

Abstract
After ischemia-reperfusion injury (IRI), kidney tubules show activated transforming growth factor β (TGF-β) signaling and increased expression of profibrotic peptides, platelet-derived growth factor-B (PDGF-B) and connective tissue growth factor (CTGF). If tubule repair after IRI is incomplete, sustained paracrine activity of these peptides can activate interstitial fibroblast progenitors and cause fibrosis. We show that lysophosphatidic acid (LPA), a ubiquitous phospholipid that is increased at sites of injury and inflammation, signals through LPA2 receptors and Gαq proteins of cultured proximal tubule cells to transactivate latent TGF-β in a Rho/Rho-kinase and αvβ6 integrin-dependent manner. Active TGF-β peptide then initiates signaling to increase the production and secretion of PDGF-B and CTGF. In a rat model of IRI, increased TGF-β signaling that was initiated early during reperfusion did not subside during recovery, but progressively increased, causing tubulointerstitial fibrosis. This was accompanied by correspondingly increased LPA2 and β6 integrin proteins and elevated tubule expression of TGF-β1, together with PDGF-B and CTGF. Treatment with a pharmacological TGF-β type I receptor antagonist suppressed TGF-β signaling, decreased the expression of β6 integrin, PDGF-B, and CTGF, and ameliorated fibrosis. We suggest that LPA-initiated autocrine signaling is a potentially important mechanism that gives rise to paracrine profibrotic signaling in injured kidney tubule cells.
See related Commentary on page 1147
Kidney tubules recovering from ischemia-reperfusion injury (IRI) exhibit increased transforming growth factor β (TGF-β) signaling1–4 that produces fibrosis.1,3–6 The expression of TGF-β and its receptors is increased in regenerating proximal tubules during recovery after IRI, suggesting the operation of an amplified autocrine signaling loop.1,7 The mechanism of initiation is unknown. However, there must be early steps that trigger TGF-β signaling which then gives rise to sustained and amplified signaling by undefined feed-forward mechanisms and cross talk with other pathways. Increased conversion of latent TGF-β to active peptide is such a required and important early step.8 TGF-β is secreted as an inactive complex with latency-associated peptide (LAP). Several physical, chemical, and enzymatic processes can convert latent TGF-β to active peptide.8–12 Among these processes, activation caused by the binding of Arg-Gly-Asp (RGD) domains in latent TGF-β1 or TGF-β3 to integrins is particularly relevant. Several integrins bind and activate TGF-β, but this action of αvβ6 integrin is restricted to epithelial cells.13
A role for the αvβ6 integrin has been shown in several disease models.10,14–17 TGF-β activation by αvβ6 contributes to lung injury and fibrosis, an action that is triggered by G-protein–coupled receptor (GPCR) ligands, lysophosphatidic acid (LPA) and thrombin.15,18 It seemed likely that a similar mechanism contributes to increased TGF-β signaling after IRI that, if sustained, causes fibrosis. αvβ6 Integrin is overexpressed in tubule epithelium of human kidneys with chronic kidney disease16,19 and contributes to renal fibrosis in mouse models of Alport syndrome and ureteral obstruction.16,17 A GPCR and integrin-mediated mechanism seems likely to account for TGF-β activation and fibrosis in these contexts, as shown for the lung.15,18 GPCR ligands reported to bear some relationship to renal injury and fibrosis and/or TGF-β signaling in kidney cells include angiotensin II, LPA, sphingosine-1-phosphate (S1P), and thrombin.17,20–28 How these ligands affect injury outcomes in kidneys or TGF-β signaling in renal cells is largely unexplored. As an exception, angiotensin II increased TGF-β production by proximal tubules through epidermal growth factor receptor transactivation and downstream signaling by extracellular signal–regulated kinase.21
We surmised that, among the myriad regenerative signals triggered by IRI, there are some with potential to transactivate TGF-β. The GPCR ligands, LPA, S1P, thrombin, adenosine, and angiotensin II, are implicated in the development of acute kidney injury after ischemia.26,29–33 Whether they are also involved in repair, as reported for LPA and thrombin in the lung,15,18 is unknown. We asked if one such ligand, LPA, transactivates TGF-β signaling in cultured tubule cells. We show that LPA activates latent TGF-β through a Gαq/11-mediated Rho and αvβ6-dependent process in proximal tubule cells, as in lung epithelium. Active TGF-β, produced in an LPA-dependent manner, then drives the secretion of profibrotic peptides, platelet-derived growth factor-B (PDGF-B) and connective tissue growth factor (CTGF). Pursuant to our earlier experiments in vivo,3,6 we found that persistently dedifferentiated tubules associated with fibrosis after IRI display increased TGF-β signaling and increased expression of PDGF-B and CTGF. Furthermore, LPA2 receptor and αvβ6 integrin proteins were markedly elevated in kidney tissue in the same time frame that TGF-β, PDGF-B, and CTGF expression was also increased. Considered in the context of our findings in cultured cells, and reported observations on lung epithelium,15,18 these observations in vivo provide suggestive, but persuasive, evidence that GPCR signaling by LPA is a possible proximate trigger for profibrotic TGF-β signaling in tubules regenerating after IRI.
Materials and Methods
Antibodies and Reagents
Antibody sources were as follows: anti-phospho-Smad2 (S465/467) from Cell Signaling, Inc. Cat. No. 3108 (Danvers, MA); anti-Smad2/3 from BD transduction, Cat. No. 610842 (San Diego, CA); and anti-Smad2 from Epitomics, Cat. No. 1736 (Burlingame, CA); αvβ6 antibodies Mab 6.3G9 and ch2A1 were produced as described previously16,34; anti-LPA2 was from Abgent, Inc., Cat. No. AP6140a (San Diego, CA); anti–TGF-β was from R&D systems, Inc., Cat. No. AB-100-NA (Minneapolis, MN) and AbD Serotec, Cat. No. AHP1734 (Raleigh, NC); anti-PDGF-B was from LS Bio, Cat. No. LS-B4182 (Seattle, WA); anti-CTGF was from Santa Cruz, Cat. No. sc14939 (Santa Cruz, CA); anti-vimentin was from Neomarkers/Thermo Scientific Lab Vision, Cat. No. MS129 (Fremont, CA). Secondary antibodies conjugated with HRP were from Jackson ImmunoResearch Labs, Cat. Nos. 715-035-152 and 715-035-150 (West Grove, PA). Secondary antibodies conjugated with Alexa dyes 488, 568 and 647 were from Invitrogen, Carlsbad, CA. Sources of other reagents were as follows: SD-208; 2-[(5-Chloro-2-fluorophenyl)pteridin-4-yl]pyridin-4-yl amine, from SCIOS, Inc., (now subsidiary of Johnson and Johnson, New Brunswick, NJ); SB431542, Ki16425 and Sphingosylphosphorylcholine from Sigma-Aldrich Cat. No. S4317, k0639 and S4275 (St. Louis, MO). Latent TGF-β1 from R&D systems, Cat. No. 299-LT (Minneapolis, MN), LPA from Enzo Life Sciences, Cat. No. LP100 (Plymouth Meeting, PA), HLZ-56 [a kind gift from Glenn D. Prestwich (University of Utah)] was also purchased from Echelon Biosciences, Cat. No. L-7416 (Salt Lake City, UT), Sphingosine-1-phosphate (S1P) from Cayman chemical, Cat. No. 62570 (Ann Arbor, MI), BIM46174 was a kind gift from Grégoire P. Prévost (IPSEN-Institut Henri Beaufour), Pertussis toxin from List biological laboratories, Cat. No. 179A (Campbell, CA), GM 6001 and GM 6001 Negative control from EMD Chemicals, Cat. Nos. 364205 and # 364210 respectively (Rockland, MA).
Cell Culture, Plasmids, and Vectors
Boston University mouse proximal tubule cells (BUMPTs; clone 306; obtained from Drs. Wilfred Lieberthal and John Schwartz, Boston University School of Medicine, Boston, MA) and human proximal convoluted tubule cells (HPCTs; obtained from Dr. Ulrich Hopfer, Case Western Reserve University, Cleveland, OH) were grown at 37°C in Dulbecco's modified Eagle's medium with 10% fetal calf serum (FCS) or in serum-free Dulbecco's modified Eagle's medium (DMEM)/Ham's F12 medium with insulin (10 μg/mL), transferrin (5 μg/mL), Na selenite (6.7 μg/mL), and dexamethasone (4 μg/mL). BUMPT cells were transfected with p3TP-Luc35 (obtained from Dr. Joan Massague, Memorial Sloan-Kettering Cancer Center, New York, NY), (CAGA)12-Luc, or 2 X ARE-Luc with pCMV-hFAST136,37 (obtained from Dr. Susumu Itoh, University of Tsukuba, Tsukuba, Japan), together with pCMV-Neo or Hygro. Stably transfected cells were selected with G418 or hygromycin and clones assayed for luciferase activity after treatment with and without TGF-β (2 ng/mL). Mouse proximal tubule cells were grown in primary culture, as previously described.3 The growth medium was modified from published formulations and contained bovine serum albumin (0.5%), insulin (10 μg/mL), epidermal growth factor (10 ng/mL), transferrin (5 μg/mL), Na selenite (6.7 μg/mL), dexamethasone (4 μg/mL), and l-ascorbic acid-2-phosphate (50 μmol/L). Cells were used at first passage. HK-2 cells (CRL-2190; ATCC, Manassas, VA) of human proximal tubular origin were grown in DMEM/Ham's F12 medium (Gibco, Invitrogen, Grand Island, NY) containing 5 mmol/L glucose, supplemented with insulin (10 μg/mL), epidermal growth factor (10 ng/mL), transferrin (5 μg/mL), Na selenite (6.7 μg/mL), and dexamethasone (4 μg/mL), supplemented with 10% FCS. G-protein C-terminal peptide minigene vectors38 (obtained from Dr. Heidi Hamm, Vanderbilt University Medical Center, Nashville, TN) were transfected using Fugene reagent. The LPA2-specific TRC shRNA-pLKO.1 plasmid construct (TRC number TRCN0000234209, clone ID NM_020028.3-1013s21c1, catalogue number NM_020028; Sigma-Aldrich) or a negative control scramble shRNA-pLKO.1 construct was cotransfected with the packaging plasmid into 293T cells using Fugene HD Reagent (catalogue number 04709705001; Roche Applied Science, Indianapolis, IN). The short hairpin RNA (shRNA) target sequence for LPA2 was as follows: 5′-CCGGTGGTCAATGCAGTGGTATATTCTCGAGAATATACCACTGCATTGACCATTTTTG-3′. Culture supernatants containing lentiviral particles were collected 48 hours after transfection and filtered through 0.45 μmol/L filters. BUMPT cells were transduced with viral supernatant in the presence of 5 μg/mL polybrene. After 48 hours, cells were replenished with fresh medium containing 1 μg/mL puromycin. Cells were maintained in puromycin medium for selection of stably transfected cells.
Growth and Wounding of Cultured Proximal Tubule Monolayer Cells
BUMPT cells were grown until confluent contact inhibition and differentiation in 21-cm2 culture dishes. The monolayer cells were wounded in concentric circular swaths with preserved strips of cells in between by using a device consisting of a flat, approximately 21-cm2 etched rubber disk with a series of alternating 0.2-mm-wide concentric grooves and 0.8-mm-wide unetched ridges. Approximately 80% to 90% of cell mass was removed from confluent BUMPT cultures by wounding. Dedifferentiation, migration, proliferation, and modulations of autocrine TGF-β signaling in the remaining cells were described.3,39 Active TGF-β in conditioned medium was measured using mink lung epithelial cells stably transfected with a truncated plasminogen activator inhibitor-1 promoter-luciferase fusion plasmid40 (obtained from Dr. Daniel Rifkin, New York University Langone School of Medicine, New York, NY).
Charcoal Treatment of FCS and Serum Lipid Extraction
Charcoal-stripped FCS (C-FCS) was prepared as previously described.41 In brief, 10 mL of FCS was incubated with 1 g of activated charcoal (Sigma-Aldrich) overnight at 4°C. After centrifugation at 2000 × g for 10 minutes, the supernatant was filtered through a 0.22 μmol/L cellulose acetate filter. Serum lipids were extracted as previously described.42 In brief, 1 mL of serum was added into 15 mL of MeOH in a glass tube. After vortex mixing and incubation on ice for 10 minutes, the mixture was centrifuged (at 10,000 × g, for 5 minutes, at room temperature), and the supernatant was collected and dried. The extract was redissolved in 6 mL of CHCl3:MeOH (1:1) and centrifuged. The supernatant was dried and saved.
Ischemia-Reperfusion Injury
Male Sprague-Dawley rats, weighing approximately 250 g body weight, were used for IRI studies, as previously described.3 The left renal vascular pedicle was clamped with aneurysm clips (Roboz Surgical Instruments Co., Gaithersburg, MD) under isoflurane anesthesia on a heated table. The pedicle was dissected, but not clamped, in controls. Right kidneys were removed in both groups. Clamps were removed after 45 minutes, and the abdominal incision was closed. Body temperature was maintained at 37°C. Blood samples were obtained before surgery and daily to measure serum creatinine by high-performance liquid chromatography.43 At 4 hours after surgery and at 12 hourly intervals, one group of rats received SD208 (60 mg/kg) by gavage. SD208, a TGF-β type I receptor antagonist,44 was suspended in 1% methylcellulose. Controls received 1% methylcellulose. Treatments were continued for 4 days.
Histological, IHC, and Immunofluorescence Analyses
Kidneys were fixed by vascular perfusion with periodic acid–lysine–paraformaldehyde and processed for histological, immunohistochemical (IHC), and immunofluorescence (IF) analyses, as previously described.3,6
Western Blot Analysis
Kidneys were placed on a metal plate cooled by melting ice, sliced, and washed with cold PBS. The outer stripe of outer medulla was dissected and flash frozen in liquid nitrogen. Frozen tissue was ground under liquid nitrogen and extracted with 2× SDS sample buffer with protease and phosphatase inhibitors. Protein was measured by BCA assay. The equivalency of protein loading was assessed by densitometry of individual lanes of Coomassie Blue–stained gels and further by loading controls. Cultured cells were extracted with SDS sample buffer with protease and phosphatase inhibitors. Procedures for SDS-PAGE and Western blot analysis were described.3
Real-Time RT-PCR
The expression of LPA receptors in BUMPT cells was determined by quantitative real-time RT-PCR. Total RNAs were purified from BUMPT cells using RNeasy Mini kit (number 74104; Qiagen, Valencia, CA) and were converted to cDNAs using a High Capacity cDNA Reverse Transcription Kit (number 4368814; Life Technologies, Carlsbad). Real-time PCR with SYBR Green (catalogue number 4309155; Life Technologies) fluorescence detection was performed using an ABI Prism 7000 ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Carlsbad, CA) with specific primers. The following primers were used for LPA receptors: LPA1, 5′-CATGGTGGCAATCTACGTCAA-3′ (sense) and 5′-AGGCCAATCCAGCGAAGAA-3′ (antisense); LPA2, 5′-TGTCTGACTGCACAGCTTGGA-3′ (sense) and 5′-CTCATGGAGTTTTCTGGTGCC-3′ (antisense); LPA3, 5′-GTACCTGAGCCCCCCATTG-3′ (sense) and 5′-AAACCCATGCGGAAACAACT-3′ (antisense); LPA4, 5′-CCTTACCAACATCTATGGGAGCAT-3′ (sense) and 5′-TGGCCAGGAAACGATCCA-3′ (antisense); and LPA5, 5′-GGGGTGCTGATGAT-3′ (sense) and 5′-TTCTGAGGGTAGTT-3′ (antisense). Other primers used were as follows: PDGF-B, 5′-CCACTCCATCCGCTCCTTT-3′ (sense) and 5′-AAGTCCAGCTCAGCCCCAT-5′ (antisense); and CTGF, 5′-AGGACTGCAGCGCGCAATGT-3′ (sense) and 5′-GAGGCCCTTGTGTGGGTCGC-3′ (antisense). Appropriate no-RT and nontemplate controls were included in the PCR, and dissociation analysis was performed at the end of each run to confirm the specificity of the reaction. Relative levels of mRNA were calculated.
Statistics
Tests were performed using paired Student t-tests and analysis of variance. Comparisons were considered significant at P < 0.05.
Results
A Subpopulation of Regenerating Proximal Tubules Show Persistently Increased Expression of TGF-β1, Nuclear Smad2, PDGF-B, and CTGF After Ischemia-Reperfusion Injury of the Kidney
By using Western blot analysis, kidneys recovering from IRI showed early increases of TGF-β1 and Smad2 phosphorylation, consistent with previous reports.1–3 Subsequently, during more prolonged reperfusion, we found that the increased TGF-β expression and Smad phosphorylation did not subside, as might be expected if repair was complete and normal tubule phenotype was restored. Instead, the signals increased substantially by 14 days (Figure 1A). These increases coincided with the development of tubulointerstitial fibrosis.3 By IHC, increased tubule expression of TGF-β at 14 days was accompanied by fibrosis (Figure 1B). By IF, dedifferentiated vimentin-positive tubules in fibrotic tissue showed increased staining and nuclear localization of Smad2 protein (Figure 1C). TGF-β signaling may stimulate the production of profibrotic peptides, such as PDGF-B and CTGF. Western blot analysis for PDGF-B was confounded by technical issues related to the presence of PDGF-B in blood platelets trapped in kidney tissue. After an early increase by 1 day of IRI, kidney expression of CTGF subsided, but not to baseline levels. The signal increased again by 14 days as fibrosis developed, concurrently with a similar increase in the expression of TGF-β (Figure 1A). By using IHC 14 days after IRI, tubules associated with fibrosis showed intense immunoreactivity for PDGF-B and CTGF (Figure 1, D and E). Treatment with the TGF-β type I receptor antagonist, SD208, did not modify the elevated expression of TGF-β that was seen 14 days after IRI (Figure 1, A and B), but instead diminished Smad2 phosphorylation (Figure 1A), the nuclear translocation of Smad2 protein (Figure 1C), and the expression of both PDGF-B and CTGF (Figure 1, D and E), and ameliorated the tubulointerstitial fibrosis that developed otherwise.3 These data indicated that TGF-β was overexpressed and TGF-β signaling was up-regulated in proximal tubule cells when renal fibrosis developed after renal IRI. However, the mechanisms by which differentiated proximal tubule cells transformed to a profibrotic phenotype expressing PDGF-B and CTGF after kidney injury were unknown.
Figure 1.

Ischemia-reperfusion injury stimulates TGF-β signaling and induces profibrotic peptide expression in renal proximal tubules. A: SDS extracts of the outer stripe of the outer medulla from normal control rats without surgery (Normal Ctrl), renal IRI with or without SD208 treatment at the indicated time after surgery, and nephrectomized control (Nephr Ctrl) at day 14 are immunoblotted for phospho-Smad2, total Smad2/3, TGF-β1, and CTGF. B, D, and E: IHC staining of TGF-β1, PDGF, and CTGF in kidneys 14 days after right unilateral nephrectomy (Nephr Cntrl), right unilateral nephrectomy, and left kidney IRI with vehicle treatment (IRI + Vehicle) or SD208 treatment (IRI + SD208). C: Double- or triple-color confocal IF 14 days after surgery. Kidneys from nephrectomy control, 14-day IRI + Vehicle, and 14-day IRI + SD208 rats are stained for Smad2 (red), vimentin (Vim; green), and DAPI (blue). Arrowheads, Vimentin-positive regenerating proximal tubules with nuclear Smad2. Scale bars: 100 μm (IHC images); 5 μm (IF images). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
LPA Is a Major Bioactive Lipid in Serum for TGF-β Signaling Stimulation
The results of our in vivo study, as shown in Figure 1, led us to investigate the possible causative factors that drive proximal tubule cells to produce and release TGF-β to increase TGF-β signaling. In view of complexities inherent to studying IRI in vivo, we used cell culture models to study these mechanisms. TGF-β signaling in cultured proximal tubule cells was autoregulated by cell density. Signaling was suppressed during confluent growth arrest, but increased again when dedifferentiation, migration, and proliferation were induced by wounding.3 By using a bioluminescent assay for active TGF-β, we investigated if the signaling modulations corresponded to variations in the levels of secreted TGF-β in growth medium. Although the assay was highly sensitive, as reported,40 the levels of activity in conditioned media were at or less than the lowest levels of detection, and there were no differences between subconfluent, confluent, or wounded cultures (data not shown). Nevertheless, inclusion of neutralizing TGF-β antibodies in the medium suppressed TGF-β signaling in subconfluent or wounded cultures, reflected by reduced activity of the TGF-β–responsive p3TP-Luc reporter and attenuated C-terminal phosphorylation of Smad23 (data not shown). These results implied that high TGF-β signaling that was constitutive in subconfluent cells or triggered by wounding of confluent cultures was caused by nascent TGF-β that was produced on the cell surface, and then was bound to the receptor before internalization by endocytosis, but was not released into the medium in active form. We inferred that neutralization of nascent TGF-β occurred on the cell surface. These considerations explained our failure to detect active TGF-β in the conditioned medium.
During normal conditions of renal perfusion with blood, proximal tubule cells are exposed to plasma and the normal variations of cellular signaling that take place under these physiological circumstances are governed, in part, by plasma-derived factors. However, after acute injury with attendant proximal tubule cell death, platelets become activated and the coagulation cascade is triggered in the injured tubule-vascular microenvironments.29 To reproduce such contexts of in vivo injury in tissue culture, we used a wound model in which most (up to approximately 90%) of confluent, quiescent, and differentiated cells was removed by crush injury and the regenerative response of remaining cells was studied in a highly reproducible manner.39 Wounding was performed in the presence or absence of serum, instead of plasma, to mimic the in vivo context of injury and activation of platelets and the coagulation cascade in the microcirculation. Wound-stimulated TGF-β signaling by confluent BUMPT cells in vitro was suppressed in serum-free medium, shown by p3TP-Luc reporter activity (Figure 2A) and decreased C-terminal phosphorylation of Smad2 (Figure 2B). Serum has abundant TGF-β, but most of the peptide is latent and complexed with large proteins.45 To identify serum factors responsible for TGF-β signaling, we fractionated serum by charcoal stripping. Wound-induced TGF-β signaling was markedly reduced in medium with stripped serum (Figure 2, C and D), and the addition of lipids extracted from serum or charcoal absorbent restored the ability of charcoal-stripped FCS to sustain wound-induced TGF-β signaling (Figure 2, C and D). Moreover, the addition of the lipid extracts to serum-starved BUMPT cells increased the C-terminal phosphorylation of Smad2 (Figure 2E) and reporter activity from p3TP-Luc (data not shown). Because amphophilic serum lipids, such as LPA and S1P, can produce pleiotropic cellular effects by activating GPCR, we tested LPA, S1P, and another serum lipid (the GPCR ligand sphingosylphosphorylcholine) for their ability to transactivate TGF-β signaling. LPA and S1P, but not sphingosylphosphorylcholine, increased the levels of phospho-Smad2 when they were added to serum-starved BUMPT cells (Figure 3A), and the addition of LPA to charcoal-stripped FCS dramatically increased its ability to stimulate Smad2 phosphorylation (Figure 2D). LPA was more potent than S1P in this respect (Figure 3, B and C), and HLZ-56, a pan-LPA receptor antagonist,46 partially, but significantly, attenuated wound-induced p3TP-Luc reporter activity in the presence of serum (Figure 3D). Therefore, we devoted further experimentation to this phospholipid.
Figure 2.

Serum lipids stimulate TGF-β signaling. A: Stably incorporated p3TP-Luc TGF-β reporter BUMPT cells (BM-Lux cells) are grown to be confluent monolayers in full medium. Luciferase activity is measured in cells that were in medium with serum (10% FCS) or without serum (FCS free) for 6 hours after wounding. Luciferase activities normalized by cell protein are shown as mean ± SE relative fold change (n = 6). *, **P < 0.01. B: Confluent quiescent BUMPT cells are wounded and incubated in DMEM with or without 10% FCS for 3 and 6 hours. SDS extracts are immunoblotted for phospho-Smad2 (S465/467) and Smad2. C: Confluent BM-Lux cells are wounded and incubated in DMEM with 10% regular FCS (R-FCS) or C-FCS with or without FCS lipid extract for 6 hours (n = 4). *P < 0.05. D: Confluent quiescent BUMPT cells are wounded and incubated in medium with R-FCS, C-FCS, FCS lipid extract, or 10 μmol/L LPA for 6 hours. SDS extracts are immunoblotted for phospho-Smad2 and Smad2. E: After overnight serum starvation in DMEM/F-12 with supplements of 5 μg/mL transferrin, 6.7 μg/mL sodium selenite, and 4 μg/mL dexamethasone, subconfluent BUMPT cells are treated with medium with 10% FCS, without FCS, or with FCS lipid extract for 1 hour. SDS extracts of cells are immunoblotted for phosphorylated and total Smad2. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure 3.
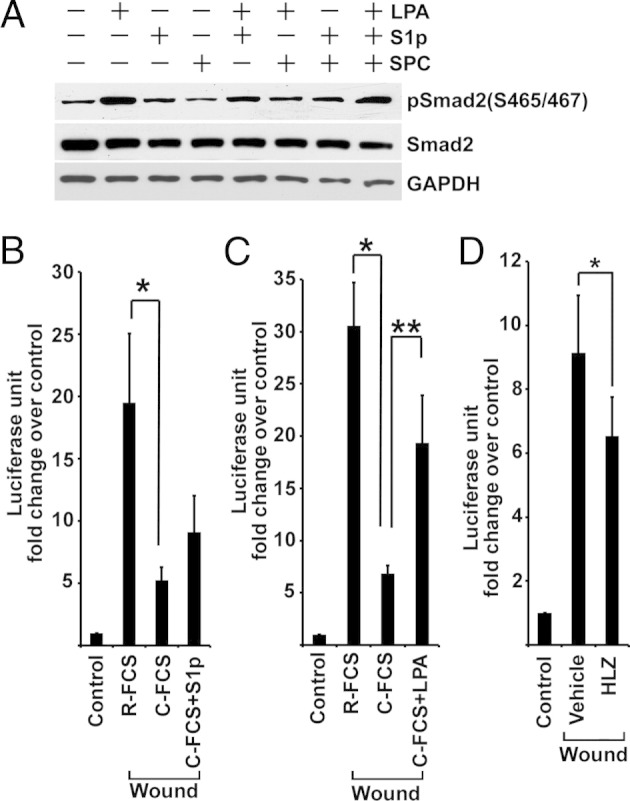
LPA is the major bioactive serum-borne lipid that stimulates TGF-β signaling. A: FCS-starved BUMPT cells are treated with 10 μmol/L LPA, 10 μmol/L S1P, or 10 μmol/L sphingosylphosphorylcholine (SPC) for 45 minutes. Cell lysates are immunoblotted for phospho-Smad2 and Smad2. B: Confluent BM-Lux cells are wounded and treated with 10% regular FCS (R-FCS) or C-FCS with or without 10 μmol/L S1P for 6 hours (n = 6). *P < 0.05. C: Confluent BM-Lux cells are wounded and treated with R-FCS or C-FCS with or without 10 μmol/L LPA for 4 hours (n = 6). *P < 0.01, **P < 0.05. D: Confluent BM-Lux cells are wounded in 10% FCS with or without 10 μmol/L HLZ-56 (HLZ) for 6 hours (n = 6). *P < 0.05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
LPA treatment of BUMPT cells increased TGF-β signaling in a dose- and time-dependent manner, shown by phosphorylation of Smad2 levels (Figure 4, A and B) and stimulation of p3TP-Luc activity (data not shown). LPA-stimulated Smad2 phosphorylation was inhibited by TGF-β type I receptor antagonist, SB431542, and by treatment with TGF-β–neutralizing antibody (Figure 4, C and D). As previously reported, Smad3 was much less abundant in BUMPT cells and Smad3 C-terminal phosphorylation was seen by using Western blot analysis only after stimulation with TGF-β.3 However, compared with the marked LPA-stimulated increase of p3TP-Luc reporter activity (Figure 4E), we observed weaker, but significant, stimulation of (CAGA)12 X Smad3 reporter activity by LPA (Figure 4F), and both activities were inhibited by TGF-β receptor antagonist, SB431542 (Figure 4, E and F). Stimulation of TGF-β signaling by LPA in BUMPT cells was confirmed by signals elicited from stably transfected Smad2-responsive 2 X ARE-Luc reporter (data not shown). Stimulation of TGF-β signaling by LPA was also demonstrated in primary cultures of mouse proximal tubule cells and in cultured human kidney proximal tubule cell lines HK2 and HPCT (Figure 5, A–C).
Figure 4.
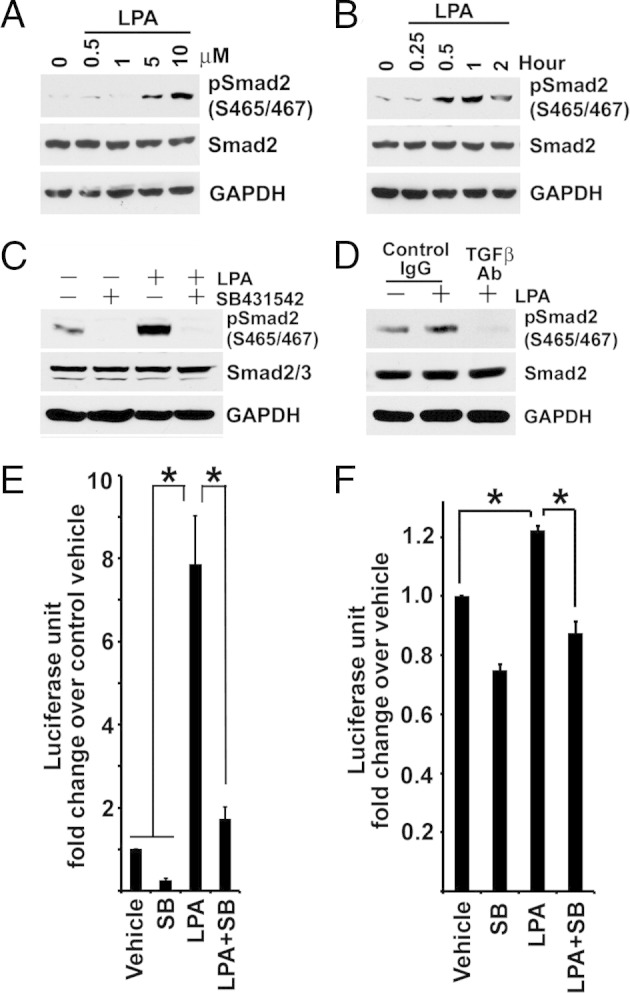
LPA stimulates TGF-β signaling in a dose- and time-dependent manner. A: Overnight FCS-starved subconfluent BUMPT cells are treated with 1.25, 2.5, 5, and 10 μmol/L LPA in FCS-free medium for 1 hour. Cell lysates are immunoblotted for phospho-Smad2 and Smad2. B: Overnight FCS-starved subconfluent BUMPT cells are treated with 10 μmol/L LPA in FCS-free medium for 15 minutes (0.25 hours), 30 minutes (0.5 hours), and 1, 2, and 6 hours. Cell lysates are immunoblotted. C: Subconfluent BUMPT cells are serum starved overnight, pre-incubated without or with 2 μmol/L SB431542 in serum-free medium, and treated with 10 μmol/L LPA for 1 hour. Cells are extracted for Western blot analysis. D: BUMPT cells grown on a membrane filter are pre-incubated with 10 μg/mL TGF-β–neutralizing antibody (TGF-β Ab) or nonimmune IgG in FCS-free medium overnight, and then treated with 10 μmol/L LPA for 1 hour. SDS extracts are immunoblotted for phospho-Smad2 and Smad2. E: Subconfluent BM-Lux cells with stably transfected p3TP-Luc are serum starved overnight, pre-incubated without or with 2 μmol/L SB431542 (SB) in serum-free medium, and then treated with 10 μmol/L LPA for 4 hours. Cells are extracted for luciferase assay (n = 7). * P < 0.01. F: Serum-starved BUMPT cells with stably incorporated (CAGA)12 X TGF-β reporter are pre-incubated with 2 μmol/L SB431542 (SB) and treated with 10 μmol/L LPA for 6 hours in serum-free medium. Luciferase activity is measured, and the values are normalized by cell protein (n = 3). *P < 0.05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure 5.
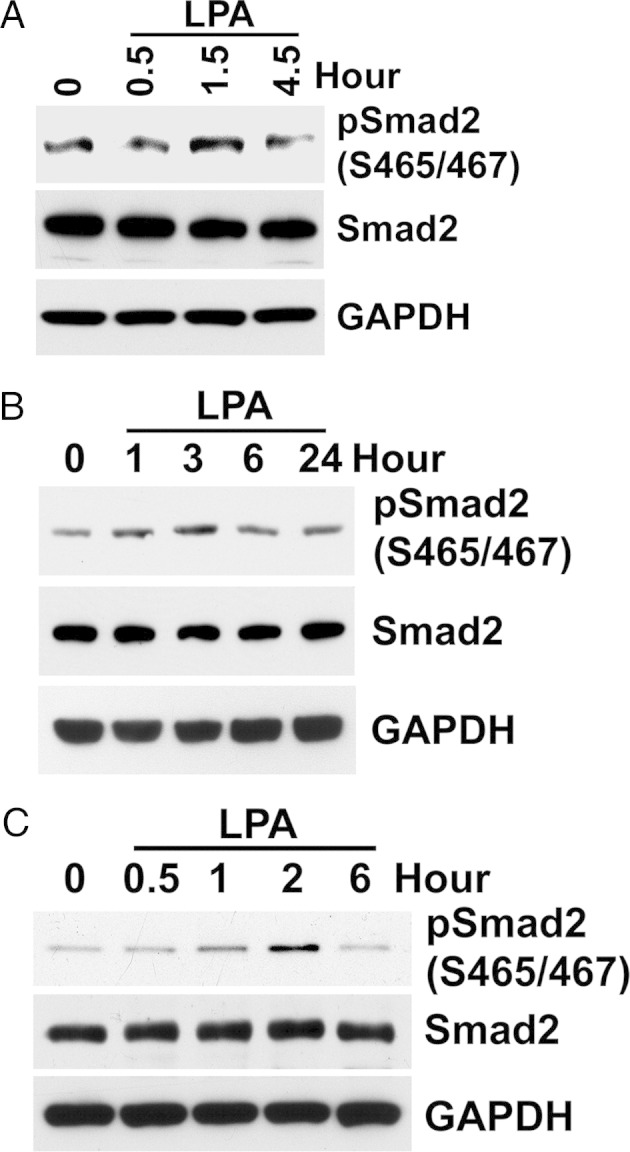
LPA stimulates TGF-β signaling in mouse proximal tubule primary cultures and cultured human proximal tubule cell lines. A: First-passage mouse primary culture of proximal tubule cells grown overnight in serum-free DMEM/F-12 with supplements of 5 μg/mL transferrin, 6.7 μg/mL sodium selenite, and 4 μg/mL dexamethasome. SDS extracts from the indicated time of 10 μmol/L LPA treatment are immunoblotted for Smad2. B and C: Subconfluent human proximal tubule cell lines, HK2 and HPCT, are treated with 10 μmol/L LPA after serum starvation overnight. Cell lysates are extracted at the indicated times and immunoblotted. As shown for BUMPT cells in Figure 4, LPA treatment results in increased phosphorylation of Smad2 in a time-dependent manner, after a variable chronology of phospho-Smad2 increase and a subsequent decrease. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
LPA-Induced TGF-β Activation Is via an LPA2 Receptor
It is known that LPA produces pleiotropic effects in cells through interactions with multiple receptors and divergent effecter pathways. We first determined that LPA-mediated TGF-β activation was mediated specifically through its receptor(s). BUMPT cells were treated with 10 μmol/L LPA, with or without the pan-LPA competitive receptor inhibitor, HLZ-56.46 In the presence of HLZ-56, LPA-induced C-terminal phosphorylation of Smad2 was markedly attenuated (Figure 6A). These impressive effects of HLZ-56 on Smad2 phosphorylation contrasted with only partial inhibition of wound-stimulated p3TP-Luc reporter activity by HLZ-56. This was likely to be explained by the responsiveness of this reporter to noncanonical as well as canonical TGF-β signaling. The noncanonical pathways could be stimulated individually, separately, and in parallel by several other ligand receptor interactions that were potentially active during cell wounding.
Figure 6.
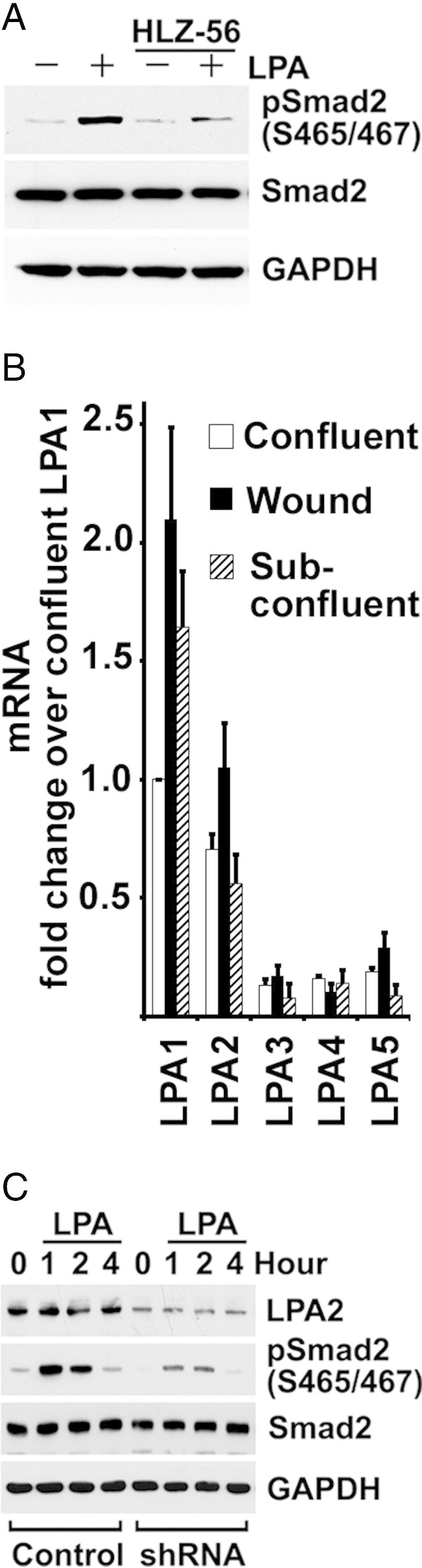
LPA-induced TGF-β activation is via the LPA2 receptor. A: Serum-starved subconfluent BUMPT cells are pre-incubated with 10 μmol/L HLZ-56 for 3 hours and treated with 10 μmol/L LPA for 1 hour. Cell lysates are subjected to Western blot analysis. B: mRNAs are extracted from confluent, subconfluent, and 6 hours of wound BUMPT cells, and analyzed by quantitative PCR. The mRNA fold change over confluent is shown as mean ± SE (n = 4). For comparisons between LPAs 1 and 2 and LPAs 3, 4, and 5, (P < 0.01). C: Subconfluent BUMPT cells with stably transfected LPA2 shRNA or scrambled control are serum starved overnight and treated with 10 μmol/L LPA in serum-free medium. Cells are extracted for Western blot analysis at indicated times. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
We measured the expression of LPA receptors in BUMPT cells by real-time RT-PCR. Although mRNAs for all receptors, LPA1 through LPA5, were expressed, LPA1 and LPA2 were predominant in subconfluent, confluent, and wounded cells (Figure 6B). Because of their relative abundance, we tested the activities of LPA1 and LPA2 receptors in BUMPT cells. Ki16425, a selective antagonist of LPA1 and LPA3 receptors,47 did not inhibit TGF-β signaling induced by LPA, as shown by phosphorylation of Smad2 levels (data not shown). Suspecting that LPA2 receptors were responsible for TGF-β activation, we tested the effects of LPA in cultures of BUMPT cells expressing shRNA specific for LPA2. Western blot analysis confirmed that BUMPT cell LPA2 protein expression was reduced after transduction with LPA2 shRNA compared with controls with scrambled shRNA (Figure 6C). LPA-induced C-terminal Smad2 phosphorylation was inhibited by LPA2 shRNA without affecting total Smad2 levels (Figure 6C), suggesting that LPA2 was the major receptor involved in LPA-induced TGF-β activation.
LPA Activates TGF-β Through a Gαq-Mediated Rho/ROCK Pathway
Next, we investigated whether LPA effects on TGF-β signaling were mediated by specific G proteins coupled to the receptors. BIM46174 is a pan inhibitor that interferes with the Gα/Gβγ protein complex by targeting Gα subunits.48 BIM46174, at a 10 μmol/L concentration, inhibited LPA-mediated TGF-β signaling, shown by Smad2 phosphorylation (Figure 7A) and by (CAGA)12 X Smad3 reporter activity (Figure 7B). LPA effects are generally coupled to Gα proteins, Gαi, Gαq, Gα12/13, and Gαs. Pertussis toxin that inhibits GPCR signaling through ADP-ribosylation of Gαi/o subunits49 had no effects on LPA-stimulated Smad2 phosphorylation or (CAGA)12 X Smad3 reporter activity (Figure 7, A and B), negating a role for Gi/Go. To determine which G-protein subunit was involved in LPA-induced TGF-β activation, Gα C-terminal minigene vectors were transfected individually into BUMPT cells. C-terminal Gα peptides selectively block G-protein binding sites on GPCR.50 LPA-induced Smad2 phosphorylation was blunted by the Gαq/11 minigene, but not by others (Figure 7, C and D). Because signal transduction by LPA via Gαq includes activation of the Rho/Rho-associated protein kinase (ROCK) and phospholipase C/protein kinase C pathways, we first examined the effects of the protein kinase C inhibitor Gö 6983 on LPA-induced TGF-β activation. Gö 6983 had no effect on LPA-induced Smad2 phosphorylation and p3TP Luc reporter activity, excluding phospholipase C/protein kinase C involvement (data not shown). We hypothesized that LPA could signal via RhoA and Rho kinase to induce TGF-β activation. When we stimulated BUMPT cells with 10 μmol/L LPA in the presence of 2 μg/mL of exoenzyme C3 transferase, a specific RhoA/B/C inhibitor, C-terminal phosphorylation of Smad2, was attenuated (Figure 7E). Moreover, we found that Y-27632, an inhibitor of ROCK,51 also attenuated the C-terminal phosphorylation of Smad2 induced by LPA (Figure 7F). Taken together, these results suggested that LPA-induced TGF-β activation was mediated through the Rho/ROCK pathway.
Figure 7.
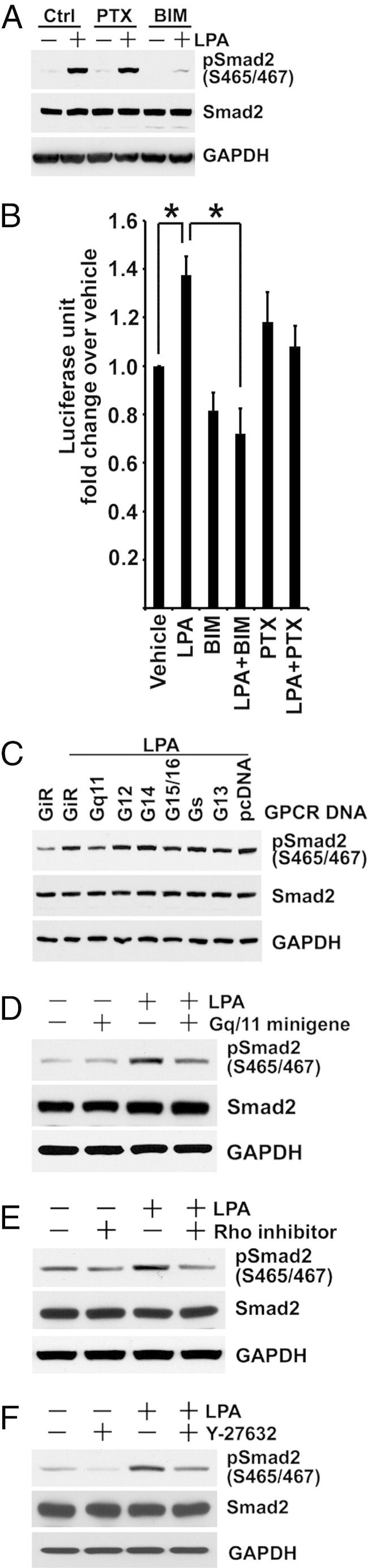
LPA induces TGF-β activation via Gαq-mediated Rho/ROCK pathway. A: After serum-starved subconfluent BUMPT cells are pre-incubated with 10 μmol/L BIM46174 (BIM) or 0.2 μg/mL pertussis toxin (PTX) for 3 hours, cells are treated with 10 μmol/L LPA for 1 hour. Cell lysates are subjected to Western blot analysis. B: BUMPT cells stably transfected with (CAGA)12 X-Luc reporter are treated as described for Figure 7A. Cell extracts after 6 hours of LPA treatment are measured for luciferase activity (n = 3). *P < 0.01. C: BUMPT cells are transfected with Gα C-terminal minigenes or a scrambled control (GiR) using Fugene HD reagent at a 2:5 ratio of DNA:FuGene. After 24 hours, cells are treated with 10 μmol/L LPA in serum-free medium for 1 hour. SDS extracts of cells are immunoblotted for Smad2. D: BUMPT cells are transfected with Gαq minigene or control. After 48 hours, cells are treated with 10 μmol/L LPA for 1 hour in serum-free medium. SDS extracts of cells are immunoblotted for Smad2. E and F: BUMPT cells are grown and serum starved as in Figure 7A. Cells are pre-incubated without or with 2 μg/mL Rho inhibitor C3 transferase or 20 μmol/L Y-27632 for 2 hours, and then treated with 10 μmol/L LPA for 1 hour. The SDS extracts of cells are immunoblotted. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
LPA Induces Integrin αvβ6-Mediated Latent TGF-β Activation
Rho/ROCK signaling has been implicated in the activation of αvβ6 integrin.18 To identify a possible integrin-dependent mechanism for LPA-induced TGF-β activation, we determined whether LPA could activate the latent form of TGF-β in BUMPT cells. In the presence of 6.5 ng/mL latent TGF-β, Smad2 C-terminal phosphorylation was increased compared with controls without latent TGF-β (Figure 8A). However, a further robust increase was induced in response to 10 μmol/L LPA, and sustained C-terminal phosphorylation was seen even after a prolonged interval of 4 hours after LPA treatment (Figure 8A). Similar observations were made using the p3TP-Luc reporter (Figure 8B). These data showed that BUMPT cells converted latent TGF-β to the active peptide under basal conditions and responded to LPA by further increasing the rate of activation. The latent TGF-β peptide, LAPβ1, contains the RGD motif that is a binding site for integrins.10 Therefore, we examined whether access to RGD binding sites governed LPA-induced TGF-β activation in BUMPT cells. Gly-Arg-Gly-Asp-Ser peptide, which blocks integrin binding to RGD sequences, decreased LPA-induced Smad2 phosphorylation compared with Gly-Arg-Gly-Glu-Ser control peptide (Figure 8C). Similarly, Arg-Gly-Asp-Leu-Gly, a peptide based on the TGF-β-LAP sequence, but not the mutant peptide Arg-Gly-Glu-Leu-Gly, inhibited LPA-stimulated Smad2 phosphorylation (Figure 8D).
Figure 8.
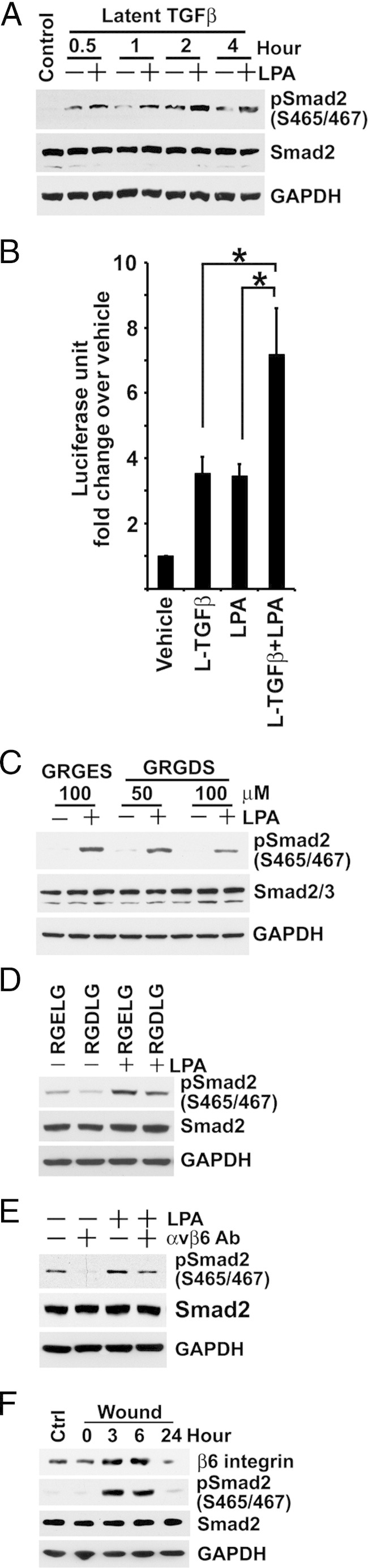
LPA induces integrin αvβ6-mediated latent TGF-β activation. A: Serum-starved subconfluent BUMPT cells are treated with 6.5 ng/mL latent TGF-β with or without 10 μmol/L LPA for the indicated times. Cell lysates are subjected to Western blot analysis for phospho-Smad2 and Smad2. B: Serum-starved subconfluent BM-Lux cells with stably transfected p3TP-Luc are extracted for luciferase activity after 6 hours of treatment with or without latent TGF-β (L-TGF-β) and LPA, as in Figure 8A (n = 5). *P < 0.05. C: Serum-starved subconfluent BUMPT cells are pre-incubated with Gly-Arg-Gly-Asp-Ser (GRGDS) or control peptide Gly-Arg-Gly-Glu-Ser (GRGES) at the indicated concentration for 3 hours, and then treated with 10 μmol/L LPA for 1 hour. SDS extracts of cells are immunoblotted for phospho-Smad2 and Smad2. D: BUMPT cells are plated and serum starved, as previously described. After cells are pre-incubated with 100 μmol/L Arg-Gly-Asp-Leu-Gly (RGDLG) or control peptide Arg-Gly-Glu-Leu-Gly (RGELG) for 3 hours, they are treated with 10 μmol/L LPA for 1 hour. SDS extracts of cells are immunoblotted for phospho-Smad2 and Smad2. E: After subconfluent BUMPT cells are incubated with 10 μg/mL neutralizing 6.3G9 αvβ6 monoclonal antibody (αvβ6 Ab) or nonimmune IgG in serum-free medium overnight, they are treated with 10 μmol/L LPA for 1 hour. SDS extracts of cells are immunoblotted for phospho-Smad2 and Smad2. F: Confluent growth-arrested BUMPT cell monolayers are wounded, and the remaining cell strips are allowed to migrate and proliferate in 10% FCS DMEM for the indicated times. SDS extracts of cells are immunoblotted under nonreducing conditions for β6 integrin with ch2A1 antibody and under reducing conditions for phospho-Smad2 and Smad2. Ctrl, confluent control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Two models have been proposed for integrin-mediated activation of latent TGF-β, which includes enzymatic cleavage of latent TGF-β1 and mechanical conformational change of latent TGF-β.9,52 Recent studies suggested that conformational change induced in the TGF-β1 prodomain by tensile forces pulling on bound αvβ6 is sufficient to release the active peptide.53 Matrix metalloproteinases are also implicated in TGF-β activation. To investigate this hypothesis, we used the general matrix metalloproteinase inhibitor, GM6001. Treatment with 5 to 50 μmol/L of GM6001 had no effect on phospho-Smad2 levels in LPA-stimulated BUMPT cells (data not shown). We also found that a protease inhibitor cocktail with 1 mmol/L aminoethylbenzenesulphonyl fluoride, 50 mmol/L ε-aminocaproic acid, 0.2 mmol/L gabexate, 0.2 mmol/L GM6001, 0.01 mmol/L leupeptin, 0.1 mmol/L D-phenylalanyl-l-prolyl-l-arginine chloromethyl ketone (PPACK), and 1 mmol/L tosyl arginine methyl ester (TAME) also could not inhibit LPA-induced TGF-β signaling (data not shown). The epithelial integrin αvβ6 could directly activate latent TGF-β1 independent of proteolytic activity by contractile cellular forces. To determine whether integrin αvβ6 was involved in LPA-induced RGD motif–mediated activation of TGF-β, we incubated BUMPT cells with 10 μg/mL neutralizing 6.3G9 anti-αvβ6 monoclonal antibody before treatment with LPA. The basal level and LPA-induced C-terminal phosphorylation of Smad2 was significantly decreased by the antibody, demonstrating that both autocrine TGF-β signaling under basal conditions and induced activation by LPA was αvβ6 integrin dependent (Figure 8E). The β6 subunit associates exclusively with the αv subunit to form the functional αvβ6 integrin. Therefore, we used an antibody against the integrin β6 subunit to evaluate protein levels of integrin αvβ6. The levels of β6 integrin protein were low in subconfluent cells and increased when cells reached high density at confluence (data not shown). Notably, the integrin was further induced substantially by 6 hours of wounding to remove approximately 90% of cell mass but returned to levels even lower than confluent nonwounded cells by 24 hours (Figure 8F), when they were again subconfluent and growing.
LPA Stimulates TGF-β–Dependent Production of PDGF-B and CTGF mRNA and Peptide
TGF-β can induce PDGF-B and CTGF expression.54–56 Consequently, we examined if LPA-stimulated TGF-β signaling could induce the production of these profibrotic peptides in cultured proximal tubule cells. Incubation of BUMPT cells with LPA led to rapidly increased expression of mRNA for PDGF-B and CTGF, increased cellular content of CTGF peptide, and substantial secretion of both peptides into the culture medium (Figure 9, A–C). These actions of LPA were substantially blunted by treatment with TGF-β type I receptor antagonist, SB431542 (Figure 9, A–C).
Figure 9.
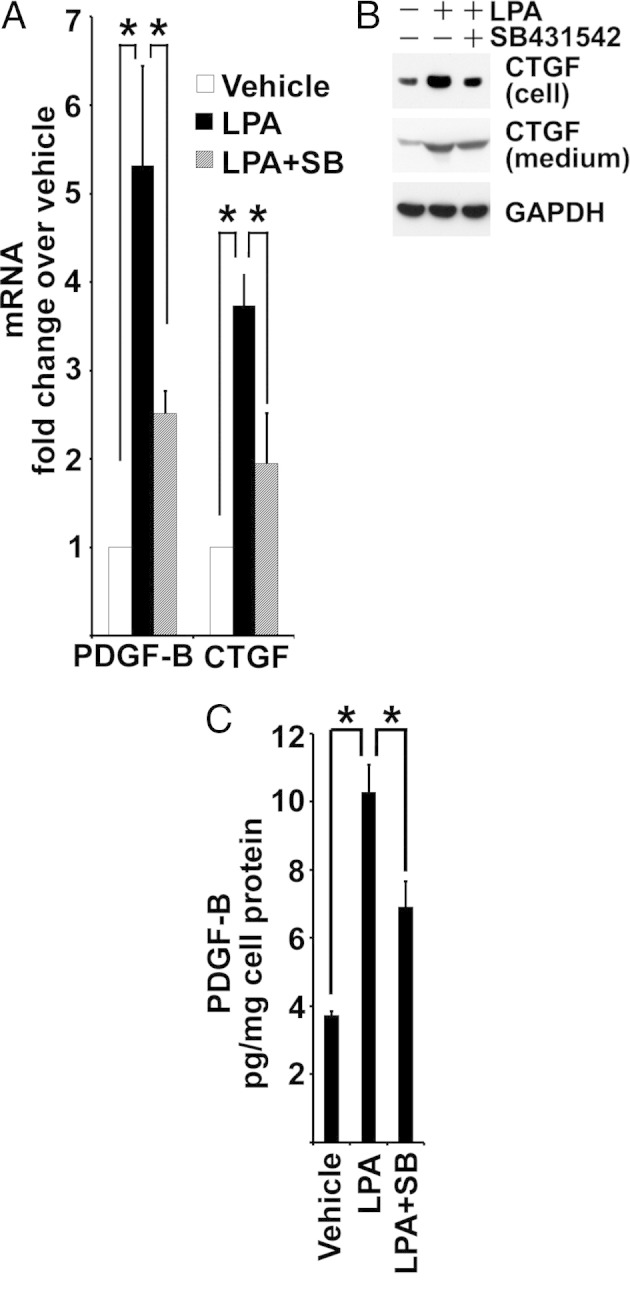
LPA stimulates TGF-β–dependent production of PDGF-B and CTGF. A: Subconfluent BUMPT cells are serum starved in DMEM/F-12 with supplements of 5 μg/mL transferrin, 6.7 μg/mL sodium selenite, and 4 μg/mL dexamethasone overnight. Cells are pre-incubated with 1 μmol/L SB431542 (SB) or vehicle for 2 hours, then treated with or without 10 μmol/L LPA for 1 hour. RNA is extracted for real-time PCR analysis (n = 4). *P < 0.05. B: Subconfluent BUMPT cells are incubated in defined medium overnight, as described for Figure 9A, in the presence of vehicle only, 10 μmol/L LPA, or 10 μmol/L LPA plus 1 μmol/L SB431542. Cells are extracted with SDS buffer. Medium is collected and concentrated using 10-kDa cutoff centrifugal filters and reconstituted with SDS buffer. Cells and medium are immunoblotted for CTGF. C: Concentrated media from BUMPT cells, as described in B, are subjected to PDGF-B analysis by enzyme-linked immunosorbent assay and normalized to total protein extracted from the cells (n = 3). *P < 0.05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
LPA2 and the αvβ6 Integrin Protein Levels Are Increased during Renal Ischemia-Reperfusion Injury in Rats
Finally, we investigated the possible relevance of LPA2 receptors and αvβ6 integrin to renal injury in vivo. Western blot analyses of SDS extracts from the outer stripe of the outer medulla of kidneys reperfused after ischemia showed augmented expression of LPA2 receptor protein by 1 day of reperfusion that increased thereafter, becoming abundant relative to the basal state by 14 days, in effect following a pattern that paralleled the increase of TGF-β and phospho-Smad2 (Figure 10). The expression of β6 integrin protein also increased rapidly by 8 hours of reperfusion, and after subsequent decline, increased again by 14 days (Figure 10). Treatment of rats with TGF-β type I receptor antagonist, SD208, had no effect on IRI-induced expression of LPA2, but decreased that of β6 integrin (Figure 10).
Figure 10.
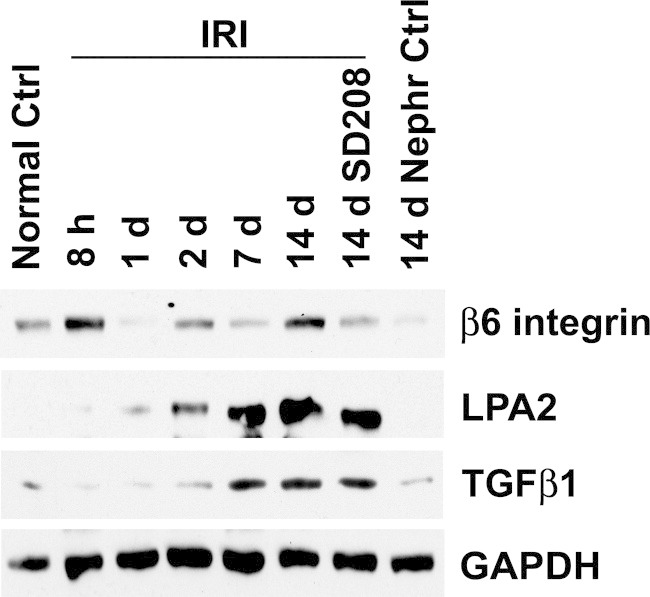
LPA2 and β6 integrin protein levels are increased during renal ischemia-reperfusion injury. Rat kidney SDS extracts, as described for Figure 1A, are immunoblotted for β6 integrin and LPA2. The TGF-β1 blot is the same as shown in Figure 1A. Ctrl, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Discussion
We demonstrate that LPA stimulates the production and secretion of fibrogenic peptides, PDGF-B and CTGF, by cultured kidney proximal tubule cells through a mechanism that transactivates TGF-β. TGF-β transactivation by LPA is mediated by LPA2 receptor signaling transduced by the G-protein, Gαq, and is dependent on αvβ6 integrin-dependent conversion of latent TGF-β to the active peptide. We also found that high TGF-β signaling and tubulointerstitial fibrosis that follows ischemic acute kidney injury is accompanied by increased LPA2 receptor and by enhancement of αvβ6 integrin expression and overproduction of PDGF-B and CTGF by tubules associated with fibrosis. All of these pathological events occurred co-incidentally within the same time frame. The critical role played by TGF-β signaling in tubule overproduction of PDGF-B and CTGF was shown by treatment with a TGF-β type I receptor antagonist that reversed the increased expression of αvβ6 integrin and fibrogenic peptide production. The net result was amelioration of fibrosis.
Experiments on cultured proximal tubule cells provided the more mechanistic information on the serial steps of LPA-induced signal transduction. In addition, these studies showed that αvβ6 integrin-mediated conversion of latent TGF-β required signaling through Rho and ROCK. The specificity of signaling steps involved in LPA-induced secretion of PDGF-B and CTGF was indicated by shRNA knockdown of LPA2, peptide minigene inhibition of Gαq, antagonism of Rho and ROCK, and blockade of LAP binding to αvβ6 integrin, TGF-β–neutralizing antibodies, and type I receptor-specific inhibition of TGF-β signaling.
PDGF-B and CTGF activate kidney pericytes and fibroblasts, causing proliferation, collagen synthesis, and fibrosis.57–59 These peptides are produced in increased amounts by tubules when they regenerate during reperfusion after ischemic injury.4,60 If repair of ischemic damage is complete, increased production of PDGF-B and CTGF should subside, provided that tubules redifferentiate to recover normal structure and function. However, a subset of tubules does not recover normally, fails to redifferentiate, and continues to signal along pathways that were initiated in response to the needs of epithelial regeneration.3,6 Unlike normal proximal tubule epithelium, such tubules without differentiated features are immunoreactive with antibodies to vimentin, an intermediate filament protein expressed by surviving cells as they dedifferentiate and proliferate after injury.6,61 These cells with the phenotype of failed differentiation show persistently high TGF-β signaling, abnormally low phosphatase and tensin homolog (PTEN) levels, and high Jun N-terminal kinase signaling.6 As our current data show, tubules with this phenotype that are surrounded by proliferating myofibroblasts and fibrosis6 continue to express high TGF-β and PDGF-B and CTGF. More important, all of these pathological alterations are ameliorated by treatment with a TGF-β type I receptor antagonist, SD2083,6 (and our current data). The production by diseased tubules of three fibrogenic peptides (TGF-β, PDGF-B, and CTGF) concurrently raises issues of precedence and relative importance of individual peptides in the pathogenesis of fibrosis. As recently indicated, TGF-β could be cooperating with both CTGF and PDGF-B to promote their fibrogenic actions.56–58
LPA mediated αvβ6 integrin-dependent transactivation of TGF-β may be relevant to the kidney pathophysiology that develops during reperfusion after IRI and fibrosis thereafter. This is suggested by increased expression of LPA2 receptor and β6 integrin during reperfusion after a time course similar to that of TGF-β1 (Figure 10). Notably, because fibrosis developed by 2 weeks of reperfusion, increases of TGF-β and phospho-Smad2, and those of LPA2 and β6 integrin, did not subside, as should happen with full recovery, but increased substantially (Figure 10). The expression of CTGF also followed a similar time course. As assessed by IHC, PDGF-B expression was also consistent with such a time scale. The temporal congruency of LPA2 and integrin expression with that of TGF-β and profibrotic peptides after IRI is circumstantial evidence, suggesting that these alterations were causally interrelated during the disease process in vivo, as demonstrated by experiments using cultured cells. Pharmacological antagonism of TGF-β receptor with SD208 did not modify the IRI-induced increase of TGF-β1 protein in the kidney. However, it decreased TGF-β signaling, assessed by Smad2 phosphorylation and nuclear localization of Smad2, corrected the elevations of PDGF-B and CTGF expression (Figure 1), and ameliorated the tubulointerstitial fibrosis caused by IRI.3,6 Together, these data provide a degree of internal consistency, suggesting that our current observations, made in vivo, were meaningful with respect to the disease process. However, we do not understand why SD208 treatment was able to reverse the IRI-induced expression of β6 integrin, but not that of the LPA2 receptor. It seems possible that increased expression of β6 integrin may occur as a consequence of the maladaptive increase of TGF-β signaling in the injured kidney because it was ameliorated by SD208. On the other hand, increased LPA2 could be related to another injury-associated mechanism that is TGF-β independent.
The actions of αvβ6 integrin to activate latent TGF-β are well recognized. However, the role played by GPCR as a proximal signaling trigger that leads to the integrin step has only recently been identified by the work of Jenkins, Sheppard, and colleagues.15,18 Protease activated receptor (PAR)1-mediated thrombin-initiated GPCR signaling led to αvβ6 integrin-dependent transactivation of TGF-β in models of acute lung injury including bleomycin-induced pulmonary damage, and LPA-initiated LPA2-mediated GPCR signaling caused αvβ6 integrin-dependent transactivation of TGF-β in bleomycin-induced pulmonary fibrosis.15,18 The signaling pathway was traced in detail by Xu et al18 from the LPA2 receptor to αvβ6 integrin via Gαq, Rho, and Rho kinase. Our study finds that the signaling mechanism is similar in proximal tubule cells and extends the observations by connecting this intricate pathway to cellular expression and secretion of fibrogenic peptides, PDGF-B and CTGF. More limited observations connect LPA-initiated TGF-β–dependent expression of CTGF in cultured myocytes.62 Although Ma et al17 identified αvβ6 integrin-mediated TGF-β–dependent fibrosis in a model of ureteral obstruction, they also found strong evidence for an integrin and TGF-β–independent mechanism; interestingly, the TGF-β–independent mechanism was initiated by another GPCR ligand, angiotensin II. More interestingly, Chen et al21 reported that the profibrotic actions of angiotensin II are related to transactivation of epidermal growth factor receptor and subsequent activation of an extracellular signal–regulated kinase–mediated pathway that increased the production of TGF-β. On the other hand, Pradere et al22 found that fibrosis in the ureteral obstruction model was related to increased production of LPA and signaling via the LPA1 receptor. Together with other evidence that has connected GPCR ligands LPA, thrombin, and angiotensin II to TGF-β production and/or renal fibrosis, we suspect that the analysis of GPCR-based signaling mechanisms that cause TGF-β activation or overproduction will be complex. This is made more difficult by multiple GPCR ligands and receptors, receptor redundancy, and interactions with other signaling mechanisms. Indeed, we cannot exclude other GPCR ligands, such as S1P, that have protective activity during the acute phase of IRI,30 which may have late effects to modify the actions of LPA. Based on these considerations, it would appear that interventions based on a single ligand or a single class of receptor are unlikely to yield conclusive information immediately relevant to the treatment of fibrosis. A full understanding of what actually happens in the diseased environment will require the detailed delineation of signaling by multiple classes of GPCR and G proteins.
Acknowledgments
We thank Daniel Rifkin for the MLEC cells with the PAI-I-luciferase construct, Grégoire Prévost for BIM46174, Glenn Prestwich for HLZ-56, John Schwartz and Wilfred Lieberthal for the BUMPT cells, Ulrich Hopfer for HPCT human proximal tubule cells, Heidi Hamm and laboratory for Gα C-terminal minigene vectors, and Drs. George Schreiner, Linda Higgins, and Satya Medicherla (formerly of SCIOS Inc., now a subsidiary of Johnson & Johnson) for SD208, which is now available from Calbiochem.
Footnotes
Supported by grants from the NIH (DK37139 to M.A.V. and DK54472 to P.S.).
CME Disclosure: The authors of this article and the planning committee members and staff have no relevant financial relationships with commercial interest to disclose.
References
- 1.Spurgeon K.R., Donohoe D.L., Basile D.P. Transforming growth factor-beta in acute renal failure: receptor expression, effects on proliferation, cellularity, and vascularization after recovery from injury. Am J Physiol Renal Physiol. 2005;288:F568–F577. doi: 10.1152/ajprenal.00330.2004. [DOI] [PubMed] [Google Scholar]
- 2.Nath K.A., Croatt A.J., Warner G.M., Grande J.P. Genetic deficiency of Smad3 protects against murine ischemic acute kidney injury. Am J Physiol Renal Physiol. 2011;301:F436–F442. doi: 10.1152/ajprenal.00162.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geng H., Lan R., Wang G., Siddiqi A.R., Naski M.C., Brooks A.I., Barnes J.L., Saikumar P., Weinberg J.M., Venkatachalam M.A. Inhibition of autoregulated TGFbeta signaling simultaneously enhances proliferation and differentiation of kidney epithelium and promotes repair following renal ischemia. Am J Pathol. 2009;174:1291–1308. doi: 10.2353/ajpath.2009.080295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang L., Besschetnova T.Y., Brooks C.R., Shah J.V., Bonventre J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543. doi: 10.1038/nm.2144. 1p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basile D.P., Martin D.R., Hammerman M.R. Extracellular matrix-related genes in kidney after ischemic injury: potential role for TGF-beta in repair. Am J Physiol. 1998;275:F894–F903. doi: 10.1152/ajprenal.1998.275.6.F894. [DOI] [PubMed] [Google Scholar]
- 6.Lan R., Geng H., Polichnowski A.J., Singha P.K., Saikumar P., McEwen D.G., Griffin K.A., Koesters R., Weinberg J.M., Bidani A.K., Kriz W., Venkatachalam M.A. PTEN loss defines a TGF-beta-induced tubule phenotype of failed differentiation and JNK signaling during renal fibrosis. Am J Physiol Renal Physiol. 2012;302:F1210–F1223. doi: 10.1152/ajprenal.00660.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basile D.P., Rovak J.M., Martin D.R., Hammerman M.R. Increased transforming growth factor-beta 1 expression in regenerating rat renal tubules following ischemic injury. Am J Physiol. 1996;270:F500–F509. doi: 10.1152/ajprenal.1996.270.3.F500. [DOI] [PubMed] [Google Scholar]
- 8.Munger J.S., Harpel J.G., Gleizes P.E., Mazzieri R., Nunes I., Rifkin D.B. Latent transforming growth factor-beta: structural features and mechanisms of activation. Kidney Int. 1997;51:1376–1382. doi: 10.1038/ki.1997.188. [DOI] [PubMed] [Google Scholar]
- 9.Jenkins G. The role of proteases in transforming growth factor-beta activation. Int J Biochem Cell Biol. 2008;40:1068–1078. doi: 10.1016/j.biocel.2007.11.026. [DOI] [PubMed] [Google Scholar]
- 10.Munger J.S., Huang X., Kawakatsu H., Griffiths M.J., Dalton S.L., Wu J., Pittet J.F., Kaminski N., Garat C., Matthay M.A., Rifkin D.B., Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 11.Crawford S.E., Stellmach V., Murphy-Ullrich J.E., Ribeiro S.M., Lawler J., Hynes R.O., Boivin G.P., Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- 12.Murphy-Ullrich J.E., Poczatek M. Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. 2000;11:59–69. doi: 10.1016/s1359-6101(99)00029-5. [DOI] [PubMed] [Google Scholar]
- 13.Nishimura S.L. Integrin-mediated transforming growth factor-beta activation, a potential therapeutic target in fibrogenic disorders. Am J Pathol. 2009;175:1362–1370. doi: 10.2353/ajpath.2009.090393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris D.G., Huang X., Kaminski N., Wang Y., Shapiro S.D., Dolganov G., Glick A., Sheppard D. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature. 2003;422:169–173. doi: 10.1038/nature01413. [DOI] [PubMed] [Google Scholar]
- 15.Jenkins R.G., Su X., Su G., Scotton C.J., Camerer E., Laurent G.J., Davis G.E., Chambers R.C., Matthay M.A., Sheppard D. Ligation of protease-activated receptor 1 enhances alpha(v) beta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J Clin Invest. 2006;116:1606–1614. doi: 10.1172/JCI27183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hahm K., Lukashev M.E., Luo Y., Yang W.J., Dolinski B.M., Weinreb P.H., Simon K.J., Chun Wang L., Leone D.R., Lobb R.R., McCrann D.J., Allaire N.E., Horan G.S., Fogo A., Kalluri R., Shield C.F., 3rd, Sheppard D., Gardner H.A., Violette S.M. Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am J Pathol. 2007;170:110–125. doi: 10.2353/ajpath.2007.060158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma L.J., Yang H., Gaspert A., Carlesso G., Barty M.M., Davidson J.M., Sheppard D., Fogo A.B. Transforming growth factor-beta-dependent and -independent pathways of induction of tubulointerstitial fibrosis in beta6(−/−) mice. Am J Pathol. 2003;163:1261–1273. doi: 10.1016/s0002-9440(10)63486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu M.Y., Porte J., Knox A.J., Weinreb P.H., Maher T.M., Violette S.M., McAnulty R.J., Sheppard D., Jenkins G. Lysophosphatidic acid induces alphavbeta6 integrin-mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q) Am J Pathol. 2009;174:1264–1279. doi: 10.2353/ajpath.2009.080160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breuss J.M., Gallo J., DeLisser H.M., Klimanskaya I.V., Folkesson H.G., Pittet J.F., Nishimura S.L., Aldape K., Landers D.V., Carpenter W., Gillett N., Sheppard D., Matthay M.A., Albelda S.M., Kramer R.H., Pytela R. Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J Cell Sci. 1995;108(Pt 6):2241–2251. doi: 10.1242/jcs.108.6.2241. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Y., Poczatek M.H., Berecek K.H., Murphy-Ullrich J.E. Thrombospondin 1 mediates angiotensin II induction of TGF-beta activation by cardiac and renal cells under both high and low glucose conditions. Biochem Biophys Res Commun. 2006;339:633–641. doi: 10.1016/j.bbrc.2005.11.060. [DOI] [PubMed] [Google Scholar]
- 21.Chen J., Chen J.K., Nagai K., Plieth D., Tan M., Lee T.C., Threadgill D.W., Neilson E.G., Harris R.C. EGFR signaling promotes TGF beta-dependent renal fibrosis. J Am Soc Nephrol. 2012;23:215–224. doi: 10.1681/ASN.2011070645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pradere J.P., Klein J., Gres S., Guigne C., Neau E., Valet P., Calise D., Chun J., Bascands J.L., Saulnier-Blache J.S., Schanstra J.P. LPA1 receptor activation promotes renal interstitial fibrosis. J Am Soc Nephrol. 2007;18:3110–3118. doi: 10.1681/ASN.2007020196. [DOI] [PubMed] [Google Scholar]
- 23.Jo S.K., Bajwa A., Awad A.S., Lynch K.R., Okusa M.D. Sphingosine-1-phosphate receptors: biology and therapeutic potential in kidney disease. Kidney Int. 2008;73:1220–1230. doi: 10.1038/ki.2008.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jo S.K., Bajwa A., Ye H., Vergis A.L., Awad A.S., Kharel Y., Lynch K.R., Okusa M.D. Divergent roles of sphingosine kinases in kidney ischemia-reperfusion injury. Kidney Int. 2009;75:167–175. doi: 10.1038/ki.2008.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xin C., Ren S., Kleuser B., Shabahang S., Eberhardt W., Radeke H., Schafer-Korting M., Pfeilschifter J., Huwiler A. Sphingosine 1-phosphate cross-activates the Smad signaling cascade and mimics transforming growth factor-beta-induced cell responses. J Biol Chem. 2004;279:35255–35262. doi: 10.1074/jbc.M312091200. [DOI] [PubMed] [Google Scholar]
- 26.Okusa M.D., Ye H., Huang L., Sigismund L., Macdonald T., Lynch K.R. Selective blockade of lysophosphatidic acid LPA3 receptors reduces murine renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2003;285:F565–F574. doi: 10.1152/ajprenal.00023.2003. [DOI] [PubMed] [Google Scholar]
- 27.Grandaliano G., Di Paolo S., Monno R., Stallone G., Ranieri E., Pontrelli P., Gesualdo L., Schena F.P. Protease-activated receptor 1 and plasminogen activator inhibitor 1 expression in chronic allograft nephropathy: the role of coagulation and fibrinolysis in renal graft fibrosis. Transplantation. 2001;72:1437–1443. doi: 10.1097/00007890-200110270-00018. [DOI] [PubMed] [Google Scholar]
- 28.Favreau F., Thuillier R., Cau J., Milin S., Manguy E., Mauco G., Zhu X., Lerman L.O., Hauet T. Anti-thrombin therapy during warm ischemia and cold preservation prevents chronic kidney graft fibrosis in a DCD model. Am J Transplant. 2010;10:30–39. doi: 10.1111/j.1600-6143.2009.02924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sevastos J., Kennedy S.E., Davis D.R., Sam M., Peake P.W., Charlesworth J.A., Mackman N., Erlich J.H. Tissue factor deficiency and PAR-1 deficiency are protective against renal ischemia reperfusion injury. Blood. 2007;109:577–583. doi: 10.1182/blood-2006-03-008870. [DOI] [PubMed] [Google Scholar]
- 30.Bajwa A., Jo S.K., Ye H., Huang L., Dondeti K.R., Rosin D.L., Haase V.H., Macdonald T.L., Lynch K.R., Okusa M.D. Activation of sphingosine-1-phosphate 1 receptor in the proximal tubule protects against ischemia-reperfusion injury. J Am Soc Nephrol. 2010;21:955–965. doi: 10.1681/ASN.2009060662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okusa M.D., Lynch K.R. Targeting sphingosine 1 phosphate receptor type 1 receptors in acute kidney injury. Drug Discov Today Dis Mech. 2007;4:55–59. doi: 10.1016/j.ddmec.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Efrati S., Berman S., Hamad R.A., Siman-Tov Y., Ilgiyaev E., Maslyakov I., Weissgarten J. Effect of captopril treatment on recuperation from ischemia/reperfusion-induced acute renal injury. Nephrol Dial Transplant. 2012;27:136–145. doi: 10.1093/ndt/gfr256. [DOI] [PubMed] [Google Scholar]
- 33.Grenz A., Bauerle J.D., Dalton J.H., Ridyard D., Badulak A., Tak E., McNamee E.N., Clambey E., Moldovan R., Reyes G., Klawitter J., Ambler K., Magee K., Christians U., Brodsky K.S., Ravid K., Choi D.S., Wen J., Lukashev D., Blackburn M.R., Osswald H., Coe I.R., Nurnberg B., Haase V.H., Xia Y., Sitkovsky M., Eltzschig H.K. Equilibrative nucleoside transporter 1 (ENT1) regulates postischemic blood flow during acute kidney injury in mice. J Clin Invest. 2012;122:693–710. doi: 10.1172/JCI60214. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Weinreb P.H., Simon K.J., Rayhorn P., Yang W.J., Leone D.R., Dolinski B.M., Pearse B.R., Yokota Y., Kawakatsu H., Atakilit A., Sheppard D., Violette S.M. Function-blocking integrin alphavbeta6 monoclonal antibodies: distinct ligand-mimetic and nonligand-mimetic classes. J Biol Chem. 2004;279:17875–17887. doi: 10.1074/jbc.M312103200. [DOI] [PubMed] [Google Scholar]
- 35.Wrana J.L., Attisano L., Carcamo J., Zentella A., Doody J., Laiho M., Wang X.F., Massague J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–1014. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- 36.Dennler S., Itoh S., Vivien D., ten Dijke P., Huet S., Gauthier J.M. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Itoh S., Thorikay M., Kowanetz M., Moustakas A., Itoh F., Heldin C.H., ten Dijke P. Elucidation of Smad requirement in transforming growth factor-beta type I receptor-induced responses. J Biol Chem. 2003;278:3751–3761. doi: 10.1074/jbc.M208258200. [DOI] [PubMed] [Google Scholar]
- 38.Gilchrist A., Li A., Hamm H.E. Design and use of C-terminal minigene vectors for studying role of heterotrimeric G proteins. Methods Enzymol. 2002;344:58–69. doi: 10.1016/s0076-6879(02)44705-2. [DOI] [PubMed] [Google Scholar]
- 39.Lan R., Geng H., Hwang Y., Mishra P., Skloss W.L., Sprague E.A., Saikumar P., Venkatachalam M.A. A novel wounding device suitable for quantitative biochemical analysis of wound healing and regeneration of cultured epithelium. Wound Repair Regen. 2010;18:159–167. doi: 10.1111/j.1524-475X.2010.00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abe M., Harpel J.G., Metz C.N., Nunes I., Loskutoff D.J., Rifkin D.B. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- 41.Lee M.J., Van Brocklyn J.R., Thangada S., Liu C.H., Hand A.R., Menzeleev R., Spiegel S., Hla T. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science. 1998;279:1552–1555. doi: 10.1126/science.279.5356.1552. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Z., Xu Y. An extremely simple method for extraction of lysophospholipids and phospholipids from blood samples. J Lipid Res. 2010;51:652–659. doi: 10.1194/jlr.D001503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuen P.S., Dunn S.R., Miyaji T., Yasuda H., Sharma K., Star R.A. A simplified method for HPLC determination of creatinine in mouse serum. Am J Physiol Renal Physiol. 2004;286:F1116–F1119. doi: 10.1152/ajprenal.00366.2003. [DOI] [PubMed] [Google Scholar]
- 44.Kapoun A.M., Gaspar N.J., Wang Y., Damm D., Liu Y.W., O'Young G., Quon D., Lam A., Munson K., Tran T.T., Ma J.Y., Murphy A., Dugar S., Chakravarty S., Protter A.A., Wen F.Q., Liu X., Rennard S.I., Higgins L.S. Transforming growth factor-beta receptor type 1 (TGFbetaRI) kinase activity but not p38 activation is required for TGFbetaRI-induced myofibroblast differentiation and profibrotic gene expression. Mol Pharmacol. 2006;70:518–531. doi: 10.1124/mol.105.021600. [DOI] [PubMed] [Google Scholar]
- 45.O'Connor-McCourt M.D., Wakefield L.M. Latent transforming growth factor-beta in serum: a specific complex with alpha 2-macroglobulin. J Biol Chem. 1987;262:14090–14099. [PubMed] [Google Scholar]
- 46.Jiang G., Xu Y., Fujiwara Y., Tsukahara T., Tsukahara R., Gajewiak J., Tigyi G., Prestwich G.D. Alpha-substituted phosphonate analogues of lysophosphatidic acid (LPA) selectively inhibit production and action of LPA. Chem Med Chem. 2007;2:679–690. doi: 10.1002/cmdc.200600280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohta H., Sato K., Murata N., Damirin A., Malchinkhuu E., Kon J., Kimura T., Tobo M., Yamazaki Y., Watanabe T., Yagi M., Sato M., Suzuki R., Murooka H., Sakai T., Nishitoba T., Im D.S., Nochi H., Tamoto K., Tomura H., Okajima F. Ki16425, a subtype-selective antagonist for EDG-family lysophosphatidic acid receptors. Mol Pharmacol. 2003;64:994–1005. doi: 10.1124/mol.64.4.994. [DOI] [PubMed] [Google Scholar]
- 48.Ayoub M.A., Damian M., Gespach C., Ferrandis E., Lavergne O., De Wever O., Baneres J.L., Pin J.P., Prevost G.P. Inhibition of heterotrimeric G protein signaling by a small molecule acting on Galpha subunit. J Biol Chem. 2009;284:29136–29145. doi: 10.1074/jbc.M109.042333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wettschureck N., Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005;85:1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- 50.Gilchrist A., Li A., Hamm H.E. G alpha COOH-terminal minigene vectors dissect heterotrimeric G protein signaling. Sci STKE. 2002;2002:pl1. doi: 10.1126/stke.2002.118.pl1. [DOI] [PubMed] [Google Scholar]
- 51.Uehata M., Ishizaki T., Satoh H., Ono T., Kawahara T., Morishita T., Tamakawa H., Yamagami K., Inui J., Maekawa M., Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 52.Giacomini M.M., Travis M.A., Kudo M., Sheppard D. Epithelial cells utilize cortical actin/myosin to activate latent TGF-beta through integrin alpha(v)beta(6)-dependent physical force. Exp Cell Res. 2012;318:716–722. doi: 10.1016/j.yexcr.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi M., Zhu J., Wang R., Chen X., Mi L., Walz T., Springer T.A. Latent TGF-beta structure and activation. Nature. 2011;474:343–349. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qi W., Chen X., Poronnik P., Pollock C.A. Transforming growth factor-beta/connective tissue growth factor axis in the kidney. Int J Biochem Cell Biol. 2008;40:9–13. doi: 10.1016/j.biocel.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 55.Taylor L.M., Khachigian L.M. Induction of platelet-derived growth factor B-chain expression by transforming growth factor-beta involves transactivation by Smads. J Biol Chem. 2000;275:16709–16716. doi: 10.1074/jbc.275.22.16709. [DOI] [PubMed] [Google Scholar]
- 56.Luft F.C. CCN2, the connective tissue growth factor. J Mol Med (Berl) 2008;86:1–3. doi: 10.1007/s00109-007-0287-x. [DOI] [PubMed] [Google Scholar]
- 57.Chen Y.T., Chang F.C., Wu C.F., Chou Y.H., Hsu H.L., Chiang W.C., Shen J., Chen Y.M., Wu K.D., Tsai T.J., Duffield J.S., Lin S.L. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011;80:1170–1181. doi: 10.1038/ki.2011.208. [DOI] [PubMed] [Google Scholar]
- 58.Chen X.M., Qi W., Pollock C.A. CTGF and chronic kidney fibrosis. Front Biosci (Schol Ed) 2009;1:132–141. doi: 10.2741/S13. [DOI] [PubMed] [Google Scholar]
- 59.Tang W.W., Ulich T.R., Lacey D.L., Hill D.C., Qi M., Kaufman S.A., Van G.Y., Tarpley J.E., Yee J.S. Platelet-derived growth factor-BB induces renal tubulointerstitial myofibroblast formation and tubulointerstitial fibrosis. Am J Pathol. 1996;148:1169–1180. [PMC free article] [PubMed] [Google Scholar]
- 60.Nakagawa T., Sasahara M., Haneda M., Kataoka H., Nakagawa H., Yagi M., Kikkawa R., Hazama F. Role of PDGF B-chain and PDGF receptors in rat tubular regeneration after acute injury. Am J Pathol. 1999;155:1689–1699. doi: 10.1016/S0002-9440(10)65484-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grone H.J., Weber K., Grone E., Helmchen U., Osborn M. Coexpression of keratin and vimentin in damaged and regenerating tubular epithelia of the kidney. Am J Pathol. 1987;129:1–8. [PMC free article] [PubMed] [Google Scholar]
- 62.Cabello-Verrugio C., Cordova G., Vial C., Zuniga L.M., Brandan E. Connective tissue growth factor induction by lysophosphatidic acid requires transactivation of transforming growth factor type beta receptors and the JNK pathway. Cell Signal. 2011;23:449–457. doi: 10.1016/j.cellsig.2010.10.019. [DOI] [PubMed] [Google Scholar]