

Abstract
The pathogenic roles of glomerular deposition of components of the complement cascade in IgA nephropathy (IgAN) are not completely clarified. To investigate the pathologic role of complement pathways in IgAN, two IgAN-prone mouse models were examined. Grouped ddY (gddY) mice showed significant high proteinuria, severe glomerular lesions, and extracellular matrix expansion compared with high serum IgA (HIGA) mice but with similar intensity of glomerular IgA deposition. Glomerular activation of the classical, lectin, and alternative pathways was demonstrated by significantly stronger staining for complement (C)3, C5b-9, C1q, C4, mannose-binding lectin (MBL)-A/C, MBL-associated serine protease-2, and factor B and properdin in gddY mice than in HIGA mice. Similarly, the serum levels of IgA-IgG2a/IgM and IgA–MBL-A/C immune complexes and polymeric IgA were significantly higher in gddY mice than in HIGA mice. Moreover, the serum levels of aberrantly glycosylated IgA characterized by the binding of Sambucus nigra bark lectin and Ricinus communis agglutinin I were significantly higher in gddY mice than in HIGA mice. This aberrancy in glycosylation was confirmed by monosaccharide compositional analysis of purified IgA using gas-liquid chromatography. This study is the first to demonstrate that aberrantly glycosylated IgA may influence the formation of macromolecular IgA including IgA-IgG immune complexes and subsequent complement activation, leading to full progression of IgAN.
IgA nephropathy (IgAN) was first reported in 1968 by Berger and Hinglais1 and is a common form of progressive primary glomerulonephritis. The major histologic characteristics of IgAN are mesangial cell proliferation and matrix expansion with granular deposition of IgA, predominantly polymeric IgA1 (pIgA1),2 and complement (C)3 in the glomerular mesangial areas. This deposition in glomeruli may provide insights into IgAN pathogenesis. In IgAN, IgA1 has aberrant glycosylation of O-glycans in the hinge region3,4 and is predominantly in the polymeric form (pIgA).2,5–7 However, the pathologic interaction between the complement cascade and pIgA has not been completely clarified in IgAN.
Three different pathways of complement activation have been described. The classical pathway is triggered by activation of the C1 complex (composed of one molecule of C1q, two molecules of C1r, and two molecules of C1s, thus forming C1qr2s2).This activation occurs when C1q binds to IgM- or IgG-antigen complexes or when C1q binds directly to the surface of a pathogen. The lectin pathway is homologous to the classical pathway but contains opsonin, mannose-binding lectin (MBL), and ficolins instead of C1q. This pathway is activated by binding of MBL to mannose residues on the pathogen surface, thereby leading to activation of MBL-associated serine proteases (MASP-1 and MASP-2). These proteases split C4 into C4a and C4b and C2 into C2a and C2b. Similar to that in the classical pathway, C4b and C2a then bind together to form C3 convertase. The alternative pathway is triggered by spontaneous C3 hydrolysis [C3(H2O)] directly due to the breakdown of the thioester bond. This change in shape allows the binding of plasma protein factor B [C3(H2O)Bb, C3 convertase]. This convertase cleaves C3 proteins into C3a and C3b, which are then capable of covalently binding to a pathogenic membrane surface. Properdin is a component of the alternative pathway. It forms complexes with C3b to stabilize the alternative C3 convertase that further cleaves more C3. In all three pathways, C3 convertase cleaves and activates component C3 by forming C3a and C3b. It also cleaves C5 into C5a and C5b. C5b initiates the membrane attack pathway by forming a membrane attack complex with other recruited complement components (C6, C7, C7, C8, and multiple C9 molecules). Membrane attack complex is the cytolytic end product of the complement cascade. It forms a transmembrane channel that causes osmotic lysis of the target cell.
We used IgAN animal models to investigate IgAN pathogenesis. The ddY mouse is an animal model of spontaneous IgAN. Glomerular lesions in this mouse include mesangial proliferation and extracellular matrix expansion with paramesangial IgA deposition that closely resemble those found in human IgAN.8 In pooled serum and glomerular elutes of 16-, 40-, and 60-week-old ddY mice, the ratio of dimeric IgA and pIgA in total IgA increases markedly with age.9 However, the IgAN incidence in commercially available ddY mice is highly variable.10 The high serum IgA (HIGA) mouse is one of the IgAN models derived from the ddY mouse.11 This mouse is an inbred strain derived from ddY mice by selective mating of animals with high serum IgA levels. No renal abnormalities manifest in the HIGA mouse until approximately 10 weeks of age, with serum IgA levels increasing dramatically thereafter and becoming elevated by 25 weeks of age. Although this mouse shows high serum IgA levels, the increase is not associated with the severity of glomerular injury and the incidence of IgAN. We have evaluated commercially available ddY mice using serial renal biopsies obtained at 20, 40, and 60 weeks of age. These mice were divided into three groups: early-onset (approximately 20 weeks), late-onset (approximately 40 weeks), and quiescent (even at 60 weeks) mice.12 In ddY mice with disease onset, the levels of serum IgA-IgG2a immune complex (IC) correlated strongly with the severity of glomerular lesions,13 although no significant correlation was observed between serum IgA levels and the incidence of glomerular injury among the groups. We bred early-onset mice in a specific pathogen–free room and established a mouse IgAN model with an almost 100% incidence of the disorder.14 This mouse strain was called grouped ddY (gddY). Although the HIGA and gddY models are derived from ddY mice, their ratios of disease onset, phenotype, and disease severity are very different. In this study, we examined these differences from the aspect of complement activation.
Materials and Methods
Mice
Eleven female gddY mice were maintained at the animal facility of Juntendo University, Tokyo, Japan. The mice were fed regular chow (Oriental Yeast Co. Ltd., Tokyo, Japan) and water ad libitum and were housed in a specific pathogen–free room. Age-matched female HIGA and BALB/c mice were maintained in an identical manner. The experimental protocol was approved by the Ethics Review Committee for Animal Experimentation of Juntendo University Faculty of Medicine.
Measurement of Urinary Albumin and Creatinine Levels
Urinary albumin levels were measured using an enzyme-linked immunosorbent assay kit (Exocell Inc., Philadelphia, PA). Using a commercial kit (Exocell Inc.), urinary creatinine levels were measured based on Jaffe's reaction with alkaline picrate. Urinary albumin was normalized to urinary creatinine.
Histologic Analysis
Kidneys were obtained after perfusion with normal saline. Renal tissue specimens for microscopic evaluation were fixed in 15% formaldehyde, embedded in paraffin, cut into 3-μm-thick sections, and then stained with H&E and PAS. The mesangial area in the renal specimen of each mouse was determined by calculating the mean nucleus-free, PAS-positive area in >20 glomeruli located in the glomerular hilus, as described previously.15
Renal specimens for immunofluorescence (IF) were mounted in OCT compound (Sakura Finetek Japan Co. Ltd., Tokyo, Japan) and stored at −80°C. The specimens were then cut into 3-μm-thick sections, fixed with acetone at −20°C for 4 minutes, and stained (IF staining) for IgA, IgG2a, C3, C4, C1q, C5b-9, MBL-A, MBL-C, MASP-2, and factor B and properdin. In brief, the sections were washed with PBS, blocked with a blocking agent (DS Pharma Biomedical Co. Ltd., Osaka, Japan) at room temperature for 30 minutes, and then incubated at room temperature for 60 minutes with the following primary antibodies: goat anti-IgA, goat anti-IgG2a (Bethyl Laboratories Inc., Montgomery, TX), rat anti-C3, rat anti-C4, rat anti-C1q, rat anti-MBL-A, rat anti-MBL-C (Hycult Biotech, Uden, The Netherlands), anti-MASP-2, anti-factor B and anti-properdin (Santa Cruz Biotechnology, Santa Cruz, CA), and anti-C5b-9 (Abcam Inc., Cambridge, MA). After three washes with PBS, the samples were incubated for 30 minutes with secondary antibodies compatible with the primary antibody to some degree, washed, and then mounted in mounting medium (Dako, Tokyo, Japan). Images were taken using a confocal laser microscope (Olympus Corp., Tokyo, Japan) with the same detector sensitivity for the same target antigen. The fluorescence-positive area was determined using ImageJ software (National Institutes of Health, Bethesda, MD) as described previously.15 For double IF staining (IgA-IgG2a, IgA–MBL-A, IgA–MBL-C, and IgA-C1q), the primary and secondary antibodies were added to the incubation mixture. Rabbit anti-mouse IgG2a was detected using Cy5-conjugated donkey anti-rabbit IgG antibody. The other primary antibodies were detected using the same secondary antibodies that were used for single IF staining.
An enzyme-labeled antibody method was used to assess type IV collagen expression in the paraffin-embedded sections. After deparaffinization and rehydration, the renal sections were washed twice for 5 minutes with PBS. The sections were treated with 0.3% H2O2 for 30 minutes to block endogenous peroxide activity. They were then incubated at room temperature for 60 minutes with anti-type IV collagen antibody (AbD Serotec MorphoSys, Oxford, UK). After reaction with the primary antibody, the sections were reacted with horseradish peroxidase–conjugated goat anti-rabbit antibody (Nichirei Biosciences, Tokyo, Japan) and then visualized by treatment with diaminobenzidine (Dako). Quantitative analysis of type IV collagen expression in the glomerular areas was performed using minor modifications of a previously described method.16 In brief, 20 random glomerular cross sections were chosen from each tissue section and examined. The examined areas were outlined, the positive staining patterns were identified, and the percentage of positive staining area in each glomerular cross section was then measured. Three nephrologists and pathologists quantified them in a blinded manner. To check the specificity of antibodies used, we examined kidneys of NOD SCID mice and MBL knockout mice as negative controls (data not shown).
Isolation of Glomeruli and Quantification of Type IV Collagen mRNA Expression
Glomeruli were isolated by a sieving technique. Total RNA was purified from the isolated glomeruli using an RNA extraction kit (Qiagen, Tokyo, Japan) according to the manufacturer's instructions. Aliquots (500 ng) of total glomerular RNA were reverse transcribed (Life Technologies, Tokyo, Japan) and were analyzed using a real-time PCR system (Life Technologies) and Power SYBR green PCR master mix (Life Technologies). The primer sequence for type IV collagen has been published previously.17 Mouse glyceraldehyde-3-phosphate dehydrogenase was chosen as the housekeeping gene. All real-time PCR measurements were performed in triplicate and included no template controls. The glomerular transcript abundance of type IV collagen was calculated by the 2−ΔΔCT method.18
Measurement of Serum IgA and Aberrantly Glycosylated IgA Levels
Serum IgA levels were measured using a sandwich enzyme-linked immunosorbent assay (ELISA) kit (Bethyl Laboratories Inc.). Aberrantly glycosylated bound serum IgA levels were measured using biotinylated Sambucus nigra bark lectin (SNA; Vector Laboratories, Burlingame, CA) and Ricinus communis agglutinin I (RCA-I; Vector Laboratories).19 Microtiter plates coated with goat anti-mouse IgA used for quantification of serum IgA were incubated with these samples, and biotinylated SNA or RCA-I was then added. Horseradish peroxidase–avidin D (Vector Laboratories) was applied, and color developed after 15 minutes of exposure to 3,3′,5,5′-tetramethylbenzidine (Becton Dickinson, Tokyo, Japan). The color development reaction was stopped by adding 2 N of H2SO4, and absorbance at 450 nm was read using a microplate reader (Nihon Molecular Devices, Tokyo, Japan). A standard curve was produced for each of the two lectins using serial dilutions of the reference serum (Bethyl Laboratories Inc.). The quantity of lectin bound to 1 g of IgA was defined as 1 U. All the samples were reduced by 9% 2-mercaptoethanol (Wako Pure Chemical Industries Ltd., Tokyo, Japan) to avoid the influence of lectin binding by other constituent molecules in IgA-IC. Western blot analysis using serum samples under the reducing condition confirmed that the IgA-IgG IC was resolved (data not shown).
Monosaccharide Compositional Analysis of IgA by Gas-Liquid Chromatography
IgA was purified from sera of gddY and HIGA mice, and the purity of the preparations was assessed by SDS-PAGE. Trichloroacetic acid-precipitated 10-μg protein for each IgA sample was analyzed by gas-liquid chromatography with sorbitol as the internal standard. The analyses were performed using a gas chromatograph (model 5890; Hewlett-Packard, Sacramento, CA) equipped with a 25-m fused silica (0.22-mm inner diameter) OV-1701 wall-coated open tubular column (Chrompack Inc., Bridgewater, NJ) and an electron capture detector. Standard sugars were used for quantification. The results are expressed relative to the internal standard and specific sugar standard, using the area under the peak in chromatograms.20,21
Measurement of Serum IC Levels
Serum IC (IgA-IgG2a, IgA–MBL-A, and IgA–MBL-C) levels were measured by sandwich ELISA using a modification of a method described previously.13 In brief, microtiter plates (Nalge Nunc International, Tokyo, Japan) were coated with anti-mouse IgA antibody. After incubation, the microtiter plates were washed with PBS containing 0.05% Tween 20 and then were blocked at room temperature for 60 minutes with 250 μL per well of blocking solution (1% blocking agent with PBS containing 0.05% Tween 20). The plates were then stored at 4°C until use. The microtiter plates coated with antibody were loaded in duplicate with diluted serum and were incubated at room temperature for 60 minutes. In the assay for the measurement of IgA-IgG2a complexes, the sample and antibody were diluted with blocking solution. For other complex assays, the samples were diluted with PBS containing 1 mmol/L CaCl2 and the antibody was diluted with blocking solution containing 1 mmol/L CaCl2. After washing with PBS containing 0.05% Tween 20, 90 μL per well of anti-IgG2a–horseradish peroxidase was used in the IgA-IgG2a complex assays. After incubation at room temperature for 60 minutes, 90 μL of 3,3′,5,5′-tetramethylbenzidine was added to each well. In the other assays, rat anti-MBL-A or rat anti-MBL-C was added to the wells and was incubated at room temperature for 60 minutes. After the plates were washed, biotinylated donkey anti-rat IgG was applied. After 60-minute incubation at room temperature, the microplates were washed five times with PBS containing 0.05% Tween 20, and horseradish peroxidase–avidin D was applied. After 60-minute incubation and further washing, the color was developed by 3,3′,5,5′-tetramethylbenzidine using the same method described for IgA-IgG2a IC. In all the assays, the color development reaction was stopped by adding 90 μL of 2 N H2SO4 to each well, and absorbance at 450 nm was read using the microplate reader. For comparison, each absorbance was adjusted for the serum IgA level.
Determination of the Molecular Weight of IgA
The molecular weight of IgA was determined by Western blot analysis using a modification of a method described previously.22 The serum was resolved by 5% to 20% gradient SDS-PAGE and was transferred onto polyvinylidene difluoride membranes (Nihon Millipore KK, Tokyo, Japan). The membranes were blocked with 4% blocking solution and then were incubated with diluted goat anti-mouse IgA at room temperature for 60 minutes. After washing in PBS, the sections were incubated with the secondary antibody that corresponded to the primary antibody used. IgA was detected by chemiluminescence using ECL (GE Healthcare, Tokyo, Japan) and radiography (Konica Minolta Healthcare, Tokyo, Japan). The samples were analyzed under nonreducing conditions.
Statistical Analysis
The data are expressed as mean ± SD. The statistical significance of the experimental observations was determined by analysis of variance, with the level of significance set at P < 0.05. Descriptive statistical analysis was performed using statistical software (IBM Japan, Tokyo, Japan).
Results
Glomerular Injury in gddY Mice Is More Severe than in HIGA Mice despite the Same Degree of IgA Deposition
The severity of glomerular injury was examined in both IgAN-prone strains of mice. The urinary albumin level in gddY mice at 11 weeks of age was considerably higher than that in age-matched HIGA mice (Figure 1A). Mesangial matrix expansion in gddY mice was also more severe than that in HIGA mice (Figure 1, B and C). Furthermore, expression of type IV collagen mRNA and protein in gddY mice was higher than that in HIGA mice (Figure 1, D–F). Although the degree of glomerular injury was very different in the two strains of mice, glomerular IgA deposition was identical in both IgAN-prone mouse strains (Figure 1, G and H).
Figure 1.

Glomerular injury in gddY mice is more severe than that in HIGA mice. A: Proteinuria at 11 weeks [albumin creatinine ratio (ACR)]. B and C: Glomerular lesions and PAS-positive areas in glomeruli of gddY mice were significantly more severe than those in glomeruli of HIGA and control BALB/c mice. D–F: Glomerular type IV collagen (Col IV) expression in each IgAN-prone mouse. Expression of Col IV mRNA and protein was significantly higher in gddY mice than in HIGA mice. G and H: Glomerular IgA deposition in each IgAN-prone mouse. No significant difference was observed in glomerular IgA deposition in gddY and HIGA mice. Eleven mice were evaluated in each group. The bars represent mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
Glomerular Complements Are More Activated by the Three Pathways in gddY Mice than in HIGA Mice
The degree of activation of complement pathways was investigated in murine IgAN. IF staining showed that glomerular deposition of C3 and C5b-9 was more abundant in gddY mice than in HIGA mice (Figure 2A). This finding indicated greater complement activation in gddY mice.
Figure 2.

Glomerular complement cascades were strongly activated in gddY mice. A: Common pathway: the positive area of glomerular C3 and C5b-9 staining in gddY mice was stronger than that in HIGA mice. B: Lectin pathway: the positive area of glomerular MBL-A, MBL-C, and MASP-2 staining was stronger in gddY mice than that in HIGA mice. C: Classical pathway: the positive area of glomerular C1q and C4 staining in gddY mice was stronger than that in HIGA mice. D: Alternative pathway: the positive area of glomerular factor B and properdin staining in gddY mice was stronger than that in HIGA mice. Eleven mice were evaluated in each group. The bars represent mean ± SD. *P < 0.01, **P < 0.001, and ***P < 0.0001.
To identify which complement pathways were involved, the key components of the three pathways were measured. The positive areas of glomerular C1q and C4 staining in gddY mice were significantly higher than those in HIGA mice (Figure 2C), indicating that the classical pathway in gddY mice was considerably more activated than that in HIGA mice. Next, the components of the lectin pathway, such as MBL-A/C and MASP-2, were analyzed in the glomeruli of both IgAN-prone strains of mice (Figure 2B). The amount of MBL-A/C deposition in gddY mice was significantly higher than that in HIGA mice. Similarly, the positive area of MASP-2 staining in gddY mice was also significantly higher than that in HIGA mice. These findings indicated that activation of the lectin pathway in glomeruli of gddY mice was higher than that in glomeruli of HIGA mice. Furthermore, the positive area of glomerular factor B and properdin staining in gddY mice was significantly higher than that in HIGA mice (Figure 2D). This finding indicated that the alternative pathway in gddY mice was also markedly more activated than that in HIGA mice.
Levels of IgA-IC in Sera and Glomeruli Are Markedly Increased in gddY Mice Compared with Levels in HIGA Mice
To assess why the classical pathway was more activated in gddY mice, the characteristics of glomerular ICs and their serum levels were investigated. Although serum IgA levels in HIGA mice were significantly higher than those in gddY mice (Figure 3A), the serum level of IgA-IgG2a IC per unit of IgA in gddY mice was significantly higher than that in HIGA mice (Figure 3B). Double IF staining showed that C1q was co-deposited with IgG2a and IgM (Figure 3, C and D). However, the positive area of glomerular IgG2a and IgM staining in gddY mice was significantly higher than that in HIGA mice (Figure 3, C and D).
Figure 3.

Levels of IgA-IgG2a IC in sera and glomerular deposition were markedly increased in gddY mice compared with HIGA mice. A: The serum IgA level was significantly higher in HIGA mice than in gddY and control mice. B: However, the serum level of IgA-IgG2a IC was significantly higher in gddY mice than in HIGA and control mice. IgG2a (C) and IgM (D) were co-localized with C1q in the glomeruli of both IgAN-prone strains of mice. The intensity of IgG2a, IgM, and C1q staining was significantly higher in gddY mice than in HIGA mice. Eleven mice were evaluated in each group. The bars represent mean ± SD. *P < 0.01, **P < 0.001, and ***P < 0.0001
Amounts of MBL Associated with Serum IC and Co-Deposited with IgA in Glomeruli Are Considerably Higher in gddY Mice than in HIGA Mice
To assess the underlying mechanism that caused greater activation of the lectin pathway in gddY mice, MBL binding to IgA-IC was investigated (Figure 4). MBL-A and MBL-C were co-localized with IgA (Figure 4, A and B). Although serum IgA levels were significantly higher in HIGA mice than in gddY mice, serum levels of MBL bound to IgA-IC in gddY mice were significantly increased compared with those in HIGA mice (Figure 4, C and D).
Figure 4.

gddY mice showed greater activation of the lectin pathway. Serum levels of IgA–MBL-A IC (A) and IgA–MBL-C IC (B) were significantly higher in gddY mice than in HIGA mice. MBL-A (C) and MBL-C (D) were co-localized with IgA in both IgAN-prone strains of mice. Eleven mice were evaluated in each group. The bars represent mean ± SD. *P < 0.01, **P < 0.001.
gddY and HIGA Mice Have pIgA, although gddY Mice Had Higher Amounts of Aberrantly Glycosylated IgA than HIGA Mice
The molecular forms of serum IgA were analyzed in two IgAN-prone strains of mice (Figure 5). The amount of high-molecular-weight IgA in HIGA mice was higher than that in gddY mice (Figure 5A), similar to the situation observed for serum IgA levels. After compensation by serum IgA levels, the amount of high-molecular-weight IgA in gddY mice was not different from that in HIGA mice (Figure 5A).
Figure 5.
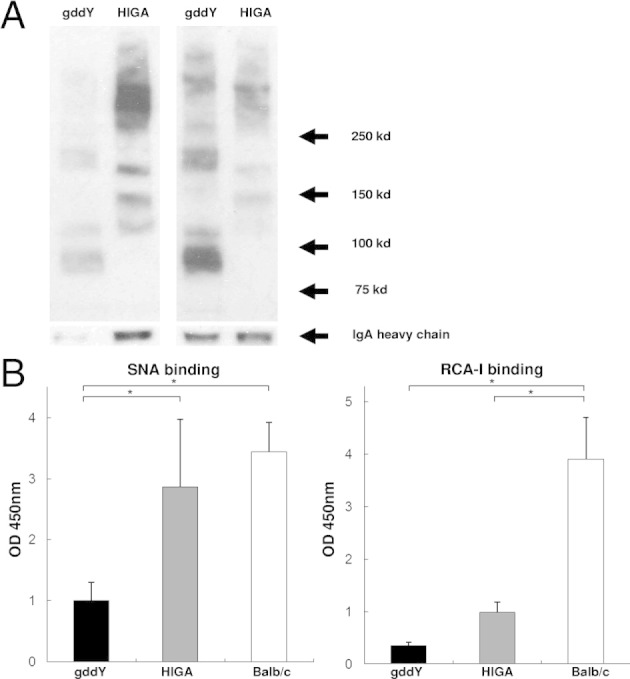
gddY mice had higher amounts of circulatory aberrantly glycosylated IgA than HIGA mice. A: Levels of serum IgA were increased in HIGA mice (left panel). However, after adjustment for serum IgA levels, high-molecular-weight IgA was more abundant in gddY mice (right panel). The loaded samples under reducing conditions were developed with anti-IgA antibody (lower panels). B: Serum IgA in gddY mice showed significantly lower binding to lectin SNA than that in HIGA and BALB/c mice and significantly lower binding to RCA-I than that in BALB/c mice. Eleven mice were evaluated in each group. The bars represent mean ± SD. *P < 0.01.
Next, the lectin-binding assays were used19 to access the glycoform of IgA in both IgAN-prone strains of mice. This assay was designed using biotinylated SNA and RCA-I that recognize terminal sialic acid (particularly Neu5Aca2-6Gal) and galactose residues, respectively. Serum IgA from gddY mice showed a significantly lower affinity to lectin SNA and RCA-I than that from HIGA mice (Figure 5B). These findings suggest that the content of terminal sialic acid (amount of binding lectin SNA) and terminal galactose (amount of binding RCA-I) on IgA in gddY mice was significantly less than that on IgA in HIGA mice.
Aberrancy in Glycosylation of Serum IgA from gddY Mice Was Confirmed by Monosaccharide Compositional Analysis
Monosaccharide compositional analysis of purified IgA from gddY and HIGA mice was determined by gas-liquid chromatography. Sugar composition was consistent with the result of a lectin ELISA using SNA and RCA-I. Specifically, monosaccharide composition revealed that IgA purified from sera of gddY mice had a lower content of galactose and sialic acid compared with that of IgA from sera of HIGA mice (Table 1).
Table 1.
Monosaccharide Composition of Murine IgA Determined by Gas-Liquid Chromatography
| Sugar | HIGA mice | gddY mice |
|---|---|---|
| Mannose | 0.485 | 0.204 |
| GlcNAc | 0.274 | 0.141 |
| GalNAc | ND | ND |
| Galactose | 0.302 | 0.148 |
| SA | 0.257 | 0.135 |
| IS | 1.000 | 1.000 |
Data are expressed as the average of two experiments relative to standard sugars and normalized to the IS, based on the area under the peak in the chromatograms. Ten micrograms of IgA was used per sample.
GlcNAc, N-acetylglucosamine; GalNAc, N-acetylgalactosamine; IS, internal standard; ND, not detected; SA, sialic acid.
Discussion
The degree of glomerular injury was more severe in gddY mice than in HIGA mice. gddY mice showed marked glomerular deposition of C5b-9 and C3 compared with HIGA mice, suggesting that in situ activation of the complement cascade was greater in gddY mice. This finding is compatible with the report that C5b-9 staining is more intense in human IgAN cases with advanced glomerular lesions than in those with minimal glomerular lesions.23 An in vitro study showed that sublytic C5b-9, which can induce injury in the mesangial cells, can induce complete DNA synthesis in these cells.24 Disease progression may, therefore, be attributed to the end products of the complement cascade, namely, C5b-9.
Using histologic analyses, this study showed activation of the three complement pathways (classical, lectin, and alternative) in glomeruli. Although C1q is not usually detected in glomeruli in human IgAN, there is evidence that cases with C1q deposition tend to become more severe.23,25–27 Furthermore, co-deposition of IgA, IgG, and IgM in glomeruli, the so-called full house pattern, is observed in association with C1q.28 In human IgAN, ICs containing pIgA1 have a high molecular mass of approximately 800 to 900 kDa.29 These ICs can pass through the larger fenestrate of endothelial cells in the glomerular capillaries (500–1000 Å) and get deposited in the mesangium.30 The present study showed that gddY mice had higher serum levels of IgA-IgG2a ICs13 than HIGA mice. In addition, glomerular depositions of IgG2a and IgM were more abundant in gddY mice than in HIGA mice. Because the classical pathway is activated mainly by IgG and IgM in ICs,31 elevation of serum and glomerular levels of IgA-IgG2a or IgA-IgM ICs in gddY mice may accelerate activation of the classical pathway in glomeruli.
Previous in vitro studies have shown that aggregated pIgA, but not monomeric IgA, can bind to MBL in a dose-dependent manner.32 This earlier report also demonstrated that pIgA activated the complement system via the lectin pathway to a greater degree than dimeric IgA, whereas no significant activation could be detected with monomeric IgA. Approximately 25% of patients with IgAN show glomerular deposition of MBL, L-ficolin, MASP, and C4d33 as proof of activation of the lectin pathway. These patients with IgAN are generally younger than those without such activation. In addition, the duration from disease onset to biopsy in such patients is shorter than that in patients without activation.34 We showed that co-deposition of MBL with IgA in glomeruli of IgAN models or with the lectin pathway in gddY mice was similar and was more activated than in HIGA mice. In addition to MBL deposition, we detected elevation in the serum levels of IgA–MBL-A/C IC in both IgAN models. However, the levels of IgA-MBL ICs in gddY mice were higher than those in HIGA mice, suggesting that serum IC consisting of aberrantly glycosylated IgA binds to MBL, thereby activating the lectin pathway in glomeruli.
The present study also demonstrated increased intensity of properdin and factor B staining in gddY mice compared with that in HIGA mice. In human IgAN, properdin and factor B are observed in glomeruli.23,35 In general, in situ deposition of properdin and factor B characterizes activation of the alternative pathway. C3b binds covalently to pathogenic membrane surfaces. The amount of C3b binding to the IgA molecule was increased by removing sialic acid and N-linked glycan chains from normal serum IgA1 and IgA2.36 There is evidence that sialylation and galactosylation of O-glycans in the hinge region of human serum IgA1 are decreased to a greater degree in patients with IgAN than in patients with non-IgAN primary glomerulonephritis and controls.37 On the basis of these studies, we suggest that aberrantly glycosylated IgA complexed with C3b and factor B are co-deposited in glomeruli. Properdin mRNA is strongly expressed in human glomeruli,38 indicating that C3bBb is stabilized by binding to properdin in situ and, thus, leads to activation of the alternative pathway. In addition to the ability of stabilization, properdin can direct complement activation more selectively by recognizing and binding to specific target surfaces referred to as the properdin-directed pathway.39,40 The other mechanism of activation of the complement cascade in this study is the ability of properdin binding to apoptotic cells38 and necrotic cells,41 initiating in situ activation of complement cascade. Although this mechanism in human IgAN is not really investigated, 18% of patients with lupus nephritis with properdin depositions on glomeruli without factor B assumed that properdin deposition might lead to properdin-directed pathway activation with disease progression.42
It is well-known that pIg or high-molecular-weight ICs have more potency for complement activation in all the pathways than do oligomeric Ig or low-molecular-weight ICs.7,43–46 One of the most important findings of this study is that the phlogogenic potency, via complement pathways leading to full progression of IgAN, is more dependent on the molecular form of IgA than on the amounts of glomerular IgA deposition. Therefore, one can speculate that the greater complement activation in gddY mice may essentially be due to a higher molecular form of IgA, including IgA-IgG IC, rather than to aberrant glycosylation of IgA itself. Indeed, to our knowledge, no study has found any direct evidence linking the aberrantly glycosylated IgA to complement activation or even the glomerular damage in human IgAN.
However, the aberrant glycosylation of IgA should importantly contribute to the high molecular formation. In human IgAN, many studies have reported serum elevation of pIgA and high-molecular-weight IgA1 levels due to self-aggregation and formation of ICs composed of aberrantly O-glycosylated IgA and anti-glycan IgG.47–50 Moreover, extracted IgA1 from mesangial deposits are also aberrantly glycosylated.3,4
Although mice do not have IgA molecules with structural analogy to human IgA1, a recent study has demonstrated the presence of O-linked glycans in the hinge region of an IgA rheumatoid factor, and its potential to induce IgAN-like glomerular lesions was associated with increased levels of O-glycosylation.51 In addition, gddY and HIGA mice carry a potential O-glycosylation acceptor site in the hinge region of IgA,14 suggesting the possibility that only a minor fraction of IgA is O-glycosylated and exhibits a pathogenic potency in gddY mice, as speculated in human IgAN. On the other hand, deficiency of β1,4-galactosylation and sialylation of N-glycan by gene targeting of β1,4-galactosyltransferese-I induces murine IgAN with serum elevation of polymeric forms of IgA and glomerular IgG co-depositions.52 In the present study, we found that gddY mice have increased levels of high-molecular-weight IgA and IgA-IC. Present lectin binding assays suggest that it may be due to the differences of oligosaccharide contents in IgA compared with HIGA mice. To further address this question, more extensive biochemical analysis of sugar contents was conducted by gas-liquid chromatography. The analysis of sugar contents revealed that purified IgA from gddY mice had lower contents of oligosaccharides compared with IgA from HIGA mice (Table 1). This finding, thus, indicates that aberrantly glycosylated IgA, whether alteration of the glycosylation site is O-glycan or N-glycan, can promote the development of macromolecular formation of IgA including IgG autoantibodies against sugar moiety on IgA in gddY mice, as in the case of human IgAN.48
Conclusion
The present study demonstrated that the classical, lectin, and alternative pathways of the complement cascade are activated in glomeruli in murine IgAN. More severe glomerular injury associated with greater activation of these pathways is observed in gddY mice. These differences between gddY and HIGA mice may be because of the degree of serum macromolecular IgA including IgA-IC with IgG/IgM or MBL-A/C. We further hypothesized that aberrantly glycosylated IgA importantly contributes to the formation of high-molecular-weight IgA and IgA-IC and subsequent complement activation, leading to full progression of IgAN (Figure 6).
Figure 6.

The hypothesis of the role of complement pathways in murine IgAN. The findings of this study in gddY and HIGA mice suggest that the degree of aberrant glycosylation of IgA may determine the degree of IC formation and IgA polymerization and, therefore, influence subsequent glomerular injury through activation of the different complement pathways.
Acknowledgments
We thank Terumi Shibata for her excellent technical assistance. We also thank Takako Ikegami, Tomomi Ikeda (Juntendo University Graduate School of Medicine), and Shinji Nakamura (BioMedical Research Center, Juntendo University Graduate School of Medicine) for their excellent technical assistance.
Footnotes
Supported in part by a research grant from the Study Group on IgA Nephropathy; a Grant-in-Aid for Progressive Renal Disease Research, Research on Intractable Disease, from the Ministry of Health, Labour, and Welfare of Japan, a Grant-in-Aid for Young Scientists (B) No. 22790804 (Y.T.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and by NIH grants DK083663, DK078244, DK082753, and DK075868 (R.B., S.H., and J.N.).
References
- 1.Berger J., Hinglais N. Intercapillary deposits of IgA-IgG. J Urol Nephrol. 1968;74:694–695. [PubMed] [Google Scholar]
- 2.Tomino Y., Sakai H., Miura M., Endoh M., Nomoto Y. Detection of polymeric IgA in glomeruli from patients with IgA nephropathy. Clin Exp Immunol. 1982;49:419–425. [PMC free article] [PubMed] [Google Scholar]
- 3.Allen A.C., Bailey E.M., Brenchley P.E., Buck K.S., Barratt J., Feehally J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: observations in three patients. Kidney Int. 2001;60:969–973. doi: 10.1046/j.1523-1755.2001.060003969.x. [DOI] [PubMed] [Google Scholar]
- 4.Hiki Y., Odani H., Takahashi M., Yasuda Y., Nishimoto A., Iwase H., Shinzato T., Kobayashi Y., Maeda K. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int. 2001;59:1077–1085. doi: 10.1046/j.1523-1755.2001.0590031077.x. [DOI] [PubMed] [Google Scholar]
- 5.Novak J., Julian B.A., Tomana M., Mestecky J. Progress in molecular and genetic studies of IgA nephropathy. J Clin Immunol. 2000;21:310–327. doi: 10.1023/a:1012284402054. [DOI] [PubMed] [Google Scholar]
- 6.Floege J., Feehally J. IgA nephropathy: recent developments. J Am Soc Nephrol. 2000;11:2395–2403. doi: 10.1681/ASN.V11122395. [DOI] [PubMed] [Google Scholar]
- 7.Van der Boog P.J., Van Kooten C., Fijter J.W., Daha M.R. Role of macromolecular IgA in IgA nephropathy. Kidney Int. 2005;67:813–821. doi: 10.1111/j.1523-1755.2005.00146.x. [DOI] [PubMed] [Google Scholar]
- 8.Imai H., Nakamoto Y., Asakura K., Miki K., Yasuda T., Miura A.B. Spontaneous glomerular IgA deposition in ddY mice: an animal model of IgA nephritis. Kidney Int. 1985;27:756–761. doi: 10.1038/ki.1985.76. [DOI] [PubMed] [Google Scholar]
- 9.Muso E., Yoshida H., Takeuchi E., Shimada T., Yashiro M., Sugiyama T., Kawai C. Pathogenic role of polyclonal and polymeric IgA in a murine model of mesangial proliferative glomerulonephritis with IgA deposition. Clin Exp Immunol. 1991;84:459–465. [PMC free article] [PubMed] [Google Scholar]
- 10.Kawaguchi S. Immunopathological study of glomerular IgA deposition in ddY mice. Acta Pathol Jpn. 1988;38:1–10. doi: 10.1111/j.1440-1827.1988.tb01067.x. [DOI] [PubMed] [Google Scholar]
- 11.Miyawaki S., Muso E., Takeuchi E., Matsushima H., Shibata Y., Sasayama S., Yoshida H. Selective breeding for high serum IgA levels from noninbred ddY mice: isolation of a strain with an early onset of glomerular IgA deposition. Nephron. 1997;76:201–207. doi: 10.1159/000190169. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki H., Suzuki Y., Yamanaka T., Hirose S., Nishimura H., Toei J., Horikoshi S., Tomino Y. Genome-wide scan in a novel IgA nephropathy model identifies a susceptibility locus on murine chromosome 10, in a region syntenic to human IGAN1 on chromosome 6q22-23. J Am Soc Nephrol. 2005;16:1289–1299. doi: 10.1681/ASN.2004030219. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki H., Suzuki Y., Aizawa M., Yamanaka T., Kihara M., Pang H., Horikoshi S., Tomino Y. Th1 polarization in murine IgA nephropathy directed by bone marrow-derived cells. Kidney Int. 2007;72:319–327. doi: 10.1038/sj.ki.5002300. [DOI] [PubMed] [Google Scholar]
- 14.Okazaki K, Suzuki Y, Otsuji M, Suzuki H, Kihara M, Kajiyama T, Hashimoto A, Nishimura H, Brown R, Hall S, Novak J, Izui S, Hirose S, Tomino Y: Development of a model of early-onset IgA nephropathy [published online July 12, 2012]. J Am Soc Nephrol [DOI] [PMC free article] [PubMed]
- 15.Kobayashi I., Nogaki F., Kusano H., Ono T., Miyawaki S., Yoshida H., Muso E. Interleukin-12 alters the physicochemical characteristics of serum and glomerular IgA and modifies glycosylation in a ddY mouse strain having high IgA levels. Nephrol Dial Transplant. 2002;17:2108–2116. doi: 10.1093/ndt/17.12.2108. [DOI] [PubMed] [Google Scholar]
- 16.Lu T.C., Wang Z.H., Feng X., Chuang P.Y., Fang W., Shen Y., Levy D.E., Xiong H., Chen N., He J.C. Knockdown of Stat3 activity in vivo prevents diabetic glomerulopathy. Kidney Int. 2009;76:63–71. doi: 10.1038/ki.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herbach N., Schairer I., Blutke A., Kautz S., Siebert A., Goke B., Wolf E., Wanke R. Diabetic kidney lesions of GIPRdn transgenic mice: podocyte hypertrophy and thickening of the GBM precede glomerular hypertrophy and glomerulosclerosis. Am J Physiol Renal Physiol. 2009;296:819–829. doi: 10.1152/ajprenal.90665.2008. [DOI] [PubMed] [Google Scholar]
- 18.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Chintalacharuvu S.R., Emancipator S.N. The glycosylation of IgA produced by murine B cells is altered by Th2 cytokines. J Immunol. 1997;159:2327–2333. [PubMed] [Google Scholar]
- 20.Renfrow M.B., Cooper H.J., Tomana M., Kulhavy R., Hiki Y., Toma K., Emmett M.R., Mestecky J., Marshall A.G., Novak J. determination of aberrant O-glycosylation in the IgA1 hinge region by electron capture dissociation Fourier transform ion cyclotron resonance mass spectrometry. J Biol Chem. 2005;280:19136–19145. doi: 10.1074/jbc.M411368200. [DOI] [PubMed] [Google Scholar]
- 21.Raska M., Takahashi K., Czernekova L., Zachova K., Hall S., Moldoveanu Z., Elliott M.C., Wilson L., Brown R., Jancova D., Barnes S., Vrbkova J., Tomana M., Smith P.D., Mestecky J., Renfrow M.B., Novak J. Glycosylation patterns of HIV-1 gp120 are cell-producing type dependent and affect antibody recognition. J Biol Chem. 2010;285:20860–20869. doi: 10.1074/jbc.M109.085472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oida E., Nogaki F., Kobayashi I., Kamata T., Ono T., Miyawaki S., Serikawa T., Yoshida H., Kita T., Muso E. Quantitative trait loci (QTL) analysis reveals a close linkage between the hinge region and trimeric IgA dominancy in a high IgA strain (HIGA) of ddY mice. Eur J Immunol. 2004;34:2200–2208. doi: 10.1002/eji.200425062. [DOI] [PubMed] [Google Scholar]
- 23.Rauterberg E.W., Lieberknecht H.M., Wingen A.M., Ritz E. Complement membrane attack (MAC) in idiopathic IgA-glomerulonephritis. Kidney Int. 1987;31:820–829. doi: 10.1038/ki.1987.72. [DOI] [PubMed] [Google Scholar]
- 24.William G.C., Jeffrey W.P., Stuart J.S. Complement (C5b-9) induces DNA synthesis in rat mesangial cells in vitro. Kidney Int. 2001;59:905–912. doi: 10.1046/j.1523-1755.2001.059003905.x. [DOI] [PubMed] [Google Scholar]
- 25.Welch T.R., McAdams J. Immunoglobulin M and C1q mesangial labeling in IgA nephropathy. Am J Kidney Dis. 1998;32:589–592. doi: 10.1016/s0272-6386(98)70021-6. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki S. Participation of complement components in glomerular deposition in IgA nephropathy. Nippon Jinzo Gakkai Shi. 1989;31:1029–1037. [PubMed] [Google Scholar]
- 27.Tomino Y. Complement system in IgA nephropathy. Tokai J Exp Clin Med. 1980;5:15–22. [PubMed] [Google Scholar]
- 28.Moriyama T., Shimizu A., Takei T., Uchida K., Honda K., Nitta K. Characteristics of immunoglobulin A nephropathy with mesangial immunoglobulin G and immunoglobulin M deposition. Nephrology. 2010;15:747–754. doi: 10.1111/j.1440-1797.2010.01296.x. [DOI] [PubMed] [Google Scholar]
- 29.Novak J., Tomana M., Matousovic K., Brown R., Hall S., Novak L., Julian B.A., Wyatt R.J., Mestecky J. IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int. 2005;67:504–513. doi: 10.1111/j.1523-1755.2005.67107.x. [DOI] [PubMed] [Google Scholar]
- 30.Matousovic K., Novak J., Julian B.A., Mestecky J. Tissue distribution and biological activities of immune complexes are determined by their size and composition. Nephrol Dial Transplant. 2010;25:1007. doi: 10.1093/ndt/gfp585. [DOI] [PubMed] [Google Scholar]
- 31.Gadjeva M.G., Rouseva M.M., Zlatarova A.S., Reid K.B., Kishore U., Kojouharova M.S. Interaction of human C1q with IgG and IgM: revisited. Biochemistry. 2000;47:13093–13102. doi: 10.1021/bi801131h. [DOI] [PubMed] [Google Scholar]
- 32.Roos A., Bouwman L.H., van Gijlswijk-Janssen D.J., Faber-Krol M.C., Stahl G.L., Daha M.R. Human IgA activates the complement system via the mannan-binding lectin pathway. J Immunol. 2001;167:2861–2868. doi: 10.4049/jimmunol.167.5.2861. [DOI] [PubMed] [Google Scholar]
- 33.Roos A., Rastaldi M.P., Calvaresi N., Oortwijn B.D., Schlagwein N., van Gijlswijk-Janssen D.J., Stahl G.L., Matsushita M., Fujita T., van Kooten C., Daha M.R. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J Am Soc Nephrol. 2006;17:1724–1734. doi: 10.1681/ASN.2005090923. [DOI] [PubMed] [Google Scholar]
- 34.Endo M., Ohi H., Ohsawa I., Fujita T., Matsushita M. Glomerular deposition of mannose-binding lectin (MBL) indicates a novel mechanism of complement activation in IgA nephropathy. Nephrol Dial Transplant. 1998;13:1984–1990. doi: 10.1093/ndt/13.8.1984. [DOI] [PubMed] [Google Scholar]
- 35.Bene M.C., Faure G.C. Composition of mesangial deposits in IgA nephropathy: complement factors. Nephron. 1987;46:219. doi: 10.1159/000184350. [DOI] [PubMed] [Google Scholar]
- 36.Nikolova E.B., Tomana M., Russell M.W. The role of the carbohydrate chains in complement (C3) fixation by solid-phase-bound human IgA. Immunology. 1994;82:321–327. [PMC free article] [PubMed] [Google Scholar]
- 37.Odani H., Hiki Y., Takahashi M., Nishimoto A., Yasuda Y., Iwase H., Shinzato T., Maeda K. Direct evidence for decreased sialylation and galactosylation of human serum IgA1 Fc O-glycosylated hinge peptides in IgA nephropathy by mass spectrometry. Biochem Biophys Res Commun. 2000;271:268–274. doi: 10.1006/bbrc.2000.2613. [DOI] [PubMed] [Google Scholar]
- 38.Song D., Zhou W., Sheerin S.H., Sacks S.H. Compartmental localization of complement component transcripts in the normal human kidney. Nephron. 1998;78:15–22. doi: 10.1159/000044876. [DOI] [PubMed] [Google Scholar]
- 39.Spitzer D., Mitchell L.M., Atkinson J.P., Hourcade D.E. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol. 2007;15:2600–2608. doi: 10.4049/jimmunol.179.4.2600. [DOI] [PubMed] [Google Scholar]
- 40.Kemper C., Mitchell L.M., Zhang L., Hourcade D.E. The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc Natl Acad Sci U S A. 2008;105:9023–9028. doi: 10.1073/pnas.0801015105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu W., Berger S.P., Trouw L.A., de Boer H.C., Schlagwein N., Mutsaers C., Daha M.R., van Kooten C. Properdin binds to late apoptotic and necrotic cells independently of C3b and regulates alternative pathway complement activation. J Immunol. 2008;180:7613–7621. doi: 10.4049/jimmunol.180.11.7613. [DOI] [PubMed] [Google Scholar]
- 42.Sato N., Ohsawa I., Nagamachi S., Ishii M., Kusaba G., Inoshita H., Toki A., Horikoshi S., Ohi H., Matsushita M., Tomino Y. Significance of glomerular activation of the alternative pathway and lectin pathway in lupus nephritis. Lupus. 2011;20:1378–1386. doi: 10.1177/0961203311415561. [DOI] [PubMed] [Google Scholar]
- 43.Gadd K.J., Reid K.B. Importance of the integrity of the inter-heavy-chain disulphide bond of rabbit IgG in the activation of the alternative pathway of human complement by the F(ab′)2 region of rabbit IgG antibody in immune aggregates. Immunology. 1981;42:75–82. [PMC free article] [PubMed] [Google Scholar]
- 44.Doekes G., Vanes L.A., Daha M.R. Influence of aggregate size on the binding and activation of the first component of human complement by soluble IgG aggregates. Immunology. 1982;45:705–713. [PMC free article] [PubMed] [Google Scholar]
- 45.Doekes G., Schouten J., Cats A., Daha M.R. Reduction of the complement activation capacity of soluble IgG aggregates and immune complexes by IgM-rheumatoid factor. Immunology. 1985;55:555–564. [PMC free article] [PubMed] [Google Scholar]
- 46.Ohsawa I., Ohi H., Tamano M., Endo M., Fujita T., Satomura A., Hidaka M., Fuke Y., Matsushita M., Fujita T. Cryoprecipitate of patients with cryoglobulinemic glomerulonephritis contains molecules of the lectin complement pathway. Clin Immunol. 2001;101:59–66. doi: 10.1006/clim.2001.5098. [DOI] [PubMed] [Google Scholar]
- 47.Kokubo T., Hiki Y., Iwase H., Tanaka A., Toma K., Hotta K., Kobayashi Y. Protective role of IgA1 glycans against IgA1 self-aggregation and adhesion to extracellular matrix proteins. J Am Soc Nephrol. 1998;9:2048–2054. doi: 10.1681/ASN.V9112048. [DOI] [PubMed] [Google Scholar]
- 48.Suzuki H., Fan R., Zhang Z., Brown R., Hall S., Julian B.A., Chatham W.W., Suzuki Y., Wyatt R.J., Moldoveanu Z., Lee J.Y., Robinson J., Tomana M., Tomino Y., Mestecky J., Novak J. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest. 2009;119:1668–1677. doi: 10.1172/JCI38468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki H., Moldoveanu Z., Hall S., Brown R., Vu H.L., Novak L., Julian B.A., Tomana M., Wyatt R.J., Edberg J.C., Alarcón G.S., Kimberly R.P., Tomino Y., Mestecky J., Novak J. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J Clin Invest. 2008;118:629–639. doi: 10.1172/JCI33189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Novak J., Julian B.A., Tomana M., Mestecky J. IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin Nephrol. 2008;28:78–87. doi: 10.1016/j.semnephrol.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Otani M., Nakata J., Kihara M., Leroy V., Moll S., Wada Y., Izui S. O-glycosylated IgA rheumatoid factor induces IgA deposits and glomerulonephritis. J Am Soc Nephrol. 2012;23:438–446. doi: 10.1681/ASN.2011070701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nishie T., Miyaishi O., Azuma H., Kameyama A., Naruse C., Hashimoto N., Yokoyama H., Narimatsu H., Wada T., Asano M. Development of immunoglobulin A nephropathy- like disease in β-1,4-galactosyltransferase-I-deficient mice. Am J Pathol. 2007;170:447–456. doi: 10.2353/ajpath.2007.060559. [DOI] [PMC free article] [PubMed] [Google Scholar]