

Abstract
Hyaluronan is activated by fragmentation and controls inflammation and fibroplasia during wound repair and diseases (eg, cancer). Hyaluronan-binding peptides were identified that modify fibrogenesis during skin wound repair. Peptides were selected from 7- to 15mer phage display libraries by panning with hyaluronan-Sepharose beads and assayed for their ability to block fibroblast migration in response to hyaluronan oligosaccharides (10 kDa). A 15mer peptide (P15-1), with homology to receptor for hyaluronan mediated motility (RHAMM) hyaluronan binding sequences, was the most effective inhibitor. P15-1 bound to 10-kDa hyaluronan with an affinity of Kd = 10−7 and appeared to specifically mimic RHAMM since it significantly reduced binding of hyaluronan oligosaccharides to recombinant RHAMM but not to recombinant CD44 or TLR2,4, and altered wound repair in wild-type but not RHAMM−/− mice. One topical application of P15-1 to full-thickness excisional rat wounds significantly reduced wound macrophage number, fibroblast number, and blood vessel density compared to scrambled, negative control peptides. Wound collagen 1, transforming growth factor β-1, and α-smooth muscle actin were reduced, whereas tenascin C was increased, suggesting that P15-1 promoted a form of scarless healing. Signaling/microarray analyses showed that P15-1 blocks RHAMM-regulated focal adhesion kinase pathways in fibroblasts. These results identify a new class of reagents that attenuate proinflammatory, fibrotic repair by blocking hyaluronan oligosaccharide signaling.
Hyaluronan (HA) is a key extracellular matrix regulator of innate immune processes, fibroblast functions, and cell migration during normal response to injury.1–8 During wound repair, HA synthesis is required for mesenchymal differentiation, including transforming growth factor β-1 (TGFβ1)-mediated differentiation of myofibroblasts, collagen production by dermal fibroblasts, and endothelial cell proliferation and migration.9–13 Wound HA is also a critical regulator of both innate immune responses during tissue repair and dendritic cell function that link adaptive immunity to innate immune responses during repair. The effects of HA on these aspects of wound healing depend on its molecular weight. For example, native high molecular weight HA (eg, >500 kDa) reduces inflammation3,14–17 and angiogenesis, and during skin excisional wound repair, increases the expression of transforming growth factor β (TGFβ3), metalloproteinase 1 and 3 (MMP1, -3), and collagen III (Col3A1) in human dermal fibroblasts.18–20 By contrast, fragmented HA that is present in intermediate sizes (50 to 400 kDa), and as smaller oligosaccharides (<20 kDa), promotes inflammation, angiogenesis, and migration of most cell types involved in tissue repair. HA fragments also strongly promote expression of collagen I (Col1A1), tissue inhibitor of metalloproteinase 1 (TIMP1), and TGFβ1.21–28 The biological differences of native HA versus fragments are due in part to their ability to activate specific signaling cascades through HA receptors such as CD44 (cluster designation 44), receptor for hyaluronan mediated motility (RHAMM; human form, HMMR) and Toll-like receptors 2 and 4 (TLR-2, -4). For example, HA fragment:HA receptor interactions activate AP-1 and NFκB-regulated signaling cascades, which culminate in the expression of genes that enhance inflammation and angiogenesis, and promote cell migration.4,29,30 Specific blockade of these HA fragments could have significant therapeutic potential for treating diseases that result from chronically high levels of proinflammatory cytokines and consequent elevation in angiogenesis and cell migration. To date, pathologies involving HA fragmentation include inflammatory bowel disease,31 arthritis,8,32 disorders leading to chronic fibrosis (eg, lung), and cancers, such as bladder, prostate, and breast cancer in which HA accumulation/fragmentation is associated with poor prognosis.1,2,33
HA-binding peptides that can interfere with the biological functions of HA have previously been designed and demonstrated to modify tissue fibrosis, wound infection, contact hypersensitivity, and melanoma metastases in experimental models providing proof of concept34–37 for the use of peptides to influence HA-driven disease processes. However, their mechanism of action is still not understood since both binding affinity to HA and sequence homology to known HA receptors are low (<25%, eg, Mummert et al38).
Previous studies in our laboratory using RHAMM-null mice demonstrated an involvement of this HA receptor in the healing of excisional dermal wounds.39 In particular, RHAMM loss blunted fibrotic repair, reduced white cell migration into wounds, and reduced myofibroblast differentiation. This study suggested that blocking RHAMM/HA interactions could have therapeutic benefits in repair and disease processes. In the present study, our goal was to develop synthetic peptides that mimic specific structural motifs found within RHAMM that are required for binding to HA and that block HA fragmentation–driven events during excisional skin wound repair, in particular inflammation and fibrogenesis. Toward this end 7-, 12-, and 15mer phage display libraries were panned with HA fragments/oligosaccharides linked to Sepharose beads. This approach led to the identification of a 15mer peptide (P15-1) that is highly homologous to the HA binding sequence of RHAMM. P15-1 preferentially binds HA oligosaccharides (<10 kDa) with a moderate affinity (kDa = 10−7 mol/L). Here, we show that this peptide specifically mimics and blocks RHAMM:HA signaling through focal adhesion kinase (FAK), resulting in the modification of wound healing responses in wild-type (eg, RHAMM+/+) but not RHAMM−/− wounds. We interpret our results to suggest that interference with RHAMM/HA fragment interactions reduces fibrogenesis and scar formation, both as a result of blunting the proinflammatory functions of macrophage subpopulations and by directly modifying specific mesenchymal properties of wound cells such as myofibroblast differentiation via blocking key signaling pathways.
Materials and Methods
Materials
Chemoenzymatically synthesized HA standards of low polydispersity and known molecular mass based on size exclusion chromatography with multiangle light scattering (Select-HA: standard mixtures HiLadder and LoLadder), as well as streptavidin-based assemblies of one to four biotin–end-labeled HA molecules (Mega-HA) were obtained from Hyalose LLC (Oklahoma City, OK) for use in agarose gel electrophoresis analyses.40 HA used for screening random phage libraries, isothermal titration calorimetry (ITC), enzyme-linked immunosorbent assay (ELISA) binding studies, and scratch wound–induced cell migration experiments had an average molecular weight of 240 kDa and was the kind gift of Hyal Pharmaceutical Corp (Mississauga, ON, Canada). Higher molecular weight HA used for ITC (500 kDa) was also the kind gift of Hyal Pharmaceutical Corp. HA oligosaccharides were prepared by partially digesting hyaluronidase with Streptococcus to generate a heterogeneous mixture of HA fragments [Oligo HA, average molecular weight (MWav): 10 kDa].40 Purified HA oligosaccharides of specific sizes that were used for stimulating cell migration were the kind gift of Seikagaku Corp. (Tokyo, Japan). All HA preparations were free of protein or nucleic acid contamination, as determined by gel electrophoresis followed by Coomassie Blue and ethidium bromide staining, respectively, and free of endotoxin as determined by an endotoxin assay (endotoxin assay kit; GenScript, Piscataway, NJ). Chondroitin 4/6 sulfate was purchased from Sigma (St. Louis, MO). Agarose (agarose NA, −mr = 0.10) was obtained from GE Healthcare (Little Chalfont, UK). Pierce Strong Anion Exchange mini spin columns and Slide-A-Lyzer mini dialysis units with a 10,000 molecular-weight cutoff were obtained from Thermo Scientific (Waltham, MA). Stains-All dye (3, 3′-dimethyl-9-methyl-4, 5, 4′, 5′-dibenzothiacarbocyanine) and bromophenol blue tracking dye were obtained from Bio-Rad Laboratories (Hercules, CA). All other chemicals were reagent grade quality, and all water was deionized. Antibodies (Ab) used were rabbit anti-rat collagen I (Biodesign International, Saco, ME), horseradish peroxidase–anti-rabbit IgG (Bio-Rad), and macrophage-3 antigen (MAC-3) (Pharmingen/BD Biosciences, San Jose, CA), arginase 1 (ARG1) (Sigma), inducible nitric oxide synthase (iNOS) (AbCam, Cambridge, UK), tenascin C (Dako, Glostrup, Denmark), αsmooth muscle actin (Sigma), and TGFβ1 (AbCam). Polymerase chain reaction (PCR) oligonucleotides were purchased from Sigma Genosys (Sigma). ELISA kit for quantification of HA, EZ-Link Sulfo-NHS-Biotinylation kit, and SYBER Green real-time PCR reagents were purchased from Echelon Biosciences (Salt Lake City, UT), Pierce Biotechnology (Fisher Scientific, Ottawa, ON, Canada), and Bio-Rad, respectively, and were used according to the manufacturers' instructions. Streptavidin/alkaline phosphatase for ELISA was purchased from Dako. Cell lines: RHAMM-overexpressing (LR21) cells, RH−/−, RhFl-rescued, and CD44−/− cells were prepared as previously described.39 Rat dermal fibroblasts were purchased from the ATCC # CRL-1213 (ATCC, Manassas, VA).
Isolation of HA-Binding Clones from Recombinant Phage Display Libraries
The 15mer phage display library was a kind gift of Chiron (Emeryville, CA) and had a complexity of approximately 1 × 1013 transformants. The 12mer library was purchased from New England BioLabs (M13 phage library, Ph.D.-12 kit; New England BioLabs, Ipswich, MA) and contained an estimated complexity of 1.2 × 109 transformants. The 7mer library (Ph.D.-7) was also purchased from New England BioLabs and contained a complexity of 2.8 × 109 transformants. Sequences from both the 7- and 12mer libraries were fused to gene 3 (pIII) proteins. The funnel for identifying HA-binding sequences from each phage display library is summarized in Supplemental Figure S1 (available at http://ajp.amjpathol.org). HA fragments (MWav = 200 kDa) were linked to Sepharose beads as previously described41 and incubated in phosphate-buffered saline (PBS) with each of the phage libraries at 4°C overnight on a shaker. Beads were recovered by centrifugation and washed four times in PBS (pH 7.4). Phage were released by incubating HA-Sepharose beads with 10 mg/mL HA (MWav = 200 kDa). The recovered phage were grown on an Escherichia coli bacterial lawn in Petri dishes for amplification and then subjected to three additional rounds of positive selection for binding to HA-Sepharose and amplification in E. coli. HA binding phage were then incubated with chondroitin sulfate (CS)-Sepharose and bound phage were discarded. The remaining phage were grown on a bacterial lawn, 75 independent phage plaques were picked from the screen of each library, and these were sequenced in an automated DNA sequencer. Sequences were aligned with each other and also with RHAMM (mouse, accession number: NP_038580.2), CD44 (mouse, accession number: CAJ18532.1), Toll-like receptor 2 (TLR2; mouse, accession number: NP_036035.3), and Toll-like receptor 4 (TLR4; mouse, accession number: NP_067272.1) using Cobalt Multiple Alignment Tool (Cobalt Constraint-based Multiple Protein Alignment Tool website, http://www.ncbi.nlm.nih.gov/tools/cobalt, last accessed March 12, 2012).
HA Pull-Down Assays
AH Sepharose 4B (0.5 g) was resuspended in 0.5 mol/L NaCl. The Sepharose gel was washed 3× with water (pH 4.5) to remove NaCl, and resuspended in 1 mL of water (pH 4.6). HA (10 kDa, 1 mg/mL) was added to the gel in a ratio of 2:1 (volume/volume) and incubated for 30 minutes at room temperature. Solid carbodiimide (EDC) powder was added to a final concentration of 0.1 mol/L. The pH was maintained at 4.5 to 5.5 for 1 hour (by addition of diluted NaOH). After 1 hour, changes observed in pH were small. The reaction was allowed to proceed for 24 hours at 4°C. The gel was washed thoroughly with water 5× (pH 4.5) and was then equilibrated with 1× PBS buffer (pH 7.6).
RHAMM-overexpressing fibroblasts were pelleted and lysed with 1 mL of radioimmunoprecipitation assay buffer [containing phenylmethanesulfonyl fluoride, 0.0002 mol/L final concentration, and Calbiochem protease inhibitors, 0.1% final concentration (Millipore, Billerica, MA)], incubated for 1 hour on ice, followed by sonication (3 × 10 seconds, with 50% pulse). The lysate was centrifuged at 13,000 × g for 15 minutes, and the supernatant fraction (protein concentration was 4 mg/mL) was used for pull-down assay.
A total of 100 μL of HA-coupled AH Sepharose beads, ratio 1:1, was mixed with 100 μL of cell lysate and incubated overnight at 4°C. The beads were centrifuged at 13,000 × g for 5 minutes and washed three times with 1× PBS (pH 7.6). The supernatant was discarded, and proteins were eluted from Sepharose beads with 50 μL of 1× sample buffer at 9°C during 10 minutes. The gel was centrifuged, and supernatant fractions were analyzed by Western blot analysis. For detection of RHAMM in pull-down samples, anti-RHAMM Ab (rabbit monoclonal, Epitomics CD 168/RHAMM; Epitomics, Burlingame, CA) was used at a dilution of 1:1000 and secondary anti-rabbit Ab at a dilution of 1:10,000. The reaction was detected by enhanced chemiluminescence.
Synthesis and Labeling of Peptides
Peptides were synthesized, purified using preparative high-performance liquid chromatography, and then characterized by electrospray ionization (ESI+) mass spectrometry. Scrambled peptides were prepared by synthesizing random sequences of the exact amino acid composition of sense peptides. Biotinylated peptides were prepared by synthesizing peptides with an additional cysteine residue at the COOH terminus and then covalently linking biotin to this residue.
In Vitro Scratch Wound Assays
Function blocking anti-RHAMM peptide Ab were prepared against sequence encoded in the N-terminus of murine RHAMM (ProSci, San Diego, CA42). Cells were plated at 7 × 105 cells/well in six-well plates using Dulbecco's modified Eagle's medium (DMEM) (Gibco BRL/Invitrogen, Carlsbad, CA) containing 10% fetal calf serum (FCS). Confluence of cell monolayers after 24 hours was between 80% and 90%. Cells were injured with either a 2-mm single-edge scraper [RHAMM-overexpressing (LR21) cells, Rh−/−, Rh-rescued, and CD44−/− mouse embryonic fibroblasts (MEF)] or a yellow pipette tip (rat dermal fibroblasts) producing one injury/well.39 Cells were washed twice with PBS and incubated for 10 to 12 hours with medium containing 30 μg/mL peptides or 30 μg/mL anti-RHAMM blocking Ab unless otherwise indicated. For quantification of HA fragment–stimulated (eg, 200 kDa) or oligosaccharide-stimulated (eg, low polydispersity, 6-kDa 30mer or more, polydisperse mixture averaging 10 kDa) cell migration, culture medium was changed to DMEM plus 1% FCS, and 50 ng/mL HA fragments/oligosaccharides were added. To block HA fragment/oligosaccharide-stimulated cell migration, 30 μg/mL peptides were added concomitantly with HA. Cell cultures were incubated for 24 hours at 37°C and 5% CO2. Following incubation, cells were stained with hematoxylin, and images were taken using a ×5 modulation objective (Carl Zeiss, Oberkochen, Germany) attached to a Zeiss Axiovert 100 inverted microscope equipped with Hoffman Modulation contrast optical filters (Modulation Optics, Greenvale, NY). The number of cells that migrated into the scratch wound was counted using 70% of the width of the scratch wound. Statistically significant (P < 0.05) differences were assessed by the unpaired Student's t-test method using Microsoft Excel software (Redmond, WA).
Preparation of Recombinant RHAMM, CD44, and TLR4 Proteins
Recombinant human CD44 was purchased from Origene (Rockville, MD), and human TLR2 and -4 recombinant proteins were purchased from Abnova (Taipei, Taiwan). Mouse recombinant glutathione S-transferase (GST) RHAMM protein was prepared as previously described.42 Recombinant protein RHAMM-COOH terminus (amino acids 706 to 766, sequence: RDSYAQLLGHQNLKQKIKHVVKLKDENSQLKSEVSKLRSQ LVKRKQNELR LQGELDKALG I, molecular weight = 7.1 kDa, pI = 10.1) was isolated from E. coli BL21 (D3) strain carrying the recombinant plasmid pPAL7-RHAMM. Bacteria were grown overnight at 37°C in liquid broth medium containing ampicillin (100 μg/mL) and 0.5% glucose, and allowed to grow to mid-log phase. Recombinant RHAMM gene expression was induced with 2 mmol/L isopropyl β-d-1-thiogalactopyranoside for 4 hours at 37°C, and bacterial cells were harvested by centrifugation at 10,000 × g for 20 minutes. Bacterial cells were re-suspended in lysis buffer [composed of 0.2 mol/L sodium phosphate, 0.2 mol/L potassium acetate, 1% triton X-100, and 0.1% protease inhibitors (pH 7.0)], sonicated (60 seconds, 10 seconds/pulse), and centrifuged (4°C, 12,000 × g, 20 minutes). The resulting supernatant was transferred to a clean tube and filtered (using a 0.45-μm filter). Purification of the eXact-tagged-recombinant RHAMM was conducted with Profinity eXact (Bio-Rad) affinity resin according to the manufacturer's protocol. For these experiments, the lysate was loaded in a gravity column packed with Profinity eXact affinity resin (4 mL of resin, column 15 × 1.5 cm) equilibrated with wash buffer [0.2 mol/L sodium phosphate (pH = 7.0)]. The column was washed with wash buffer to eliminate impurities, and recombinant RHAMM was eluted with elution buffer [composed of 0.2 mol/L sodium phosphate, 0.1 mol/L sodium fluoride (pH = 7.0)]. The protein was then dialyzed and concentrated, using a Millipore filter (cutoff: 3 kDa) in a buffer consisting of 0.2 mol/L sodium phosphate and 0.2 mol/L potassium acetate (pH 7.0). The purity of the isolated protein was verified on one-dimensional sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Identification of RHAMM was performed and confirmed with anti-RHAMM Ab by Western blot analysis. The purity of recombinant RHAMM was assessed by high-performance liquid chromatography and mass spectrometry analysis (electrospray ionization) of the molecular weight. Purity ranged from 80% to 90%, and the major contaminant was GST. Therefore, GST alone was used in binding assays as a negative control for GST-RHAMM.
Binding of Peptide 15-1 to HA in an ELISA
ELISA plates (Nalge Nunc, Penfield, NY) were coated overnight with 20 μg of HA oligosaccharides/well, prepared as a testicular hyaluronidase digestion, and resulting in an MWav = 10 kDa (see above) or purified 4mer to 50mer HA oligosaccharides (kind gift of Seikagaku). Negative control wells were incubated with PBS. Excess HA was removed by washing with PBS three times for 5 minutes. Nonspecific binding sites were covered by incubation of the ELISA plate with 3% bovine serum albumin/PBS for 1 hour at room temperature followed by three 5-minute PBS washes. For biotinylation of peptides, a commercially available biotinylation kit43 was used. A total of 10 mL of peptide solution (2 μg/mL in PBS) was mixed with 10 mmol/L Sulfo-NHS-Biotin (Fisher Scientific) and incubated for 3 hours at room temperature. Excess biotin was removed by passage over a gel filtration column. The biotinylated peptide was diluted 1:3 and added to the HA-coated ELISA plates (100 μL of biotinylated peptide/well) followed by a 1-hour incubation at room temperature and three 5-minute wash steps with PBS. HA-coated positive control wells were incubated with 1:50 diluted biotinylated HA detection probe from Echelon Biosciences. For detection of biotinylated peptides that were bound to HA, ELISA plates were incubated with a 1:1000 dilution (100 μL/well) of streptavidin-phosphatase (Dako) for 1 hour at room temperature followed by three 5-minute PBS washes. Next, 100 μL/well of 3,3-diaminobenzidine (DAB)/buffer mixture were added, and the plates were incubated for 10 minutes at room temperature in the dark or until a color reaction was visible. The light absorption was then measured with a Bio-Rad 3550 plate reader at 405 nm.
Isothermal Calorimetry
A buffer solution of 0.9 mmol/L HA (MWav = 10 kDa, in disaccharide units molecular weight 401 g/mol) was injected into a 0.6 mmol/L peptide 15-1 solution in 1 mol/L 2-(N-morpholino)ethanesulfonic acid (pH 6.0) at 25°C. Experimental conditions: 28 × 10 μL injections, 300 to 400 seconds of spacing, 1.0062 × g, 60 seconds as an initial delay, 10 mcal/second as the reference power.44
Excisional Skin Wounds
All animal experiments followed the guidelines of the local animal use committee (University of Western Ontario protocol #2009–60). Three-month-old Sprague Dawley rats (female) were anesthetized and wounded by lifting the back skin from the underlying muscle, pinching the left and right sides of the skin together as one sheet then placing punch biopsies through both sides at once to generate two excisional wounds with each biopsy punch. Three 4-mm of such punch biopsies resulted in 6 separate wounds in rats. The wounds were filled with 100 μL of a sterile solution of 1% collagen type 1 gel (pH 7.2) containing either PBS, HA-binding peptides, or scrambled peptide, which contained the same amino acids as the HA-binding peptides but in a random order. After application of the 1% collagen solutions to the wounds, the collagen was allowed to gel for 20 minutes. At various time points after wounding, the wounds were recovered from the euthanized rats with an 8-mm punch biopsy. Three- to six-month-old wild-type and RHAMM−/− mice were wounded as described for rats, with the exception that only one full-thickness punch biopsy/mouse was used, resulting in two wounds/mouse. Peptide treatment and wound isolation were done as described for rats.
Quantification of Wound Contraction
Wound edges were traced at different times after wounding using a transparent plastic sheet. Wound traces were then scanned in, and the wound area45,46 was determined by image analysis using Image J software (NIH, Bethesda, MD, http://rsb.info.nih.gov/ij, last accessed April 22, 2010).
Cytokine Array
Rats were wounded as described above. Twenty-four hours after wounding, wounds were removed and placed on six-well cell culture plates in DMEM plus 10% FBS plus antibiotic and antimycotic agents.47 P15-1 or scrambled control peptide was added to the cultures (50 μg/mL). At days 3 and 7, culture medium was removed, and cytokine profile was determined using Proteome Profiler Rat Cytokine Array Panel A array kit (R&D Systems, Minneapolis, MN) following manufacturer's instructions.
Isolation and Purification of HA from Skin Excisional Wounds
Skin samples, 60 to 100 mg, were digested overnight at 55°C in 500 μL of 0.15 mol/L Tris, 0.15 mol/L NaCl, 0.01 mol/L CaCl2, 5 mmol/L deferoxamine mesylate (pH 8.3), 7 U of proteinase K/mg tissue, 0.1% SDS, followed by centrifugation at 18,000 × g for 5 minutes. Dry NaCl was added to the supernatant to increase salt concentration to 2 mol/L. An equal volume of chloroform was added, and the samples were mixed by shaking. Samples were spun at 18,000 × g for 10 minutes. The upper, aqueous phase was removed and dialyzed to 0.1 mol/L NaCl for 2 hours using MINI Dialysis Units 7000 MWCO (Thermo Scientific). Samples were mixed with four volumes of ethanol and stored overnight at −20°C. Samples were spun at 18,000 × g for 20 minutes at 4°C. Pellets were dissolved in 100 μL of 50 mmol/L Tris (pH 8), 20 mmol/L NaCl plus 50 U of Benzonase followed by 4 hours incubation at 37°C. NaCl concentration was increased to 2 mol/L, and samples were extracted with one volume of chloroform. Aqueous and organic phases were separated by 5 minutes of centrifugation, and the aqueous phase was dialyzed against 0.1 mol/L NaCl for 2 hours. HA was precipitated with four volumes of ethanol at −20°C overnight followed by 30 minutes of centrifugation at 18,000 × g and 4°C. Control samples were redissolved in 30 μL of water. Peptide-treated samples were redissolved in 100 μL of water, boiled 5 minutes, and then retreated with Benzonase, chloroform extraction, and precipitation with ethanol before being dissolved in 30 μL of water. Crude HA was further purified by anion exchange chromatography. Each HA sample was dissolved in 20 μL of water and then mixed with additional water and 2 mol/L NaCl to reach a final volume of 400 μL in 0.1 mol/L NaCl. The entire sample was loaded on a prewashed anion exchange mini spin column, and spun at 2000 × g for 5 minutes. The column was washed with 400 μL of 0.1 mol/L NaCl. Three protocols were used in elution of the HA from the column. Protocol 1 used for the day 0 control sample: the column was eluted twice with 400-μL aliquots of 0.7 mol/L NaCl. Protocol 2 used for peptide-treated samples: the column was eluted twice with 400-μL aliquots of 0.4 mol/L NaCl, then twice with 400-μL aliquots of 0.7 mol/L NaCl. In protocol 2, some non-HA contaminants and some very low molecular mass HA were eluted with the 0.4 mol/L NaCl, and then larger HA was eluted with 0.7 mol/L NaCl. Protocol 3 used for days 5 and 7 control samples: the anion exchange column was first eluted with 0.2 mol/L NaCl to remove some non-HA contaminants, but no HA, and then eluted with 0.7 mol/L NaCl as for the day 0 sample. (Protocol 3 was developed during the course of this study, and will be the preferred protocol for the future.) Following each elution protocol, the HA-containing eluate was dialyzed overnight against water, and then dried using a centrifugal vacuum concentrator. Quantitative analysis of the densitometric data from electrophoretic analysis was performed as has been described.48 The area under the densitometric profile for each sample was calculated and normalized to the weight of the tissue sample from which HA had been isolated.
Agarose Gel Electrophoresis
The agarose gel electrophoresis method used in this study was based on the method of Lee and Cowman,49 as adapted to a mini-gel format and optimized by Cowman and colleagues.50 Agarose gels (0.5% w/v) were prepared in Tris/borate/EDTA buffer [100 mmol/L Tris-borate, 1 mmol/L EDTA (pH 8.3)]. Approximately, 3.2 μg of HA isolated from each skin sample was purified by ion exchange chromatography as described above, then each fraction dissolved in 15 μL of deionized water. A 3-μL aliquot of loading buffer (0.02% bromophenol blue, 2 mol/L sucrose in 1× Tris/borate/EDTA) was added to each sample. Select-HA standards were prepared in water and loaded at 0.05 to 0.2 μg per HA component. Gels were pre-electrophoresed for 20 minutes at 40 V, and then loaded and electrophoresed at 40 V for 3.5 hours. The gel was stained overnight in 0.005% Stains-All in 50% ethanol, then destained in 10% ethanol solution. The gel was scanned using a GE Healthcare ImageScanner III running LabScan v.6. Quantitative analysis of the calibrated image was accomplished using ImageQuant TL software.
RNA Extraction from Wounds
Punch rat skin biopsies were homogenized for 1 minute in 1 mL of Trizol, and total RNA was extracted by using Trizol Reagent kit (Gibco BRL). The amount of total RNA concentration was measured with a NanoDrop ND 1000 spectrophotometer (Thermo Scientific).
PCR
PCR amplification was performed using Platinum TaqDNA polymerase (Gibco BRL) and specific primers for β-actin, collagen I, collagen III, ED-1, and regulated on activation normal T cell expressed and secreted (RANTES). PCR conditions were as follows: β-actin. Primer 1: 5′-CTCTTTGATGTCACGCACGATTTC-3′, primer 2: 5′-GTGGGCCGCTCTAGGCACCAA-3′; 45 seconds at 94°C, 45 seconds at 60°C, 45 seconds at 72°C, 25 cycles. Collagen I. Primer 1: 5′-CGATGTCGCTATCCAGCTGA-3′, primer 2: 5′-AGTCCGAATTCCTGGTCTGG-3′; 45 seconds at 94°C, 45 seconds at 60°C, 45 seconds at 70°C, 20 cycles. Collagen III. Primer 1: 5′-GGGACACTCGGGAGAGATAC-3′, primer 2: 5′-AACAATCAGTCAGCCATCTAC-3′; 45 seconds at 94°C, 45 seconds at 50°C, 45 seconds at 72°C, 25 cycles. ED-1. Primer 1: 5′-CGATGGCAGGACAGTAGTCGC-3′, primer 2: 5′-AAGGCTGCTGTTGAAAGGACG-3′; 45 seconds at 94°C, 45 seconds at 59°C, 45 seconds at 72°C, 29 cycles. RANTES. Primer 1: 5′-AAGATCTCTGCAGCTGCATC-3′, primer 2: 5′-ACACTTGGCGGTTCCTTCGA-3′; 45 seconds at 94°C, 45 seconds at 59°C, 45 seconds at 72°C, 29 cycles.
RT-PCR
For the synthesis of oligo-dT–primed cDNA, 2 μg of total RNA, 1 μg of oligo dT,28 and Moloney Murine Leukemia Virus Reverse Transcriptase (Gibco BRL) or SuperScript II Reverse Transcriptase (Invitrogen) were used. Following 1-hour incubation at 37°C, the reaction was stopped by heating at 95°C for 5 minutes, and 2 μL of room temperature reaction mixture was used for each PCR reaction. PCR amplification was performed using a SYBER Green real-time PCR kit (Bio-Rad) following the manufacturer's instructions with the modification that the PCR reaction was downscaled to 25 μL.
Immunohistochemistry
Rat and mouse wounds were collected using an 8-mm punch biopsy, and recovered punch biopsies were fixed in freshly prepared 3.5% paraformaldehyde and processed for serial paraffin sections. Five-micron sections were obtained and deparaffinized by passage through an alcohol series. Sections were stained with Masson's trichrome by the pathology department at the Hospital for Sick Children in Toronto. Sections were stained for MAC-3, TGFβ1, tenascin, smooth muscle actin, ARG1, and iNOS using UltraVision LP detection system (Thermo Scientific).
Quantification of Immunohistochemistry
Masson's trichrome, tenascin C, and collagen I and -III stained tissue section images were taken with air objectives (×4, NA 0.16; ×20, NA 0.7; Olympus, Tokyo, Japan) on a microscope (AX70 Provis; Olympus) with a color camera (Cooke SensiCam CCD imaging; PCO-TECH, Romulus, MI) and Image-Pro Plus 4.5.1.2.9 (Media Cybernetics, Rockville, MD). Image J software was used for staining quantification. Color deconvolution plugin DAB-HE was used to separate blue and brown pixels. After application of the threshold function, positive pixels were counted.
Wound Hydroxyproline Content
Weighed granulation tissue was hydrolyzed in 6 N HCl for 24 hours at 110°C, evaporated to dryness at 110°C, and then dissolved in acetate buffer (pH 6) (25 g citric acid, 6 mL of glacial acetic acid, 60 g of sodium acetate 3H2O, 17 mg of sodium hydroxide to 500 mL of ddH2O) to 0.05 mg/mL. Two microliters of the sample was added to 98 μL of acetate buffer (pH 6). One milliliter of chloramine solution (1.4 g Chloramine-T, 10 mL of n-propanol, 10 mL of ddH2O, 80 mL of acetate buffer) was added to each tube and mixed. Tubes were incubated at room temperature for 20 minutes. One microliter of Ehrlich solution (15 g of Ehrlich, 62 mL of n-propanol, 25 mL of 70% perchloric acid, 12 mL of ddH2O) was added to each tube and mixed. Tubes were incubated at 65°C for 15 minutes. Absorbance was measured against a blank at 550 nm.
Western Blot Analyses for Detection of Collagen I in Cultured Rat Dermal Fibroblasts
Cells were plated at 7 × 105 cells/well in six-well plates in complete growth medium (DMEM plus 10% FCS; Gibco/BRL). Cell monolayers were scraped twice in perpendicular orientations with a gel comb producing three 2-mm scratch wounds. To remove cell debris, cell monolayers were washed with complete growth medium. Growth medium containing 30 μg/mL peptides was added, and cell cultures were incubated for the indicated times at 37°C and 5% CO2. Cells were lysed in 250 mL of radioimmunoprecipitation assay buffer containing 1:100 diluted proteinase inhibitor cocktail (Sigma). Protein concentration was determined by Lowry protein assay kit (Cytoskeleton, Denver, CO) following the manufacturer's instructions. Twenty micrograms of protein was resolved over an 8% SDS-PAGE gel followed by protein transfer to a nitrocellulose membrane. Nitrocellulose membranes were incubated for 1 hour at room temperature in 5% skim milk/Tris-buffered saline/0.1% NP-40 to block nonspecific protein binding sites. Membranes were then incubated overnight at 4°C with Tris-buffered saline/1% skim milk/0.1% NP-40 containing 1:250 diluted collagen I Ab. Next, membranes were washed with Tris-buffered saline/0.1% NP-40 15 minutes at room temperature followed by a 30-minute incubation with 1:4000 diluted horseradish peroxidase–coupled anti-rabbit secondary Ab. Next, membranes were washed three times 15 minutes with Tris-buffered saline/0.1% NP-40 at room temperature followed by protein detection with ECL Plus kit (Amersham/GE Healthcare) according to the manufacturer's instructions.
Collagen Gel Contraction
Collagen 1 gel solution was prepared following the manufacturer's instructions. In brief, 800 μL of ice-cold collagen 1 solution was mixed with 100 μL of 0.1 mol/L NaOH and 100 μL of 10× PBS. Four hundred microliters/well of this mixture was spread on cell culture six-well plates and allowed to form a gel at 37°C. A total of 25,000 cells/well were seeded onto the collagen 1 gel. After 24 hours incubation at 37°C, 5% CO2, collagen gels were released from the plastic surface using a spatula, and returned to the incubator. Ten hours later, gels were fixed with 3.7% paraformaldehyde and the gel area measured by image analysis using Image J. Primary mouse MEF and rat dermal fibroblasts were used for these studies.
Time-Lapse Cinemicrography
To quantify random motility, cells were plated at low density in T-25 cell culture flasks. Cells were serum starved by culture in defined medium (DMEM, insulin/transferrin, antibiotics/antimycotics) at 37°C for 24 hours. Cell migration was stimulated by addition of 10% FCS or Oligo HA (50 to 500 ng/mL) immediately before filming. Time-lapse images were acquired every 10 minutes using a Hamamatsu CCD digital camera (Hamamatsu Photonics, Hamamatsu City, Japan) attached to a Zeiss Axiovert 100 inverted microscope equipped with heated stage. Random cell migration was quantified by tracing nuclear displacement over a 6- to 12-hour period using image analysis software Northern Exposure 2.9 (Empix Imaging, Mississauga, ON, Canada).
Microarray Analysis
Subconfluent cultures of RHAMM-transfected (LR21) and parental 10T1/2 cells were serum starved overnight in defined culture medium (DMEM, 4 μg/mL insulin, 8 μg/mL transferrin). This allowed isolation of mRNA that is modified in expression as a result of RHAMM overexpression and not the consequence of RHAMM + serum or HA supplements. Total RNA was isolated using TRIzol Reagent (Invitrogen) as per the manufacturer's instructions. RNA samples were further purified using the QIAgen RNAeasy kit (Qiagen, Valencia, CA) as per the manufacturer's instructions. After purification, RNA concentration was determined by absorbance at 260 nm. Three biological replicates were used. All gene chips were processed at the London Regional Genomics Centre (Robarts Research Institute, London, ON, Canada). RNA quality was assessed using Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) and the RNA 6000 Nano kit (Caliper Life Sciences, Hopkinton, MA). Biotinylated complimentary RNA (cRNA) was prepared from 10 μg of total RNA as per the Affymetrix GeneChip Technical Analysis manual (Affymetrix, Santa Clara, CA). Double-stranded cDNA was synthesized using SuperScript II (Invitrogen) and oligo primers. Biotin-labeled cRNA was prepared by cDNA in vitro transcription using the BioArray High-Yield RNA Transcript Labeling kit (Enzo Biochem, New York, NY) incorporating biotinylated UTP and CTP. Labeled cRNA (10 μg) was hybridized to Mouse Genome 430 2.0 GeneChips for 16 hours at 45°C as described in the Affymetrix Technical Analysis manual (Affymetrix). GeneChips were stained with streptavidin-phycoerythrin, followed by an Ab solution and a second streptavidin-phycoerythrin solution, with all liquid handling performed by a GeneChip Fluidics Station 450. GeneChips were scanned with the Affymetrix GeneChip Scanner 3000 (Affymetrix). Signal Intensities for genes were generated using GCOS1.3 (Affymetrix) using default values for the Statistical Expression algorithm parameters and a target signal of 150 for all probe sets and a normalization value of 1. Gene-level data were generated using the RMA preprocessor in GeneSpring GX 7.3.1. (Agilent Technologies). Data were then transformed (measurements less than 0.01 were set to 0.01) and normalized per chip to the 50th percentile and per gene to control samples. To determine the effect of RHAMM overexpression on gene expression, LR21cells were compared to parental 10T1/2 cells. GeneSpring was used to generate fold changes in gene expression between the two cell lines by applying a t-test with Bonferroni multiple testing correction with a significance cutoff of 0.05. The genes with expression change greater or equal to twofold were considered for further analysis. Signaling pathways were identified by Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA).
The consequence of P15-1 peptide on protein expression was determined using immunohistochemistry analysis of P15-1 versus scrambled peptide wounds and microarray analysis of P15-1–treated subconfluent dermal fibroblasts versus scrambled peptide–treated fibroblasts. Immunohistochemistry analyses are described above, and microarray analyses were processed as above. Gene lists were queried with Ingenuity Pathway Analysis, and identification of signaling/functional networks was used to identify key signaling proteins that are RHAMM/P15-1 regulated.
FAK Signaling
Fibroblasts were cultured to subconfluence (60%), and then serum starved overnight as described above. Ten-kDa HA (10 μg/mL) was added to cultures, and cells were lysed 10 to 15 minutes later. FAK was immunoprecipitated with anti-FAK Ab (BD Biosciences, Mississauga, ON, Canada), immunoprecipitates were separated on SDS-PAGE and transferred to a nitrocellulose membrane. Immunoprecipitated FAK phosphorylated on tyrosine was detected using AG10 monoclonal antibody (Cell Signaling Technology, Danver, MA).
Statistics
Statistical data analysis was performed using Excel software (Microsoft). A two-tailed Student's t-test was used to assess significance, and P ≤ 0.05 was considered significant.
Results
RHAMM Binds Preferentially to HA Fragments and Is Required for HA Fragment–Promoted Fibroblast Motility
Previous studies suggest that RHAMM binds to HA fragments and that this interaction plays a role in apical airway microtubule and endothelial cell microfilament-based motility.27,51 We extended these studies and assessed whether RHAMM also binds to HA fragments/oligosaccharides when expressed on fibroblasts and whether this results in increased motility. As shown in Figure 1A, HA fragments (intermediate, 220 kDa) and oligosaccharides (10 kDa) both bound to RHAMM from fibroblast lysates. However, the binding capacity of RHAMM for HA appeared to be higher for oligosaccharides than intermediate-sized polymers. The effect of these two HA sizes on fibroblast random motility was measured (Figure 1B). Both the 220-kDa and 10-kDa HA stimulated cell motility significantly above negative control cells receiving only defined medium. Stimulation by both sizes of HA was similar to that of FCS supplements, which were used as a positive control in this experiment. To assess the role of RHAMM in this motility response, the random motility of RHAMM−/− fibroblasts in response to FCS, 220-kDa HA, and 10-kDa HA was also measured. As shown in Figure 1B, loss of RHAMM negated a significant response to both FCS and 10-kDa HA, but not to 220-kDa HA. Collectively, these results suggested that RHAMM preferentially binds to HA oligosaccharides (eg, 10 kDa) and is required for a motogenic response to this size of HA. Using this information, we next developed an approach for isolating HA binding peptides that mimic RHAMM.
Figure 1.

A motogenic response to HA fragments requires RHAMM. A: RHAMM binds to 220-kDa and 10-kDa HA. Lysates from RHAMM overexpressing mesenchymal cells were incubated with 220-kDa and 10-kDa HA coupled to Sepharose beads. Proteins bound to beads were resolved on SDS-PAGE, and RHAMM was detected by Western blot analysis. Arrows indicate bands that represent full-length RHAMM (RHAMMFL) and a processed (truncated) RHAMMTR that is commonly expressed after an injury. B: Wild-type and RHAMM−/− MEFs were seeded sparsely on tissue culture plastic and serum starved in defined medium. Cell migration was stimulated with 10% FCS (positive control), 220-kDa HA, or 10-kDa HA and quantified by real-time cinemicrography. Values represent mean ± SD of 50 cells. *P < 0.001 between wild-type and RHAMM−/− cells, †P < 0.001 versus defined medium.
Isolation of HA Binding Peptides
HA binding peptides have previously been reported, but these neither share common sequence patterns nor exhibit strong resemblances to known HA binding sequences.34–37,52 We developed a combined unbiased/rational method for identifying HA binding peptides from phage display libraries using polydisperse HA, which included the motogenic 220-kDa and 10-kDa polymers as bait. Phage display libraries were of three different peptide lengths, which we considered might influence the size of bound HA (see the flow diagram in Supplemental Figure S1 at http://ajp.amjpathol.org). To identify phage display peptides that bind to HA and not to the Sepharose backing, a step in the isolation procedures was included in which binding of peptides to HA beads was competed by an excess of unlabeled, 10-kDa HA (10 mg/mL) (see Supplemental Figure S1 at http://ajp.amjpathol.org). HA binding phage, which also bound to CS-Sepharose, were discarded. Phage display peptide sequences were aligned using BLAST programs53 to determine their relatedness to each other and also to known HA receptor-binding regions.1,54–58 Since RHAMM binds to HA fragments and oligosaccharides, we focused on characterizing phage peptide sequences that resembled the protein sequence of this HA receptor. Peptides that were related to one another and that also aligned to the HA binding sequence of RHAMM were synthesized and tested for their ability to affect response to injury in fibroblast culture assays.
Screening the 7- and 12mer libraries yielded higher numbers of unique peptide sequences (15 and 14, respectively) than the 15mer library, which resulted in recovery of only 2 unique sequences (see Supplemental Figure S1 at http://ajp.amjpathol.org). Screening of the 7mer library yielded the most sequence heterogeneity: only 1 sequence of 15 was obtained three times, with the remaining sequences obtained only once. There were four groups of sequences isolated from the 12mer library, two of which were represented only once, whereas the others were isolated four and seven times, respectively (see Supplemental Figure S1 at http://ajp.amjpathol.org; and data not shown). Screening the 15mer library resulted in the least sequence heterogeneity since one sequence was obtained 47 times, whereas the other was isolated 18 times (see Supplemental Figure S1 at http://ajp.amjpathol.org).
BLAST analyses of HA binding peptides isolated from the 7- and 12mer libraries showed that they were partially homologous to one another, often sharing four to five contiguous amino acids. The 7- and 12mer sequences contained a high percentage of hydrophobic residues (50% to 70%) in comparison to the residues of 15mer peptides (<35%), in this way resembling a previously reported 12mer peptide (Pep-1).38 Only 1 of 15 7mer and 3 of 14 12mer peptides aligned to the HA binding region of RHAMM, but these exhibited very low overall homology to this protein (eg, <25%, data not shown). By contrast, the two 15mer HA binding sequences, which were similar to one another, both aligned to the HA binding region of RHAMM with a sequence homology of 67% (most frequent sequence) and 33%, respectively. Furthermore, the most frequently isolated clone, STMMSRSHKTRSHHV (designated peptide 15-1), aligned to a sequence within the RHAMM HA binding region that contains basic amino acids essential for binding to HA and that form a BX7B motif59 (see Supplemental Figure S1 at http://ajp.amjpathol.org). None of the peptides aligned within the HA binding region of either CD44 or LYVE-1, and none showed significant alignment (eg, >25%) to TLR2 or TLR4 protein sequence. These results suggested that the size of peptide influenced selection of peptides that resemble biological HA receptors. HA binding phage peptides isolated from the 7-, 12-, and 15mer libraries were synthesized and tested for their ability to inhibit HA-stimulated fibroblast migration.
Peptide 15-1 Reduces HA-Dependent Fibroblast Migration in a Dose-Dependent Manner, and This Effect Requires RHAMM Expression
The effect of the above HA binding peptides on fibroblast migration was measured using scratch wound tests. A previously isolated 12mer, HA binding peptide Pep-1, was also included in this screen since it blocked migration of Langerhan's cells,38 reduced experimental metastases of melanoma cells,60 and decreased focal contacts of squamous esophageal carcinoma cells.61 Most HA binding peptides had a slight inhibitory or stimulatory effect on fibroblast migration responding to FCS in scratch wound assays (data not shown). However, P15-1 caused the largest significant decrease in fibroblast migration (data not shown). We therefore selected this peptide for further characterization. A dose-response analysis showed that P15-1 inhibited random fibroblast motility in a concentration-dependent manner between 0.1 and 10 μg/mL, reaching a maximum inhibition when added to subconfluent cells at 10 μg/mL (see Supplemental Figure S2A at http://ajp.amjpathol.org).
To verify that P15-1 blocked HA oligosaccharide-dependent migration of rat dermal fibroblasts, the effect of P15-1 and its scrambled counterpart, used as a negative control, were added to scratch-wounded fibroblast monolayers (Figure 2A). HA oligosaccharides (500 ng/mL, 10-kDa polydisperse mixture) or purified 4-, 6-, and 30-monosaccharide HA oligosaccharides (4-, 6-, 30-mer HA) were added together with either P15-1 or scrambled peptide (10 μg/mL) at the time the monolayers were wounded. A similar stimulation of migration was obtained for both the sized 30mer HA oligosaccharides and 10-kDa polydisperse mixture (Figure 2A shows stimulation with 30mer HA), but 4- and 6mer oligosaccharides were not stimulatory in this assay (data not shown). These results indicated that P15-1 prevented dermal fibroblast migration in response to the 30mer HA, whereas the scrambled peptide did not. The blocking ability of P15-1 peptide was similar to a function-blocking anti-RHAMM Ab (Figure 2B).
Figure 2.

P15-1 blocks HA oligosaccharide–induced migration in RHAMM-expressing fibroblasts. A: Confluent monolayers of rat dermal fibroblasts were scratch wounded using disposable, plastic Pipetman tips (Gilson, Middleton, WI). Cells were incubated with culture medium containing 500 ng of HA oligosaccharides (10 kDa, sized 30mer HA) plus either 50 μg/mL P15-1 or scrambled control peptide (Scr Pep). Twenty-four hours later, cell migration was quantified by counting cells that migrated into the scratch wound. P15-1 significantly reduces the number of cells that have migrated into the wound in comparison to scrambled peptide controls. Values represent the mean ± SD of three culture wells. ***P < 0.001. B: Confluent monolayers of RHAMM-expressing MEF were scratch wounded using pipette tips. Cells were incubated with medium containing anti-RHAMM Ab (30 μg/mL), P15-1 (30 μg/mL), or Scr Pep (30 μg/mL). In the control experiments, cells were incubated with medium alone. After 11 to 14 hours, cells that migrated into the scratch wound were counted. P15-1 significantly decreased (***P < 0.001) migration of RHAMM-expressing MEF, and to the same extent as RHAMM-blocking Ab (*P < 0.05). Scrambled control peptide and Isotype-matched IgG control (IgG) had no effect. Values represent mean ± SD of three culture wells. C: Effect of P15-1 and scrambled control peptide on scratch wound–induced migration of wild-type, CD44−/−, and RHAMM−/− MEFs. Peptide treatment and cell migration quantification were performed as described in B. The graph shows that whereas the migration of wild-type and CD44−/− MEF were significantly inhibited by P15-1, it had no effect on migration of RHAMM−/− MEF. Values represent mean ± SD of nine culture wells.
We next assessed whether the ability of P15-1 to block motility in response to 30mer or 10-kDa (polydisperse mixture) HA required expression of RHAMM or CD44. Wild-type, CD44−/−, and RHAMM−/− MEF were used for these experiments.39 The migration of both wild-type MEF, which expressed both CD44 and RHAMM, and CD44−/− MEF, which expressed RHAMM, into scratch wounds was blocked by P15-1, but not by scrambled P15-1 sequence (Figure 2C). By contrast, the migration of RHAMM−/− MEF was unaffected by P15-1 (Figure 2C). These results suggested that P15-1 reduced dermal fibroblast migration by mimicking RHAMM HA binding sequences and competitively blocking HA fragment/RHAMM interactions. To directly demonstrate this ability of P15-1, we assessed the degree to which it blocks recombinant RHAMM:HA interactions.
Peptide 15-1 Binds Directly to 10-kDa HA Oligosaccharides and Competes with Their Binding to Recombinant RHAMM
We first determined whether we could detect binding of P15-1 to HA and whether this binding was HA size dependent. We therefore quantified binding of P15-1 to native HA (MWav = 500 kDa) and HA oligosaccharides with MWav = 10 kDa, using ITC. The binding curve for the P15-1 interaction with HA oligosaccharides was sigmoidal and saturable, therefore permitting Scatchard analyses (Figure 3A). Analyses indicated a binding constant (kDa) of 10−7 mol/L, an affinity that is approximately six times higher than the affinity of the previously reported 12mer HA binding peptide Pep-138 for HA oligosaccharides. Interestingly, the kDa of P15-1 for native HA was <106, indicating a much higher affinity of this peptide to oligosaccharides (data not shown), thus resembling the preference of RHAMM for oligosaccharides relative to high molecular weight HA (eg, Figure 1A). ELISA confirmed the binding of P15-1 to 10-kDa HA oligosaccharides (Figure 3B). High-performance liquid chromatography analysis of this HA, which had been generated by partial digestion with testicular hyaluronidase, had an MWav of 10 kDa but was polydisperse in its size distribution, ranging from 6mers to 40-kDa fragments. We therefore also assessed the binding of P15-1 to purified, sized 30 HA monosaccharide species (30mer, approximately 10 kDa) since P15-1 effectively blocked the ability of this oligosaccharide to promote fibroblast migration as noted in Figure 2A. ELISA confirmed that P15-1 bound to the 30mer HA (data not shown). These data suggested that P15-1, like RHAMM, binds preferentially to HA oligosaccharides.
Figure 3.

Peptide 15-1 binds to HA oligosaccharides and blocks their interaction with recombinant RHAMM protein. A: The graph shows quantification of the interaction between P15-1 and HA oligosaccharides by ITC. P15-1 bound to HA oligosaccharides (avg. 10 kDa) with a binding constant of kDa = 10−7 mol/L. A 0.9 mmol/L solution of HA was injected into MES (pH 6.0) containing 0.6 mmol/L peptide 15-1, and the energy released on interaction with P15-1 was determined by ITC. Binding curves of two independent experiments (red or black) are depicted. B: HA oligosaccharide:P15-1 interactions were confirmed with ELISA. ELISA plates were coated with HA oligosaccharides (1 mg/mL), followed by incubation with either biotinylated P15-1 or Scr Pep (50 μg/mL). PBS alone was used as control. Bound biotinylated peptides were detected by streptavidin-horseradish peroxidase. Peptide 15-1 binds to HA oligosaccharides. Values represent the mean ± SD of three ELISA wells. C: ELISA plates were coated with 1 μg/mL recombinant RHAMM followed by incubation with either 10 μg/mL 30mer HA or HA oligosaccharides (10 kDa) together with P15-1 or Scr Pep (1 μg/mL). Bound HA was detected with biotinylated aggrecan followed by streptavidin-horseradish peroxidase. P15-1 reduces RHAMM/HA binding. Values represent the mean ± SD of n = 3. *P < 0.001.
To determine whether P15-1 competitively blocked binding of HA oligosaccharides to recombinant RHAMM protein, we designed ELISA assays to detect displacement of HA fragments from purified recombinant GST-RHAMM protein, using the polydisperse HA oligosaccharide mixture (10-kDa oligosaccharides) for these assays. P15-1, but not scrambled peptide, significantly reduced the binding of HA oligosaccharides to recombinant GST-RHAMM compared to the PBS control (Figure 3C). Neither HA nor P15-1 bound to GST alone (data not shown). HA fragments also bound to recombinant CD44 and TLR4 protein in these ELISA assays, but P15-1 did not affect binding to these HA receptors (data not shown). Higher molecular weight HA (>200 kDa) also bound to recombinant RHAMM in ELISA, but P15-1 was less effective in reducing binding of this HA size than the above fragments (data not shown). Collectively, these results confirmed that P15-1 bound directly to HA and with highest affinity to HA oligosaccharides. Results also suggested that P15-1 blocked HA:RHAMM, but not HA:CD44 or HA:TLR4, interactions.
We, and others, have previously shown that RHAMM expression is required for robust inflammation and fibroplasia during repair of adult excisional skin wounds and that RHAMM function is related to HA fragmentation/oligosaccharide production.27,39,51,62,63 We, therefore, chose this model to assess whether P15-1 blocked HA-induced cellular functions in vivo. We used excisional skin injury as a model of acute fibrotic repair in adults64 since, although loose-skinned animals such as rats do not develop cutaneous scars after injury to the extent that tight-skinned animals do, fibrosis markers including robust inflammation, collagen I production, fibroplasia, and differentiation into myofibroblasts are easily detected in rodent models.64–67
Wound HA Accumulation and Fragmentation Increases during Repair and Is Maximal between Days 3 and 7
Fibrogenic repair, which is typical of most adult response-to-injury processes, is initiated by rapid hemostasis and consequent release of factors from platelets and serum, including hyaluronidase 2,68 which collectively initiate a robust inflammatory response.1,69,70 This in turn promotes vigorous fibroplasia and scar formation, particularly in the deeper dermal regions.71 HA fragments are key regulators of innate immunity and both fibroblast and endothelial migration/proliferation/differentiation,1,4,72 which collectively contribute to wound fibrosis. We first verified that HA production and fragmentation occurred in excisional skin wounds in a reproducible and temporally regulated manner (Figure 4). Separation of purified, wound HA on agarose gels showed that high molecular weight HA (ca. 3050 to 4570 kDa) was evident in biopsied skin [day 0 (D0)], and there was very little evidence of HA fragmentation. By day 1 (D1) after injury, the high molecular weight HA was decreased, and smaller HA fragments (eg, between 30 and 3050 kDa) appeared. These continued to increase at day 3 (D3), reached a maximum at day 7 (D7), and then began to decrease at day 14 (D14). Much smaller HA oligosaccharides (eg, >10 kDa) were evident in day 5 and 7 wounds, which was when RHAMM expression was maximal.39 Estimates of the total amount of HA in each identically treated sample showed that the amount of HA was also highest at day 7 in wounds treated with scrambled peptide, and in control wounds treated with PBS/collagen alone, but was relatively constant with time in P15-1–treated wounds (data not shown). We therefore next determined the consequence of P15-1 on excisional skin wound repair.
Figure 4.

HA is fragmented in excisional wounds. Agarose gel electrophoretic analysis of molecular weight distribution for HA isolated from full-thickness excisional wounds 0 to 14 days (D) after wounding. HA was visualized in the electrophoretic gels by Stains-All dye. Mainly high molecular weight HA (3050 to 4570 kDa) was present at day 0, but a broad distribution of HA sizes appeared by day 3, and were prominently observed at day 7. HA molecular weight distribution was measured by densitometry of agarose gels, and results are normalized to the weight of tissue used for HA isolation. Arrow indicates bands representing 10-kDa Oligo-HA, used for experimental analyses.
P15-1 Reduces Inflammation, Angiogenesis, and Fibroplasia/Fibrosis in Excisional Skin Wounds
Since the hyaluronan fragments accumulating in excisional wounds were in the MW range of those reported to promote expression of proinflammatory cytokines by macrophages,4 we assessed whether P15-1 modified properties of these cells. Macrophages are a key wound cell type required for re-establishing tissue integrity and in particular control the initiation and resolution of the inflammatory phase of tissue repair.73,74 Their functions vary with each phase of repair and depend on status of activation, number, differentiation, and plasticity.75–79 Macrophage depletion from wounds can therefore have widely differing effects on the course of repair, depending on the extent to which they are depleted, the timing of depletion, and whether or not depletion affects all or subsets of macrophages.
We first assessed whether P15-1 affected macrophage influx into wounds by quantifying expression of ED-1 mRNA, which provides a sensitive measure of the rat homolog of CD68 expressed on wound macrophages in their early stages of differentiation.80 We also measured Mac3 protein expression since this is a marker for activated macrophages present in wounds.81 The amount of ED-1 PCR product was determined following administration of 0.1 to 100 μg/wound site of P15-1 and was standardized against cellular β-actin levels. The reduction of this standardized ED-1 transcript by P15-1 was presented as a ratio of the total ED-1 transcript present in day 1 wounds treated with collagen gel only (Figure 5A). P15-1 decreased the presence of ED-1–positive macrophages in day 1 wounds (Figure 5A), confirming our previous preliminary demonstration that P15-1 induced decreased ED-1 protein expression in wounds detected by immunohistochemistry.82 Further, the decrease in wound ED-1 mRNA expression was dose dependent, varying from 40% to 100% inhibition (0.1 to 100 μg/wound site); Mac3 protein expression, which was quantified by counting the number of Mac-3–positive cells per field in wounds (see Supplemental Figure S2B at http://ajp.amjpathol.org) was reduced by P15-1 by approximately 45%. On the basis of these results, we used 30 to 50 μg of P15-1 in subsequent experiments to determine its consequences to repair outcome.
Figure 5.

Peptide 15-1 reduces macrophage influx into excisional skin wounds and TGFβ1 production. A: Full-thickness excisional wounds were covered with either vehicle alone or vehicle containing different concentrations (100 ng/mL to 100 μg/wound site) of P15-1. Wound tissue was isolated 24 hours later. Expression of the macrophage-specific marker ED-1 was analyzed by PCR. Amplification of β-actin transcripts was used as loading control. Amplification products were separated on an agarose gel. The ED-1 amplification product was quantified by densitometry and corrected for equal loading by calculating the ratio ED-1:β-actin control. P15-1 decreased the amount of wound ED-1 in a dose-dependent manner. Values are mean ± SD. n = 3. B: Full-thickness excisional skin wounds were treated with either P15-1 or scrambled peptide (Scr Pep; 50 μg/wound site). Wound sections were stained with iNos- or Arg1-specific Ab. Positive cells were counted. P15-1 treatment reduced infiltration of proinflammatory (iNos+) macrophages into wounds. All graphs represent results of n = 3 wounds, five areas/wound. Values are mean ± SD. n = 15. *P < 0.001. C: Full-thickness excisional wounds were treated with vehicle alone, P15-1 (50 μg/wound site), or scrambled peptide (50 μg/wound site). Day 1 and day 3 wound sections were stained with TGFβ1-specific Ab as described in Materials and Methods. Microscopic images were taken at an original magnification of ×60, and TGFβ1+ cells (arrows) were counted. The number of TGFβ1+ cells is reduced in P15-1–treated wounds at days 1 and 3 after wounding. Graphs represent mean ± SD. n = 30. *P < 0.01.
Macrophages recruited to wounds are highly plastic but have been loosely grouped as proinflammatory (classically activated) or prohealing (alternatively activated) macrophages.75,83 We next compared the consequences of P15-1 on macrophages polarized to a proinflammatory state versus those polarized to a prohealing state using iNOS84 and Arg1, respectively.85,86 As shown in Figure 5, B and C, at days 3 and 5, P15-1 strongly reduced wound iNOS+ but had little effect on Arg+ other than a slight stimulation (P < 0.05) of this marker at day 5 after wounding. Representative immunohistochemistry images of iNOS+ and Arg1+ macrophages are shown in Supplemental Figure S3 at http://ajp.amjpathol.org). To identify the predominant proinflammatory pathways that were blocked by P15-1, we used Proteome Profiler to determine the relative levels of proinflammatory cytokines produced by activated macrophages in wounds treated with P15-1 or the scrambled peptide control (see Supplemental Figure S4 at http://ajp.amjpathol.org). Surprisingly, P15-1 had no detectable effect on the 16 proinflammatory cytokines in this profile, which included IL6, RANTES, and IL1B. Although not conclusive, these results suggested that P15-1 may have a selective effect on specific proinflammatory cytokines produced by macrophages in early stages of wound repair. Since depleting early proinflammatory macrophages has previously been shown to reduce TGFβ1 production,76 since genetic deletion of RHAMM reduces fibroplasia in excisional wounds,39 and since both HA and RHAMM are required for TGFβ signaling,13,87–89 we reasoned that P15-1 treatment might selectively alter wound TGFβ1 levels. Indeed, treatment of wounds with P15-1 significantly reduced TGFβ1 levels when compared to scrambled peptide controls (Figure 5C).
TGFβ1 is essential for the phase of repair following inflammation, called tissue formation, which involves fibrogenesis, neovascularization, and transient scar formation typified by increased collagen production and appearance of myofibroblasts.90–92 Selective reduction of macrophages during the inflammation stage of wound repair results in modification of processes characteristic of the tissue formation phase, in particular, reducing wound TGFβ1, fibrogenesis, neovascularization, myofibroblast differentiation, and contraction. By contrast, depletion of macrophages during the tissue formation stage of wound repair has much more serious consequences to repair and results in severe hemorrhage as well as lack of wound maturation and closure.76 Based on our results showing a reduction in proinflammatory macrophages and wound TGFβ1 levels, we predicted that processes occurring during the tissue formation phase of wound repair might be most strongly affected by P15-1.
By day 7 in collagen gel alone or scrambled peptide–treated control wounds, re-epithelialization was almost complete, the clot was intact, and fibroblasts as well as endothelial cells had migrated into the granulation tissue so that both fibroplasia and angiogenesis were abundant, as shown by wound cross sections stained with Masson's trichrome (see Supplemental Figure S5 at http://ajp.amjpathol.org, and Figure 6). Both the density of fibroblasts and blood vessels was significantly reduced by P15-1 treatment compared to wounds treated with collagen gels alone (Figure 6; see also Supplemental Figure S5 at http://ajp.amjpathol.org) or scrambled peptide controls (data not shown), whereas re-epithelialization appeared to be enhanced (Supplemental Figure S5 at http://ajp.amjpathol.org, and data not shown). Higher magnification images clearly showed reduced fibroblast and blood vessel density resulting from P15-1 treatment (Figure 6A). As expected by the blunted fibroplasia, wound collagen was also significantly decreased by P15-1 as measured by wound hydroxylproline content at day 7 (Figure 7A), PCR of collagen I mRNA of day 1 wounds (Figure 7B), and image analysis of Masson's trichrome staining (see Supplemental Figure S5 at http://ajp.amjpathol.org). To determine whether the effect of P15-1 on wound collagen production was a direct effect like its antimigration properties or an indirect consequence of dampened inflammation, we repeated these experiments using rat dermal fibroblasts in culture. P15-1 or scrambled peptide was added to fibroblast monolayers that had received multiple scratch wounds, and collagen I expression was detected at various times after wounding using Western blot analyses. β-actin was used as a loading control (Figure 8B). As shown in Figure 8A, P15-1 strongly reduced collagen I expression 24, 72, and 96 hours after wounding. These results suggested that P15-1 directly inhibits collagen I production in dermal fibroblasts. We next assessed the consequences of P15-1 on wound myofibroblast differentiation in vivo using smooth muscle actin as a marker, as shown in Figure 9, A and B. P15-1 reduced the expression of smooth muscle actin in day 5 wounds. Consistent with this observation, P15-1 also significantly reduced wound contraction at day 5 post injury compared to untreated or scrambled peptide–treated control wounds. To determine whether these effects of P15-1 on wound contraction were direct or indirect, peptide was added to collagen lattice contraction assays imbedded with dermal fibroblasts. As shown in Figure 9C, P15-1 significantly inhibited collagen lattice contraction, showing that P15-1 has the potential to directly modulate myofibroblast activity in addition to fibroblast collagen production. TGFβ1 regulates expression of tenascin C, a matricellular protein, which like RHAMM is expressed in postnatal tissue only in wounds or disease.93,94 Tenascin C plays a complex role in wound remodeling and, for example, can both enhance and limit dermal fibroblast migration.95 High tenascin expression in rodent skin is associated with modified adult skin repair, in particular a reduction in wound scarring and an increase in regenerative healing.93,96 Since P15-1 reduced markers associated with fibrosis/scarring and since genetic deletion of RHAMM modified wound tenascin C expression,39 we assessed whether this peptide altered wound tenascin C expression. As shown in Figure 10, P15-1 significantly increased tenascin C expression at day 5 but had no effect at the other time points.
Figure 6.

Peptide 15-1 reduces fibroblast density and blood vessel density in granulation tissue. A: Full-thickness day 7 excisional wounds were treated with collagen vehicle alone (PBS control), scrambled peptide in collagen vehicle (image not shown), or P15-1 in collagen vehicle (P15-1, 50 μg/wound site). Cross sections of day 7 wounds were stained with Masson's trichrome, allowing differentiation of collagen (blue), muscle, and cells.71 Angiogenesis and fibrogenesis was reduced in P15-1–treated wounds at day 7. B: Granulation tissue fibroblasts and blood vessels were counted in wound sections. P15-1 significantly decreased the numbers of fibroblasts and blood vessels in granulation tissue (day 7) compared to both PBS and scrambled peptide (scr pep) controls. The effects of scrambled peptide on fibroblasts and blood vessels were not significantly different from vehicle control. Graphs represent the mean ± SD. n = 6 wounds. *P < 0.001.
Figure 7.
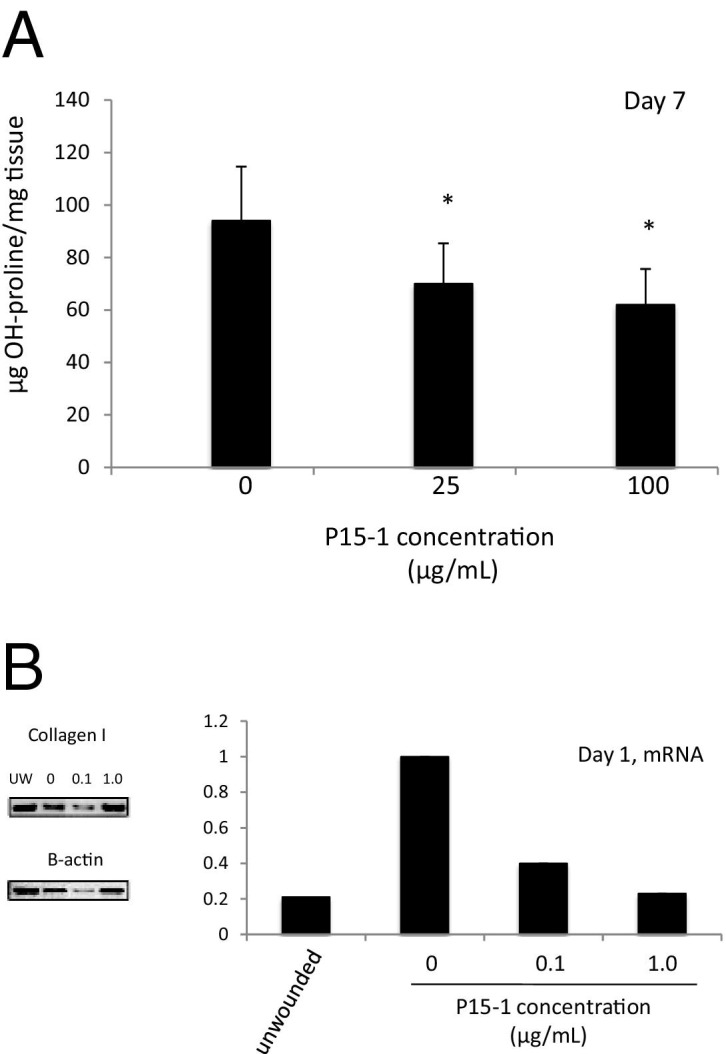
P15-1 inhibits collagen I accumulation within wounds. A: Full-thickness excisional wounds were treated with collagen vehicle alone (0 μg/mL P15-1 concentration) or P15-1 + collage vehicle (25, 100 μg/wound site). At day 7, wounds were isolated, and the OH-proline content was analyzed as described in Materials and Methods. P15-1 significantly reduced wound collagen at day 7 after wounding at both concentrations (*P < 0.01). Each column represents the mean ± SD. n = 6. B: Full-thickness excisional wounds were treated with either collagen vehicle alone or P15-1 in collagen vehicle (100 ng/mL to 1 μg/wound site). Wound tissue mRNA was isolated 24 hours after wounding, and collagen I transcripts were amplified by RT PCR. Amplification of β-actin transcripts was used as loading control. Amplification products were separated on an agarose gel and quantified by densitometry. The collagen I amplification products were corrected for equal loading by calculating the ratio collagen I/β-actin control. Collagen I expression in control wounds were set to one. P15-1 decreased collagen I mRNA expression 24 hours after wounding. The values represent typical results that were replicated three separate times.
Figure 8.
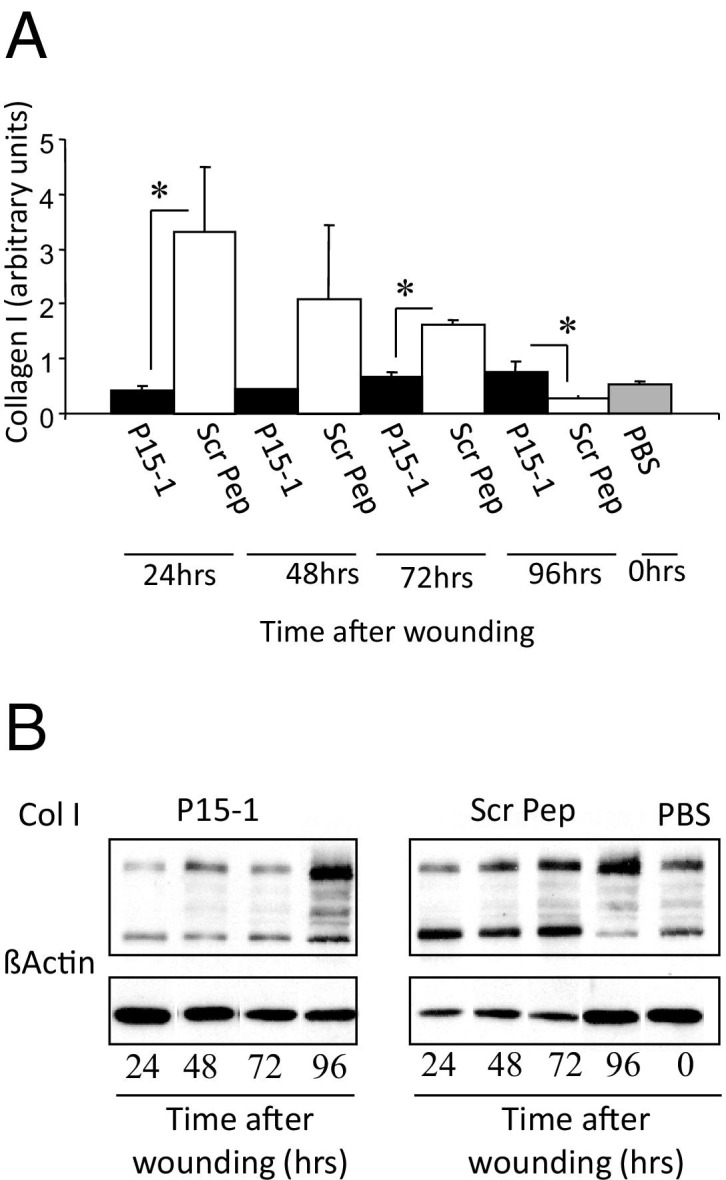
Peptide 15-1 reduces in vitro collagen production of cultured dermal fibroblasts. Monolayers of rat dermal fibroblasts were scratched multiple times using a plastic comb. A: Cells were incubated for the indicated times in medium containing either peptide 15-1 (30 μg/mL) or scrambled control peptide (Scr Pep; 30 μg/mL). B: Collagen I production was quantified by Western blot analysis. β-actin was used as loading control. Protein band intensities were determined by densitometry, and the ratio of Col I/β-actin was calculated. P15-1 detectably reduced collagen I protein levels at 24, 48, and 72 hours after its addition. Values represent the mean ± SD. n = 3. *P < 0.001.
Figure 9.

P15-1 reduces wound contraction and smooth muscle actin in day 5 wounds. A: Full-thickness excisional wounds were treated with collagen vehicle alone or collagen vehicle plus P15-1 peptide. Wounds were harvested at day 5 post injury, and cross sections were stained with smooth muscle actin–specific Ab. A: Panel depicts representative images of collagen vehicle–treated control wounds and P15-1 in collagen vehicle–treated wounds. Smooth muscle actin staining was reduced in P15-1–treated wounds. B: Wound contraction was reduced in P15-1–treated wounds at day 5 after wounding. Full-thickness excisional wounds were either treated with collagen vehicle plus P15-1, collagen vehicle plus scrambled control peptide, or left untreated. Wound contraction was measured by tracing wound edges on days 3, 5, and 7 after wounding and quantification of the wound area by image analysis. The graph depicts percentage wound contraction. n = 6 wounds/treatment. C: P15-1 inhibits collagen gel contraction in culture. Collagen I gel contraction by human foreskin fibroblasts was analyzed as described in Materials and Methods. Graph depicts the gel area ± SD of n = 4 replicates/experiment. *P < 0.01.
Figure 10.
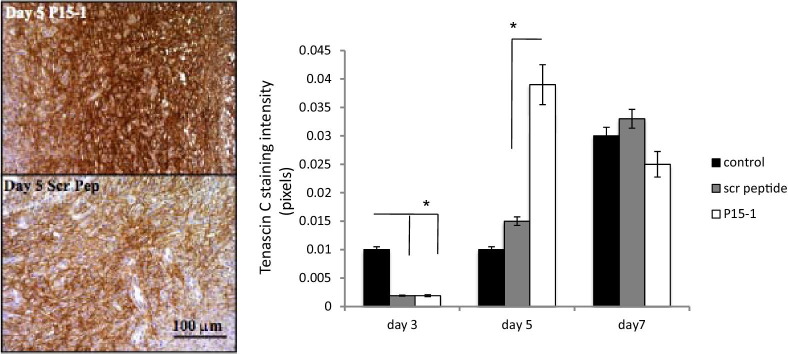
Peptide 15-1 increases wound tenascin C protein. Full-thickness excisional wounds were covered with collagen vehicle alone (image not shown), P15-1 in collagen gel (50 μg/wound site), or scrambled control peptide in collagen gel (50 μg/wound site). Sections were stained with tenascin C–specific Ab as described in Materials and Methods. Microscopic images were taken, and tenascin C staining intensity was quantified by image analysis using Image J. P15-1 increased tenascin C protein expression in wounds at day 5 after injury. Graphs represent the mean ± SD. n = 6 wounds. *P < 0.001.
P15-1 did not appear to delay wound resolution in terms of resolution of granulation tissue, fibrogenesis or re-appearance of normal skin architecture (data not shown). Therefore, our collective results suggested that P15-1 was dampening “fibrosis” and transient scar formation during the tissue formation phase of skin wound repair partly as a result of a selective effect of P15-1 on proinflammatory macrophage function, in particular production of wound TGFβ1 and partly by direct blocking of RHAMM function in fibroblasts.
P15-1 Does Not Alter Excisional Skin Wound Repair of RHAMM−/− Mice
Our data showing that P15-1 exhibited sequence homology with RHAMM HA binding regions, blocked the binding of HA oligosaccharides to recombinant RHAMM, blocked migration of RHAMM-expressing, but not RHAMM−/−, fibroblasts and had similar effects on excisional skin wound properties to RHAMM loss, suggested that P15-1 acted as a RHAMM peptide mimic. To more conclusively assess this possibility, we compared the effects of P15-1 on excisional skin wounds of wild-type and RHAMM−/− mice. We reasoned that if P15-1 acts as a specific RHAMM antagonist (eg, RHAMM mimetic peptide), we would observe an effect of the peptide on wild-type, but not RHAMM−/−, wounds. Since we showed that P15-1 had an easily measured effect on the production of TGFβ1 and tenascin C accumulation in wounds, we examined the effects of P15-1 on these properties. As shown in Figure 11A, P15-1 significantly reduced TGFβ1 staining in wild-type wounds. As expected from the blunted fibrosis/fibrogenesis of RHAMM−/− wounds, TGFβ1 staining was less than in wild-type wounds. Nevertheless, P15-1 treatment did not significantly alter levels of this cytokine in RHAMM−/− wounds compared to scrambled peptide controls. We have previously reported that wound tenascin C protein levels are increased in RHAMM−/− relative to wild-type wounds.39 Here, we show that P15-1 similarly increased tenascin C expression in wild-type wounds but had no such detectable effect on RHAMM−/− wounds (Figure 11B). Collectively, these results provided strong evidence that P15-1 behaves as a RHAMM peptide mimetic and likely blocks HA:RHAMM signaling through a competitive antagonism.
Figure 11.

P15-1 does not affect TGFβ1 or tenascin C levels in RHAMM−/− wounds. A and B: Full-thickness excisional wounds of wild-type and RHAMM−/− mice were treated with collagen vehicle plus either P15-1 or scrambled control peptide. A: Cross sections of day 3 wounds were stained with TGFβ1-specific Ab. The graph depicts the number of positively stained cells per microscopic field ± SD. n = 4. *P < 0.001. B: Cross sections of day 5 wounds were stained with tenascin C–specific Ab. A representative image of n = 4 is shown.
P15-1 Blocks a RHAMM Signal Guiding Differentiation of Myofibroblasts
To further dissect the molecular mechanisms by which P15-1 affected wound fibrosis, we used microarray analyses to assess the consequences of RHAMM overexpression and P15-1 inhibition on gene expression. We used cultured fibroblasts for these studies because we showed here that P15-1 acted directly on fibroblasts by a RHAMM-mediated mechanism. We analyzed the consequences of RHAMM overexpression in MEF in serum-free medium as described in Materials and Methods to isolate the effect of elevating the expression of this gene from growth factor effects. The consequences of P15-1 on mRNA expression of cultured RHAMM expressing MEF responding to wound injuries were also analyzed. The relationship of genes that were altered as a consequence of both RHAMM expression and P15-1 exposure (see Supplemental Table S1 at http://ajp.amjpathol.org) were probed using Ingenuity Pathway Analysis. As shown in Figure 12A, pathway analysis identified a functional network that affected dermal, connective tissue, and inflammatory diseases and that featured a canonical FAK signaling pathway. Pathway components of this network are important genes controlling myofibroblast differentiation and include RHAMM/HMMR, TGFβ1, smooth muscle actin (acta 2), tenascin C (Tnc), and collagen I (Col1).97 Since the results of these unbiased analyses are consistent with our evidence that P15-1 appears to block fibroblast/myofibroblast differentiation, we determined whether P15-1 alters FAK tyrosine phosphorylation status, an important posttranslational mechanism for regulating the activity of this kinase. We, and others, have previously shown that FAK undergoes cycles of phosphorylation/dephosphorylation that are required for focal adhesion turnover and appropriate dynamic timing of signaling.98–102 Our results showed that P15-1 promoted hypertyrosine phosphorylation of FAK in comparison to treatment with HA oligosaccharides alone, and thus, P15-1 has the potential of disrupting appropriate signaling through FAK (Figure 12B). These results are similar to previous evidence showing that FAK activity is required for TGFβ1-induced myofibroblast differentiation,103–106 that blocking the HA binding ability of RHAMM results in both loss of cell migration and an increase in phosphorylation of FAK on tyrosine,102 and that blocking Ras-regulated pathways via HA oligosaccharide promoted ERK1,2 activity26 results in hyperphosphorylation of FAK and consequent inhibition of cellular functions such as migration and differentiation.107–109 Collectively, our results suggested that P15-1 directly modifies the myofibroblast phenotype in excisional skin wounds as depicted in the model (Figure 12C).
Figure 12.

RHAMM-regulated signaling pathways blocked by P15-1. A: Protein or mRNA expression of genes, which are subject to regulation by both RHAMM and P15-1 (see Materials and Methods), were identified by performing immunohistochemistry of wound beds or microarray analysis of RHAMM transfected versus parental fibroblasts, and P15-1–treated versus scrambled peptide–treated RHAMM-expressing fibroblasts. Genes that were altered in their expression by P15-1 (but not by negative controls), as well as by RHAMM transfection, were identified and analyzed for functional networks/canonical signaling pathways using Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA). Analyses identified the top functional network (shown in A) as one associated with connective tissue disorders, dermatological diseases, and inflammation. The most significant molecular and cellular functions predicted by these associations included cell cycle (P < 3.7 × 10−7) and cell-to-cell signaling and interaction (P < 3.9 × 10−7). The top canonical signaling pathway was FAK (focal adhesion kinase) signaling (P = 2.5 × 10−7) as shown in A. Genes that have previously been linked to RHAMM (red box, identified as HMMR) are marked in orange, and those that have been shown to be directly or indirectly modified through an action on signaling pathways are marked in solid (direct) or dashed (indirect) red lines. Genes identified in the present study to be regulated by RHAMM and P15-1 on the FAK signaling pathway are pink, and their relationships to each other are indicated by solid or dashed pink lines. B: Based on the analysis in A, the consequence of P15-1 on FAK tyrosine phosphorylation status was assessed following exposure to HA oligosaccharides (10 kDa) as described in Materials and Methods. The control, scrambled peptide, resulted in light phosphorylation of FAK, whereas by contrast, P15-1 treatment strongly and significantly increased FAK phosphorylation at all doses tested (P < 0.001). This was sustained over time and was associated with inability of focal adhesions, which contain large signaling complexes linked to the microenvironment, to turnover and signal appropriately (data not shown). C: Literature searches of the FAK pathway shown in A regulate myofibroblast differentiation within wounds (see Discussion). The consequences of RHAMM, HA oligosaccharides and blocking of these interactions by P15-1 are modeled in C. HA oligosaccharides bind to cell surface RHAMM, which in turn regulates signaling in focal adhesions through FAK, thus contributing to myofibroblast differentiation/migration. P15-1 is proposed to compete for RHAMM in binding to HA oligosaccharides thus blocking appropriate RHAMM signaling through FAK/ERK1. This aberrant signaling block by P15-1 results in a downstream decrease in Tgfβ1, Acta2, Col1, and Col3 expression and an increase in Tnc expression with a consequent loss of myofibroblast phenotype.
Discussion
HA deposition is transiently increased following acute response to injury of most tissues1,5,72,98,110,111 and performs complex and molecular weight–dependent functions during repair. High molecular weight or native HA is present in all layers of uninjured skin and is, as we have confirmed here, increased following excisional injury in a time-dependent manner. High molecular weight HA (eg, >500 kDa) performs a number of functions during repair including promoting keratinocyte migration, regulating tissue edema, suppressing vascularization and inflammation, and controlling myofibroblast differentiation.112–114 Following tissue injury, both oxygen free radicals generated by injury and hyaluronidases released by pathogens and hemostatic events (eg, platelet aggregation) at the wound site degrade HA to produce fragments (eg, 50 to 400 kDa) and oligosaccharides (ca. <25 kDa). Intermediate HA fragments (50 to 400 kDa) stimulate keratinocyte activation and proliferation, as well as fibroblast proliferation observed during repair, act as innate alloimmune agonists, promote neoangiogenesis, control inflammatory cascades, and also contribute to myofibroblast differentiation.1,4,72 HA oligosaccharides (eg, <10 kDa) may perform dual roles during repair and disease processes because, on the one hand, they promote fibroblast migration/proliferation, angiogenesis, and increased expression of wound proinflammatory cytokines,1,3,26 but on the other hand, can block HA/HA receptor interactions that promote survival pathways.2 Collectively, these results suggest that a complex interplay exists amongst high molecular weight, intermediate molecular weight, and small HA oligosaccharides, which collectively regulate wound repair and resolution. These same HA MW forms appear to play key roles in disease initiation and progression, in particular those diseases in which chronic inflammation is involved.1,2,4
Here, we identify a screening process for isolating HA receptor peptide mimics, which block the activity of low MW HA. Specifically, we describe isolation of a 15mer peptide, which acts as a RHAMM peptide mimic: since it resembles sequences within the HA binding region of RHAMM, it effectively and selectively competes with recombinant RHAMM for HA oligosaccharides, it has similar consequences to wound repair as genetic depletion of RHAMM,39 and requires RHAMM expression to have a blocking effect. Our results are consistent with previous reports that RHAMM binds to low molecular weight (eg, 15 to 40 kDa) fragments and oligosaccharides.26,51,63,115,116
Our results suggest that P15-1 may primarily disrupt the interaction of HA oligosaccharides with RHAMM and that this particularly affects the inflammatory functions of proinflammatory macrophages and differentiation of fibroblasts into a myofibroblast subtype. Thus, P15-1 binds with higher affinity to a mixture of HA oligosaccharides than to native HA, as detected by ITC. Function blocking data further suggest that P15-1 may bind to a specific size range of HA oligosaccharides that are proinflammatory and fibrogenic. For example, although P15-1 blocks migration in the presence of HA oligosaccharide mixtures, it maximally affects this cell function in response to a purified 30-monosaccharide oligosaccharide. However, further studies are required to determine the precise range of HA fragment and oligosaccharide sizes that P15-1 binds to.
Our data suggest that the HA oligosaccharide:RHAMM interactions, which are blocked by P15-1, directly modify signaling pathways in wound fibroblasts and thus alter myofibroblast differentiation. Thus, we show that RHAMM and P15-1 regulated genes group into a function network and FAK signaling pathway that is commonly seen in dermal diseases, connective tissue disorders, and inflammatory diseases. The top signaling network is a FAK-regulated pathway that includes RHAMM (HMMR), TGFβ1, ERK1, PDGFBB, Col1, Col 3, Tnc, and Nfkb. RHAMM has previously been linked to PDGFBB function,117 specifically activates ERK1,42 and is required for TGFβ1-promoted fibroblast migration in culture88 and angiogenesis via TGFβR1/CD44 complexes.89 HA, TGFβ1, and FAK97,103–106,118 are now considered to be partners that coordinate myofibroblast differentiation. The ability of P15-1 to block myofibroblast differentiation is associated with the promotion of FAK tyrosine phosphorylation. Ligands that activate cell migration/gene expression through Ras appear to dephosphorylate FAK and facilitate turnover of the signaling structures within focal adhesions: reagents that block dephosphorylation of FAK in this signaling context also block cell migration and focal adhesion–mediated signaling.107–109 Whether or not P15-1 directly modifies signaling to affect macrophage polarization and trafficking into the wounds was not addressed in this study but remains for future investigation.
A number of reports have described the isolation of HA binding peptides that range in size from 7- to 12mer,33–36,52,53 and these peptides exhibit biological activity consistent with their reported HA binding activity. Some of these peptides share properties known to be required for binding to HA, such as clusters of basic amino acids,34,35,52 and one 12mer exhibits weak homology with TSG-6,53 but they do not exhibit strong homology with the primary sequence of known HA binding regions in HA receptors or binding proteins.38 Our studies predict that a 15mer may represent the minimal peptide size required for isolation of HA receptor peptide mimics with homology to small HA binding regions such as RHAMM. We also predict that larger peptides or multimers of peptides will be required for competitively dislodging HA from the bigger HA binding groove of proteins with link module-like HA binding regions, such as CD44.
Aberrant wound repair is an increasing burden on health care due to the aging population and rise of obesity-related diseases such as diabetes.119 Both chronic wounds and fibrotic repair of wounds cause significant morbidity and mortality within these patient groups.69,120–122 Chronic wounds and excessive scarring of wounds are both driven by prolonged inflammation, which affects the proliferative and remodeling phases of the response-to-injury process.69,73,111,123,124 Considerable evidence using transgenic mouse models supports an essential role for macrophages in acute wound repair of adult tissues.64,65 Macrophages release factors that affect keratinocytes, endothelial cells, and fibroblasts. For example, depletion of macrophages from the inflammatory stage of tissue response to injury results in reduced granulation tissue formation, scarring, and compromised re-epithelialization of the wound. The depletion of macrophages mid-repair during tissue granulation tissue formation results in severe vascular defects and hemorrhaging of the wound. In both instances, wounds do not complete remodeling and do not close completely.76,77 By contrast, the role of dermal fibroblasts, which are clearly a heterogeneous cell population of papillary, reticular fibroblasts, fibrocytes, and pericytes, in excisional wound repair is not yet as well understood.
HA and at least two of its receptors, CD44 and RHAMM, are known to regulate immunity during tissue repair,1,4,16,125–127 in particular, innate immune cells such as macrophages, and fibrogenesis. Loss of CD44 results in prolonged macrophage infiltration into bleomycin-injured lung tissue, and persistence/activation of this cell type is considered to be largely responsible for the unremitting inflammation and subsequent tissue destruction following tissue injury.4 Our previous results show that genetic deletion of RHAMM also disrupts signals coordinating innate immunity and fibrogenesis. For example, excisional wounds of RHAMM−/− mouse skin exhibit prolonged neutrophil survival but reduced macrophage accumulation.39 Wounds of such mice also exhibit reduced fibroplasia and aberrant mesenchymal differentiation. Our collective evidence, particularly the lack of P15-1 effect on RHAMM−/− wounds, indicate P15-1 is a RHAMM mimic. Our results further predict that the consequence of RHAMM loss on inflammation and fibroplasia results, at least in part, from aberrant HA oligosaccharide signaling.
Although interventions that promote wound repair have been known for centuries, these are largely devoid of drug-based therapies, and promoting wound repair to resolve ulcers and reduce fibrotic repair to attenuate scarring continue to be major clinical problems. Drugs that affect sensitivity to the TGFβ family of fibrotic cytokines are in phase I and II clinical trials, and blocking CX43 function/expression is in the pre-clinical phase of assessment for treatment of chronic and fibrotic wounds.93,119 Clearly, there is need for additional targets that can on the one hand reduce tissue fibrosis and on the other hand promote a regenerative mechanism during normal and pathological wound repair. Our results suggest that targeting the interactions between HA and cell receptors, which blunt inflammation and fibrogenesis via an effect on FAK signaling, is a feasible method for fine tuning the extent of fibrosis versus regeneration within acute wounds.
Footnotes
Supported in part by a grant from the NIH/National Cancer Institute (5R01CA119092 to J.B.M. and E.A.T.). Financial assistance from the Ontario Institute for Cancer Research (L.G.L., E.A.T.), Canadian Breast Cancer Foundation (E.A.T.), and the Western Innovation Fund of the University of Western Ontario (C.T., L.G.L). M.C. and R.A. were supported by the Research Fund for Neurodegeneration and Inflammation at Polytechnic Institute of New York University.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at http://dx.doi.org/10.1016/j.ajpath.2012.06.036.
Contributor Information
Jim B. McCarthy, Email: mccar001@umn.edu.
Eva A. Turley, Email: Eva.Turley@lhsc.on.ca.
Supplementary data
Fragmented HA was covalently linked to Sepharose beads that were subsequently used for the screening of 7-, 12-, and 15mer phage display libraries that were eluted with free HA. Isolated phage clones were grown on a bacterial lawn, and the entire selection procedure was repeated. HA binding clones that also bound to chondroitin sulfate-Sepharose beads were excluded from further analyses. HA binding clones were picked and sequenced. Sequences were aligned with known HA binding regions from HA receptors and extracellular HA binding proteins. Only peptides P15-1 and P15-2 showed partial alignment with a known HA binding region, and that was to the HA binding domain of RHAMM. The HA binding sequences of RHAMM are highlighted with boxes, and known amino acids involved in HA binding that were identified by mutational analyses or computer modeling are marked green. P15-1 and P15-2 align to the second HA binding region of RHAMM.
P15-1 blocks motility of fibroblasts in a concentration-dependent manner and inhibits trafficking of macrophages. A: The random motility of RHAMM-expressing fibroblasts was quantified by real-time cinemicrographic analyses. Addition of different P15-1 concentrations to the culture medium during migration analysis inhibited random cell migration in a dose-dependent manner. The graph depicts cell migration speed ± SD of n = 50 cells. B: Full-thickness excisional skin wounds were treated with either P15-1 (50 μg/mL) or scrambled peptide (50 μg/mL). Cross sections of day 3 wounds were stained with a macrophage specific Ab, Mac-3. P15-1 significantly decreased the appearance of Mac-3 cells in day 3 wounds. The graph depicts the number of positive cells/microscopic field ± SD. n = 36 cells, 3 wounds. *P < 0.001.
Full-thickness excisional skin wounds were treated with either P15-1 (50 μg/mL) or scrambled control peptide (50 μg/mL). Cross sections of day 3 and 5 wounds were stained with iNOS-specific Ab. Representative images from n = 6 wounds are shown.
Proinflammatory cytokine protein expression by P15-1 and control skin wounds ex vivo. Proteome Profiler was used to determine the levels of cytokines released by wound cells in the presence or absence of P15-1. Supernatant medium was collected and processed for presence of cytokines according to the manufacturer's instructions. P15-1 had no detectable effect on the cytokines profiled in this assay other than a slight increase in L-selection. Values are the average of two duplicates.
Cross section of full-thickness excisional wounds treated with collagen vehicle alone (PBS control) or collagen vehicle plus P15-1 were stained with Masson's trichrome, allowing discrimination between collagen (blue) and cells.71Solid arrows indicate wound edges, and dotted arrows indicate blood vessels. Inserts show images of the entire wound site at ×4 original magnification. P15-1 decreased collagen accumulation within the wounds, as detected by image analysis. Values are the mean ± SD. n = 6 wounds. P < 0.001.
References
- 1.Veiseh M., Turley E.A. Hyaluronan metabolism in remodeling extracellular matrix: probes for imaging and therapy of breast cancer. Integr Biol (Camb) 2011;3:304–315. doi: 10.1039/c0ib00096e. [DOI] [PubMed] [Google Scholar]
- 2.Toole B.P., Slomiany M.G. Hyaluronan: a constitutive regulator of chemoresistance and malignancy in cancer cells. Semin Cancer Biol. 2008;18:244–250. doi: 10.1016/j.semcancer.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang D., Liang J., Noble P.W. Hyaluronan in tissue injury and repair. Annu Rev Cell Dev Biol. 2007;23:435–461. doi: 10.1146/annurev.cellbio.23.090506.123337. [DOI] [PubMed] [Google Scholar]
- 4.Jiang D., Liang J., Noble P.W. Hyaluronan as an immune regulator in human diseases. Physiol Rev. 2011;91:221–264. doi: 10.1152/physrev.00052.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Volpi N., Schiller J., Stern R., Soltes L. Role, metabolism, chemical modifications and applications of hyaluronan. Curr Med Chem. 2009;16:1718–1745. doi: 10.2174/092986709788186138. [DOI] [PubMed] [Google Scholar]
- 6.Burdick J.A., Prestwich G.D. Hyaluronic acid hydrogels for biomedical applications. Adv Mater. 2011;23:H41–H56. doi: 10.1002/adma.201003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lennon F.E., Singleton P.A. Role of hyaluronan and hyaluronan-binding proteins in lung pathobiology. Am J Physiol Lung Cell Mol Physiol. 2011;301:L137–L147. doi: 10.1152/ajplung.00071.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murano E., Perin D., Khan R., Bergamin M. Hyaluronan: from biomimetic to industrial business strategy. Nat Prod Commun. 2011;6:555–572. [PubMed] [Google Scholar]
- 9.Meran S., Thomas D., Stephens P., Martin J., Bowen T., Phillips A., Steadman R. Involvement of hyaluronan in regulation of fibroblast phenotype. J Biol Chem. 2007;282:25687–25697. doi: 10.1074/jbc.M700773200. [DOI] [PubMed] [Google Scholar]
- 10.Simpson RM, Wells A, Thomas D, Stephens P, Steadman R, Phillips A: Aging fibroblasts resist phenotypic maturation because of impaired hyaluronan-dependent CD44/epidermal growth factor receptor signaling, Am J Pathol 176:1215–1228 [DOI] [PMC free article] [PubMed]
- 11.Simpson R.M., Meran S., Thomas D., Stephens P., Bowen T., Steadman R., Phillips A. Age-related changes in pericellular hyaluronan organization leads to impaired dermal fibroblast to myofibroblast differentiation. Am J Pathol. 2009;175:1915–1928. doi: 10.2353/ajpath.2009.090045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Webber J., Jenkins R.H., Meran S., Phillips A., Steadman R. Modulation of TGFbeta1-dependent myofibroblast differentiation by hyaluronan. Am J Pathol. 2009;175:148–160. doi: 10.2353/ajpath.2009.080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Webber J., Meran S., Steadman R., Phillips A. Hyaluronan orchestrates transforming growth factor-beta1-dependent maintenance of myofibroblast phenotype. J Biol Chem. 2009;284:9083–9092. doi: 10.1074/jbc.M806989200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jameson J.M., Cauvi G., Sharp L.L., Witherden D.A., Havran W.L. Gammadelta T cell-induced hyaluronan production by epithelial cells regulates inflammation. J Exp Med. 2005;201:1269–1279. doi: 10.1084/jem.20042057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki Y., Yamaguchi T. Effects of hyaluronic acid on macrophage phagocytosis and active oxygen release. Agents Actions. 1993;38:32–37. doi: 10.1007/BF02027210. [DOI] [PubMed] [Google Scholar]
- 16.Johnson P., Ruffell B. CD44 and its role in inflammation and inflammatory diseases. Inflamm Allergy Drug Targets. 2009;8:208–220. doi: 10.2174/187152809788680994. [DOI] [PubMed] [Google Scholar]
- 17.Khan A.I., Kerfoot S.M., Heit B., Liu L., Andonegui G., Ruffell B., Johnson P., Kubes P. Role of CD44 and hyaluronan in neutrophil recruitment. J Immunol. 2004;173:7594–7601. doi: 10.4049/jimmunol.173.12.7594. [DOI] [PubMed] [Google Scholar]
- 18.Ferguson E.L., Roberts J.L., Moseley R., Griffiths P.C., Thomas D.W. Evaluation of the physical and biological properties of hyaluronan and hyaluronan fragments. Int J Pharm. 2011;420:84–92. doi: 10.1016/j.ijpharm.2011.08.031. [DOI] [PubMed] [Google Scholar]
- 19.Schimizzi A.L., Massie J.B., Murphy M., Perry A., Kim C.W., Garfin S.R., Akeson W.H. High-molecular-weight hyaluronan inhibits macrophage proliferation and cytokine release in the early wound of a preclinical postlaminectomy rat model. Spine J. 2006;6:550–556. doi: 10.1016/j.spinee.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 20.David-Raoudi M., Tranchepain F., Deschrevel B., Vincent J.C., Bogdanowicz P., Boumediene K., Pujol J.P. Differential effects of hyaluronan and its fragments on fibroblasts: relation to wound healing. Wound Repair Regen. 2008;16:274–287. doi: 10.1111/j.1524-475X.2007.00342.x. [DOI] [PubMed] [Google Scholar]
- 21.Taylor K.R., Trowbridge J.M., Rudisill J.A., Termeer C.C., Simon J.C., Gallo R.L. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–17084. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 22.Gariboldi S., Palazzo M., Zanobbio L., Selleri S., Sommariva M., Sfondrini L., Cavicchini S., Balsari A., Rumio C. Low molecular weight hyaluronic acid increases the self-defense of skin epithelium by induction of beta-defensin 2 via TLR2 and TLR4. J Immunol. 2008;181:2103–2110. doi: 10.4049/jimmunol.181.3.2103. [DOI] [PubMed] [Google Scholar]
- 23.Kaya G., Tran C., Sorg O., Hotz R., Grand D., Carraux P., Didierjean L., Stamenkovic I., Saurat J.H. Hyaluronate fragments reverse skin atrophy by a CD44-dependent mechanism. PLoS Med. 2006;3:e493. doi: 10.1371/journal.pmed.0030493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cordero A., Leon-Dorantes G., Pons-Guiraud A., Di Pietro A., Asensi S.V., Walkiewicz-Cyraska B., Litvik R., Turlier V., Mery S., Merial-Kieny C. Retinaldehyde/hyaluronic acid fragments: a synergistic association for the management of skin aging. J Cosmet Dermatol. 2011;10:110–117. doi: 10.1111/j.1473-2165.2011.00552.x. [DOI] [PubMed] [Google Scholar]
- 25.Barnes L., Tran C., Sorg O., Hotz R., Grand D., Carraux P., Didierjean L., Stamenkovic I., Saurat J.H., Kaya G. Synergistic effect of hyaluronate fragments in retinaldehyde-induced skin hyperplasia which is a Cd44-dependent phenomenon. PLoS One. 2010;5:e14372. doi: 10.1371/journal.pone.0014372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao F., Liu Y., He Y., Yang C., Wang Y., Shi X., Wei G. Hyaluronan oligosaccharides promote excisional wound healing through enhanced angiogenesis. Matrix Biol. 2010;29:107–116. doi: 10.1016/j.matbio.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Gao F., Yang C.X., Mo W., Liu Y.W., He Y.Q. Hyaluronan oligosaccharides are potential stimulators to angiogenesis via RHAMM mediated signal pathway in wound healing. Clin Invest Med. 2008;31:E106–E116. doi: 10.25011/cim.v31i3.3467. [DOI] [PubMed] [Google Scholar]
- 28.Voelcker V., Gebhardt C., Averbeck M., Saalbach A., Wolf V., Weih F., Sleeman J., Anderegg U., Simon J. Hyaluronan fragments induce cytokine and metalloprotease upregulation in human melanoma cells in part by signalling via TLR4. Exp Dermatol. 2008;17:100–107. doi: 10.1111/j.1600-0625.2007.00638.x. [DOI] [PubMed] [Google Scholar]
- 29.Lees V.C., Fan T.P., West D.C. Angiogenesis in a delayed revascularization model is accelerated by angiogenic oligosaccharides of hyaluronan. Lab Invest. 1995;73:259–266. [PubMed] [Google Scholar]
- 30.Slevin M., Kumar S., Gaffney J. Angiogenic oligosaccharides of hyaluronan induce multiple signaling pathways affecting vascular endothelial cell mitogenic and wound healing responses. J Biol Chem. 2002;277:41046–41059. doi: 10.1074/jbc.M109443200. [DOI] [PubMed] [Google Scholar]
- 31.de la Motte C.A. Hyaluronan in intestinal homeostasis and inflammation: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol. 2011;301:G945–G949. doi: 10.1152/ajpgi.00063.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balazs E.A. Viscosupplementation for treatment of osteoarthritis: from initial discovery to current status and results. Surg Technol Int. 2004;12:278–289. [PubMed] [Google Scholar]
- 33.Sironen R.K., Tammi M., Tammi R., Auvinen P.K., Anttila M., Kosma V.M. Hyaluronan in human malignancies. Exp Cell Res. 2011;317:383–391. doi: 10.1016/j.yexcr.2010.11.017. [DOI] [PubMed] [Google Scholar]
- 34.Zhang L.S., Greyner H.J., Mummert M.E., Petroll W.M. Development of a hyaluronan bioconjugate for the topical treatment of melanoma. J Dermatol Sci. 2009;55:56–59. doi: 10.1016/j.jdermsci.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaleski K.J., Kolodka T., Cywes-Bentley C., McLoughlin R.M., Delaney M.L., Charlton B.T., Johnson W., Tzianabos A.O. Hyaluronic acid binding peptides prevent experimental staphylococcal wound infection. Antimicrob Agents Chemother. 2006;50:3856–3860. doi: 10.1128/AAC.00082-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savani R.C., Hou G., Liu P., Wang C., Simons E., Grimm P.C., Stern R., Greenberg A.H., DeLisser H.M., Khalil N. A role for hyaluronan in macrophage accumulation and collagen deposition after bleomycin-induced lung injury. Am J Respir Cell Mol Biol. 2000;23:475–484. doi: 10.1165/ajrcmb.23.4.3944. [DOI] [PubMed] [Google Scholar]
- 37.Lee J.C., Greenwich J.L., Zhanel G.G., Han X., Cumming A., Saward L., McLoughlin R.M. Modulation of the local neutrophil response by a novel hyaluronic acid-binding peptide reduces bacterial burden during staphylococcal wound infection. Infect Immun. 2010;78:4176–4186. doi: 10.1128/IAI.01364-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mummert M.E., Mohamadzadeh M., Mummert D.I., Mizumoto N., Takashima A. Development of a peptide inhibitor of hyaluronan-mediated leukocyte trafficking. J Exp Med. 2000;192:769–779. doi: 10.1084/jem.192.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tolg C., Hamilton S.R., Nakrieko K.A., Kooshesh F., Walton P., McCarthy J.B., Bissell M.J., Turley E.A. Rhamm-/- fibroblasts are defective in CD44-mediated ERK1,2 motogenic signaling, leading to defective skin wound repair. J Cell Biol. 2006;175:1017–1028. doi: 10.1083/jcb.200511027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Armand G., Balazs E.A., Meyer K., Reyes M. Isolation and characterization of ichthyosan from tuna vitreous. Connect Tissue Res. 1983;11:21–33. doi: 10.3109/03008208309015008. [DOI] [PubMed] [Google Scholar]
- 41.Turley E.A., Austen L., Vandeligt K., Clary C. Hyaluronan and a cell-associated hyaluronan binding protein regulate the locomotion of ras-transformed cells. J Cell Biol. 1991;112:1041–1047. doi: 10.1083/jcb.112.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tolg C., Hamilton S.R., Morningstar L., Zhang J., Zhang S., Esguerra K.V., Telmer P.G., Luyt L.G., Harrison R., McCarthy J.B., Turley E.A. RHAMM promotes interphase microtubule instability and mitotic spindle integrity through MEK1/ERK1/2 activity. J Biol Chem. 2010;285:26461–26474. doi: 10.1074/jbc.M110.121491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bergin B.J., Pierce S.W., Bramlage L.R., Stromberg A. Oral hyaluronan gel reduces post operative tarsocrural effusion in the yearling Thoroughbred. Equine Vet J. 2006;38:375–378. doi: 10.2746/042516406777749218. [DOI] [PubMed] [Google Scholar]
- 44.Tribet C., Diab C., Dahmane T., Zoonens M., Popot J.L., Winnik F.M. Thermodynamic characterization of the exchange of detergents and amphipols at the surfaces of integral membrane proteins. Langmuir. 2009;25:12623–12634. doi: 10.1021/la9018772. [DOI] [PubMed] [Google Scholar]
- 45.Uhl E., Barker J.H., Bondar I., Galla T.J., Leiderer R., Lehr H.A., Messmer K. Basic fibroblast growth factor accelerates wound healing in chronically ischaemic tissue. Br J Surg. 1993;80:977–980. doi: 10.1002/bjs.1800800812. [DOI] [PubMed] [Google Scholar]
- 46.Mack J.A., Feldman R.J., Itano N., Kimata K., Lauer M., Hascall V.C., Maytin E.V. Enhanced inflammation and accelerated wound closure following tetraphorbol ester application or full-thickness wounding in mice lacking hyaluronan synthases Has1 and Has3. J Invest Dermatol. 2012;132:198–207. doi: 10.1038/jid.2011.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fromes Y., Liu J.M., Kovacevic M., Bignon J., Wdzieczak-Bakala J. The tetrapeptide acetyl-serine-aspartyl-lysine-proline improves skin flap survival and accelerates wound healing. Wound Repair Regen. 2006;14:306–312. doi: 10.1111/j.1743-6109.2006.00125.x. [DOI] [PubMed] [Google Scholar]
- 48.Cowman M.K., Chen C.C., Pandya M., Yuan H., Ramkishun D., LoBello J., Bhilocha S., Russell-Puleri S., Skendaj E., Mijovic J., Jing W. Improved agarose gel electrophoresis method and molecular mass calculation for high molecular mass hyaluronan. Anal Biochem. 2011;417:50–56. doi: 10.1016/j.ab.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 49.Lee H.G., Cowman M.K. An agarose gel electrophoretic method for analysis of hyaluronan molecular weight distribution. Anal Biochem. 1994;219:278–287. doi: 10.1006/abio.1994.1267. [DOI] [PubMed] [Google Scholar]
- 50.Bhilocha S., Amin R., Pandya M., Yuan H., Tank M., LoBello J., Shytuhina A., Wang W., Wisniewski H.G., de la Motte C., Cowman M.K. Agarose and polyacrylamide gel electrophoresis methods for molecular mass analysis of 5- to 500-kDa hyaluronan. Anal Biochem. 2011;417:41–49. doi: 10.1016/j.ab.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manzanares D., Monzon M.E., Savani R.C., Salathe M. Apical oxidative hyaluronan degradation stimulates airway ciliary beating via RHAMM and RON. Am J Respir Cell Mol Biol. 2007;37:160–168. doi: 10.1165/rcmb.2006-0413OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amemiya K., Nakatani T., Saito A., Suzuki A., Munakata H. Hyaluronan-binding motif identified by panning a random peptide display library. Biochim Biophys Acta. 2005;1724:94–99. doi: 10.1016/j.bbagen.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 53.Rodi D.J., Agoston G.E., Manon R., Lapcevich R., Green S.J., Makowski L. Identification of small molecule binding sites within proteins using phage display technology. Comb Chem High Throughput Screen. 2001;4:553–572. doi: 10.2174/1386207013330779. [DOI] [PubMed] [Google Scholar]
- 54.Jackson D.G. Immunological functions of hyaluronan and its receptors in the lymphatics. Immunol Rev. 2009;230:216–231. doi: 10.1111/j.1600-065X.2009.00803.x. [DOI] [PubMed] [Google Scholar]
- 55.Cherr G.N., Yudin A.I., Overstreet J.W. The dual functions of GPI-anchored PH-20: hyaluronidase and intracellular signaling. Matrix Biol. 2001;20:515–525. doi: 10.1016/s0945-053x(01)00171-8. [DOI] [PubMed] [Google Scholar]
- 56.Day A.J., Prestwich G.D. Hyaluronan-binding proteins: tying up the giant. J Biol Chem. 2002;277:4585–4588. doi: 10.1074/jbc.R100036200. [DOI] [PubMed] [Google Scholar]
- 57.Kiani C., Chen L., Wu Y.J., Yee A.J., Yang B.B. Structure and function of aggrecan. Cell Res. 2002;12:19–32. doi: 10.1038/sj.cr.7290106. [DOI] [PubMed] [Google Scholar]
- 58.Ziebell M.R., Prestwich G.D. Interactions of peptide mimics of hyaluronic acid with the receptor for hyaluronan mediated motility (RHAMM) J Comput Aided Mol Des. 2004;18:597–614. doi: 10.1007/s10822-004-5433-8. [DOI] [PubMed] [Google Scholar]
- 59.Yang B., Yang B.L., Savani R.C., Turley E.A. Identification of a common hyaluronan binding motif in the hyaluronan binding proteins RHAMM, CD44 and link protein. EMBO J. 1994;13:286–296. doi: 10.1002/j.1460-2075.1994.tb06261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mummert M.E., Mummert D.I., Ellinger L., Takashima A. Functional roles of hyaluronan in B16-F10 melanoma growth and experimental metastasis in mice. Mol Cancer Ther. 2003;2:295–300. [PubMed] [Google Scholar]
- 61.Twarock S., Tammi M.I., Savani R.C., Fischer J.W. Hyaluronan stabilizes focal adhesions, filopodia, and the proliferative phenotype in esophageal squamous carcinoma cells. J Biol Chem. 2010;285:23276–23284. doi: 10.1074/jbc.M109.093146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benitez A., Yates T.J., Lopez L.E., Cerwinka W.H., Bakkar A., Lokeshwar V.B. Targeting hyaluronidase for cancer therapy: antitumor activity of sulfated hyaluronic acid in prostate cancer cells. Cancer Res. 2011;71:4085–4095. doi: 10.1158/0008-5472.CAN-10-4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lokeshwar V.B., Selzer M.G. Differences in hyaluronic acid-mediated functions and signaling in arterial, microvessel, and vein-derived human endothelial cells. J Biol Chem. 2000;275:27641–27649. doi: 10.1074/jbc.M003084200. [DOI] [PubMed] [Google Scholar]
- 64.Fang R.C., Mustoe T.A. Animal models of wound healing: utility in transgenic mice. J Biomater Sci Polym Ed. 2008;19:989–1005. doi: 10.1163/156856208784909327. [DOI] [PubMed] [Google Scholar]
- 65.Benavides F., Oberyszyn T.M., VanBuskirk A.M., Reeve V.E., Kusewitt D.F. The hairless mouse in skin research. J Dermatol Sci. 2009;53:10–18. doi: 10.1016/j.jdermsci.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wong V.W., Sorkin M., Glotzbach J.P., Longaker M.T., Gurtner G.C. Surgical approaches to create murine models of human wound healing. J Biomed Biotechnol. 2011;969618:2011. doi: 10.1155/2011/969618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lindblad W.J. Considerations for selecting the correct animal model for dermal wound-healing studies. J Biomater Sci Polym Ed. 2008;19:1087–1096. doi: 10.1163/156856208784909390. [DOI] [PubMed] [Google Scholar]
- 68.de la Motte C., Nigro J., Vasanji A., Rho H., Kessler S., Bandyopadhyay S., Danese S., Fiocchi C., Stern R. Platelet-derived hyaluronidase 2 cleaves hyaluronan into fragments that trigger monocyte-mediated production of proinflammatory cytokines. Am J Pathol. 2009;174:2254–2264. doi: 10.2353/ajpath.2009.080831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shaw T.J., Kishi K., Mori R. Wound-associated skin fibrosis: mechanisms and treatments based on modulating the inflammatory response. Endocr Metab Immune Disord Drug Targets. 2010;10:320–330. doi: 10.2174/1871530311006040320. [DOI] [PubMed] [Google Scholar]
- 70.Stramer B.M., Mori R., Martin P. The inflammation-fibrosis link?: A Jekyll and Hyde role for blood cells during wound repair. J Invest Dermatol. 2007;127:1009–1017. doi: 10.1038/sj.jid.5700811. [DOI] [PubMed] [Google Scholar]
- 71.Wang J., Dodd C., Shankowsky H.A., Scott P.G., Tredget E.E. Deep dermal fibroblasts contribute to hypertrophic scarring. Lab Invest. 2008;88:1278–1290. doi: 10.1038/labinvest.2008.101. [DOI] [PubMed] [Google Scholar]
- 72.Itano N. Simple primary structure, complex turnover regulation and multiple roles of hyaluronan. J Biochem. 2008;144:131–137. doi: 10.1093/jb/mvn046. [DOI] [PubMed] [Google Scholar]
- 73.Delavary B.M., van der Veer W.M., van Egmond M., Niessen F.B., Beelen R.H. Macrophages in skin injury and repair. Immunobiology. 2011;216:753–762. doi: 10.1016/j.imbio.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 74.Kondo T., Ishida Y. Molecular pathology of wound healing. Forensic Sci Int. 2010;203:93–98. doi: 10.1016/j.forsciint.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 75.Daley J.M., Brancato S.K., Thomay A.A., Reichner J.S., Albina J.E. The phenotype of murine wound macrophages. J Leukoc Biol. 2010;87:59–67. doi: 10.1189/jlb.0409236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lucas T., Waisman A., Ranjan R., Roes J., Krieg T., Muller W., Roers A., Eming S.A. Differential roles of macrophages in diverse phases of skin repair. J Immunol. 2010;184:3964–3977. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 77.Mirza R., DiPietro L.A., Koh T.J. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol. 2009;175:2454–2462. doi: 10.2353/ajpath.2009.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goren I., Allmann N., Yogev N., Schurmann C., Linke A., Holdener M., Waisman A., Pfeilschifter J., Frank S. A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am J Pathol. 2009;175:132–147. doi: 10.2353/ajpath.2009.081002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martin P., D'Souza D., Martin J., Grose R., Cooper L., Maki R., McKercher S.R. Wound healing in the PU.1 null mouse—tissue repair is not dependent on inflammatory cells. Curr Biol. 2003;13:1122–1128. doi: 10.1016/s0960-9822(03)00396-8. [DOI] [PubMed] [Google Scholar]
- 80.Juniantito V., Izawa T., Yamamoto E., Murai F., Kuwamura M., Yamate J. Heterogeneity of macrophage populations and expression of galectin-3 in cutaneous wound healing in rats. J Comp Pathol. 2011;145:378–389. doi: 10.1016/j.jcpa.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 81.Jadus M.R., Irwin M.C., Irwin M.R., Horansky R.D., Sekhon S., Pepper K.A., Kohn D.B., Wepsic H.T. Macrophages can recognize and kill tumor cells bearing the membrane isoform of macrophage colony-stimulating factor. Blood. 1996;87:5232–5241. [PubMed] [Google Scholar]
- 82.Savani R.C., Khalil N., Turley E.A. Hyaluronan receptor antagonists alter skin inflammation and fibrosis following injury. Proc West Pharmacol Soc. 1995;38:131–136. [PubMed] [Google Scholar]
- 83.Mirza R., Koh T.J. Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine. 2011;56:256–264. doi: 10.1016/j.cyto.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 84.Lee R.H., Efron D., Tantry U., Barbul A. Nitric oxide in the healing wound: a time-course study. J Surg Res. 2001;101:104–108. doi: 10.1006/jsre.2001.6261. [DOI] [PubMed] [Google Scholar]
- 85.Park N.Y., Lim Y. Short term supplementation of dietary antioxidants selectively regulates the inflammatory responses during early cutaneous wound healing in diabetic mice. Nutr Metab (Lond) 2011;8:80. doi: 10.1186/1743-7075-8-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weller R. Nitric oxide: a key mediator in cutaneous physiology. Clin Exp Dermatol. 2003;28:511–514. doi: 10.1046/j.1365-2230.2003.01365.x. [DOI] [PubMed] [Google Scholar]
- 87.Amara F.M., Entwistle J., Kuschak T.I., Turley E.A., Wright J.A. Transforming growth factor-beta1 stimulates multiple protein interactions at a unique cis-element in the 3′-untranslated region of the hyaluronan receptor RHAMM mRNA. J Biol Chem. 1996;271:15279–15284. doi: 10.1074/jbc.271.25.15279. [DOI] [PubMed] [Google Scholar]
- 88.Samuel S.K., Hurta R.A., Spearman M.A., Wright J.A., Turley E.A., Greenberg A.H. TGF-beta 1 stimulation of cell locomotion utilizes the hyaluronan receptor RHAMM and hyaluronan. J Cell Biol. 1993;123:749–758. doi: 10.1083/jcb.123.3.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Park D., Kim Y., Kim H., Kim K., Lee Y.S., Choe J., Hahn J.H., Lee H., Jeon J., Choi C., Kim Y.M., Jeoung D. Hyaluronic acid promotes angiogenesis by inducing RHAMM-TGFbeta receptor interaction via CD44-PKCdelta. Mol Cells. 2012;33:563–574. doi: 10.1007/s10059-012-2294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Diegelmann R.F., Evans M.C. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci. 2004;9:283–289. doi: 10.2741/1184. [DOI] [PubMed] [Google Scholar]
- 91.Levesque M., Villiard E., Roy S. Skin wound healing in axolotls: a scarless process. J Exp Zool B Mol Dev Evol. 2010;314:684–697. doi: 10.1002/jez.b.21371. [DOI] [PubMed] [Google Scholar]
- 92.Seifert A.W., Monaghan J.R., Voss S.R., Maden M. Skin regeneration in adult axolotls: a blueprint for scar-free healing in vertebrates. PLoS One. 2012;7:e32875. doi: 10.1371/journal.pone.0032875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Harty M., Neff A.W., King M.W., Mescher A.L. Regeneration or scarring: an immunologic perspective. Dev Dyn. 2003;226:268–279. doi: 10.1002/dvdy.10239. [DOI] [PubMed] [Google Scholar]
- 94.Wehrhan F., Rodel F., Grabenbauer G.G., Amann K., Bruckl W., Schultze-Mosgau S. Transforming growth factor beta 1 dependent regulation of Tenascin-C in radiation impaired wound healing. Radiother Oncol. 2004;72:297–303. doi: 10.1016/j.radonc.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 95.Trebaul A., Chan E.K., Midwood K.S. Regulation of fibroblast migration by tenascin-C. Biochem Soc Trans. 2007;35:695–697. doi: 10.1042/BST0350695. [DOI] [PubMed] [Google Scholar]
- 96.Wong J.W., Gallant-Behm C., Wiebe C., Mak K., Hart D.A., Larjava H., Hakkinen L. Wound healing in oral mucosa results in reduced scar formation as compared with skin: evidence from the red Duroc pig model and humans. Wound Repair Regen. 2009;17:717–729. doi: 10.1111/j.1524-475X.2009.00531.x. [DOI] [PubMed] [Google Scholar]
- 97.Wight T.N., Potter-Perigo S. The extracellular matrix: an active or passive player in fibrosis. Am J Physiol Gastrointest Liver Physiol. 2011;301:G950–G955. doi: 10.1152/ajpgi.00132.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Buchanan E.P., Longaker M.T., Lorenz H.P. Fetal skin wound healing. Adv Clin Chem. 2009;48:137–161. doi: 10.1016/s0065-2423(09)48006-5. [DOI] [PubMed] [Google Scholar]
- 99.Zhao J., Guan J.L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- 100.Zhao X., Guan J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011;63:610–615. doi: 10.1016/j.addr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hall C.L., Turley E.A. Hyaluronan: rHAMM mediated cell locomotion and signaling in tumorigenesis. J Neurooncol. 1995;26:221–229. doi: 10.1007/BF01052625. [DOI] [PubMed] [Google Scholar]
- 102.Hall C.L., Yang B., Yang X., Zhang S., Turley M., Samuel S., Lange L.A., Wang C., Curpen G.D., Savani R.C., Greenberg A.H., Turley E.A. Overexpression of the hyaluronan receptor RHAMM is transforming and is also required for H-ras transformation. Cell. 1995;82:19–26. doi: 10.1016/0092-8674(95)90048-9. [DOI] [PubMed] [Google Scholar]
- 103.Mimura Y., Ihn H., Jinnin M., Asano Y., Yamane K., Tamaki K. Constitutive phosphorylation of focal adhesion kinase is involved in the myofibroblast differentiation of scleroderma fibroblasts. J Invest Dermatol. 2005;124:886–892. doi: 10.1111/j.0022-202X.2005.23701.x. [DOI] [PubMed] [Google Scholar]
- 104.Liu S., Xu S.W., Kennedy L., Pala D., Chen Y., Eastwood M., Carter D.E., Black C.M., Abraham D.J., Leask A. FAK is required for TGFbeta-induced JNK phosphorylation in fibroblasts: implications for acquisition of a matrix-remodeling phenotype. Mol Biol Cell. 2007;18:2169–2178. doi: 10.1091/mbc.E06-12-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lagares D., Busnadiego O., Garcia-Fernandez R.A., Kapoor M., Liu S., Carter D.E., Abraham D., Shi-Wen X., Carreira P., Fontaine B.A., Shea B.S., Tager A.M., Leask A., Lamas S., Rodriguez-Pascual F. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2012;64:1653–1664. doi: 10.1002/art.33482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Thannickal V.J., Lee D.Y., White E.S., Cui Z., Larios J.M., Chacon R., Horowitz J.C., Day R.M., Thomas P.E. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- 107.Piotrowicz R.S., Damaj B.B., Hachicha M., Incardona F., Howell S.B., Finlayson M. A6 peptide activates CD44 adhesive activity, induces FAK and MEK phosphorylation, and inhibits the migration and metastasis of CD44-expressing cells. Mol Cancer Ther. 2011;10:2072–2082. doi: 10.1158/1535-7163.MCT-11-0351. [DOI] [PubMed] [Google Scholar]
- 108.Zheng Y., Lu Z. Paradoxical roles of FAK in tumor cell migration and metastasis. Cell Cycle. 2009;8:3474–3479. doi: 10.4161/cc.8.21.9846. [DOI] [PubMed] [Google Scholar]
- 109.Zheng Y., Yang W., Xia Y., Hawke D., Liu D.X., Lu Z. Ras-induced and extracellular signal-regulated kinase 1 and 2 phosphorylation-dependent isomerization of protein tyrosine phosphatase (PTP)-PEST by PIN1 promotes FAK dephosphorylation by PTP-PEST. Mol Cell Biol. 2011;31:4258–4269. doi: 10.1128/MCB.05547-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Slevin M., Krupinski J., Gaffney J., Matou S., West D., Delisser H., Savani R.C., Kumar S. Hyaluronan-mediated angiogenesis in vascular disease: uncovering RHAMM and CD44 receptor signaling pathways. Matrix Biol. 2007;26:58–68. doi: 10.1016/j.matbio.2006.08.261. [DOI] [PubMed] [Google Scholar]
- 111.Namazi M.R., Fallahzadeh M.K., Schwartz R.A. Strategies for prevention of scars: what can we learn from fetal skin. Int J Dermatol. 2011;50:85–93. doi: 10.1111/j.1365-4632.2010.04678.x. [DOI] [PubMed] [Google Scholar]
- 112.Robert L., Robert A.M., Renard G. Biological effects of hyaluronan in connective tissues, eye, skin, venous wall: Role in aging. Pathol Biol (Paris) 2010;58:187–198. doi: 10.1016/j.patbio.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 113.Verdier-Sevrain S., Bonte F. Skin hydration: a review on its molecular mechanisms. J Cosmet Dermatol. 2007;6:75–82. doi: 10.1111/j.1473-2165.2007.00300.x. [DOI] [PubMed] [Google Scholar]
- 114.Liao Y.H., Jones S.A., Forbes B., Martin G.P., Brown M.B. Hyaluronan: pharmaceutical characterization and drug delivery. Drug Deliv. 2005;12:327–342. doi: 10.1080/10717540590952555. [DOI] [PubMed] [Google Scholar]
- 115.Kouvidi K., Berdiaki A., Nikitovic D., Katonis P., Afratis N., Hascall V.C., Karamanos N.K., Tzanakakis G.N. Role of receptor for hyaluronic acid-mediated motility (RHAMM) in low molecular weight hyaluronan (LMWHA)-mediated fibrosarcoma cell adhesion. J Biol Chem. 2011;286:38509–38520. doi: 10.1074/jbc.M111.275875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Matou-Nasri S., Gaffney J., Kumar S., Slevin M. Oligosaccharides of hyaluronan induce angiogenesis through distinct CD44 and RHAMM-mediated signalling pathways involving Cdc2 and gamma-adducin. Int J Oncol. 2009;35:761–773. doi: 10.3892/ijo_00000389. [DOI] [PubMed] [Google Scholar]
- 117.Zhang S., Chang M.C., Zylka D., Turley S., Harrison R., Turley E.A. The hyaluronan receptor RHAMM regulates extracellular-regulated kinase. J Biol Chem. 1998;273:11342–11348. doi: 10.1074/jbc.273.18.11342. [DOI] [PubMed] [Google Scholar]
- 118.Greenberg R.S., Bernstein A.M., Benezra M., Gelman I.H., Taliana L., Masur S.K. FAK-dependent regulation of myofibroblast differentiation. FASEB J. 2006;20:1006–1008. doi: 10.1096/fj.05-4838fje. [DOI] [PubMed] [Google Scholar]
- 119.Fan K., Tang J., Escandon J., Kirsner R.S. State of the art in topical wound-healing products. Plast Reconstr Surg. 2011;127(Suppl 1):44S–59S. doi: 10.1097/PRS.0b013e3181fbe275. [DOI] [PubMed] [Google Scholar]
- 120.Toriseva M., Kahari V.M. Proteinases in cutaneous wound healing. Cell Mol Life Sci. 2009;66:203–224. doi: 10.1007/s00018-008-8388-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Park H., Copeland C., Henry S., Barbul A. Complex wounds and their management. Surg Clin North Am. 2010;90:1181–1194. doi: 10.1016/j.suc.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 122.Bran G.M., Goessler U.R., Hormann K., Riedel F., Sadick H. Keloids: current concepts of pathogenesis (review) Int J Mol Med. 2009;24:283–293. doi: 10.3892/ijmm_00000231. [DOI] [PubMed] [Google Scholar]
- 123.Rodero M.P., Khosrotehrani K. Skin wound healing modulation by macrophages. Int J Clin Exp Pathol. 2010;3:643–653. [PMC free article] [PubMed] [Google Scholar]
- 124.Shih B., Garside E., McGrouther D.A., Bayat A. Molecular dissection of abnormal wound healing processes resulting in keloid disease. Wound Repair Regen. 2010;18:139–153. doi: 10.1111/j.1524-475X.2009.00553.x. [DOI] [PubMed] [Google Scholar]
- 125.Sloane J.A., Blitz D., Margolin Z., Vartanian T. A clear and present danger: endogenous ligands of Toll-like receptors. Neuromolecular Med. 2010;12:149–163. doi: 10.1007/s12017-009-8094-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gill S., Wight T.N., Frevert C.W. Proteoglycans: key regulators of pulmonary inflammation and the innate immune response to lung infection. Anat Rec (Hoboken) 2010;293:968–981. doi: 10.1002/ar.21094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Menezes G.B., McAvoy E.F., Kubes P. Hyaluronan, platelets, and monocytes: a novel pro-inflammatory triad. Am J Pathol. 2009;174:1993–1995. doi: 10.2353/ajpath.2009.081138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fragmented HA was covalently linked to Sepharose beads that were subsequently used for the screening of 7-, 12-, and 15mer phage display libraries that were eluted with free HA. Isolated phage clones were grown on a bacterial lawn, and the entire selection procedure was repeated. HA binding clones that also bound to chondroitin sulfate-Sepharose beads were excluded from further analyses. HA binding clones were picked and sequenced. Sequences were aligned with known HA binding regions from HA receptors and extracellular HA binding proteins. Only peptides P15-1 and P15-2 showed partial alignment with a known HA binding region, and that was to the HA binding domain of RHAMM. The HA binding sequences of RHAMM are highlighted with boxes, and known amino acids involved in HA binding that were identified by mutational analyses or computer modeling are marked green. P15-1 and P15-2 align to the second HA binding region of RHAMM.
P15-1 blocks motility of fibroblasts in a concentration-dependent manner and inhibits trafficking of macrophages. A: The random motility of RHAMM-expressing fibroblasts was quantified by real-time cinemicrographic analyses. Addition of different P15-1 concentrations to the culture medium during migration analysis inhibited random cell migration in a dose-dependent manner. The graph depicts cell migration speed ± SD of n = 50 cells. B: Full-thickness excisional skin wounds were treated with either P15-1 (50 μg/mL) or scrambled peptide (50 μg/mL). Cross sections of day 3 wounds were stained with a macrophage specific Ab, Mac-3. P15-1 significantly decreased the appearance of Mac-3 cells in day 3 wounds. The graph depicts the number of positive cells/microscopic field ± SD. n = 36 cells, 3 wounds. *P < 0.001.
Full-thickness excisional skin wounds were treated with either P15-1 (50 μg/mL) or scrambled control peptide (50 μg/mL). Cross sections of day 3 and 5 wounds were stained with iNOS-specific Ab. Representative images from n = 6 wounds are shown.
Proinflammatory cytokine protein expression by P15-1 and control skin wounds ex vivo. Proteome Profiler was used to determine the levels of cytokines released by wound cells in the presence or absence of P15-1. Supernatant medium was collected and processed for presence of cytokines according to the manufacturer's instructions. P15-1 had no detectable effect on the cytokines profiled in this assay other than a slight increase in L-selection. Values are the average of two duplicates.
Cross section of full-thickness excisional wounds treated with collagen vehicle alone (PBS control) or collagen vehicle plus P15-1 were stained with Masson's trichrome, allowing discrimination between collagen (blue) and cells.71Solid arrows indicate wound edges, and dotted arrows indicate blood vessels. Inserts show images of the entire wound site at ×4 original magnification. P15-1 decreased collagen accumulation within the wounds, as detected by image analysis. Values are the mean ± SD. n = 6 wounds. P < 0.001.