcal gene deletions of PIK1, RUB1, and ENA1 interfere with the ability of C. neoformans to disseminate or establish growth in extrapulmonary sites, either delaying (PIK1, RUB1) or completely eliminating (ENA1) extrapulmonary disease in mice.
Table 1.
Effect of Cryptococcal PIK1, RUB1, and ENA1 Deletions on the Extrapulmonary Dissemination of C. neoformans
| Organ and strain | Days post-infection |
Death | ||
|---|---|---|---|---|
| 7 | 14 | 21 | ||
| Spleen | ||||
| H99 | 0 | 1.23 ± 0.33 | 3.37 ± 0.34 | 3.37 ± 0.5 |
| pik1Δ | 0 | 0 | 0 | 3.58 ± 0.3 |
| rub1Δ | 0 | 0 | 0 | 2.42 ± 0.22⁎† |
| ena1Δ | 0 | 0 | 0 | |
| Brain | ||||
| H99 | 0.68 ± 0.43 | 2.48 ± 0.82 | 4.99 ± 0.94 | 5.92 ± 0.26 |
| pik1Δ | 0 | 0 | 0 | 5.61 ± 0.41 |
| rub1Δ | 0 | 0 | 0.33 ± 0.33 | 3.75 ± 0.52⁎† |
| ena1Δ | 0.32 ± 0.32 | 0 | 0 | |
Brains and spleens of the mice infected with pik1Δ, rub1Δ, ena1Δ mutants, and the wild-type strain H99 were harvested at days 7, 14, 21, postinfection and at death for analysis of fungal burden. Data are expressed as the mean log colony-forming units (CFU) ± SEM (5 or more mice per group at each time point).
P < 0.05 compared to H99 group.
P < 0.05 compared to pik1Δ group.
Figure 8.

Effect of cryptococcal PIK1, RUB1, and ENA1 deletions on the development of brain pathologies. Brains were collected at day 21 from H99-infected mice (A) and at day 60 from pik1Δ-infected (B), rub1Δ-infected (C), and ena1Δ-infected (D) mice and processed for histology (H&E and mucicarmine stains). Mice infected with pik1Δ or rub1Δ show similar pathological lesions in their brains as H99-infected mice, with large lesions in the brain containing proliferating fungi in these mice (A–C); (D) ena1Δ-infected mice without brain pathology.
Discussion
This study shows that expression of cryptococcal genes PIK1, RUB1, and ENA1 profoundly affects host immune responses in the lungs, extrapulmonary dissemination, and survival of C. neoformans–infected mice. Of the three cryptococcal genes compared here, expression of ENA1 most profoundly promoted fungal virulence through the induction of a nonprotective immune response; RUB1 exerted intermediate effects, and PIK1 expression had the least pronounced effect on pathogenic properties of the yeast. These outcomes expand on and clarify the previously reported effects of these mutants displayed in several acute screening assays.24 We demonstrate that the major factor shaping the impact of these genes on the “overall” virulence composite of Cryptococcus is modulation of the adaptive immune responses and macrophage activation status. We conclude that increasingly used acute screening assays, although valuable, have some limitations in predicting the overall effect of putative virulence genes on host defenses and the clinical outcomes in the infected mammalian host.
Our data show that the level of virulence attenuation of the mutants measured as improved mice survival precisely matches the changes in kinetics of pulmonary yeast growth, dissemination, and lung and brain histopathology. These outcomes are consistent with analyzed genes promoting pathogenic fitness of the organism within the local environments present in the infected host,23 as well as modulating the immunophenotype that develops in infected mice. Deletion of PIK1, RUB1, and ENA1 each differentially affected the balance of immune polarization hallmark expression, including: Th1/Th2 cytokine profiles; level of pulmonary eosinophilia and neutrophilia; and the pulmonary macrophage activation status. The effects of these genes on the development of pathological lesions in the lungs reflected the changes in immune polarization. Such immunomodulatory effects of cryptococcal virulence genes have been reported for cryptococcal urease17 and phospholipase B.20 The present study demonstrates that “immunomodulatory mechanisms” can be linked with a wider group of cryptococcal virulence genes, and that the level and/or the type of immunomodulation induced by each factor largely defines the overall impact of each gene on the course of cryptococcal disease.
We conclude that a broader scope of immunological analyses is needed to establish a full impact of each fungal factor. For example, Th1-promoting cytokine IFN-γ has a well-established role in protection of the infected host to C. neoformans, whereas Th2-driving IL-4 is known to be detrimental4–6,9,16,31,41–43; however, our study shows that narrowing analysis to just these two parameters may be insufficient. On one hand, the comparison of these cytokine responses in mice infected with H99 versus PIK1, RUB1, or ENA1 mutants demonstrated that each of these factors contributed to a shifted IL-4/IFN-γ ratio toward a Th2 response (Figure 6) and showed changes proportional to the virulence composite displayed by strains H99, pik1Δ, and rub1Δ. However, ena1Δ and rub1Δ show a different trend, with the former having the strongest effect on fungal virulence, but a lesser effect on IFN-γ and IL-4 production (Figures 1 and 6). The strongest effect of the ENA1 gene on cryptococcal virulence can be attributed to its additional role in reducing production of TNF-α, which is essential for immune protection against C. neoformans.44 The simultaneous increase in expression of both TNF-α and IFN-γ in response to cryptococcal ENA1deletion provides a composite immunological explanation for the most prominent role of ENA1 in H99 virulence among the factors analyzed in this study.
Among various factors analyzed here, the readout of M1/M2 polarization—ARG1/iNOS expression ratio—was the most precise predictor of virulence potential displayed by these cryptococcal strains in vivo. Our data demonstrate that cryptococcal expression of PIK1, RUB1, and ENA1 genes promote, to a varying degree, the development of a nonprotective M2 pulmonary macrophage phenotype. Deletions of each of these genes resulted in a dramatic switch from the M2-polarized macrophages in H99-infected mice to a protective M1 phenotype, with ENA1 gene deletion resulting in the most M1-skewed phenotype, followed by RUB1 and lastly by PIK1 deletion. This result supports the notion that relative contributions of M1 and M2 phenotypes in the macrophages, shaped by the pulmonary cytokine environment, is the major factor affecting cryptococcal clearance dynamics.6,9,10,45
Previous studies testing the fitness of various cryptococcal strains using the in vitro macrophage assays proposed that the level of cryptococcal resistance to the fungicidal effect of macrophages in vitro correlated well with the mortality of mice infected with these strains.28–30 Our studies further clarified this point, demonstrating that macrophage fungicidal assay in vitro is a good preliminary estimate of decreased virulence; however, it is not a precise indicator of the attenuation level displayed by each strain in vivo. Consistent with our previous report of significantly decreased survival of pik1Δ, rub1Δ, and ena1Δ mutants in the presence of IFN-γ activated macrophages in vitro.24 we observed significantly decreased virulence displayed by these strains in our mouse model (Figure 1). However, the attenuation levels displayed by these strains in the macrophage assay in vitro,24 as compared to our outcomes in vivo, varied significantly. The differences in outcomes of the “macrophage effect” of these various virulence genes in vivo and in vitro can be explained by differences in immunological microenvironments. This is consistent with the current understanding that macrophage phenotype functions as an “ultimate readout” of integrated immunological signals, which results from various interactions between the yeast, macrophage, and all other components of the immune system.6,9–12 The in vitro assay can capture only some of these interactions and cannot account for the dynamic changes that occur during the immune response over time. This view is further supported by analysis of histopathological lesions observed 60 dpi in rub1Δ-infected lungs. The evident hallmarks of M2 (accumulation of both YM1/2 crystals and live C. neoformans in macrophages) suggest that macrophage activation status has not remained M1 as observed at 21 dpi (Figure 8). The variable level of rub1Δ control by the mice is reflected by its pulmonary growth dynamics, which appeared more static at times (from days 3 to 7 and days 14 to 21) in contrast with steadily increasing H99 and pik1Δ fungal burdens.
Comparison of our previously reported results to our present in vivo infection data provides insight as to the predictive capacity of other screening assays, including tests of virulence in Caenorhabditis elegans infection and growth within human CSF in vitro.24 In the C. elegans infection assay used to identify fungal virulence factors, especially those affecting the innate immune system,26,46–48 pik1Δ and rub1Δ were attenuated.24 However, the ena1Δ strain showed wild-type virulence,24 contrasting with its rapid decrease in pulmonary fungal load postinoculation in mice (Figure 2C, day 3), which can be attributed to the innate host defenses. Since C. neoformans typically causes death by CNS infection, microbial growth in CSF would also be expected to correspond to the ability of the fungus to cause mortality. However, of all three strains preselected for their decreased ability to survive in CSF, only ena1Δ was unable to establish an infection in the brain of infected mice.24 Despite their attenuation in CSF in vitro,24 pik1Δ and rub1Δ were able to disseminate into the brain and cause mortality in our murine model, although with a delay compared to H99 infection. One explanation for this discrepancy is differences in timing, which could allow for development of compensatory mechanisms in this yeast during prolonged infection. The extended time the mutants persisted in the lung environment could promote the development of compensatory mechanisms, which eventually enhanced yeast survival within the CNS. C. neoformans has a strong potential for phenotypic and genotypic switching, which are important components of cryptococcal virulence.49–54
In terms of connection between the functions of PIK1, RUB1, and ENA1 genes in cryptococcal physiology and their effects leading to enhanced virulence, capsule formation, melanin production, and the ability to grow at 37°C are well established as traits necessary for virulence. In particular, melanin, and capsule polysaccharides can have immunomodulatory effects.13,55,56 However, Lee et al.24 found that ena1Δ and pik1Δ strains show normal phenotypes for these traits, suggesting that the link for these two strains lies in other virulence pathways. The rub1Δ strain did show partial temperature sensitivity and reduced melanin production.24 Thus, the effects of RUB1 on immune response and virulence could, to a certain degree, be related to these virulence traits. However, up to this point, the connection between the function of PIK1, RUB1, and ENA1 genes in cryptococcal physiology and their effects leading to the enhanced virulence remains undefined and warrants future studies.
In conclusion, we demonstrate that the virulence genes PIK1, RUB1, and ENA1 have significant, yet differential, roles in the pathogenesis of pulmonary and disseminated cryptococcosis. We demonstrate that cryptococcal PIK1, RUB1, and ENA1 promote changes in the immune responses of the host, including alterations of Th polarization and macrophage activation status. In contrast with previously reported screenings in which pik1Δ, rub1Δ, and ena1Δ mutant attenuations were differently ranked with various assays and/or analyzed time points,24 the in vivo analysis in the mouse model yielded results that were consistent across all host features, in terms of immune polarization, macrophage activation status, pulmonary growth, CNS dissemination, and mortality. Thus, screenings of cryptococcal genes for virulence potential in simplified assays need to be followed by analysis in a complex mammalian system to determine their full impact on different aspects of pathogenesis and the composite virulence of C. neoformans in the infected host.
Acknowledgments
We thank Dr. Yanmei Zhang, Zachary Hadd, James Mossner, and Stuart Zeltzer, who helped in different parts of this project and were supported by the Undergraduate Research Opportunity Program.
Footnotes
Supported in part by Merit Review grant I01 BX000656 (M.A.O.) and Career Development Award (J.J.O.) from the Department of Veterans Affairs, Public Health Service grants R01-AI28388, and R01-AI73896 (J.R.P.). T32-HL07749-19 training grant (Pulmonary and Critical Care Medicine) supported M.J.D., and the Undergraduate Research Opportunity Program supported undergraduate students (D.M.L., D.L.M., Z.H., J.M., S.Z.) during their work in our laboratory.
X.H. and D.M.L. contributed equally to this work.
References
- 1.Huffnagle G.B., Lipscomb M.F., Lovchik J.A., Hoag K.A., Street N.E. The role of CD4+ and CD8+ T cells in the protective inflammatory response to a pulmonary cryptococcal infection. J Leukoc Biol. 1994;55:35–42. doi: 10.1002/jlb.55.1.35. [DOI] [PubMed] [Google Scholar]
- 2.Kawakami K., Koguchi Y., Qureshi M.H., Yara S., Kinjo Y., Miyazato A., Nishizawa A., Nariuchi H., Saito A. Circulating soluble CD4 directly prevents host resistance and delayed-type hypersensitivity response to Cryptococcus neoformans in mice. Microbiol Immunol. 2000;44:1033–1041. doi: 10.1111/j.1348-0421.2000.tb02600.x. [DOI] [PubMed] [Google Scholar]
- 3.Traynor T.R., Kuziel W.A., Toews G.B., Huffnagle G.B. CCR2 expression determines T1 versus T2 polarization during pulmonary Cryptococcus neoformans infection. J Immunol. 2000;164:2021–2027. doi: 10.4049/jimmunol.164.4.2021. [DOI] [PubMed] [Google Scholar]
- 4.Jain A.V., Zhang Y., Fields W.B., McNamara D.A., Choe M.Y., Chen G.H., Erb-Downward J., Osterholzer J.J., Toews G.B., Huffnagle G.B., Olszewski M.A. Th2 but not Th1 immune bias results in altered lung functions in a murine model of pulmonary Cryptococcus neoformans infection. Infect Immun. 2009;77:5389–5399. doi: 10.1128/IAI.00809-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen G.H., McNamara D.A., Hernandez Y., Huffnagle G.B., Toews G.B., Olszewski M.A. Inheritance of immune polarization patterns is linked to resistance versus susceptibility to Cryptococcus neoformans in a mouse model. Infection and immunity. 2008;76:2379–2391. doi: 10.1128/IAI.01143-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardison S.E., Ravi S., Wozniak K.L., Young M.L., Olszewski M.A., Wormley F.L., Jr. Pulmonary infection with an interferon-gamma-producing Cryptococcus neoformans strain results in classical macrophage activation and protection. Am J Pathol. 2010;176:774–785. doi: 10.2353/ajpath.2010.090634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hernandez Y., Arora S., Erb-Downward J.R., McDonald R.A., Toews G.B., Huffnagle G.B. Distinct roles for IL-4 and IL-10 in regulating T2 immunity during allergic bronchopulmonary mycosis. J Immunol. 2005;174:1027–1036. doi: 10.4049/jimmunol.174.2.1027. [DOI] [PubMed] [Google Scholar]
- 8.Huffnagle G.B., Traynor T.R., McDonald R.A., Olszewski M.A., Lindell D.M., Herring A.C., Toews G.B. Leukocyte recruitment during pulmonary Cryptococcus neoformans infection. Immunopharmacology. 2000;48:231–236. doi: 10.1016/s0162-3109(00)00222-8. [DOI] [PubMed] [Google Scholar]
- 9.Arora S., Olszewski M.A., Tsang T.M., McDonald R.A., Toews G.B., Huffnagle G.B. Effect of cytokine interplay on macrophage polarization during chronic pulmonary infection with Cryptococcus neoformans. Infect Immun. 2011;79:1915–1926. doi: 10.1128/IAI.01270-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muller U., Stenzel W., Kohler G., Werner C., Polte T., Hansen G., Schutze N., Straubinger R.K., Blessing M., McKenzie A.N., Brombacher F., Alber G. IL-13 induces disease-promoting type 2 cytokines, alternatively activated macrophages and allergic inflammation during pulmonary infection of mice with Cryptococcus neoformans. J Immunol. 2007;179:5367–5377. doi: 10.4049/jimmunol.179.8.5367. [DOI] [PubMed] [Google Scholar]
- 11.Arora S., Hernandez Y., Erb-Downward J.R., McDonald R.A., Toews G.B., Huffnagle G.B. Role of IFN-gamma in regulating T2 immunity and the development of alternatively activated macrophages during allergic bronchopulmonary mycosis. J Immunol. 2005;174:6346–6356. doi: 10.4049/jimmunol.174.10.6346. [DOI] [PubMed] [Google Scholar]
- 12.Loke P., Nair M.G., Parkinson J., Guiliano D., Blaxter M., Allen J.E. IL-4 dependent alternatively-activated macrophages have a distinctive in vivo gene expression phenotype. BMC Immunol. 2002;3:7. doi: 10.1186/1471-2172-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mansour M.K., Vyas J.M., Levitz S.M. Dynamic virulence: real-time assessment of intracellular pathogenesis links Cryptococcus neoformans phenotype with clinical outcome. MBio. 2011;2 doi: 10.1128/mBio.00217-11. e00217-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alanio A., Desnos-Ollivier M., Dromer F. Dynamics of Cryptococcus neoformans-macrophage interactions reveal that fungal background influences outcome during cryptococcal meningoencephalitis in humans. MBio. 2011;2 doi: 10.1128/mBio.00158-11. e00158-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olszewski M.A., Zhang Y., Huffnagle G.B. Mechanisms of cryptococcal virulence and persistence. Future Microbiol. 2010;5:1269–1288. doi: 10.2217/fmb.10.93. [DOI] [PubMed] [Google Scholar]
- 16.Olszewski M.A., Noverr M.C., Chen G.H., Toews G.B., Cox G.M., Perfect J.R., Huffnagle G.B. Urease expression by Cryptococcus neoformans promotes microvascular sequestration, thereby enhancing central nervous system invasion. Am J Pathol. 2004;164:1761–1771. doi: 10.1016/S0002-9440(10)63734-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osterholzer J.J., Surana R., Milam J.E., Montano G.T., Chen G.H., Sonstein J., Curtis J.L., Huffnagle G.B., Toews G.B., Olszewski M.A. Cryptococcal urease promotes the accumulation of immature dendritic cells and a non-protective T2 immune response within the lung. Am J Pathol. 2009;174:932–943. doi: 10.2353/ajpath.2009.080673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi M., Li S.S., Zheng C., Jones G.J., Kim K.S., Zhou H., Kubes P., Mody C.H. Real-time imaging of trapping and urease-dependent transmigration of Cryptococcus neoformans in mouse brain. J Clin Invest. 2010;120:1683–1693. doi: 10.1172/JCI41963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noverr M.C., Williamson P.R., Fajardo R.S., Huffnagle G.B. CNLAC1 is required for extrapulmonary dissemination of Cryptococcus neoformans but not pulmonary persistence. Infect Immun. 2004;72:1693–1699. doi: 10.1128/IAI.72.3.1693-1699.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noverr M.C., Cox G.M., Perfect J.R., Huffnagle G.B. Role of PLB1 in pulmonary inflammation and cryptococcal eicosanoid production. Infect Immun. 2003;71:1538–1547. doi: 10.1128/IAI.71.3.1538-1547.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kronstad J.W., Attarian R., Cadieux B., Choi J., D'Souza C.A., Griffiths E.J., Geddes J.M., Hu G., Jung W.H., Kretschmer M., Saikia S., Wang J. Expanding fungal pathogenesis: cryptococcus breaks out of the opportunistic box. Nat Rev Microbiol. 2011;9:193–203. doi: 10.1038/nrmicro2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blackstock R., Buchanan K.L., Cherniak R., Mitchell T.G., Wong B., Bartiss A., Jackson L., Murphy J.W. Pathogenesis of Cryptococcus neoformans is associated with quantitative differences in multiple virulence factors. Mycopathologia. 1999;147:1–11. doi: 10.1023/a:1007041401743. [DOI] [PubMed] [Google Scholar]
- 23.Panepinto J.C., Williamson P.R. Intersection of fungal fitness and virulence in Cryptococcus neoformans. FEMS Yeast Res. 2006;6:489–498. doi: 10.1111/j.1567-1364.2006.00078.x. [DOI] [PubMed] [Google Scholar]
- 24.Lee A., Toffaletti D.L., Tenor J., Soderblom E.J., Thompson J.W., Moseley M.A., Price M., Perfect J.R. Survival defects of Cryptococcus neoformans mutants exposed to human cerebrospinal fluid result in attenuated virulence in an experimental model of meningitis. Infect Immun. 2010;78:4213–4225. doi: 10.1128/IAI.00551-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mylonakis E., Ausubel F.M., Perfect J.R., Heitman J., Calderwood S.B. Killing of Caenorhabditis elegans by Cryptococcus neoformans as a model of yeast pathogenesis. Proc Natl Acad Sci U S A. 2002;99:15675–15680. doi: 10.1073/pnas.232568599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mylonakis E., Casadevall A., Ausubel F.M. Exploiting amoeboid and non-vertebrate animal model systems to study the virulence of human pathogenic fungi. PLoS Pathog. 2007;3:e101. doi: 10.1371/journal.ppat.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu O.W., Chun C.D., Chow E.D., Chen C., Madhani H.D., Noble S.M. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell. 2008;135:174–188. doi: 10.1016/j.cell.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cox G.M., Harrison T.S., McDade H.C., Taborda C.P., Heinrich G., Casadevall A., Perfect J.R. Superoxide dismutase influences the virulence of Cryptococcus neoformans by affecting growth within macrophages. Infect Immun. 2003;71:173–180. doi: 10.1128/IAI.71.1.173-180.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feldmesser M., Tucker S., Casadevall A. Intracellular parasitism of macrophages by Cryptococcus neoformans. Trends Microbiol. 2001;9:273–278. doi: 10.1016/s0966-842x(01)02035-2. [DOI] [PubMed] [Google Scholar]
- 30.Liu L., Tewari R.P., Williamson P.R. Laccase protects Cryptococcus neoformans from antifungal activity of alveolar macrophages. Infect Immun. 1999;67:6034–6039. doi: 10.1128/iai.67.11.6034-6039.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y., Wang F., Tompkins K.C., McNamara A., Jain A.V., Moore B.B., Toews G.B., Huffnagle G.B., Olszewski M.A. Robust Th1 and Th17 immunity supports pulmonary clearance but cannot prevent systemic dissemination of highly virulent Cryptococcus neoformans H99. Am J Pathol. 2009;175:2489–2500. doi: 10.2353/ajpath.2009.090530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blackstock R., Murphy J.W. Role of interleukin-4 in resistance to Cryptococcus neoformans infection. Am J Respir Cell Mol Biol. 2004;30:109–117. doi: 10.1165/rcmb.2003-0156OC. [DOI] [PubMed] [Google Scholar]
- 33.Huffnagle G.B., Chen G.H., Curtis J.L., McDonald R.A., Strieter R.M., Toews G.B. Down-regulation of the afferent phase of T cell-mediated pulmonary inflammation and immunity by a high melanin-producing strain of Cryptococcus neoformans. J Immunol. 1995;155:3507–3516. [PubMed] [Google Scholar]
- 34.Hesse M., Modolell M., La Flamme A.C., Schito M., Fuentes J.M., Cheever A.W., Pearce E.J., Wynn T.A. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-arginine metabolism. J Immunol. 2001;167:6533–6544. doi: 10.4049/jimmunol.167.11.6533. [DOI] [PubMed] [Google Scholar]
- 35.Milam J.E., Erb-Downward J.R., Chen G.H., Osuchowski M.F., McDonald R., Chensue S.W., Toews G.B., Huffnagle G.B., Olszewski M.A. CD11c+ cells are required to prevent progression from local acute lung injury to multiple organ failure and death. Am J Pathol. 2010;176:218–226. doi: 10.2353/ajpath.2010.081027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milam J.E., Herring-Palmer A.C., Pandrangi R., McDonald R.A., Huffnagle G.B., Toews G.B. Modulation of the pulmonary type 2 T-cell response to Cryptococcus neoformans by intratracheal delivery of a tumor necrosis factor alpha-expressing adenoviral vector. Infect Immun. 2007;75:4951–4958. doi: 10.1128/IAI.00176-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Punturieri A., Alviani R.S., Polak T., Copper P., Sonstein J., Curtis J.L. Specific engagement of TLR4 or TLR3 does not lead to IFN-β-mediated innate signal amplification and STAT1 phosphorylation in resident murine alveolar macrophages. J Immunol. 2004;173:1033–1042. doi: 10.4049/jimmunol.173.2.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glantz S.A. 6th ed. McGraw-Hill; New York: 2005. Primer of Biostatistics. [Google Scholar]
- 39.Vecchiarelli A., Pietrella D., Dottorini M., Monari C., Retini C., Todisco T., Bistoni F. Encapsulation of Cryptococcus neoformans regulates fungicidal activity and the antigen presentation process in human alveolar macrophages. Clin Exp Immunol. 1994;98:217–223. doi: 10.1111/j.1365-2249.1994.tb06128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindell D.M., Moore T.A., McDonald R.A., Toews G.B., Huffnagle G.B. Generation of antifungal effector CD8+ T cells in the absence of CD4+ T cells during Cryptococcus neoformans infection. J Immunol. 2005;174:7920–7928. doi: 10.4049/jimmunol.174.12.7920. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y., Wang F., Bhan U., Huffnagle G.B., Toews G.B., Standiford T.J., Olszewski M.A. TLR9 signaling is required for generation of the adaptive immune protection in Cryptococcus neoformans-infected lungs. Am J Pathol. 2010;177:754–765. doi: 10.2353/ajpath.2010.091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schop J. Protective immunity against cryptococcus neoformans infection. Mcgill J Med. 2007;10:35–43. [PMC free article] [PubMed] [Google Scholar]
- 43.Koguchi Y., Kawakami K. Cryptococcal infection and Th1-Th2 cytokine balance. Int Rev Immunol. 2002;21:423–438. doi: 10.1080/08830180213274. [DOI] [PubMed] [Google Scholar]
- 44.Huffnagle G.B., Toews G.B., Burdick M.D., Boyd M.B., McAllister K.S., McDonald R.A., Kunkel S.L., Strieter R.M. Afferent phase production of TNF-alpha is required for the development of protective T cell immunity to Cryptococcus neoformans. J Immunol. 1996;157:4529–4536. [PubMed] [Google Scholar]
- 45.Goldman D., Cho Y., Zhao M., Casadevall A., Lee S.C. Expression of inducible nitric oxide synthase in rat pulmonary Cryptococcus neoformans granulomas. Am J Pathol. 1996;148:1275–1282. [PMC free article] [PubMed] [Google Scholar]
- 46.Fuchs B.B., Mylonakis E. Using non-mammalian hosts to study fungal virulence and host defense. Curr Opin Microbiol. 2006;9:346–351. doi: 10.1016/j.mib.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 47.Tang R.J., Breger J., Idnurm A., Gerik K.J., Lodge J.K., Heitman J., Calderwood S.B., Mylonakis E. Cryptococcus neoformans gene involved in mammalian pathogenesis identified by a Caenorhabditis elegans progeny-based approach. Infect Immun. 2005;73:8219–8225. doi: 10.1128/IAI.73.12.8219-8225.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aballay A., Ausubel F.M. Caenorhabditis elegans as a host for the study of host-pathogen interactions. Curr Opin Microbiol. 2002;5:97–101. doi: 10.1016/s1369-5274(02)00293-x. [DOI] [PubMed] [Google Scholar]
- 49.Garcia-Hermoso D., Dromer F., Janbon G. Cryptococcus neoformans capsule structure evolution in vitro and during murine infection. Infect Immun. 2004;72:3359–3365. doi: 10.1128/IAI.72.6.3359-3365.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guerrero A., Fries B.C. Phenotypic switching in Cryptococcus neoformans contributes to virulence by changing the immunological host response. Infect Immun. 2008;76:4322–4331. doi: 10.1128/IAI.00529-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Desnos-Ollivier M., Patel S., Spaulding A.R., Charlier C., Garcia-Hermoso D., Nielsen K., Dromer F. Mixed infections and in vivo evolution in the human fungal pathogen Cryptococcus neoformans. MBio. 2010;1 doi: 10.1128/mBio.00091-10. e00091-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blasi E., Brozzetti A., Francisci D., Neglia R., Cardinali G., Bistoni F., Vidotto V., Baldelli F. Evidence of microevolution in a clinical case of recurrent Cryptococcus neoformans meningoencephalitis. Eur J Clin Microbiol Infect Dis. 2001;20:535–543. doi: 10.1007/s100960100549. [DOI] [PubMed] [Google Scholar]
- 53.Fries B.C., Casadevall A. Serial isolates of Cryptococcus neoformans from patients with AIDS differ in virulence for mice. J Infect Dis. 1998;178:1761–1766. doi: 10.1086/314521. [DOI] [PubMed] [Google Scholar]
- 54.Pietrella D., Fries B., Lupo P., Bistoni F., Casadevall A., Vecchiarelli A. Phenotypic switching of Cryptococcus neoformans can influence the outcome of the human immune response. Cell Microbiol. 2003;5:513–522. doi: 10.1046/j.1462-5822.2003.00297.x. [DOI] [PubMed] [Google Scholar]
- 55.Mednick A.J., Nosanchuk J.D., Casadevall A. Melanization of Cryptococcus neoformans affects lung inflammatory responses during cryptococcal infection. Infect Immun. 2005;73:2012–2019. doi: 10.1128/IAI.73.4.2012-2019.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fonseca F.L., Nohara L.L., Cordero R.J., Frases S., Casadevall A., Almeida I.C., Nimrichter L., Rodrigues M.L. Immunomodulatory effects of serotype B glucuronoxylomannan from Cryptococcus gattii correlate with polysaccharide diameter. Infect Immun. 2010;78:3861–3870. doi: 10.1128/IAI.00111-10. [DOI] [PMC free article] [PubMed] [Google Scholar]