

Abstract
The receptor for advanced glycation end products (RAGE) is a multiligand receptor that has been shown to contribute to the pathogenesis of diabetes, atherosclerosis, and neurodegeneration. However, its role in asthma and allergic airway disease is largely unknown. These studies use a house dust mite (HDM) mouse model of asthma/allergic airway disease. Respiratory mechanics were assessed and compared between wild-type and RAGE knockout mice. Bronchovascular architecture was assessed with quantitative scoring, and expression of RAGE, immunoglobulins, and relevant cytokines was assessed by standard protein detection methods and/or quantitative RT-PCR. The absence of RAGE abolishes most assessed measures of pathology, including airway hypersensitivity (resistance, tissue damping, and elastance), eosinophilic inflammation, and airway remodeling. IL-4 secretion, isotype class switching, and antigen recognition are intact in the absence of RAGE. In contrast, normal increases in IL-5, IL-13, eotaxin, and eotaxin-2 production are abrogated in the RAGE knockouts. IL-17 indicates complex regulation, with elevated baseline expression in RAGE knockouts, but no induction in response to allergen. Treatment of WT mice with an inhibitor of RAGE markedly reduces inflammation in the HDM model, suggesting that RAGE inhibition may serve as a promising therapeutic strategy. Finally, the results in the HDM model are recapitulated in an ovalbumin model of asthma, suggesting that RAGE plays a role in asthma irrespective of the identity of the allergens involved.
The receptor for advanced glycation end products (RAGE) is a multiligand receptor first identified as a potential mediator in diabetes.1 RAGE has many additional ligands, including S100 proteins, HMGB1, amyloid β, and heparin.2,3 The current paradigm maintains that membrane RAGE (mRAGE) signaling is proinflammatory, whereas soluble RAGE (sRAGE), a secreted form of RAGE, is generally anti-inflammatory because it scavenges proinflammatory ligands.4 RAGE transcript and protein are predominantly expressed in the lung,5 and specifically by pulmonary type I alveolar epithelial cells,6 suggesting that RAGE has an important role in lung pathophysiology.
Recent studies suggest that RAGE contributes to pulmonary disease; RAGE knockout mice are protected against hyperoxia-induced lung injury7 and exhibit attenuated responses to bacterial pneumonia.8 RAGE also appears to have an important role in pulmonary fibrosis, but this depends on the type of injury model used to generate the fibrotic response.6,9–11 RAGE expression is altered in these models of disease, for example, the appearance of sRAGE in the bronchoalveolar lavage fluid (BALF) in a mouse model of pneumonia and loss of RAGE expression in models of pulmonary fibrosis.
Asthma/allergic airway disease (AAD) is a main inflammatory condition of modern industrial societies, seen with increasing frequency throughout the developing world. Bronchodilators and corticosteroids remain the mainstays of therapy, but they are ineffective or inadequate for some groups of patients. Novel therapies that exploit auxiliary mechanisms of disease and that incur fewer side effects than chronic corticosteroid treatment are greatly needed. To date, there appears to have been no studies of RAGE in animal models of asthma. A few studies in humans have suggested that there is an increase in the levels of RAGE ligands HMGB112 and S100A8/A913 in samples from patients with asthma compared with controls, suggesting that RAGE may contribute to asthma/AAD pathogenesis. Although one recent study suggested that sRAGE is increased concomitantly with HMGB1 in patients with asthma,12 another suggested a decrease in sRAGE and no change in HMGB1 in patients with neutrophilic asthma.14 Apart from the potential inconsistency between the latter two results, those studies have not provided mechanistic insight as to the role of mRAGE versus sRAGE in asthma, nor have they elucidated how cytokines and chemokines key to allergic disease are differentially regulated in the presence or absence of RAGE.
The present study used a house dust mite (HDM) antigen sensitization/challenge model of asthma/AAD to directly assess the role of RAGE in asthma/AAD. An advantage of this model is that sensitization and challenge are effected by intranasal HDM extract application in the absence of adjuvant. Moreover, HDM antigen has been identified as a key contributor to asthma pathogenesis in humans and is a known trigger of acute asthmatic exacerbations.15,16 To strengthen the applicability of the results and to further clarify mechanism, analogous experiments were also performed in an ovalbumin model of AAD to ensure that any effects of RAGE are not specific to the model investigated. Wild-type (WT) C57BL/6 strain mice subjected to these models develop airway hyperresponsiveness to methacholine challenge; bronchial, vascular, and interstitial eosinophilia; goblet cell hyperplasia with mucus hypersecretion; and elevated titers of IgE, the immunoglobulin class most closely associated with allergic disease.
Materials and Methods
Animals and Reagents
WT male C57BL/6 mice were purchased from Taconic (Hudson, NY). Founder RAGE knockout (RAGE KO) mice were provided by Dr. A. Bierhaus (University of Heidelberg), and from these mice a breeding colony was initiated.6,11,17 These mice are congenic with the C57BL/6 background. RAGE KO mice were age- and sex-matched to WT mice for each experiment. In all cases mice were housed in the animal care facility of the University of Pittsburgh, and experimental protocols were approved by the University's Institutional Animal Care and Use Committee. HDM extract was obtained from Greer Laboratories (Lenoir, NC). Mouse serum albumin (MSA) was purchased from Sigma (St. Louis, MO). sRAGE was purified from mouse lung tissue, and endotoxin was removed with a Detoxi-Gel column (Thermo-Fisher) as described previously.2,18 Purity was confirmed by SDS-PAGE and Coomassie Brilliant Blue staining. Specific binding of the purified sRAGE to a known RAGE ligand, HMGB1, was assessed as described elsewhere19 to ensure ligand binding was intact in the purified protein.
Models of Allergic Airway Inflammation/Asthma
AAD/asthma was induced in mice with the use of one of three protocols. In the first protocol, 8-week-old male WT or RAGE KO mice were treated intranasally (i.n.) four times per week for 7 weeks with 40 μg of HDM extract in 25 μL of saline. Control mice were treated with saline vehicle alone. Mice were sacrificed 48 hours after the last treatment. In the second protocol, 8-week-old male WT C57BL/6 mice were treated i.n. five times per week for 3 weeks with one of the following six treatments [in each case in 25 μL of phosphate-buffered saline (PBS)]: saline control, 25 μg of MSA alone, 25 μg of mouse sRAGE alone, 40 μg of HDM extract alone, 40 μg of HDM extract with 25 μg of MSA, or 40 μg of HDM extract with 25 μg of sRAGE. The third protocol used ovalbumin as the sensitization/challenge antigen. Sensitization was effected by intraperitoneal treatment with 50 μg of ovalbumin (Sigma) with 2 mg of aluminum hydroxide gel (Brenntag, Reading, PA) in 0.5 mL of saline on days 0 and 7. Control mice were given alum with saline alone. Intranasal challenge commenced at day 14 with 10 μg of ovalbumin in 25 μL of saline (or saline alone, in controls), continuing on alternate days for a total of three treatments per week for 3 weeks. Mice were sacrificed 24 hours after the last treatment. Pulmonary function was assayed with a flexiVent apparatus as described elsewhere.20 Briefly, mice were anesthetized with pentobarbital, underwent tracheotomy and cannula placement, and were coupled to the flexiVent ventilator apparatus (SCIREQ, Montreal, QC). Mice were ventilated with a 0.2 mL tidal volume and positive end-expiratory pressure of 3 cm of H2O. Pressure and volume were measured and were fit by multiple linear regression to a linear model of the lung. Methacholine was delivered via a nebulizer; after each dose, the response was measured by applying 2-second perturbations at 10-second intervals for a total of 3 minutes. Dose response curves were then determined for each of three parameters that measured lung function.
Serum and BALF Preparation and Studies
After pulmonary function testing, the mice were exsanguinated, and sera were prepared with the use of serum separator tubes (Becton Dickinson, Franklin Lakes, NJ). Saline (0.8 mL) was instilled in the lungs via the trachea and withdrawn. BALF cell counts were obtained, and cytospin slides were prepared for differential cell counting. Slides were stained with Diff-Quik (Siemens, Washington, DC) and air dried, and Permount coverslips (Fisher, Pittsburgh, PA) were placed on the slides. The relative percentages of monocytes, eosinophils, neutrophils, and lymphocytes were determined by counting five high-power fields. After centrifugation, BALF supernatant fluid and sera were frozen at −80°C for future studies.
qRT-PCR
RNA was prepared from whole lung with the use of an RNeasy Mini Kit (Qiagen, Valencia, CA), per the manufacturer's instructions. Reverse transcription was performed with Moloney murine leukemia virus reverse transcriptase (Applied Biosystems, Foster City, CA) in a Techne thermal cycler (Bibby Scientific US, Burlington, NJ). Quantitative RT-PCR (qRT-PCR) was performed with universal PCR buffer and TaqMan primer/probe assay reagent (Applied Biosystems) with primers for RAGE (Mm00545815 m1), IL-5 (Mm00439646 m1), IL-13 (Mm00505403 m1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control (Mm99999915 g1). The following sequence was performed on an ABI Prism 7300 machine (Applied Biosystems): 50°C (2 minutes), 95°C (10 minutes), and then 40 cycles of 95°C (15 seconds) followed by 60°C (1 minute). The fold change in RAGE, IL-5, or IL-13 mRNA expression, compared with GAPDH mRNA housekeeping control, was determined with the ΔΔCt method.
Immunoblotting
Lung homogenates were prepared by homogenizing and sonicating mouse lungs in cold CHAPS (3[lsqb[(3-cholamidopropyl)dimethylammonio]-propanesulfonic acid) buffer with protease inhibitors (150 mmol/L NaCl, 50 mmol/L Tris-HCl, 10 mmol/L CHAPS, 100 μmol/L 3,4-dichloroisocoumarin, 10 μmol/L E-64, 2 mmol/L ortho-phenanthroline, pH 7.4), followed by protein extraction over several hours at 4°C. Total protein content in lung homogenates was determined with the Bradford method. Ten micrograms of lung homogenate protein, 65 μL of undiluted BALF, or 65 μL of 1:10 diluted (in PBS) serum samples, were analyzed by SDS-PAGE and immunoblotting as described previously.21 After blocking in 5% milk, membranes were incubated in 1:5000 primary rabbit anti-RAGE polyclonal antibody (GenScript, Piscataway, NJ) followed by 1:5000 secondary horseradish peroxidase anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA). For studies that explored the antigen binding profile of serum IgG from sensitized and challenged mice, 100 μg of HDM extract was separated by SDS-PAGE under reducing (50 mmol/L dithiothreitol) and nonreducing (no dithiothreitol) conditions, then transferred to membranes and blocked in 5% milk. Membranes were then incubated in 1:20 diluted pooled sera from sensitized and challenged WT or RAGE KO mice followed by 1:5000 secondary horseradish peroxidase anti-mouse IgG antibody (Jackson ImmunoResearch). Membranes were developed with enhanced chemiluminescent detection (Thermo-Fisher, Waltham, MA). Loading control was performed with Ponceau S staining for BALF and serum and by stripping and reprobing with an antibody against β-actin (Sigma) on lung homogenate blots.
Immunofluorescence Microscopy
Paraffin-embedded inflation-fixed lungs were sectioned at a 5-μm thickness, mounted on SuperFrost Plus slides (Fisher), air dried, melted, deparaffinized with xylenes, and rehydrated with graded ethanol solutions. Antigen retrieval was performed in 0.2 N HCl with 1 mg/mL pepsin (Fisher) at 37°C for 10 minutes. Between each step, three 5-minute washes with PBS or PBS containing Tween-20 (PBST) were performed. Sections were blocked with 2% bovine serum albumin in PBST, whereas primary and secondary antibody treatments were performed in 0.5% bovine serum albumin in PBST. To detect RAGE, 1:500 diluted goat polyclonal antibody in serum raised against full-length mouse sRAGE (GenScript) was used, followed by 1:500 diluted donkey anti-goat antibody conjugated to cyanine 3 (Jackson ImmunoResearch). Control preimmune serum from the same animal in which the RAGE antibody was raised and additionally RAGE KO lung sections were used to confirm specificity. Nuclei were stained with a brief application of Hoechst stain (10 mg/mL). Sections were coverslipped with gelvatol and examined by an Olympus IX71 inverted microscope (Olympus, Tokyo, Japan). All images were processed with ImageJ software version 1.44o (NIH, Bethesda, MD).22 Images were background subtracted as individual colors, followed by color channels being merged. In all cases a rolling ball radius of 50.0 pixels was used.
H&E and PAS Staining
Lungs were inflation fixed with 10% neutral-buffered formalin, paraffin embedded, cut into sections 5 μm thick, stained with H&E or PAS (Research Histology Services, University of Pittsburgh), and examined by light microscopy All images were processed with ImageJ software version 1.44o. Images were light background subtracted. In all cases a rolling ball radius of 50.0 pixels was used.
IgG1, IgE, Eotaxin, Eotaxin-2, IL-4, IL-5, IL-17, and IL-13 ELISA
Eotaxin, eotaxin-2 (both from Abcam, Cambridge, MA), IL-4, IL-5 (both from BD Biosciences, San Jose, CA), IL-17 (R&D Systems, Minneapolis, MN), and IL-13 (eBioscience, San Diego, CA) enzyme-linked immunosorbent assays (ELISAs) were performed per manufacturer's instructions, using neat or diluted BALF in all cases except for IL-17 ELISA, which used neat lung homogenate. HDM-specific IgG1 ELISA was performed by coating absorbent plates with 2 μg/mL HDM extract in coating buffer (100 mmol/L NaHCO3, 30 mmol/L Na2CO3, pH 9.5) overnight at 4°C. Between each step, washes were performed with PBST. After blocking with 1% bovine serum albumin, 1:100 diluted serum or neat lung homogenate (in CHAPS buffer) was applied and incubated at 4°C overnight with agitation. Detection was performed by treating with 1:1000 diluted goat anti-mouse IgG1 conjugated to horseradish peroxidase (Jackson ImmunoResearch), and then o-phenylenediamine dihydrochloride (OPD) substrate (Sigma). Plates were read at 450 nm, and data were recorded as absolute absorbances. Total IgE ELISA was performed with an OptEIA IgE ELISA kit (BD Biosciences) per the manufacturer's instructions. Serum samples were diluted 1:20 in assay diluent. For HDM-specific ELISA, HDM extract was first biotinylated with the use of 6-((biotinoyl)amino)hexanoic acid, sulfosuccinimidyl ester (biotin-X, SSE) (Invitrogen, Carlsbad, CA). Then, free biotin was removed with a Sephadex G-25 quick spin column (Roche, Indianapolis, IN). Plates were incubated overnight at 4°C with capture antibody (OptEIA IgE ELISA kit), then application of 1:5 diluted (in assay diluent) serum overnight at 4°C with agitation. Biotinylated HDM extract was applied to detect HDM-specific IgE. This was followed by streptavidin conjugated to horseradish peroxidase (OptEIA IgE ELISA kit), and then OPD substrate. The plates were read at 450 nm, and data were recorded as absolute absorbances.
Histologic Scoring
H&E-stained lung sections from C57BL/6 mice were examined by a board-certified pathologist (T.D.O.) who had been blinded to the identity of the samples. The total number of bronchovascular bundles in each lung section was counted, and inflammation was scored. Inflammation (if any) in each bronchovascular bundle was graded as 0 (none), 1 (mild), 2 (moderate), or 3 (severe). The results are expressed as the percentage of all bronchovascular bundles that involve any inflammatory infiltrates, and, in addition, as an average severity score obtainable by dividing the pan-section sum of the bronchovascular bundle inflammation scores by the total number of bronchovascular bundles in that section.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism version 5.0 (GraphPad Software Inc., San Diego, CA), and quantitative results were expressed as means ± SEMs. Statistical significance was determined with two-way analysis of variance and, when appropriate, unpaired Student's t-test. A P value < 0.05 was considered to be significant.
Results
To investigate whether RAGE expression was altered in mice sensitized and challenged with HDM extract compared with vehicle-treated controls, qRT-PCR and immunoblotting on whole lung homogenates were performed. Although there was a modest decrease in RAGE transcript expression in the lungs of mice treated with HDM extract compared with saline-treated controls (Figure 1A), this difference was not statistically significant. No change was observed in the overall expression level of RAGE protein or in the relative proportions of sRAGE versus two different isoforms of membrane-bound RAGE, xRAGE,23 and mRAGE (Figure 1B). Finally, sRAGE could not be detected in BALF or serum of either sensitized/challenged or naive mice (Figure 1C).
Figure 1.

Pulmonary RAGE expression and localization is not altered in AAD/asthma in mice. A: qRT-PCR of whole lung homogenates of mice treated i.n. with HDM extract or saline control (mean ± SEM of the fold change in RAGE signal normalized to the GAPDH signal). B: Immunoblot of RAGE and β-actin (loading control) in whole lung homogenates of mice treated with HDM extract or saline control (top panel). Summary of normalized RAGE-to-actin signal intensity ratios for each RAGE isoform, with saline-treated controls set arbitrarily to 1.0 (mean ± SEM) (bottom panel). C: Immunoblot probing for RAGE in BALF and serum samples of mice treated with HDM extract or saline control. Ponceau S stain serves as loading control. D: RAGE immunofluorescence staining (red) in lung sections of mice treated with HDM extract or saline control. Cells and bronchial and vascular structure were identified by nuclear staining (blue), whereas underlying alveolar basement membrane was identified by collagen IV immunofluorescence staining (green). Scale bar = 100 μm.
Because RAGE is abundantly expressed by type I alveolar epithelial cells, it was thought that subtle changes in RAGE expression in other cell types more closely associated with asthma/AAD, such as bronchial epithelial cells or inflammatory cells, might be undetectable by whole lung expression assays. Immunofluorescence microscopy studies of lung sections performed to address this question showed no appreciable change in bronchial or vascular RAGE expression in response to HDM extract sensitization and challenge, and expression in type I alveolar epithelial cells appeared unaltered (Figure 1D).
Expression of a pathogenetic mediator need not be altered in a disease state, particularly when the mediator is a receptor with multiple, potentially mutually antagonistic, roles. Therefore, to explore whether the absence of RAGE dampens or augments asthma/AAD, RAGE KO mice on the C57BL/6 background were subjected to the HDM extract sensitization/challenge protocol, alongside WT mice for comparison. Pulmonary function testing showed significant alterations in airway function in WT mice treated with HDM extract, consistent with an asthmatic profile (Figure 2A). These changes in responsiveness to methacholine challenge were evident in the parameters corresponding to large airway resistance (Rn), small airway tissue damping (G), and tissue elastance (H), respectively. The modest change in Rn, compared with what is often seen in studies in BALB/c mice, was consistent with findings of previous studies in the C57BL/6 strain.24 The picture with the Rn parameter was somewhat complex, with a slightly elevated responsiveness to methacholine in naive RAGE KO mice compared with WT counterparts, but no difference between saline- versus HDM extract-treated RAGE KO mice was seen. Because there were no observable differences in airway or parenchymal architecture in RAGE KO mice, it is likely that any intrinsic changes in methacholine responsiveness in this strain are due to a minor effect of RAGE (directly or by compensatory gene expression) on airway smooth muscle function (see Discussion for the potential role of IL-17). However, it is important to note that RAGE KO mice treated with HDM extract showed G and H parameters indistinguishable from those of naive WT and RAGE KO mice.
Figure 2.

Mice lacking RAGE do not develop airway hyperresponsiveness, eosinophilia, or increased mucin production in response to HDM extract treatment. In all cases results are expressed as means ± SEMs (n = 6 to 10 per strain/treatment group; *P < 0.05 versus comparison). A: Airway responsiveness to methacholine challenge: Rn (Newtonian resistance), G (tissue damping), and H (tissue elastance). B: Representative Romanowsky stain of BALF cells of WT or RAGE KO mice treated with HDM extract (top panel). Scale bar = 6 μm. Summary of cell counts and differentials in BALF of WT or RAGE KO mice treated with HDM extract or saline control (bottom panels). C and D: Representative H&E stain (C) or PAS stain (D) (red shows mucin) of lung sections of WT or RAGE KO mice treated with HDM extract or saline control. Scale bar = 30 μm.
Modified Romanowsky staining of BALF cells showed markedly elevated cell counts and eosinophilia in the allergic WT mice compared with controls (Figure 2B). Histologic evaluation of lung sections indicated peribronchial, perivascular, and interstitial eosinophilia in lungs of allergic WT mice by H&E staining (Figure 2C), whereas PAS staining showed goblet cell hyperplasia and elevated expression of mucin in numerous bronchi of the allergic WT mice compared with controls (Figure 2D). In contrast to WT mice, RAGE KO mice had essentially no inflammatory infiltrates and no elevated mucin expression or goblet cell hyperplasia. These results suggested that RAGE plays a vital role in the key tissue changes observed in asthma/AAD, including airway hyperresponsiveness, eosinophilic peribronchial infiltrates, mucus hypersecretion, and airway remodeling.
To determine whether the humoral immune response to HDM extract sensitization and challenge was altered in RAGE KO mice, ELISA was performed on sera and lung homogenates of WT and RAGE KO mice to evaluate the levels of HDM-specific and total immunoglobulins. Interestingly, significantly elevated levels of HDM-specific IgG1 in serum and lung homogenate of both WT and RAGE KO mice that had been sensitized and challenged with HDM extract were observed (Figure 3A). To test whether there were broad differences between the HDM antigen binding profiles of serum IgG from allergen-primed and -challenged WT versus RAGE KO mice, HDM extracts separated by SDS-PAGE and transferred to membranes were probed with sera from sensitized and challenged WT and RAGE KO mice (Figure 3B), indicating no differences in antigen binding profiles between the two strains. Furthermore, the levels of both HDM-specific IgE and total IgE were significantly elevated in the sera of both WT and RAGE KO mice sensitized and challenged with HDM extract, compared with their naive counterparts (Figure 3C). IL-4 is a key T lymphocyte cytokine involved in driving CD4+ T helper cell differentiation into T helper type 2 (Th2) cells, as well as in directing B-cell isotype class switching to IgE synthesis.25 To assess how the absence of RAGE affected the production of this cytokine in response to HDM extract sensitization and challenge, ELISA was performed on BALF samples. The IL-4 response was found to be intact in RAGE KO mice, as there was no significant difference between RAGE KO and WT mice in the expression of this cytokine in response to allergen (Figure 3D). Although not a classic cytokine of the Th2 immune response, IL-17 has been suggested to be a molecular mediator relevant to asthma, particularly to subtypes with a prominent neutrophilic component.26,27 Interestingly, IL-17 appeared to have significantly heightened baseline expression in RAGE KO mice compared with WT mice. However, although the WT mice showed induction of IL-17 expression in response to HDM extract sensitization and challenge, the RAGE KO mice did not (Figure 3E).
Figure 3.

Lack of RAGE does not alter the immunoglobulin response to HDM or affect the synthesis and secretion of the Th2 cytokine IL-4. In all cases results are expressed as means ± SEMs (n = 3 to 10 per strain/treatment group; *P < 0.05 versus comparison). A: ELISA results show levels of HDM antigen-specific IgG1 antibodies in sera and lung homogenates of WT or RAGE KO mice treated with HDM extract or saline control. B: Immunoblot shows HDM antigen binding by serum IgG from WT or RAGE KO mice treated with HDM extract. C: ELISA results show levels of total IgE or HDM antigen-specific IgE antibodies in sera of WT or RAGE KO mice treated with HDM extract or saline control. D: ELISA results show levels of IL-4 in BALF of WT or RAGE KO mice treated with HDM extract or saline control. E: ELISA results show levels of IL-17 in lung homogenate of WT or RAGE KO mice treated with HDM extract or saline control. DTT, dithiothreitol.
In asthma/AAD, eosinophils are thought to contribute to airway hyperresponsiveness28 and airway remodeling observed in the disease.29 Eotaxins are the main class of chemokines involved in the selective recruitment of eosinophils to the tissues. Thus, to determine whether RAGE mediates its effects in the HDM mouse model of asthma/AAD by inducing eotaxin and/or eotaxin-2 secretion, ELISA was performed on BALF of WT and RAGE KO mice sensitized/challenged with HDM extract or with saline control. The levels of neither eotaxin (Figure 4A) nor eotaxin-2 (Figure 4B) in RAGE KO mice treated with HDM extract were significantly changed from baseline, whereas WT mice thus treated exhibited elevated levels of both chemokines.
Figure 4.
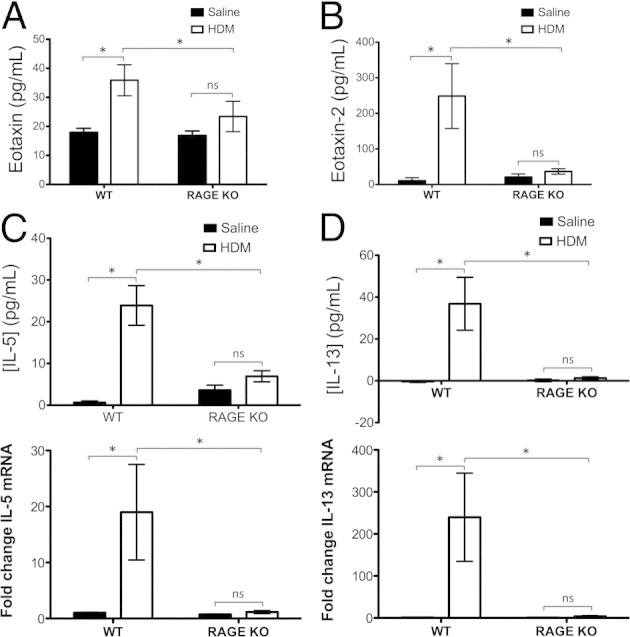
Lack of RAGE abrogates eosinophil-recruiting chemokines eotaxin and eotaxin-2 as well as Th2 cytokines IL-5 and IL-13. In all cases, results are expressed as means ± SEMs (n = 3 to 10 per strain/treatment group; *P < 0.05 versus comparison). A and B: ELISA results show levels of eotaxin (A) and eotaxin-2 (B) in BALF of WT or RAGE KO mice treated with HDM extract or saline control. C and D: ELISA results (top panels) show levels of IL-5 (C) and IL-13 (D) protein in BALF, whereas qRT-PCR results (bottom panels) show levels of IL-5 (C) and IL-13 (D) transcript in whole lung homogenate of WT or RAGE KO mice treated with HDM extract or saline control.
IL-5 is a key Th2 cytokine involved in eosinophil proliferation, maturation, survival, function, and recruitment to tissue.30,31 IL-13, another key Th2 cytokine, acts in a complementary fashion to IL-5 in that it stimulates changes in tissue structure and physiology, such as airway hyperresponsiveness to methacholine challenge, goblet cell hyperplasia, and mucus hypersecretion.32 In addition, IL-13 plays a key role in inducing expression of the eotaxins.33–36 ELISA on BALF and qRT-PCR on whole lung mRNA were performed to assess whether these two key cytokines are dysregulated in RAGE KO mice receiving HDM extract. Although expression of IL-5 (Figure 4C) and IL-13 (Figure 4D) protein and mRNA were significantly elevated in WT mice sensitized and challenged with allergen, these cytokines were not elevated in allergen-challenged RAGE KO mice.
Numerous studies of RAGE in animal models of inflammatory disease have suggested that sRAGE has an anti-inflammatory and generally salutary effect, likely by virtue of its sequestration of RAGE ligands away from the proinflammatory membrane isoform.37–39 To determine which of the two isoforms, mRAGE or sRAGE, was driving the allergic process in the HDM mouse model, and to explore whether sRAGE has a therapeutic effect (as has been seen in many other disease models) in WT C57BL/6 mice sensitized and challenged with HDM extract, sRAGE purified from mouse lungs (Figure 5A) was i.n. coadministered alongside HDM extract. In addition, to rule out a nonspecific protein effect, an equal mass of MSA was coadministered with HDM extract in a separate group of mice. Asthma/AAD as reflected by total BALF cell and BALF eosinophil counts was markedly reduced in mice that had received sRAGE alongside HDM extract compared with mice that had received either HDM extract alone or MSA alongside HDM extract (Figure 5B). H&E-stained lung sections (Figure 5C) were scored by a pathologist blinded to treatment group for the total number of bronchovascular bundles, the number of bundles showing inflammatory infiltrates, and a scoring of the severity of the inflammation in each bundle. The percentage of bronchovascular bundles involving inflammation and the overall average inflammation severity score were both significantly reduced in mice given sRAGE alongside HDM extract compared with the two other HDM extract sensitized/challenged groups, mirroring the BALF cell count data (Figure 5D).
Figure 5.

sRAGE treatment decreases HDM-induced pulmonary inflammation. In all cases, results are expressed as means ± SEMs (WT HDM, n = 3; all other treatment groups, n = 4; *P < 0.05 versus comparison). A: SDS-PAGE separation and Coomassie Brilliant Blue staining of 5 μg of MSA or mouse sRAGE. B: Total cell and eosinophil counts and differentials in BALF of WT mice treated with HDM extract versus saline control, in each case alone or alongside MSA or sRAGE treatment. C: Representative H&E stain of lung sections of WT mice treated with HDM extract or saline control alongside no additional protein, MSA, or sRAGE. Scale bar = 30 μm. D: Bronchovascular bundle inflammation in mice treated with HDM extract or saline, with or without MSA or sRAGE treatment. Results are expressed as percentage of airways involved or average severity across all bronchovascular bundles (0 = no inflammatory infiltrate, 1 = mild inflammatory infiltrate, 2 = moderate inflammatory infiltrate, and 3 = severe inflammatory infiltrate).
A key question of these studies was to what degree, if any, the role of RAGE in murine models of asthma/AAD depended on the identity of the epitopes driving the immune response. An ovalbumin model of asthma/AAD was used to address this question, because the antigen profile and sensitization mechanism differ markedly from that of the HDM model. Sensitization was effected with the use of aluminum hydroxide-adsorbed ovalbumin administered to the peritoneal compartment. Challenge proceeded i.n., as in the HDM model. Pulmonary function testing indicated alterations in the Rn, G, and H parameters in WT mice treated with ovalbumin compared with mice treated with saline, analogous to findings in the HDM model (Figure 6A). Although the change in the Rn parameter did not reach statistical significance, the changes in the G and H parameters reached statistical significance. BALF cell counts were elevated in allergic WT mice, and eosinophilia was notable (Figure 6B); peribronchial, perivascular, and interstitial eosinophilic inflammation (Figure 6C) and mucin overexpression (Figure 6D) were evident in allergic WT mice compared with controls. RAGE KO mice have unaltered airway function and no or minimal BALF eosinophilia, inflammatory infiltrates, or mucus hypersecretion. These data suggested that RAGE's role in murine asthma/AAD is independent of the epitopes involved.
Figure 6.

Mice lacking RAGE do not develop airway hyperresponsiveness, eosinophilia, or increased mucin production in response to ovalbumin treatment. Results are expressed as means ± SEMs (n = 8 to 10 per strain/treatment group; *P < 0.05 versus comparison). A: Airway responsiveness to methacholine challenge, as indicated by the Rn, G, and H parameters. B: Representative BALF cells of WT or RAGE KO mice treated with ovalbumin extract (top panel). Scale bar = 6 μm. Summary of BALF data of WT or RAGE KO mice treated with ovalbumin or saline control (bottom panels). C and D: Representative H&E stain (C), or PAS stain (D) of lung sections of WT or RAGE KO mice treated with ovalbumin or saline control. Scale bar = 30 μm.
Discussion
The results of this study suggest that RAGE is a key mediator in the disease mechanisms that eventuate in airway hyperresponsiveness, mucus hypersecretion and airway remodeling, and pulmonary eosinophilia in an HDM mouse model of asthma/AAD. Taking into consideration the great difference in response of the WT versus RAGE KO mice to HDM extract sensitization/challenge, it is perhaps surprising that RAGE levels and localization in the pulmonary compartment appear to be essentially unchanged between allergen- and vehicle control-treated groups. This might invite the suggestion that it is not RAGE itself, but rather a protein whose expression is tightly linked to RAGE, that is responsible for the effects seen in RAGE KO mice. Although this possibility cannot be definitively excluded, given the observation that treatment with sRAGE (a decoy for RAGE ligands) specifically and markedly reduces bronchovascular inflammation in response to HDM extract sensitization/challenge suggests that (lack of) RAGE itself, rather than a compensatory process in KO animals, is responsible for the effect seen. Furthermore, the sRAGE treatment studies indicate that manipulation of the RAGE signaling axis via administration of sRAGE, antagonistic antibodies, or small molecule inhibitors, may have promise in the treatment of asthma/AAD.
The data suggest that innate and adaptive immune response mechanisms that drive humoral immunity are intact in the absence of RAGE, including antigen recognition, processing, and presentation; B-lymphocyte production of immunoglobulin; and isotype class switching, this latter phenomenon being driven by IL-4. Although T lymphocytes are thought to be the main source of IL-4, IL-5, and IL-13, other cell types have been linked to the production of one or more members of this triad. Thus, IL-4 induction in the absence of the other two canonical Th2 cytokines could be reflective of either uncoupling in the cytokine response at the T-cell level or the recruitment of other cell types that may produce IL-4, such as macrophages40 or basophils.41 The fact that immunoglobulin production in response to antigens occurs in the absence of RAGE, despite the lack of other features associated with AAD, is consistent with prior studies suggesting that B-cell deficiency (and hence immunoglobulin production) does not affect the physiologic and pathologic changes seen in response to antigen sensitization and challenge in an ovalbumin mouse model of asthma/AAD.42 Furthermore, the fact that in the presence of high levels of IgE the RAGE KO mice do not develop airway hypersensitivity, eosinophilia, or remodeling would seem to indicate that IgE alone is not playing a central mechanistic role in this model of asthma, although it must be noted that changes in airway physiology were assayed with graded doses of methacholine rather than allergen.
IL-17 has been suggested to play a role in allergy and asthma; in the HDM model of asthma/AAD, IL-17 is up-regulated in response to allergen sensitization and challenge in WT mice.43 That RAGE KO mice showed no IL-17 induction in response to HDM extract, and furthermore showed heightened baseline expression of IL-17 in the absence of antigen, suggests that RAGE plays a role in IL-17 regulation. RAGE may actively inhibit the secretion of IL-17, directly or by disrupting precursor cytokines, such as IL-23. Conversely, the absence of RAGE may lead to compensatory induction of other factors that in turn drive the IL-23/IL-17 axis. Although IL-17 has been shown to play a role in neutrophilic asthma,44 it has also been shown to be a negative regulator of established allergic asthma.45,46 The baseline elevation of IL-17 in RAGE KO mice may thus impede the initiation of a primary asthmatic response. Moreover, it may account for the subtle differences in airway responsiveness to methacholine challenge between naive saline-treated WT and RAGE KO mice (RAGE KO mice treated with saline showed an elevated Rn parameter compared with WT mice thus treated), as a number of studies have indicated a role for IL-17 in promoting airway hyperresponsiveness.47–50 As an asthmatic response is not initiated in the RAGE KO mice treated with HDM extract (as evidenced by pulmonary function parameters, histology, and cytokine profiling), it is not particularly surprising that in this strain there was no consequent up-regulation in IL-17 from baseline in response to allergen.
The cytokine and chemokine data presented herein show that canonical molecular mediators of eosinophil recruitment and airway remodeling, such as IL-5 and IL-13, are indeed abrogated in the absence of RAGE, suggesting that RAGE likely acts as a proximal mediator in one or more proinflammatory pathways that eventuate in the altered physiology and histology seen in this model of asthma/AAD. Because of its multivalency, RAGE has for some time been considered by many to be a pathogen recognition receptor. To date, however, no exogenous ligand has been discovered. Studies in the ovalbumin model of asthma suggest that RAGE KO mice are resistant to the tissue and physiological changes seen in WT mice treated with antigen, notably hypersensitivity to methacholine challenge, pulmonary eosinophilic inflammation, and airway remodeling. Given the substantial differences in the sensitization process and the disparate immune epitopes involved between the HDM and ovalbumin models, and taking into consideration the integrity of the immunoglobulin response against HDM antigens in RAGE KO mice, it is reasonable to conclude that RAGE acts downstream of antigen recognition; thus, its ligands in asthma/AAD are likely to be endogenous and not derived from the initial antigenic stimulus. Finally, although RAGE is predominantly expressed on alveolar type I epithelial cells, some studies have suggested that it is expressed on hematopoietic cells and endothelium as well. Characterization of the cell type(s) responsible for the effect seen in these studies (not necessarily those with the greatest expression), as well as elucidation of the molecular pathways between RAGE and the T-cell cytokines IL-5 and IL-13, will constitute key aims of future studies.
Acknowledgment
We thank Angelika Bierhaus for providing our laboratory with the RAGE KO mouse strain.
Footnotes
Supported by NIH grants 5R21HL095495-02 (T.D.O.) 1T32HL094295-01A2 (P.S.M.).
Portions of this work have been presented in the doctoral dissertation of P.S.M.
References
- 1.Neeper M., Schmidt A.M., Brett J., Yan S.D., Wang F., Pan Y.C., Elliston K., Stern D., Shaw A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- 2.Hanford L.E., Enghild J.J., Valnickova Z., Petersen S.V., Schaefer L.M., Schaefer T.M., Reinhart T.A., Oury T.D. Purification and characterization of mouse soluble receptor for advanced glycation end products (sRAGE) J Biol Chem. 2004;279:50019–50024. doi: 10.1074/jbc.M409782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sparvero L.J., Asafu-Adjei D., Kang R., Tang D., Amin N., Im J., Rutledge R., Lin B., Amoscato A.A., Zeh H.J., Lotze M.T. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med. 2009;7:17. doi: 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramasamy R., Yan S.F., Schmidt A.M. RAGE: therapeutic target and biomarker of the inflammatory response–the evidence mounts. J Leukoc Biol. 2009;86:505–512. doi: 10.1189/jlb.0409230. [DOI] [PubMed] [Google Scholar]
- 5.Brett J., Schmidt A.M., Yan S.D., Zou Y.S., Weidman E., Pinsky D., Nowygrod R., Neeper M., Przysiecki C., Shaw A. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–1712. [PMC free article] [PubMed] [Google Scholar]
- 6.Englert J.M., Hanford L.E., Kaminski N., Tobolewski J.M., Tan R.J., Fattman C.L., Ramsgaard L., Richards T.J., Loutaev I., Nawroth P.P., Kasper M., Bierhaus A., Oury T.D. A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol. 2008;172:583–591. doi: 10.2353/ajpath.2008.070569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reynolds P.R., Schmitt R.E., Kasteler S.D., Sturrock A., Sanders K., Bierhaus A., Nawroth P.P., Paine R., III, Hoidal J.R. Receptors for advanced glycation end-products targeting protect against hyperoxia-induced lung injury in mice. Am J Respir Cell Mol Biol. 2010;42:545–551. doi: 10.1165/rcmb.2008-0265OC. [DOI] [PubMed] [Google Scholar]
- 8.Ramsgaard L., Englert J.M., Manni M.L., Milutinovic P.S., Gefter J., Tobolewski J., Crum L., Coudriet G.M., Piganelli J., Zamora R., Vodovotz Y., Enghild J.J., Oury T.D. Lack of the receptor for advanced glycation end-products attenuates E. coli pneumonia in mice. PLoS One. 2011;6:e20132. doi: 10.1371/journal.pone.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Englert J.M., Kliment C.R., Ramsgaard L., Milutinovic P.S., Crum L., Tobolewski J.M., Oury T.D. Paradoxical function for the receptor for advanced glycation end products in mouse models of pulmonary fibrosis. Int J Clin Exp Pathol. 2011;4:241–254. [PMC free article] [PubMed] [Google Scholar]
- 10.He M., Kubo H., Ishizawa K., Hegab A.E., Yamamoto Y., Yamamoto H., Yamaya M. The role of the receptor for advanced glycation end-products in lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1427–L1436. doi: 10.1152/ajplung.00075.2007. [DOI] [PubMed] [Google Scholar]
- 11.Ramsgaard L., Englert J.M., Tobolewski J., Tomai L., Fattman C.L., Leme A.S., Kaynar A.M., Shapiro S.D., Enghild J.J., Oury T.D. The role of the receptor for advanced glycation end-products in a murine model of silicosis. PLoS One. 2010;5:e9604. doi: 10.1371/journal.pone.0009604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe T., Asai K., Fujimoto H., Tanaka H., Kanazawa H., Hirata K. Increased levels of HMGB-1 and endogenous secretory RAGE in induced sputum from asthmatic patients. Respir Med. 2011;105:519–525. doi: 10.1016/j.rmed.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 13.Halayko A.J., Ghavami S. S100A8/A9: a mediator of severe asthma pathogenesis and morbidity? Can J Physiol Pharmacol. 2009;87:743–755. doi: 10.1139/Y09-054. [DOI] [PubMed] [Google Scholar]
- 14.Sukkar M.B., Wood L.G., Tooze M., Simpson J.L., McDonald V.M., Gibson P.G., Wark P.A. Soluble RAGE is deficient in neutrophilic asthma and chronic obstructive pulmonary disease. Eur Respir J. 2012;39:721–729. doi: 10.1183/09031936.00022011. [DOI] [PubMed] [Google Scholar]
- 15.Gavett S.H., Koren H.S. The role of particulate matter in exacerbation of atopic asthma. Int Arch Allergy Immunol. 2001;124:109–112. doi: 10.1159/000053685. [DOI] [PubMed] [Google Scholar]
- 16.Thomas W.R., Hales B.J., Smith W.A. House dust mite allergens in asthma and allergy. Trends Mol Med. 2010;16:321–328. doi: 10.1016/j.molmed.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 17.Constien R., Forde A., Liliensiek B., Grone H.J., Nawroth P., Hammerling G., Arnold B. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis. 2001;30:36–44. doi: 10.1002/gene.1030. [DOI] [PubMed] [Google Scholar]
- 18.Englert J.M., Ramsgaard L., Valnickova Z., Enghild J.J., Oury T.D. Large scale isolation and purification of soluble RAGE from lung tissue. Protein Expr Purif. 2008;61:99–101. doi: 10.1016/j.pep.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demling N., Ehrhardt C., Kasper M., Laue M., Knels L., Rieber E.P. Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. 2006;323:475–488. doi: 10.1007/s00441-005-0069-0. [DOI] [PubMed] [Google Scholar]
- 20.Alcorn J.F., Rinaldi L.M., Jaffe E.F., van Loon M., Bates J.H., Janssen-Heininger Y.M., Irvin C.G. Transforming growth factor-beta1 suppresses airway hyperresponsiveness in allergic airway disease. Am J Respir Crit Care Med. 2007;176:974–982. doi: 10.1164/rccm.200702-334OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bury A.F. Analysis of protein and peptide mixtures - evaluation of 3 sodium dodecyl sulfate-polyacrylamide gel-electrophoresis buffer systems. J Chromatogr. 1981;213:491–500. [Google Scholar]
- 22.Abramoff M.D., Magalhaes P.J., Ram S.J. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 23.Gefter J.V., Shaufl A.L., Fink M.P., Delude R.L. Comparison of distinct protein isoforms of the receptor for advanced glycation end-products expressed in murine tissues and cell lines. Cell Tissue Res. 2009;337:79–89. doi: 10.1007/s00441-009-0791-0. [DOI] [PubMed] [Google Scholar]
- 24.Alcorn J.F., Ckless K., Brown A.L., Guala A.S., Kolls J.K., Poynter M.E., Irvin C.G., van der Vliet A., Janssen-Heininger Y.M. Strain-dependent activation of NF-kappaB in the airway epithelium and its role in allergic airway inflammation. Am J Physiol Lung Cell Mol Physiol. 2010;298:L57–L66. doi: 10.1152/ajplung.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryan J.J. Interleukin-4 and its receptor: essential mediators of the allergic response. J Allergy Clin Immunol. 1997;99:1–5. doi: 10.1016/s0091-6749(97)70293-8. [DOI] [PubMed] [Google Scholar]
- 26.Aujla S.J., Alcorn J.F. T(H)17 cells in asthma and inflammation. Biochim Biophys Acta. 2011;1810:1066–1079. doi: 10.1016/j.bbagen.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 27.Besnard A.G., Togbe D., Couillin I., Tan Z., Zheng S.G., Erard F., Le Bert M., Quesniaux V., Ryffel B. Inflammasome-IL-1-Th17 response in allergic lung inflammation. J Mol Cell Biol. 2012;4:3–10. doi: 10.1093/jmcb/mjr042. [DOI] [PubMed] [Google Scholar]
- 28.Lee J.J., Dimina D., Macias M.P., Ochkur S.I., McGarry M.P., O'Neill K.R., Protheroe C., Pero R., Nguyen T., Cormier S.A., Lenkiewicz E., Colbert D., Rinaldi L., Ackerman S.J., Irvin C.G., Lee N.A. Defining a link with asthma in mice congenitally deficient in eosinophils. Science. 2004;305:1773–1776. doi: 10.1126/science.1099472. [DOI] [PubMed] [Google Scholar]
- 29.Humbles A.A., Lloyd C.M., McMillan S.J., Friend D.S., Xanthou G., McKenna E.E., Ghiran S., Gerard N.P., Yu C., Orkin S.H., Gerard C. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–1779. doi: 10.1126/science.1100283. [DOI] [PubMed] [Google Scholar]
- 30.Kopf M., Brombacher F., Hodgkin P.D., Ramsay A.J., Milbourne E.A., Dai W.J., Ovington K.S., Behm C.A., Kohler G., Young I.G., Matthaei K.I. IL-5-deficient mice have a developmental defect in CD5+ B-1 cells and lack eosinophilia but have normal antibody and cytotoxic T cell responses. Immunity. 1996;4:15–24. doi: 10.1016/s1074-7613(00)80294-0. [DOI] [PubMed] [Google Scholar]
- 31.Lee N.A., McGarry M.P., Larson K.A., Horton M.A., Kristensen A.B., Lee J.J. Expression of IL-5 in thymocytes/T cells leads to the development of a massive eosinophilia, extramedullary eosinophilopoiesis, and unique histopathologies. J Immunol. 1997;158:1332–1344. [PubMed] [Google Scholar]
- 32.Wills-Karp M., Luyimbazi J., Xu X., Schofield B., Neben T.Y., Karp C.L., Donaldson D.D. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 33.Matsukura S., Stellato C., Georas S.N., Casolaro V., Plitt J.R., Miura K., Kurosawa S., Schindler U., Schleimer R.P. Interleukin-13 upregulates eotaxin expression in airway epithelial cells by a STAT6-dependent mechanism. Am J Respir Cell Mol Biol. 2001;24:755–761. doi: 10.1165/ajrcmb.24.6.4351. [DOI] [PubMed] [Google Scholar]
- 34.Moore P.E., Church T.L., Chism D.D., Panettieri R.A., Jr, Shore S.A. IL-13 and IL-4 cause eotaxin release in human airway smooth muscle cells: a role for ERK. Am J Physiol Lung Cell Mol Physiol. 2002;282:L847–L853. doi: 10.1152/ajplung.00245.2001. [DOI] [PubMed] [Google Scholar]
- 35.Pope S.M., Fulkerson P.C., Blanchard C., Akei H.S., Nikolaidis N.M., Zimmermann N., Molkentin J.D., Rothenberg M.E. Identification of a cooperative mechanism involving interleukin-13 and eotaxin-2 in experimental allergic lung inflammation. J Biol Chem. 2005;280:13952–13961. doi: 10.1074/jbc.M406037200. [DOI] [PubMed] [Google Scholar]
- 36.Wenzel S.E., Trudeau J.B., Barnes S., Zhou X., Cundall M., Westcott J.Y., McCord K., Chu H.W. TGF-beta and IL-13 synergistically increase eotaxin-1 production in human airway fibroblasts. J Immunol. 2002;169:4613–4619. doi: 10.4049/jimmunol.169.8.4613. [DOI] [PubMed] [Google Scholar]
- 37.Goova M.T., Li J., Kislinger T., Qu W., Lu Y., Bucciarelli L.G., Nowygrod S., Wolf B.M., Caliste X., Yan S.F., Stern D.M., Schmidt A.M. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol. 2001;159:513–525. doi: 10.1016/S0002-9440(10)61723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Y., Yan S.S., Colgan J., Zhang H.P., Luban J., Schmidt A.M., Stern D., Herold K.C. Blockade of late stages of autoimmune diabetes by inhibition of the receptor for advanced glycation end products. J Immunol. 2004;173:1399–1405. doi: 10.4049/jimmunol.173.2.1399. [DOI] [PubMed] [Google Scholar]
- 39.Wendt T.M., Tanji N., Guo J., Kislinger T.R., Qu W., Lu Y., Bucciarelli L.G., Rong L.L., Moser B., Markowitz G.S., Stein G., Bierhaus A., Liliensiek B., Arnold B., Nawroth P.P., Stern D.M., D'Agati V.D., Schmidt A.M. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol. 2003;162:1123–1137. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pouliot P., Turmel V., Gelinas E., Laviolette M., Bissonnette E.Y. Interleukin-4 production by human alveolar macrophages. Clin Exp Allergy. 2005;35:804–810. doi: 10.1111/j.1365-2222.2005.02246.x. [DOI] [PubMed] [Google Scholar]
- 41.van Panhuys N, Prout M, Forbes E, Min B, Paul WE, Le Gros G: Basophils are the major producers of IL-4 during primary helminth infection. J Immunol 186:2719–2728 [DOI] [PMC free article] [PubMed]
- 42.MacLean J.A., Sauty A., Luster A.D., Drazen J.M., De Sanctis G.T. Antigen-induced airway hyperresponsiveness, pulmonary eosinophilia, and chemokine expression in B cell-deficient mice. Am J Respir Cell Mol Biol. 1999;20:379–387. doi: 10.1165/ajrcmb.20.3.3291. [DOI] [PubMed] [Google Scholar]
- 43.Lajoie S., Lewkowich I.P., Suzuki Y., Clark J.R., Sproles A.A., Dienger K., Budelsky A.L., Wills-Karp M. Complement-mediated regulation of the IL-17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat Immunol. 2010;11:928–935. doi: 10.1038/ni.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Al-Ramli W., Prefontaine D., Chouiali F., Martin J.G., Olivenstein R., Lemiere C., Hamid Q. T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol. 2009;123:1185–1187. doi: 10.1016/j.jaci.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 45.Moreira A.P., Cavassani K.A., Ismailoglu U.B., Hullinger R., Dunleavy M.P., Knight D.A., Kunkel S.L., Uematsu S., Akira S., Hogaboam C.M. The protective role of TLR6 in a mouse model of asthma is mediated by IL-23 and IL-17A. J Clin Invest. 2011;121:4420–4432. doi: 10.1172/JCI44999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schnyder-Candrian S., Togbe D., Couillin I., Mercier I., Brombacher F., Quesniaux V., Fossiez F., Ryffel B., Schnyder B. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med. 2006;203:2715–2725. doi: 10.1084/jem.20061401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Durrant D.M., Gaffen S.L., Riesenfeld E.P., Irvin C.G., Metzger D.W. Development of allergen-induced airway inflammation in the absence of T-bet regulation is dependent on IL-17. J Immunol. 2009;183:5293–5300. doi: 10.4049/jimmunol.0803109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kudo M., Melton A.C., Chen C., Engler M.B., Huang K.E., Ren X., Wang Y., Bernstein X., Li J.T., Atabai K., Huang X., Sheppard D. IL-17A produced by alphabeta T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med. 2012;18:547–554. doi: 10.1038/nm.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park S.J., Lee K.S., Kim S.R., Min K.H., Moon H., Lee M.H., Chung C.R., Han H.J., Puri K.D., Lee Y.C. Phosphoinositide 3-kinase delta inhibitor suppresses interleukin-17 expression in a murine asthma model. Eur Respir J. 2010;36:1448–1459. doi: 10.1183/09031936.00106609. [DOI] [PubMed] [Google Scholar]
- 50.Wilson R.H., Whitehead G.S., Nakano H., Free M.E., Kolls J.K., Cook D.N. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med. 2009;180:720–730. doi: 10.1164/rccm.200904-0573OC. [DOI] [PMC free article] [PubMed] [Google Scholar]