

Abstract
In Alzheimer disease (AD), deposition of neurofibrillary tangles and loss of synapses in the neocortex and limbic system each correlate strongly with cognitive impairment. Tangles are composed of misfolded hyperphosphorylated tau proteins; however, the link between tau abnormalities and synaptic dysfunction remains unclear. We examined the location of tau in control and AD cortices using biochemical and morphologic methods. We found that, in addition to its well-described axonal localization, normal tau is present at both presynaptic and postsynaptic terminals in control human brains. In AD, tau becomes hyperphosphorylated and misfolded at both presynaptic and postsynaptic terminals, and this abnormally posttranslationally modified tau is enriched in synaptoneurosomal fractions. Synaptic tau seems to be hyperphosphorylated and ubiquitinated, and forms stable oligomers resistant to SDS denaturation. The accumulation of hyperphosphorylated tau oligomers at human AD synapses is associated with increased ubiquitinated substrates and increased proteasome components, consistent with dysfunction of the ubiquitin-proteasome system. Our findings suggest that synaptic hyperphosphorylated tau oligomers may be an important mediator of the proteotoxicity that disrupts synapses in AD.
Alzheimer disease (AD) is the most common neurodegenerative disorder in the elderly, and affects primarily the neocortex and the limbic system, with complex pathophysiologic features that include tau inclusions (neurofibrillary tangles, neuropil threads, and dystrophic neurites), β-amyloid inclusions (plaques and cerebral amyloid angiopathy), loss of neurons and synapses, astrogliosis, microglial activation, and inflammation.1,2 Among these features, synaptic loss3,4 and neurofibrillary tangle deposition5,6 seem to best correlate with cognitive decline.
Neurofibrillary and synaptic loss are correlated in clinicopathologic studies of AD7; however, whether this is a co-occurrence of parallel pathologic processes or synaptic loss is more directly related to alterations in tau biology is uncertain.8,9 We, therefore, used new biochemical and morphologic techniques to address the issue of tau protein accumulation in synapses in the adult human brain and in AD.
Normal tau is an abundant microtubule-associated protein that has been described as predominantly localized in axons in mature neurons.10 In AD, abnormally folded and hyperphosphorylated tau (p-tau) accumulates in axons, dendrites, and somas.11,12 In contrast to these long-held generalizations, recent reports have suggested that tau is also normally present in dendritic spines, where it interacts with postsynaptic density (PSD) proteins such as Fyn kinase.13
We hypothesized that tau may pathologically accumulate at synaptic sites in AD because it has been recently suggested that tau can be present in postsynaptic locales in normal mice,13 ubiquitinated tau accumulates in the brain in AD,14,15 and a major site of protein ubiquitination and proteasome-mediated degradation is at presynaptic and postsynaptic structures.16,17 By isolating synaptic terminals, we observed that, in control brains, tau is present at both presynaptic and postsynaptic terminals. In contrast, in synaptoneurosomes isolated from brains in AD, p-tau can form stable SDS-resistant oligomers that accumulate on both sides of the synapse, showing synaptic enrichment when compared with the cytoplasm. The accumulation of p-tau at the synapse mirrors the accumulation of ubiquitinated proteins in the same fraction, as well as the accumulation of proteasomes and related chaperones, which suggests that tau aggregates are associated with impaired proteolysis mediated by the ubiquitin-proteasome system (UPS).18
Materials and Methods
Reagents
Protease inhibitor (cOmplete tablet) was purchased from Roche Applied Science (Roche Diagnostics Corp., Indianapolis, IN). Phosphatase inhibitor cocktails 2 and 3 were purchased from Sigma-Aldrich Corp. (St. Louis, MO) and used in a 1:1 combination. Mouse monoclonal antibodies PHF1 (pS396/pS404 tau), CP13 (pS202 tau), and DA9 (total tau) were gifts of Peter Davies (Albert Einstein College of Medicine, Bronx, NY). Rabbit anti–total tau (A20024) was purchased from Dako Denmark A/S (Glostrup, Denmark); rabbit anti-PSD95 (No. 2507) from Cell Signaling Technology, Inc. (Danvers, MA); mouse anti-MBP (SMI-99P) from Covance, Inc. (Princeton, NJ); mouse anti-actin (A4700), rabbit anti-actin (A5060), and mouse anti-tubulin β3 (T8660) from Sigma-Aldrich; mouse anti-synaptophysin (AB8049), mouse anti-VCP (AB19444), and rabbit anti-VDAC (AB34726) from Abcam Inc. (Cambridge, MA); mouse anti-GAPDH (MAB374), rabbit anti-histone H3 (05-928), and rabbit anti-Myc (06-549) from Millipore Corp. (Billerica, MA); and rabbit anti-ubiquitin conjugates (UG9510), mouse anti-α7 (20S subunit, PW8110), and mouse anti-Rpt1 (26S subunit, PW8852) from Enzo Clinical Laboratories, Inc. (Farmingdale, NY).
Human Subjects
Brains from human subjects with a diagnosis of AD or no cognitive deficits were obtained through the Massachusetts Alzheimer's Disease Research Center and Massachusetts General Hospital Neuropathology Department. All donor tissue was obtained in accord with local and national institutional review board regulations. Characteristics of controls and subjects with AD used for quantitative analyses are given in Table 1.
Table 1.
Characteristics of Control and AD-Affected Brains Used in Quantitative Studies
| Case No. | Age (years) | Sex | Clinical Diagnosis | Disease Duration (years) | ApoE Genotype | PMI (hours) | Braak Stage | Experiment |
|---|---|---|---|---|---|---|---|---|
| C1 | 89 | F | Control | NA | 2/3 | 13 | 2 | Figure 4 |
| C2 | 91 | F | Control | NA | 3/3 | 19 | 2 | Figure 4 |
| C3 | 71 | M | Control | NA | NA | 5 | 0 | Figure 4 |
| C4 | 87 | M | Control | NA | NA | 36 | 1 | Figure 4 |
| C5 | 80 | F | Control | NA | 2/4 | 54 | 1 | Figure 7 |
| C6 | 76 | M | Control | NA | 3/4 | 48 | 1 | Figure 7 |
| C7 | 85 | M | Control | NA | 3/3 | 24 | 2 | Figure 7 |
| C8 | 57 | F | Control | NA | 3/3 | 13 | 0 | Figure 7 |
| C9 | 74 | F | Control | NA | 3/3 | 24 | 1 | Figure 7 |
| C10 | 88 | F | Control | NA | 3/3 | 20 | 2 | Figure 7 |
| AD1 | 83 | F | AD | 13 | 3/4 | 12 | 5 | Figure 4 |
| AD2 | 82 | M | AD | 6 | 3/4 | 7 | 6 | Figure 4 |
| AD3 | 91 | F | AD | 14 | 3/4 | 9 | 5 | Figure 4 |
| AD4 | 95 | M | AD | NA | 3/3 | 11 | 6 | Figure 4 |
| AD5 | 85 | F | AD | 4 | 3/4 | 10 | 5 | Figure 5 |
| AD6 | 73 | F | AD | 19 | 3/3 | 14 | 5 | Figure 5 |
| AD7 | 84 | F | AD | 16 | 3/4 | 12 | 5 | Figure 5 |
| AD8 | 65 | M | AD | 8 | 3/3 | 21 | 5 | Figures 7, 8 |
| AD9 | 84 | F | AD | 7 | 3/3 | 7 | 6 | Figures 7, 8 |
| AD10 | 75 | F | AD | 5 | 3/3 | 26 | 6 | Figures 7, 8 |
| AD11 | 92 | F | AD | 9 | 3/3 | 4 | 6 | Figures 7, 8 |
| AD12 | 93 | M | AD | 17 | 3/3 | 6 | 5 | Figures 7, 8 |
| AD13 | 74 | M | AD | 11 | 3/3 | 15 | 5 | Figures 7, 8 |
| AD14 | 92 | M | AD | 22 | 4/4 | 12 | 5 | Figures 5, 7, 8 |
| AD15 | 68 | F | AD | 11 | 4/4 | 20 | 6 | Figures 7, 8 |
| AD16 | 80 | F | AD | 12 | 4/4 | 11 | 6 | Figures 7, 8 |
| AD17 | 71 | F | AD | 17 | 4/4 | 14 | 6 | Figures 7, 8 |
| AD18 | 74 | M | AD | 17 | 4/4 | 20 | 5 | Figures 7, 8 |
| AD19 | 89 | M | AD | 10 | 4/4 | 10 | 5 | Figures 7, 8 |
F = female; M = male; AD = Alzheimer disease; NA = not applicable or not available; PMI = postmortem interval.
Subcellular Fractionation and Protein Extraction
Cortical gray matter (200 to 300 mg) taken from frozen human brains was gently ground in a Potter-Elvehjem homogenizer with 1.5 mL ice-cold buffer A (25 mmol/L HEPES [pH 7.5], 120 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, and 2 mmol/L CaCl2) supplemented with 2 mmol/L dithiothreitol (DTT), protease inhibitors, and phosphatase inhibitors. The homogenate was passed through two layers of 80-μm nylon filters (Millipore) to remove tissue debris, and a 200-μL aliquot was saved. The saved aliquot was mixed with 200 μL water and 70 μL 10% SDS, passed through a 27-gauge needle, and boiled for 5 minutes to prepare the total extract.
To prepare filtered synaptoneurosomes as described by Hollingsworth et al,19 the remainder of the homogenate was passed through a 5-μm Supor membrane filter (Pall Corp., Port Washington, NY) to remove large organelles and nuclei, and centrifuged at 1000 × g for 5 minutes. The pellet was washed once with buffer A and centrifuged again, yielding the synaptoneurosome pellet. Supernatant from the first centrifugation was clarified via centrifugation at 100,000 × g for 1 hour to obtain the cytosol fraction. Cytosolic extract was prepared by adding 1.5% SDS and boiling for 5 minutes. Synaptoneurosome pellets were extracted using 0.5 mL buffer B (50 mmol/L Tris [pH 7.5], 1.5% SDS, and 2 mmol/L DTT), and were boiled for 5 minutes.
Sucrose Flotation Gradient
Flotation sucrose gradient for isolating synaptic terminals was modified from previously published procedures.20 Ice-cold sucrose solutions of 0.3, 0.93, and 1.2 mol/L were prepared using 10 mmol/L HEPES (pH 7.5). The synaptoneurosome pellet was resuspended in 1.2 mmol/L sucrose and transferred to a centrifuge tube, and then overlaid with 1.2, 0.93, and 0.3 mol/L sucrose to form a three-layer discontinuous gradient. After centrifugation at 60,000 × g for 2 hours, synaptic terminals were collected from the 0.93/1.2-mol/L interface, and myelin from the 0.3/0.93-mol/L interface, and both were diluted with water to about 0.3 mol/L final sucrose concentration. The pellet was also resuspended in 0.3 mol/L sucrose. All three fractions were centrifuged at 20,000 × g for 20 minutes, and the pellets were extracted using buffer B and boiled for 5 minutes.
Triton Extraction
The synaptoneurosome pellet was resuspended in ice-cold buffer C [50 mmol/L Tris (pH 7.5), 150 mmol/L NaCl, 2% Triton X-100, and protease inhibitors]. After 20 minutes of incubation in a rotating tube, the mixture was centrifuged at 100,000 × g for 1 hour. The collected supernatant was supplemented with 1% SDS and boiled for 5 minutes. The pellet was extracted using buffer B and boiled for 5 minutes.
Gel Electrophoresis and Immunoblotting
SDS-denatured protein extracts were clarified via centrifugation at 20,000 × g for 15 minutes, followed by bicinchoninic acid assays (Pierce Protein Biology, Fisher Scientific, Inc., Rockford, IL) to determine protein concentrations. Extracts were boiled again for 3 minutes after adding 5X sample buffer (250 mmol/L Tris [pH 7.5], 5% SDS, 400 mmol/L DTT, 50% glycerol, and 0.2% Orange G). Samples were resolved via SDS-PAGE using Bis-Tris 4% to 12% gels (Invitrogen Corp., Carlsbad, CA), and were transferred to low-fluorescence polyvinylidene difluoride or nitrocellulose membranes for immunoblotting, detected using an Odyssey laser scanner (LI-COR Biosciences, Inc., Lincoln, NE). Blocking buffer, stripping buffer, and secondary antibodies were purchased from LI-COR, and were used according to the manufacturer's protocols.
Ubiquitin Pull-Down and Tau Immunoprecipitation
Synaptoneurosome pellets were resuspended in buffer D (20 mmol/L Tris [pH 7.5], 150 mmol/L NaCl, 0.2% SDS, and 4 mmol/L DTT), boiled for 5 minutes, and cleared via centrifugation at 15,000 × g for 10 minutes. The supernatant was mixed with an equal volume of buffer E (20 mmol/L Tris [pH 7.5], 150 mmol/L NaCl, 2% Triton X-100, and 1% sodium deoxycholate) to neutralize SDS. The mixture was pre-cleared via incubation using Protein G Sepharose (GE Healthcare, Pittsburg, PA) at 4°C for 1 hour, followed by overnight incubation with S5a UIM agarose conjugate (UW9820; Enzo Clinical Laboratories) or protein G sepharose (control). After centrifugation at 1000 × g for 2 minutes, the supernatant was collected as the flow-through fraction. The resin was washed three times with cold buffer F (20 mmol/L Tris [pH 7.5], 150 mmol/L NaCl, and 1% Triton X-100) and boiled for 5 minutes with 1.5X SDS-PAGE sample buffer to elute captured ubiquitinated proteins. In immunoprecipitation experiments, the pre-cleared extract was incubated overnight at 4°C with rabbit anti–total tau or anti-Myc (control), followed by incubation with protein G sepharose for 2 hours. After centrifugation at 1000 × g for 2 minutes, the supernatant was collected as the flow-through fraction. The resin was washed three times with cold buffer F and boiled for 5 minutes with 1.5X SDS-PAGE sample buffer to elute captured proteins. Western blots of immunoprecipitation proteins were detected via enhanced chemiluminescence using TrueBlot Ultra anti-mouse horseradish peroxidase conjugate (eBioscience, Inc., San Diego, CA) or Clean-Blot horseradish peroxidase conjugate (against rabbit primary antibodies; Pierce) to minimize signals from denatured IgG.
Immunostaining of Synaptoneurosomes
Synaptoneurosome pellets were resuspended in ice-cold buffer A, passed through 27-gauge needles, and mixed with an equal volume of 2% paraformaldehyde in PBS-MC (1 mmol/L MgCl2 and 1 mmol/L CaCl2) in Lab-Tek II CC2 pre-coated chamber slides (Nunc, Rochester, NY). After 10 minutes of settling at 4°C, synaptoneurosomes became fixed and attached to the glass surface, and were washed three times using PBS-MC (room temperature from this point on). Synaptoneurosomes were permeabilized using 0.05% Triton X-100 in PBS-MC with 3% bovine serum albumen (BSA), and washed three times. Slides were blocked using 4% normal goat serum and 3% BSA in PBS-MC for 30 minutes and then incubated with primary antibodies diluted in PBS-MC with 3% BSA for 2 hours, followed by three washes. Secondary antibodies diluted in PBS-MC with 3% BSA were incubated for 1 hour, followed by three washes. The slide was mounted with a No. 1.5 glass coverslip and Prolong Gold Antifade reagent (Invitrogen).
Primary antibodies for immunostaining included guinea pig anti-vGlut1 (Millipore AB590, 1:150), chicken anti-MAP2 (Abcam AB5392, 1: 100), goat anti-PSD95 (Abcam AB12093, 1:100), DA9 (1:150), and PHF1 (1:80). Fluorescent secondary donkey antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA) and used at 1:100 dilutions (anti-guinea pig DyLight 649, anti-chicken Cy3, anti-goat Alexa 488, and anti-mouse Alexa 488).
Image Acquisition and Analysis
Fluorescent and brightfield images of immunostained synaptoneurosomes were acquired using an Axio Imager Z epifluorescence microscope (Carl Zeiss AG, Oberkochen, Germany) equipped with a 63X oil immersion objective (numerical aperture, 1.40). Images were deconvolved using the Iterative Deconvolution plug-in (by Bob Dougherty, OptiNav, Inc.) in ImageJ software (version 1.44). Synaptic protein co-localization was determined via manual analysis on randomly selected areas from wide-field images.
Transmission Electron Microscopy
Synaptoneurosome pellets were fixed in 2% glutaraldehyde and 2% paraformaldehyde in PBS overnight at 4°C, rinsed, post-fixed in 1% osmium tetroxide, and embedded in LR White resin (Electron Microscopy Sciences, Hatfield, PA). Images were acquired using a transmission electron microscope equipped with an ATM digital camera (JEOL1011; JEOL USA, Inc., Peabody, MA).
Data and Statistical Analyses
Western blots were quantified via densitometry using the gel analysis function in ImageJ software (version 1.44). Statistical tests (paired t-test, Mann-Whitney test, two-way analysis of variance, and linear regression) were performed using statistical software (GraphPad version 5.03; Prism Software Corp., La Jolla, CA).
Results
p-tau Oligomers Accumulate at Synapses in AD
Proteins from control and AD cortical homogenates were extracted using 1.5% SDS and analyzed using SDS-PAGE. In total protein extracts, control brains exhibited only monomeric nonhyperphosphorylated tau (50 to 65 kDa). AD-affected brains contained tau species that were hyperphosphorylated (reactive against p-tau antibodies CP1321 and PHF122) and migrated as a smear (Figure 1). Fast-migrating species (15 to 50 kDa) represented truncated p-tau; slow-migrating species (65 to several hundred kDa) represented p-tau oligomers,23 which were stably misfolded and resistant to SDS denaturation and reducing agents.
Figure 1.
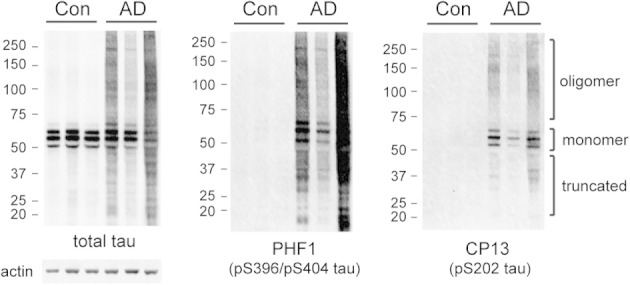
Total SDS-soluble extracts of human AD cortices show p-tau (detected using PHF1 and CP13) accumulation compared with controls. Actin serves as loading control.
To understand the subcellular localization of tau proteins in AD, we separated cortical tissue homogenates into cytosolic and synaptic (synaptoneurosome) fractions. The synaptoneurosome fraction, when examined under electron microscopy, showed well-preserved synaptic structures. In both control and AD synaptoneurosomes, no fibrillar protein aggregates were detected under electron microscopy (Figure 2A). Biochemical analysis of tau in control samples showed that nonphosphorylated tau was more enriched in the cytosol than in synaptoneurosomes, consistent with its expected primary distribution in the axonal cytoplasm. In contrast, analysis of AD samples revealed that p-tau accumulated to high levels in AD synaptoneurosomes, but remained surprisingly absent in cytosolic extracts (Figure 2B). Considered together, our data suggest that p-tau can form nonfibrillar aggregates associated with the synaptoneurosomal pellet, containing SDS-resistant oligomers.
Figure 2.
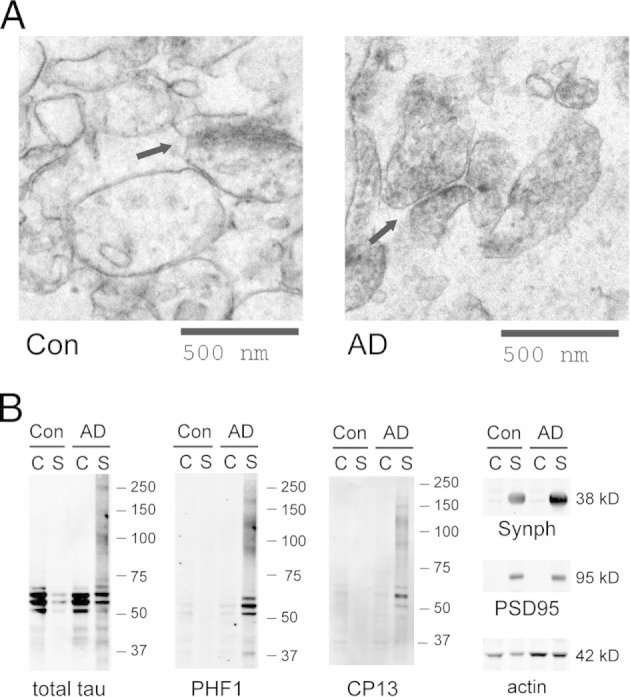
A: Synaptoneurosomes from human cortices examined under electron microscopy show well-preserved synaptic structures (arrows). No protein fibrils were observed in control or AD samples. B: In AD-affected brains, p-tau oligomers (detected using PHF1 and CP13) accumulate in the synaptoneurosome (S) but not in the cytosol (C). Synaptophysin (Synph) and PSD95 serve as synaptic markers. Images are representative of three independent experiments.
To examine whether the p-tau oligomers observed in AD synaptoneurosomes were associated with synapses rather than originating from other contaminants such as tau inclusions, we used sucrose gradient to further purify synaptic terminals (Figure 3A). One potential concern was that large nonfibrillar tau aggregates could have co-precipitated with synaptoneurosomes during low-speed centrifugation (1000 × g). Because large protein assemblies are more dense than lipid-rich vesicles, we chose a flotation gradient to purify synaptic terminals. Flotation gradient–purified synaptic terminals showed great reductions in organelle contaminants such as nuclei and myelin (Figure 3B). In control brains, they contained low but clearly detectable levels of nonphosphorylated monomeric tau, suggesting a synaptic localization of tau and confirming recent reports of a normal role for tau at the synapse.13,24 In AD-affected brains, they contained high levels of p-tau including aggregate species (Figure 3C), which suggested that hyperphosphorylation and oligomerization are specific to AD synapses.
Figure 3.

A: Human cortical synaptoneurosomes separated by sucrose flotation gradient to collect highly purified synaptic terminals. B: The purity is confirmed by immunoblotting against organelle markers: synaptophysin (Synph; presynaptic), PSD95 (postsynaptic), myelin basic protein (MBP), mitochondrial voltage-dependent anion channel (VDAC), histone H3, actin, and β3 tubulin. Total (T) and synaptoneurosomal (S) extracts are also loaded for comparison. C: Highly purified synaptic terminals still contain high levels of p-tau including oligomeric species. Images are representative of three independent experiments.
Tau is Localized to Both Presynaptic and Postsynaptic Terminals in Control and AD-Affected Brains
Biochemical analysis did not demonstrate whether tau and p-tau were localized to presynaptic or postsynaptic terminals; therefore, we developed a new immunostaining protocol to determine this. To visualize isolated synaptic terminals, we spread a dilute solution of synaptoneurosomes onto a glass slide and fixed it lightly in place (see Materials and Methods). The fixed permeabilized synaptoneurosomes were immunostained for vesicular glutamate transporter 1 (vGlut1) as a presynaptic marker, and microtubule-associated protein 2 (MAP2) as a postsynaptic marker (Figure 4A). MAP2 is a major microtubule-binding protein in dendrites, but is also present at lower levels inside dendritic spines and at the PSD.25,26 We confirmed the postsynaptic localization of MAP2 by co-staining with PSD95 (Figure 4D), and noted that PSD95 and vGlut1 often seemed to be so close as to almost overlap, whereas MAP2 and vGlut1 seemed most frequently to be adjacent but distinct puncta; we, therefore, chose to use MAP2 to mark postsynaptic sites to enable more straightforward identification of the presynaptic or postsynaptic localization of other proteins including tau.
Figure 4.

A: Control brain synaptoneurosomes fixed on glass slides were immunostained against presynaptic marker vGlut1, postsynaptic marker MAP2, and total tau (DA9 antibody). Magnifications of boxed areas represent tau immunoreactivity at individual synapses. Top, postsynaptic, bottom, pre and postsynaptic. B: Tau proteins are also detected at presynaptic and postsynaptic terminals from brains affected by AD. C: Quantification of the percentage of synaptic terminals positive for tau in control (4 cases, 400 presynapses, 276 postsynapses) and AD temporal cortices (4 cases, 400 presynapses, 287 postsynapses). No significant difference was found by two-way analysis of variance. Error bars represent SEM. D: Co-localization of PSD95 and MAP2 at postsynaptic sites. Scale bars = 1 μm (A–D).
Using an antibody (DA9) that recognizes both phosphorylated and nonphosphorylated forms of tau, we detected tau in many vGlut1-positive presynaptic puncta (55.3%) and also in most MAP2-positive postsynaptic sites (70.2%) in control human brains. In AD-affected brains, we detected tau at 63.3% of presynaptic sites and 70.1% of postsynaptic sites (Figure 4, B and C). With two-way analysis of variance, we did not detect a significant difference in tau distribution between presynaptic and postsynaptic sites or between control and AD-affected brains.
Under electron microscopy, approximately 80% of the presynaptic terminals isolated from rat brains contain visible microtubules.27 Inasmuch as tau is an abundant microtubule-binding protein in axons, our identification of tau in 55.3% of the presynapses is reasonable, and possibly an underestimate. With use of the same criteria, neighboring postsynaptic structures from control brains exhibit 70.2% tau labeling, which is consistent with recent reports that indicated which tau may also have a postsynaptic localization.13
p-tau is Localized to Both Presynaptic and Postsynaptic Sites and Co-Sediments with PSD Markers
Immunocytochemistry using PHF1 antibody did not detect p-tau in control synapses, but labeled a substantial percentage of AD synapses: 20.8% of vGlut1-positive presynaptic puncta and 32.0% of MAP2-positive postsynaptic puncta (Figure 5). These data support the hypothesis that normal tau is already present at both presynaptic and postsynaptic terminals in normal brains and that, during the pathologic progression of AD, tau misfolding and hyperphosphorylation occur on both sides of the synapse.
Figure 5.

A: p-Tau proteins stained using PHF1 antibody are detectable in synaptoneurosomes in brains affected by AD but not in control brains B: p-Tau localizes to both presynaptic (vGlut1) and postsynaptic (MAP2) terminals. Scale bar = 1 μm. C: Postsynapses have higher tau-positive percentages than do presynapses in AD temporal cortices (4 cases, 400 presynapses, 291 postsynapses). Error bars represent SEM. *P < 0.05, paired t-test.
To understand the properties of synaptic p-tau oligomers, we extracted synaptoneurosomes using 2% Triton X-100 to solubilize membrane and free cytoplasmic proteins, and the remaining insoluble material was collected as the crude PSD pellet, which retained nearly all of the PSD95 protein and most of the actin and microtubule cytoskeleton (Figure 6). Most of the normal tau protein in control human synapses was extractable using Triton, and the small amount of tau retained in the crude PSD pellet was consistent with mouse studies that co-immunoprecipitated tau and PSD95.13 In contrast, in AD synaptoneurosomes, essentially all p-tau species remained in the Triton-insoluble pellet, indicating that they were associated with the PSD and the cytoskeleton fraction, which supports our immunostaining data that they accumulate in dendritic spines. The crude PSD pellet also exhibited an enrichment of ubiquitinated proteins and ubiquitin-binding 26S proteasomes, but not ubiquitin-nonbinding 20S proteasomes.
Figure 6.
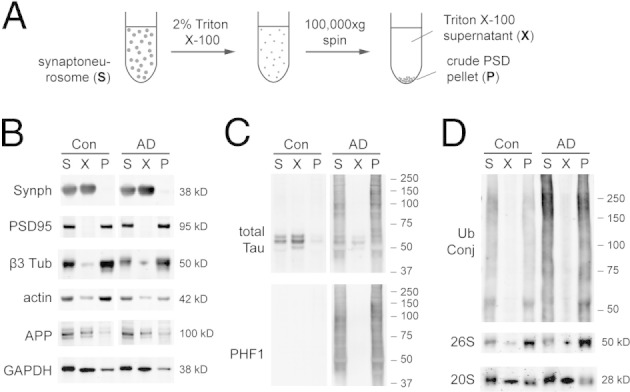
A: Synaptoneurosomes separated into the Triton-extractable fraction and the crude PSD pellet. Cytosolic protein GAPDH and membrane proteins such as synaptophysin (Synph) and amyloid precursor protein (APP) are mostly Triton extractable. B: The crude PSD pellet retains most of the PSD95 protein and cytoskeleton proteins (actin and β3 tubulin). C: Nonphosphorylated normal tau is mostly Triton extractable, but disease-associated p-tau is retained in the crude PSD pellet. D: The pellet is also enriched in ubiquitin conjugates (Ub Conj) and Rpt1 subunit of 26S proteasome, but not α7 subunit of 20S proteasome. Images are representative of three independent experiments.
p-tau Accumulation, but Not Aβ Accumulation, is Associated with Altered Protein Turnover at Synapses
The accumulation of p-tau oligomers at AD synapses implies that the protein quality control system is overwhelmed. The primary proteolytic mechanism for both normal and abnormal proteins at the synapse is the UPS.28 To examine whether there is UPS impairment at the synapse in AD, we measured the levels of ubiquitin-protein conjugates (proteasome substrates) in total and synaptic extracts. The synaptic extracts showed significant increases in ubiquitin conjugates in AD-affected brains compared with control brains (P = 0.02; Figure 7A). This was accompanied by significant increases in synaptic 26S proteasomes and valosin-containing protein (VCP), a chaperone that delivers substrates to proteasomes (Figure 7B). The importance of VCP in the neuronal defense against misfolded proteins is reflected in its known mutations that lead to frontotemporal dementia and amyotrophic lateral sclerosis.29 The simultaneous increase in proteasome substrates, proteasomes, and related chaperones suggested reduced UPS efficiency at AD synapses.
Figure 7.
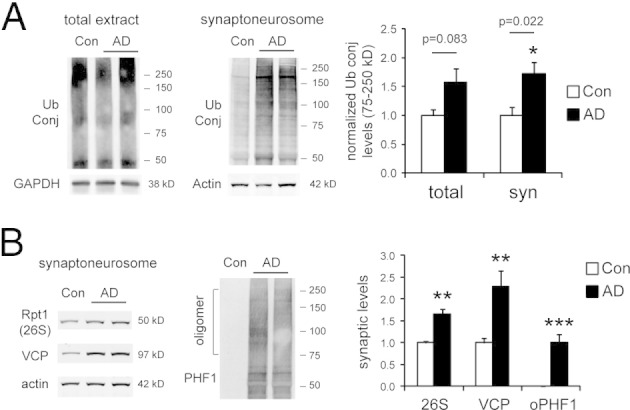
A: In brains affected by AD, ubiquitin conjugate (Ub Conj) levels are significantly elevated in synaptoneurosomal extracts (6 controls, 12 AD cases, frontal cortex). GAPDH and actin are loading controls. In AD synaptoneurosomal extracts, Rpt1 (26S proteasome subunit) and valosin-containing protein (VCP) also show significant increases. B: We also quantified oligomeric p-tau detected using PHF1 antibody (oPHF1) in the 75- to 250-kDa range, which is dramatically increased in AD synaptoneurosomes. Error bars represent SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Mann-Whitney test.
Next, we examined the correlation between p-tau accumulation and UPS alteration in AD synaptoneurosomes. We found that p-tau oligomer levels positively correlated with ubiquitin conjugate levels (P = 0.007; Figure 8A) and proteasome levels (P = 0.02; Figure 8B), and to a lesser extent with VCP (P = 0.08; Figure 8C). Previously, we have also quantified the synaptoneurosomal enrichment of Aβ monomers and oligomers (2-3mer) in these AD cases using SDS-PAGE and immunoblotting.30 We, therefore, compared the Aβ measures previously reported from these same cases with the tau and UPS-related measures presented herein. We found that within the AD group, the levels of presumably extracellular Aβ monomers or oligomers did not correlate with the levels of intracellular ubiquitin conjugates, proteasomes, or p-tau oligomers (Figure 8, D–I). These results suggest that at AD synapses, UPS impairment and tau abnormalities may progress together; however, the involvement of Aβ species is much less clear because of lack of correlation.
Figure 8.
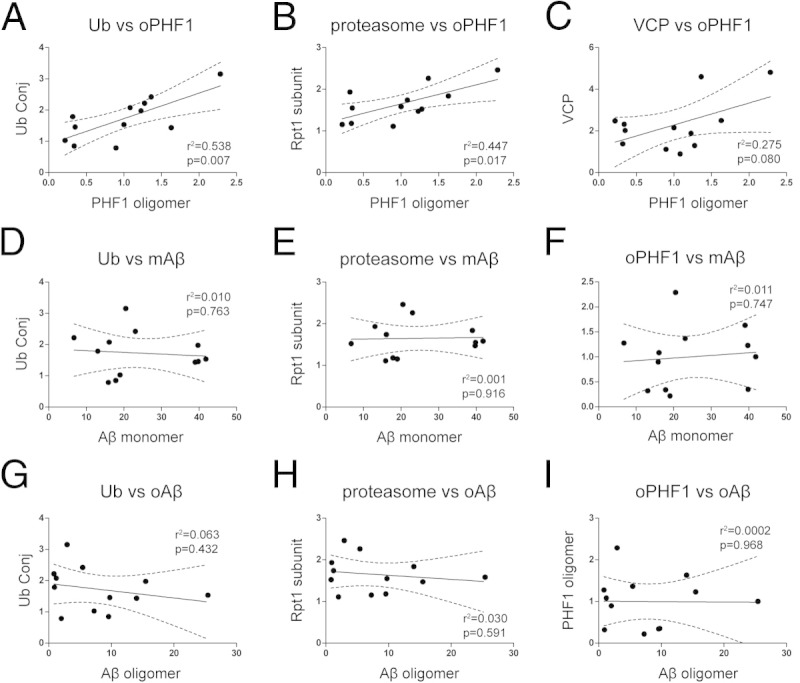
Correlations of various protein measurements in synaptoneurosomes in AD (12 cases, characterized in Figure 7). A and B: The levels of PHF1-reactive p-tau oligomers (oPHF1, 75 to 250 kDa) positively correlate with the levels of ubiquitin conjugates (Ub Conj, 75 to 250 kDa) and Rpt1 (subunit of 26S proteasome). C: There is a trend toward a significant correlation between oligomeric PHF1 and valosin-containing protein. Monomeric Aβ (mAβ) (D–F) and oligomeric Aβ (oAβ, dimers and trimers) (G–I) do not correlate with ubiquitin conjugates, proteasomes, and oligomeric PHF1. Linear regression is shown with 95% confidence intervals (dashed lines).
Synaptic p-tau at AD Synapses is Ubiquitinated and Interacts with the 26S Proteasome
The co-enrichment of misfolded tau, ubiquitin conjugates, and 26S proteasomes in the crude PSD pellet (Figure 6) provides a hint that the accumulation of ubiquitinated tau at synapses may sequester proteasomes through direct physical interaction. To verify that synaptic tau in AD-affected brains is ubiquitinated and capable of interacting with proteasomes, we performed a ubiquitin pull-down experiment using denatured synaptoneurosome extracts (Figure 9A). The capture resin used agarose conjugated with the ubiquitin-interacting motif of S5a subunit in the 26S proteasome. Among the pool of captured proteins, which carry ubiquitin moieties with high affinity for 26S proteasomes, we detected high levels of p-tau species including oligomers. This suggested that a tau molecule can be simultaneously hyperphosphorylated and ubiquitinated at AD synapses, in addition to being misfolded and aggregated. We were also able to detect ubiquitin moieties on immunoprecipitated tau from denatured synaptoneurosome extracts of AD-affected brains (Figure 9B). Immunoprecipitation also revealed that ubiquitinated tau comprises only a small fraction of ubiquitinated proteins at synapses, and hence the accumulation of ubiquitinated proteins in AD is suggestive of general UPS dysfunction. In control brain synaptoneurosomes, tau ubiquitination was below the detection limit of our methods, which suggests that ubiquitinated tau is efficiently degraded by UPS under normal conditions.
Figure 9.
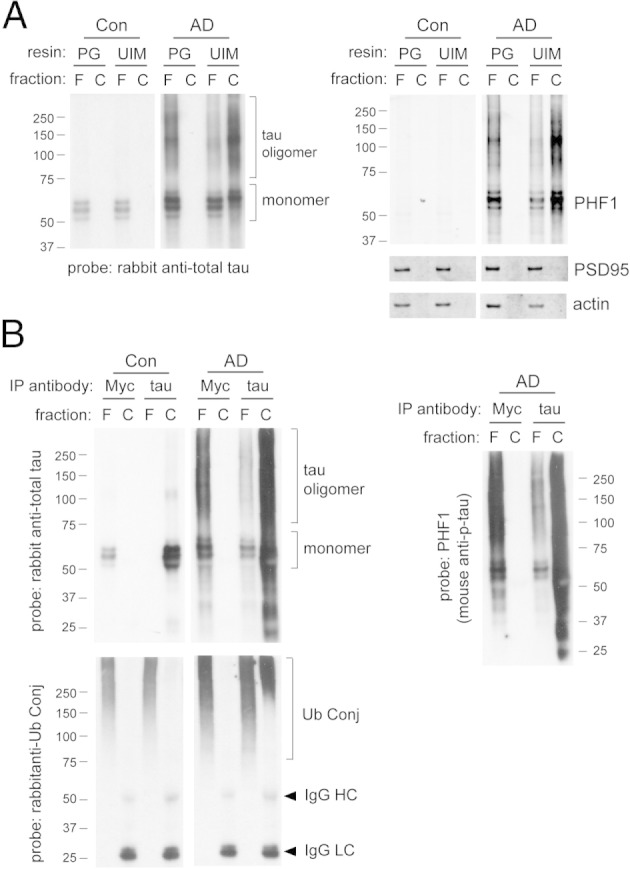
Synaptic p-tau oligomers in AD are ubiquitinated. A: Ubiquitinated proteins from denatured synaptoneurosome extracts were isolated using agarose conjugated with the ubiquitin-interacting motif (UIM) of 26S proteasome subunit S5a. Control resin was protein G (PG)–agarose conjugates. The captured (C) fraction and 20% of the corresponding flow-through (F) fraction were loaded for SDS-PAGE analysis. In AD-affected brains, p-tau (PHF1-reactive) becomes ubiquitinated and captured by UIM resin. PSD95 and actin serve as negative controls for ubiquitin pull-down. B: Tau immunoprecipitation (IP) using rabbit anti–total tau and protein G–agarose conjugates was performed using denatured synaptoneurosomal extract. Rabbit anti-Myc was the control antibody for IP. The captured (C) fraction and 20% of the corresponding flow-through (F) fraction were loaded for SDS-PAGE analysis. In AD-affected brains, synaptic tau oligomers isolated by IP are reactive against antibodies for ubiquitin conjugates (Ub Conj) and p-tau (PHF1). Cross-reactivity of secondary antibodies with denatured IgG heavy chain (HC) or light chain (LC) is sometimes observed. Images are representative of two independent experiments.
Discussion
There is growing evidence that synaptic toxicity in AD may be mediated, at least in part, by smaller, more diffusible forms of misfolded Aβ and tau aggregates, generally termed “oligomers.”9,30–32 The objective of the present study was to examine the localization and biochemical properties of synapse-associated tau oligomers. To do so, we developed a new method to biochemically isolate synaptoneurosomal preparations from human brains and also to then visualize that preparation of remarkably intact synaptic structures by smearing them on a microscope slide. Both techniques suggested that tau is present at both presynaptic and postsynaptic sites and that abnormal tau dramatically accumulates at the synapse in AD.
Oligomeric forms of tau including SDS-stable tau aggregates, likely formed from noncovalent interactions,23,33 are readily observed when AD-affected brain homogenates are extracted using SDS and analyzed via SDS-PAGE under reducing conditions. Typically, tau produces a “smear” pattern in Western blots (Figure 1). By performing subcellular fractionation before SDS extraction, we found that p-tau oligomers are also present at high levels at AD synapses (Figures 2 and 3). Immunocytochemistry experiments further revealed that p-tau accumulates in both presynaptic and postsynaptic terminals in AD (Figure 5). Finally, we noted that increased synaptic oligomeric tau co-occurs with evidence of UPS dysfunction (Figures 7 and 8), which suggests the possibility that tau accumulation is related to an underlying defect in synaptic UPS capability, which may more generally contribute to the accumulation of pathologic proteins at the synapse.
Potential Causes of Synaptic p-tau Oligomers
Our identification of p-tau at AD synapses supports similar observations by other groups using different methods. Applying immunohistochemistry to fixed, AD-affected brains, p-tau has been observed at thorny excrescences (large spine clusters) on CA3 hippocampal dendrites.34 Using flow cytometry, another study found p-tau in 32.3% of AD synaptosomes,35 which is in agreement with our data.
What causes the accumulation of p-tau oligomers at AD synapses? Two general possibilities are that tau is normally present at both presynaptic and postsynaptic sites, and becomes hyperphosphorylated, ubiquitinated, and possibly misfolded in situ where it accumulates, or that posttranslationally modified tau is transported to synaptic sites, where it accumulates. We detected normal tau in most presynaptic and postsynaptic terminals in control brains (Figure 4). We also found that synaptic p-tau aggregates are “sticky”; that is, they, remain bound to PSD and cytoskeleton after Triton extraction (Figure 6). To translocate to dendritic spines, misfolded p-tau with sticky properties originating from axons or somas would have to be transported over long distances to remote confined spaces, which seems unlikely. Thus, we favor the hypothesis that tau misfolding and hyperphosphorylation occurs in situ at both presynaptic and postsynaptic structures.
This model would also imply impairment in the local degradation of tau. We further explored this possibility because the UPS has recently been shown to have a major role in degradation of both neuronal tau36–38 and synaptic components.28,39 We found evidence of accumulation of misfolded, posttranslationally modified tau (hyperphosphorylated and ubiquitinated) at synapses, with parallel increases in ubiquitinated proteins and UPS components. It is likely that tau misfolding and UPS impairment may form a vicious cycle at synapses. Misfolded ubiquitinated tau inhibits proteasomes,15,18 which in turn may inhibit the normal turnover of tau, to favor the formation of additional misfolded species.
Potential Toxic Effects of p-tau Oligomers
From study of postmortem brain tissues, it is difficult to ascertain whether p-tau oligomers are toxic to synapses. We noted, however, that numerous studies have demonstrated that misfolded, aggregation-prone proteins are toxic to neurons in general, including abnormal tau.8,40,41 The toxic gain-of-function of misfolded proteins is generally attributed both to the exposure of sticky, hydrophobic surfaces on the misfolded protein and to the disruption of protein quality control systems.42,43 Our present observations are consistent with both features: synaptic p-tau seems to be sticky and is associated with itself and with the PSD and cytoskeleton in a detergent-resistant fashion, and p-tau oligomers also correlate well with the accumulation of proteasome substrates. We found p-tau oligomers to be ubiquitinated and to interact directly with the ubiquitin-interacting motif of 26S proteasomes (the docking site of ubiquitinated substrates on proteasomes), potentially acting as poor substrates that impede the proteolytic enzyme.15,18 It is also conceivable that SDS-resistant oligomers at synapses can act as seeds of aggregation and spread tauopathy to other cellular compartments, or even across synapses, as implicated in animal models of tauopathy.44,45
Synaptic tau in the Context of Other AD Lesions
The observation that misfolded tau accumulates at the synapse is reminiscent of the observation that oligomeric Aβ also seems to accumulate at the synapse.30,46 We were intrigued to find that Aβ species (both monomers and oligomers) did not correlate with p-tau accumulation or UPS impairment, whereas the latter two correlated well (Figure 8). This pattern of correlation seems to correspond to the subcellular compartmentalization of various proteins and proteolytic systems. Intracellular nonmembrane proteins such as tau are degraded by proteasomes37,38; therefore, tau and UPS changes are expected to be correlated. In contrast, extracellular Aβ is degraded by extracellular proteases such as neprilysin and insulin-degrading enzyme, whereas intracellular Aβ is membrane associated and may, therefore, be degraded by lysosomal proteases.47
Our data imply that there may be an Aβ-independent pathologic progression at AD synapses that involves both tau misfolding and UPS impairment. This parallels our earlier findings that Aβ accumulates and plateaus in the early stage of AD and that there is a lack of correlation between Aβ species and late-stage disease progression characterized by synaptic loss and NFT deposition.7
In conclusion, in the present study, we devised reliable methods to detect tau proteins at human synapses, leading to the demonstration that tau is a normal postsynaptic protein in addition to its traditional localization as a presynaptic and axonal protein. In AD, we identified p-tau oligomers as a potential synaptotoxic species that accumulates in both presynaptic and postsynaptic terminals. The accumulation of abnormal tau species (misfolded, hyperphosphorylated, and ubiquitinated) parallels evidence of UPS dysfunction at the synapse. A better understanding of the subcellular localization of abnormal tau species and their properties may provide improved understanding of the molecular mechanisms of synaptic dysfunction and loss that are believed to underlie cognitive impairments in AD.
Acknowledgments
We thank Marian DiFiglia and the Philly Dake Electron Microscopy Center for access to the transmission electron microscope, Karlotta Fitch for assistance with brain tissues, and Peter Davies for anti-tau antibodies PHF1, CP13, and DA9.
Footnotes
Supported by an Alzheimer Research Fellowship from the American Health Assistance Foundation (H.C.T.); Fundacion Alfonso Martin Escudero (Madrid, Spain) (A.S.P.); and grants AG033670 (T.S.J.), and AG08487 and P50 AG005134 from NIH (B.T.H.).
Current address of H.C.T., Department of Chemistry, National Taiwan University, Taipei, Taiwan; and of T.H., Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan.
References
- 1.Serrano-Pozo A., Frosch M.P., Masliah E., Hyman B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braak E., Griffing K., Arai K., Bohl J., Bratzke H., Braak H. Neuropathology of Alzheimer's disease: what is new since A. Alzheimer? Eur Arch Psychiatry Clin Neurosci. 1999;249(Suppl 3):14–22. doi: 10.1007/pl00014168. [DOI] [PubMed] [Google Scholar]
- 3.Terry R.D., Masliah E., Salmon D.P., Butters N., DeTeresa R., Hill R., Hansen L.A., Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 4.DeKosky S.T., Scheff S.W. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 5.Arriagada P.V., Marzloff K., Hyman B.T. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer's disease. Neurology. 1992;42:1681–1688. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- 6.Giannakopoulos P., Herrmann F.R., Bussiere T., Bouras C., Kovari E., Perl D.P., Morrison J.H., Gold G., Hof P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 7.Ingelsson M., Fukumoto H., Newell K.L., Growdon J.H., Hedley-Whyte E.T., Frosch M.P., Albert M.S., Hyman B.T., Irizarry M.C. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 8.Morris M., Maeda S., Vossel K., Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spires-Jones T.L., Stoothoff W.H., de Calignon A., Jones P.B., Hyman B.T. Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci. 2009;32:150–159. doi: 10.1016/j.tins.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Dotti C.G., Banker G.A., Binder L.I. The expression and distribution of the microtubule-associated proteins tau and microtubule-associated protein 2 in hippocampal neurons in the rat in situ and in cell culture. Neuroscience. 1987;23:121–130. doi: 10.1016/0306-4522(87)90276-4. [DOI] [PubMed] [Google Scholar]
- 11.Avila J., Lucas J.J., Perez M., Hernandez F. Role of tau protein in both physiological and pathological conditions. Physiol Rev. 2004;84:361–384. doi: 10.1152/physrev.00024.2003. [DOI] [PubMed] [Google Scholar]
- 12.Brandt R., Hundelt M., Shahani N. Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim Biophys Acta. 2005;1739:331–354. doi: 10.1016/j.bbadis.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 13.Ittner L.M., Ke Y.D., Delerue F., Bi M., Gladbach A., van Eersel J., Wolfing H., Chieng B.C., Christie M.J., Napier I.A., Eckert A., Staufenbiel M., Hardeman E., Gotz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 14.Cripps D., Thomas S.N., Jeng Y., Yang F., Davies P., Yang A.J. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem. 2006;281:10825–10838. doi: 10.1074/jbc.M512786200. [DOI] [PubMed] [Google Scholar]
- 15.Morishima-Kawashima M., Hasegawa M., Takio K., Suzuki M., Titani K., Ihara Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron. 1993;10:1151–1160. doi: 10.1016/0896-6273(93)90063-w. [DOI] [PubMed] [Google Scholar]
- 16.Bingol B., Schuman E.M. Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature. 2006;441:1144–1148. doi: 10.1038/nature04769. [DOI] [PubMed] [Google Scholar]
- 17.Yi J.J., Ehlers M.D. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev. 2007;59:14–39. doi: 10.1124/pr.59.1.4. [DOI] [PubMed] [Google Scholar]
- 18.Keck S., Nitsch R., Grune T., Ullrich O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer's disease. J Neurochem. 2003;85:115–122. doi: 10.1046/j.1471-4159.2003.01642.x. [DOI] [PubMed] [Google Scholar]
- 19.Hollingsworth E.B., McNeal E.T., Burton J.L., Williams R.J., Daly J.W., Creveling C.R. Biochemical characterization of a filtered synaptoneurosome preparation from guinea pig cerebral cortex: cyclic adenosine 3′:5′-monophosphate-generating systems, receptors, and enzymes. J Neurosci. 1985;5:2240–2253. doi: 10.1523/JNEUROSCI.05-08-02240.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lathia D., Wesemann W. Serotonin uptake and release by biochemically characterized nerve endings isolated from rat brain by concomitant flotation and sedimentation centrifugation. J Neural Transm. 1975;37:111–126. doi: 10.1007/BF01663628. [DOI] [PubMed] [Google Scholar]
- 21.Duff K., Knight H., Refolo L.M., Sanders S., Yu X., Picciano M., Malester B., Hutton M., Adamson J., Goedert M., Burki K., Davies P. Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol Dis. 2000;7:87–98. doi: 10.1006/nbdi.1999.0279. [DOI] [PubMed] [Google Scholar]
- 22.Greenberg S.G., Davies P., Schein J.D., Binder L.I. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem. 1992;267:564–569. [PubMed] [Google Scholar]
- 23.Watanabe A., Takio K., Ihara Y. Deamidation and isoaspartate formation in smeared tau in paired helical filaments: Unusual properties of the microtubule-binding domain of tau. J Biol Chem. 1999;274:7368–7378. doi: 10.1074/jbc.274.11.7368. [DOI] [PubMed] [Google Scholar]
- 24.Roberson E.D., Scearce-Levie K., Palop J.J., Yan F., Cheng I.H., Wu T., Gerstein H., Yu G.Q., Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 25.Morales M., Fifkova E. Distribution of MAP2 in dendritic spines and its colocalization with actin: an immunogold electron-microscope study. Cell Tissue Res. 1989;256:447–456. doi: 10.1007/BF00225592. [DOI] [PubMed] [Google Scholar]
- 26.Caceres A., Binder L.I., Payne M.R., Bender P., Rebhun L., Steward O. Differential subcellular localization of tubulin and the microtubule-associated protein MAP2 in brain tissue as revealed by immunocytochemistry with monoclonal hybridoma antibodies. J Neurosci. 1984;4:394–410. doi: 10.1523/JNEUROSCI.04-02-00394.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gordon-Weeks P.R., Burgoyne R.D., Gray E.G. Presynaptic microtubules: organisation and assembly/disassembly. Neuroscience. 1982;7:739–749. doi: 10.1016/0306-4522(82)90079-3. [DOI] [PubMed] [Google Scholar]
- 28.Tai H.C., Schuman E.M. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci. 2008;9:826–838. doi: 10.1038/nrn2499. [DOI] [PubMed] [Google Scholar]
- 29.van Langenhove T., van der Zee J., van Broeckhoven C. The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Ann Med. 2012 doi: 10.3109/07853890.2012.665471. http://dx.doi.org/10.3109/07853890.2012.665471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koffee R.M., Hashimoto T., Tai H.C., Kay K.R., Serrano-Pozo A., Joyner D., Hou S., Kopeikina K., Frosch M.P., Lee V.M., Holtzman D.M., Hyman B.T., Spires-Jones T.L. Apolipoprotein E4 effects in Alzheimer disease are mediated by synaptotoxic oligomeric amyloid-beta. Brain. 2012;135:2155–2168. doi: 10.1093/brain/aws127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benilova I., Karran E., De Strooper B. The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 32.Shankar G.M., Li S., Mehta T.H., Garcia-Munoz A., Shepardson N.E., Smith I., Brett F.M., Farrell M.A., Rowan M.J., Lemere C.A., Regan C.M., Walsh D.M., Sabatini B.L., Selkoe D.J. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lasagna-Reeves C.A., Castillo-Carranza D.L., Sengupta U., Sarmiento J., Troncoso J., Jackson G.R., Kayed R. Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. FASEB J. 2012;26:1946–1959. doi: 10.1096/fj.11-199851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blazquez-Llorca L., Garcia-Marin V., Merino-Serrais P., Avila J., DeFelipe J. Abnormal tau phosphorylation in the thorny excrescences of CA3 hippocampal neurons in patients with Alzheimer's disease. J Alzheimers Dis. 2011;26:683–698. doi: 10.3233/JAD-2011-110659. [DOI] [PubMed] [Google Scholar]
- 35.Fein J.A., Sokolow S., Miller C.A., Vinters H.V., Yang F., Cole G.M., Gylys K.H. Co-localization of amyloid beta and tau pathology in Alzheimer's disease synaptosomes. Am J Pathol. 2008;172:1683–1692. doi: 10.2353/ajpath.2008.070829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dickey C.A., Yue M., Lin W.L., Dickson D.W., Dunmore J.H., Lee W.C., Zehr C., West G., Cao S., Clark A.M., Caldwell G.A., Caldwell K.A., Eckman C., Patterson C., Hutton M., Petrucelli L. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J Neurosci. 2006;26:6985–6996. doi: 10.1523/JNEUROSCI.0746-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tseng B.P., Green K.N., Chan J.L., Blurton-Jones M., LaFerla F.M. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008;29:1607–1618. doi: 10.1016/j.neurobiolaging.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J.Y., Liu S.J., Li H.L., Wang J.Z. Microtubule-associated protein tau is a substrate of ATP/Mg(2+)-dependent proteasome protease system. J Neural Transm. 2005;112:547–555. doi: 10.1007/s00702-004-0196-x. [DOI] [PubMed] [Google Scholar]
- 39.Bingol B., Sheng M. Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron. 2011;69:22–32. doi: 10.1016/j.neuron.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 40.Congdon E.E., Duff K.E. Is tau aggregation toxic or protective? J Alzheimers Dis. 2008;14:453–457. doi: 10.3233/jad-2008-14415. [DOI] [PubMed] [Google Scholar]
- 41.Pritchard S.M., Dolan P.J., Vitkus A., Johnson G.V. The toxicity of tau in Alzheimer disease: turnover, targets and potential therapeutics. J Cell Mol Med. 2011;15:1621–1635. doi: 10.1111/j.1582-4934.2011.01273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 43.Ross C.A., Poirier M.A. Opinion: what is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Biol. 2005;6:891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- 44.Liu L., Drouet V., Wu J.W., Witter M.P., Small S.A., Clelland C., Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Calignon A., Polydoro M., Suarez-Calvet M., William C., Adamowicz D.H., Kopeikina K.J., Pitstick R., Sahara N., Ashe K.H., Carlson G.A., Spires-Jones T.L., Hyman B.T. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shankar G.M., Bloodgood B.L., Townsend M., Walsh D.M., Selkoe D.J., Sabatini B.L. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saido T., Leissring M.A. Proteolytic degradation of amyloid beta-protein. Cold Spring Harbor Perspect Med. 2012;2:a006379. doi: 10.1101/cshperspect.a006379. [DOI] [PMC free article] [PubMed] [Google Scholar]