

Summary
Background
Upon contact with an appropriate surface, factor XII (FXII) undergoes autoactivation or cleavage by kallikrein. Zn2+ is known to facilitate binding of FXII and the cofactor, high molecular weight kininogen (HK), to anionic surfaces.
Objectives
To investigate whether transition metals immobilized on liposome surfaces can initiate coagulation via the contact pathway.
Methods & Results
Liposomes containing a metal ion-chelating lipid (DOGS-NTA) were prepared by membrane extrusion (20% DOGS-NTA, 40% phosphatidylcholine, 10% phosphatidylserine, and 30% phosphatidylethanolamine). Ni2+ immobilized on such liposomes accelerated clotting in normal, but not FXI- or FXII-deficient plasma. Results were comparable to a commercial aPTT reagent. Charging such liposomes with other transition metals revealed differences in their procoagulant capacity, with Ni2+> Cu2+> Co2+ and Zn2+. Plasma could be depleted of FXI, FXII and HK by adsorption with Ni2+-containing beads, resulting in delayed clot times. Consistent with this, FXI, FXII and HK bound to immobilized Ni2+ or Cu2+ with high affinity as determined by surface plasmon resonance. In the presence of Ni2+-bearing liposomes, Km and kcat values derived for autoactivation of FXII and prekallikrein, as well as for activation of FXII by kallikrein or prekallikrein by FXIIa, were similar to literature values in the presence of dextran sulfate.
Conclusions
Immobilized Ni2+ and Cu2+ bind FXII, FXI and HK with high affinity and stimulate activation of the contact pathway, driving FXII-mediated coagulation. Activation of the contact system by immobilized transition metals may have implications during pathogenic infection or in individuals exposed to high levels of pollution.
Keywords: Contact pathway, Factor XII, Factor XI, Metal ions, Coagulation, Thrombin
Introduction
In this study we investigate the role of immobilized transition metal ions in triggering the contact pathway of blood clotting, with possible pathophysiologic consequences.
Initiation of the contact pathway involves reciprocal proteolytic activation of prekallikrein (PK) and factor XII (FXII) to the active serine proteases, kallikrein and factor XIIa (FXIIa). FXIIa then activates factor XI (FXI) by limited proteolysis. The non-enzymatic cofactor, high molecular weight kininogen (HK), facilitates the initiation of the contact pathway by assembling PK and FXI on the activating surface. The product of these surface-bound reactions, factor XIa (FXIa), propagates the clotting cascade, ultimately leading to thrombin generation and fibrin formation.
Activators of the contact pathway commonly feature an anionic polymer or surface to which FXII and HK bind directly and PK indirectly. In vitro, the contact pathway is triggered when plasma is exposed to artificial surfaces like kaolin or glass [1,2], artificial anionic polymers such as dextran sulfate, or liposomes containing high levels of sulfatide or phosphatidylinositol phosphate [3-7]. Recent studies have identified potential physiological activators of this pathway including RNA [8], collagen [9] and platelet polyphosphate [10,11]. Activation of portions of the contact pathway can occur on cell surfaces as part of the kallikrein/kinin system [12].
Certain transition metal ions—most particularly Zn2+—are reported to participate in activation of the contact pathway. Zn2+ and Cu2+, but not Co2+, accelerates FXII activation induced by dextran sulfate [13], while sulfatide-mediated activation of FXII and prekallikrein in the presence of HK is markedly accelerated by Zn2+ [14]. Zn2+ also enhances binding of HK to platelet surfaces [15] and has been reported to induce a conformational change in both HK and FXII [14,16,17], potentially altering their susceptibility to cleavage. The low molecular weight compound, ellagic acid, was originally thought to be the exception to the rule that potent contact activators are anionic polymers or surfaces. However, it was subsequently discovered that only insoluble complexes of ellagic acid are capable of triggering the contact pathway [18]. Furthermore, these insoluble complexes must include transition metal ions in order to exhibit procoagulant activity, with Cu2+ being more effective than Zn2+, Co2+ or Fe3+ (Ni2+ was not tested) [18].
In this study we report that transition metals in addition to Zn2+ (most particularly, Ni2+ and Cu2+) interact strongly with FXI, FXII and HK. Furthermore, immobilizing Ni2+, Cu2+, Co2+ or Zn2+ on the surface of phospholipid bilayers via metal-chelating lipids potently stimulates FXII-mediated coagulation.
Materials & Methods
Reagents
Reagents were from the following suppliers: citrated pooled normal human plasma (PNP) and factor-deficient plasmas, George King Bio-Medical (Overland Park, KS); HK-deficient plasma, Affinity Biologicals (Ancaster, Ontario, Canada); egg phosphatidylcholine (PC), porcine brain phosphatidylserine (PS), bovine liver phosphatidylethanolamine (PE), 1,2-dioleoyl-sn-glycero-3-[(N(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] ammonium salt (DOGS-NTA), and the same lipid as the nickel salt (DOGS-NTA-Ni), Avanti Polar Lipids (Alabaster, AL); D-Pro-Phe-Arg-p-nitroanilide, Bachem (Torrance, CA); STA-PTT-Automate 5, Diagnostica Stago (Parsippany, NJ); human FXII, α-FXIIa, PK, kallikrein and HK, Enzyme Research Laboratories (South Bend, IN); FXI and corn trypsin inhibitor, Haematologic Technologies (Essex Junction, VT); soybean trypsin inhibitor and polybrene (hexadimethrine bromide), Sigma-Aldrich; dextran sulfate (average MW, 500,000), Fisher; bovine serum albumin (BSA), Calbiochem; Ni SepharoseTM 6 Fast Flow, Amersham Biosciences; and NTA (nitrilotriacetic acid) sensor chips, BIAcore (GE Healthcare, Piscataway, NJ).
Preparation of Liposomes
Liposomes were prepared via membrane extrusion as described [19] but without BSA. Some liposomes were prepared using Bio-Beads [20]; both methods gave comparable results. Unless otherwise stated, “Ni-NTA-liposomes” contained 20% DOGS-NTA-Ni, 40% PC, 10% PS, and 30% PE; and “NTA-liposomes” had the same lipid composition except that DOGS-NTA (no Ni2+) replaced DOGS-NTA-Ni. Lipid compositions are reported as mol% and liposome concentrations as molar concentration of total lipid.
Clotting Assays
Activated partial thromboplastin time (aPTT) clotting assays were performed in an ST4 coagulometer (Diagnostica Stago). Plasma (50 μl) was incubated at 37°C with 50 μl liposomes (in 20 mM HEPES-NaOH pH 7.5, 0.1% BSA, 0.1% NaN3) for various times, followed by 25 mM CaCl2 (50 μl). Some assays used a commercial aPTT reagent (STA-PTT-Automate 5) according to the manufacturer’s instructions. Prothrombin time (PT) clotting assays were performed by incubating plasma (50 μl) with 0.1 ng/ml relipidated tissue factor (50 μl) [20] for 2 min at 37°C before adding 25 mM CaCl2 (50 μl).
Depletion of Contact Factors from Plasma
Ni SepharoseTM 6 Fast Flow beads were washed thrice in H2O and resuspended in 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 0.02 % NaN3. Beads were collected in a Zebra™ spin column (Pierce) and resuspended in 730 μl pooled normal plasma before incubating on a rotator for 30 min at ambient temperature. Plasma was collected by centrifugation.
Chromogenic Activity Assays
FXII activation by kallikrein
Initial rates of FXII activation were quantified at 37°C by incubating FXII (200 nM), kallikrein (200 pM), ± HK (600 nM) and either dextran sulfate (25 μg/ml) or liposomes (100 μM) in 20 mM HEPES-NaOH pH 7.5, 150 mM NaCl, 0.01% NaN3, 0.1% BSA (HBSA). At various times, 10 μl aliquots were removed to wells of a ultra-low binding polystyrene plate (Corning) containing 90 μl 1.1% Triton X-100, 5.5 μg/ml polybrene, 55.5 μg/ml soybean trypsin inhibitor, 50 mM HEPES-NaOH pH 7.5, 50 mM NaCl, 0.1% BSA. 1 mM D-Pro-Phe-Arg-p-nitroanilide (100 μl) was added, and change in A405 was monitored in a multiwell spectrophotometer.
FXII autoactivation
FXII autoactivation was analyzed as described [6] with the following changes. Varying concentrations of FXII (0.25 to 2 μM) were incubated with 100 μM liposomes in HBSA. At various times, 10 μl aliquots were mixed with 90 μl 1.1 % Triton X-100, 50 mM HEPES-NaOH pH 7.5, 50 mM NaCl, 0.1% BSA. Chromogenic substrate hydrolysis was quantified as for FXII activation. Data were analyzed as described [6] to determine Km and kcat.
PK activation by FXIIa
Initial rates of PK activation were quantified at 37°C by incubating PK (50 nM), FXIIa (20 pM), ± HK (600 nM) and either dextran sulfate (25 μg/ml) or liposomes (100 μM) in HBSA. At various times, 10 μl aliquots were mixed with 90 μl 1.1% Triton X-100, 5.5 μg/ml polybrene, 14 μg/ml corn trypsin inhibitor (CTI), 50 mM HEPES-NaOH pH 7.5, 50 mM NaCl, 0.1% BSA. Chromogenic substrate hydrolysis was quantified as for FXII activation.
PK autoactivation
Experiments were performed as described for PK activation by FXIIa, but without FXIIa. Data were analyzed as described [7] to derive apparent second-order rate constants.
Surface Plasmon Resonance
Binding studies were conducted in a BIAcore 3000. NTA sensor chips were treated with 300 mM EDTA pH 8 for 1 min at 20 μl/min to remove bound metal ions, after which the surface was charged with NiSO4 or CuSO4 at 25 μM for 1 min at 20 μl/min, until a response of approximately 50 resonance units (RU) was achieved. An uncharged NTA channel was the control surface. Various concentrations of FXI, FXII, PK or HK in 10 mM HEPES-NaOH pH 7.4, 150 mM NaCl, 0.005% P-20 were flowed over chips at 10 μl/min until a steady-state was reached. A positive control, soluble tissue factor with a hexahistidine tag [21] was included. Maximal (steady-state) RU levels were plotted versus protein concentration, to which the single-site ligand binding equation was fitted. NTA chips were regenerated with 300 mM EDTA (20 μl/min for 1 min).
Results
Liposomes containing Ni2+ are strongly procoagulant & stimulate contact activation
We prepared liposomes containing Ni2+ bound to the nickel-chelating lipid, NTA-DOGS, along with PC, PS and PE (termed Ni-NTA-liposomes), and also liposomes with the same composition but without bound Ni2+ (NTA-liposomes). Recalcifying citrated PNP in the presence of NTA-liposomes resulted in clot times of >300 s, but these clot times were dramatically shortened with Ni-NTA-liposomes (30 s, P<0.001). We obtained the shortest clotting times when liposomes contained 20% DOGS-NTA-Ni, 40% PC, 10% PS, and 30% PE (although PE was not an absolute requirement; data not shown). These data suggest that Ni2+ bound to NTA-liposomes triggers clotting in plasma, potentially via the contact pathway. The initial stages of contact activation are relatively slow and calcium-independent, so aPTT assays are typically conducted by preincubating plasma with a contact activator for 2 to 10 min before adding CaCl2. Preincubating Ni-NTA-liposomes with citrated normal plasma prior to addition of CaCl2 dramatically shortened the clotting time, with a maximal effect at 4-5 min (Fig. 1). In contrast, no change in clot time was observed in FXI- or FXII-deficient plasma (Fig. 1), consistent with Ni-NTA-liposomes acting via the contact pathway.
FIGURE 1. Ni-NTA-liposomes are strongly procoagulant.
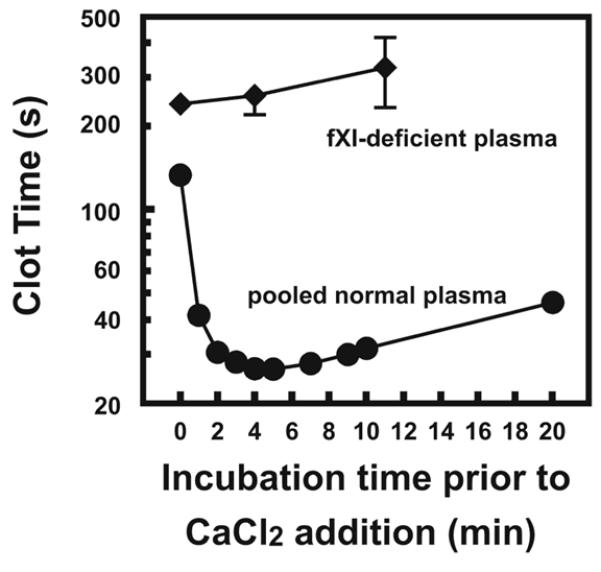
100 μM Ni-NTA-liposomes were incubated with PNP (circles) or FXI-deficient plasma (diamonds) for various times at 37°C before initiation of clotting with 25 mM CaCl2. Clot times of > 500 s were obtained with FXII-deficient plasma and are not shown. Data are mean ± SEM (n = 3).
Clot times with Ni-NTA-liposomes were compared to a commercial aPTT reagent using a panel of factor-deficient plasmas (Fig. 2). Ni-NTA-liposomes behaved similarly to the commercial aPTT reagent, with prolonged clotting times evident in plasma deficient in the intrinsic pathway (FVIII, FIX, FXI, FXII, PK and HK), and the common pathway (FV, FX). Clot times were not prolonged with FVII-deficient plasma, as FVII functions exclusively via the tissue factor pathway.
FIGURE 2. Clotting initiated by Ni-NTA-liposomes proceeds via the contact pathway.
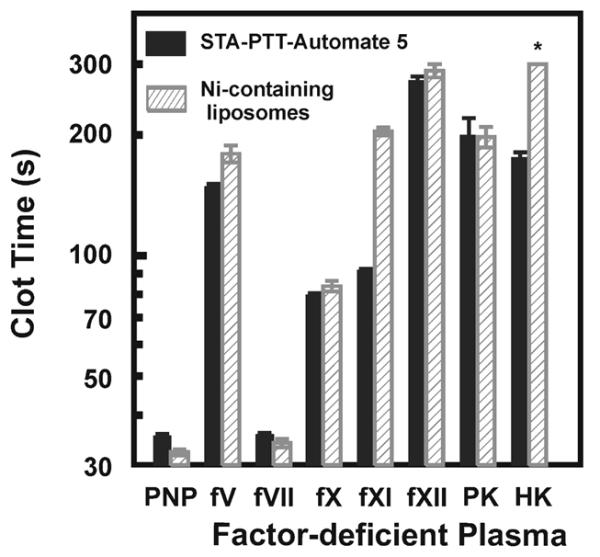
Ni-NTA-liposomes (50 μM; hatched bars) or the commercial aPTT reagent, STA-PTT-Automate 5, (solid bars) were incubated with PNP or plasma deficient in the indicated clotting factors at 37°C for 180 s, after which clotting was triggered by addition of 25 mM CaCl2. Clot times > 300 s are indicated with an asterisk. Data are mean ± SEM (n = 3).
Immobilized Cu2+, Co2+ and Zn2+ also promote contact activation
NTA-liposomes supported a plasma clot time of 423 s, similar to that with liposomes lacking NTA-DOGS altogether (> 300 s), indicating that the NTA moiety itself does not initiate clotting via the contact pathway. NTA-liposomes were then loaded with different metal ions, including Cu2+, Co2+, Zn2+, Fe2+, Cd2+, Cr2+, Ag+, or Mn2+, to examine whether they could trigger contact activation. Ni2+-containing liposomes were the most procoagulant; however, Cu2+, Co2+ and Zn2+ also significantly shortened clot times of normal plasma (Fig. 3). The other metal salts analyzed (FeSO4, CdCl2, CrCl2, AgNO3 or MnCl2) yielded clot times in excess of 300 s (data not shown), indicating little or procoagulant activity.
FIGURE 3. Ni2+, Cu2+, Co2+ and Zn2+ are procoagulant when bound to NTA-liposomes.
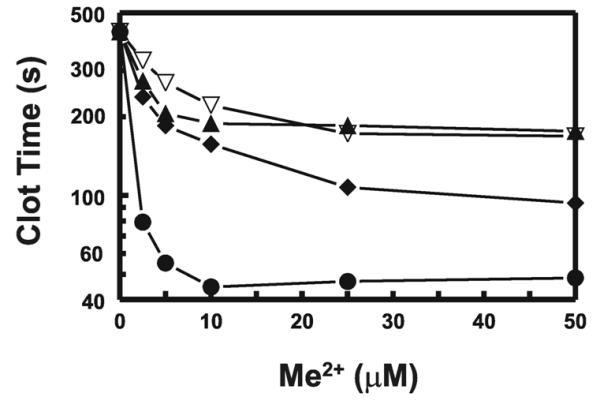
The indicated concentrations of NiSO4 (closed circles), CuSO4 (closed diamonds), CoCl2 (closed triangles), or ZnCl2 (open, inverted triangles) were incubated with 100 μM NTA-liposomes. aPTT clot times with PNP were then determined as described in Fig. 2. The final DOGS-NTA concentration in these preparations is 15 μM. Assuming half the lipids are exposed to the outer leaflet of the bilayers, then roughly 7.5 μM DOGS-NTA would be available to chelate transition metal ions. Data are mean ± SEM (n = 3).
Binding of contact pathway proteins to immobilized transition metals
We hypothesized that Ni-NTA-liposomes must bind to one or more proteins in the contact pathway to promote reciprocal activation of FXII and PK. We investigated the ability of immobilized Ni2+ to bind clotting factors by incubating PNP with Ni-Sepharose beads, removing the beads by centrifugation, and analyzing the plasma in PT and aPTT clotting assays. PT clot times were similar in untreated and treated PNP, indicating that the factors involved in the extrinsic and final common pathways of coagulation were not altered by incubation with Ni-Sepharose. In contrast, aPTT clot times were significantly prolonged in Ni-Sepharose-adsorbed PNP (Fig. 4A), indicating that one or more of the contact proteins was depleted by exposure to immobilized Ni2+. When Ni-Sepharose-adsorbed PNP was mixed 1:1 with factor-deficient plasmas and analyzed in aPTT assays, significantly prolonged clot times were observed using FXI-, FXII-, or HK-deficient plasmas, but nearly normal clot times were obtained using FVIII-, FIX-, FX- or PK-deficient plasmas (Fig. 4B). Thus, Ni-Sepharose depleted the plasma of FXI, FXII, and HK, suggesting that these three proteins directly interact with immobilized Ni2+.
FIGURE 4. Ni-Sepharose beads deplete plasma of specific contact pathway proteins.
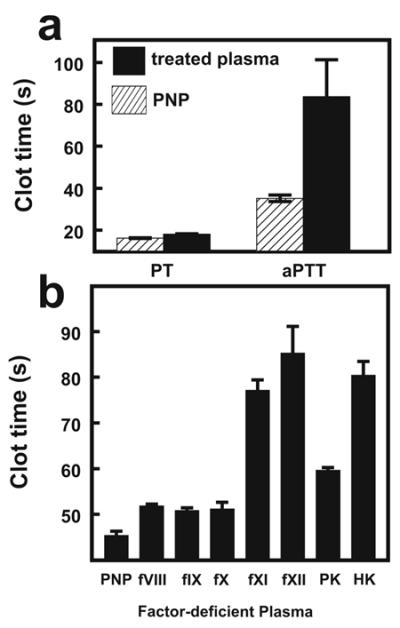
PNP pre-adsorbed with Ni-Sepharose beads was used in PT and aPTT assays (the latter with the commercial aPTT reagent, STA-PTT-Automate 5). (A) Comparison of clotting times in Ni-Sepharose-treated plasma (solid bars) and untreated plasma (PNP; hatched bars). (B) aPTT assays were performed with 1:1 mixtures of Ni-Sepharose-treated plasma and either PNP or plasma deficient in the indicated clotting factor. Data are mean ± SEM (n = 3).
Direct binding of contact pathway proteins to immobilized transition metal ions was investigated using surface plasmon resonance. NTA sensor chips were preloaded with Ni2+ or Cu2+ and purified FXI, FXII, PK or HK was flowed over the bound metal ions. Binding data were analyzed by plotting the maximal (steady-state) RU signal versus protein concentration, to which the single site ligand binding equation was fitted to derive Kd values [22]. Control experiments were performed in which proteins were flowed over an NTA surface in the absence of Ni2+ or Cu2+, and the values in Table 1 represent the difference in this binding. FXI, FXII and HK bound to Ni2+ and Cu2+ with high affinity (Table 1). PK was unable to bind to Cu2+ and showed a reduced capacity to bind to Ni2+, with a Kd value orders of magnitude higher than the other contact proteins. A positive control of soluble tissue factor with hexahistidine tag [21] also bound to immobilized Ni2+, but intriguingly the affinity was approximately 10-fold weaker than the Kd values observed for binding of FXI, FXII or HK. These results indicate that immobilized Ni2+ and Cu2+ stimulate coagulation by directly interacting with several of the proteins important in the initiation and propagation of the contact pathway.
Table 1.
Binding of contact factors to immobilized Ni2+ and Cu2+
| Ni2+ | Cu2+ | |
|---|---|---|
|
|
||
| Kd, nM | Kd, nM | |
| FXI | 8.03 ± 0.43 | 1.22 ± 0.13 |
| FXII | 38.75 ± 4.36 | 4.92 ± 0.92 |
| PK | 1091.5 ± 344.7 | NBa |
| HK | 29.82 ± 3.99 | 5.73 ± 1.28 |
| sTF | 198.23 ± 58.4 | NDb |
Ni2+ or Cu2+ was immobilized on NTA sensor chips and binding affinities were determined using surface plasmon resonance. Data are mean ± SEM (n = 3).
NB, negligible binding.
ND, not determined.
Immobilized Ni2+ supports activation and autoactivation of FXII and PK
The ability of Ni-NTA-liposomes to promote activation of FXII and PK was quantified. Autoactivation of FXII in the presence of Ni-NTA-liposomes was comparable to reported literature values in the presence of dextran sulfate [6] (Table 2). NTA-liposomes were unable to support FXII autoactivation, with rates similar to the no-surface control (Table 2). Activation of FXII by kallikrein in the absence or presence of HK was also investigated with Ni-NTA-liposomes or dextran sulfate as the activating surface. In the presence of HK, FXII activation by kallikrein was strongly promoted by Ni-NTA-liposomes, but not by NTA-liposomes (Table 2). In the absence of HK, NTA-liposomes alone weakly stimulated FXII activation by kallikrein, but this was dramatically accelerated upon incorporation of Ni+ (Table 2).
Table 2.
Kinetics of FXII activation
| FXII autoactivation (−HK) |
FXII activation by kallikrein | ||||
|---|---|---|---|---|---|
| Activating surface | Km | k cat | kcat/Km | initial rate (+HK) | initial rate (−HK) |
| μM | min−1 | × 103 M−1 s−1 | nM·min−1nM−1 | nM·min−1nM−1 | |
| dextran sulfate | 7.5a | 2.0a | 4.0a | 114 ± 14 | 257 ± 20 |
| Ni-NTA-liposomes | 2.6 ± 0.3 | 1.4 ± 0.2 | 8.7 ± 1.1 | 48 ± 5 | 471 ± 20 |
| NTA-liposomes | NAb | NA | NA | NA | 119 ± 20 |
| No surface | NA | NA | NA | NA | NA |
Data are mean ± SEM (n = 3).
Literature values from Tankersley and Finlayson.
NA, negligible activity.
Ni-NTA-liposomes also strongly supported PK autoactivation in the presence of HK, with apparent rate constants comparable to those of dextran sulfate (Table 3). No autoactivation of PK was observed with Ni-NTA-liposomes or dextran sulfate in the absence of HK, consistent with its role in facilitating binding of PK to a surface (Table 3). NTA-liposomes alone also stimulated autoactivation of PK, but at a 10-fold lower rate. This is potentially explained by the anionic nature of the NTA-DOGS lipid in the absence of bound transition metal ions. Ni-NTA-liposomes and dextran sulfate showed comparable ability to stimulate activation of PK by FXIIa both in the absence and presence of HK (Table 3). However, significant kallikrein activity was detected with NTA-liposomes alone and in the no-surface control.
Table 3.
Kinetics of PK activation
| PK autoactivation | PK activation by FXIIa | |||
|---|---|---|---|---|
|
|
||||
| Activating surface | k2,app (+HK) | k2,app (−HK) | rate (+HK) | rate (−HK) |
| × 105 M−1s−1 | × 105 M−1s−1 | nM·min−1nM−1 | nM·min−1nM−1 | |
| dextran sulfate | 2.9 ± 0.25 | NAa | 445 ± 87 | 374 ± 58 |
| Ni-NTA-liposomes | 2.8 ± 0.04 | NA | 545 ± 43 | 366 ± 67 |
| NTA-liposomes | 1.0 ± 0.10 | NA | 310 ± 20 | 348 ± 45 |
| No surface | NA | NA | 29 ± 9 | 126 ± 65 |
Data are mean ± SEM (n = 3).
NA, negligible activity.
Discussion
Here we show that immobilization of transition metals (particularly Ni2+ and Cu2+) on metal-chelating lipids can initiate activation of the contact pathway. Metal-chelating lipids such as DOGS-NTA-Ni have primarily been used to create two-dimensional crystals of oligohistidine-tagged recombinant proteins for electron crystallography [23]. We previously showed that attaching a C-terminal oligohistidine tag to the isolated ectodomain of tissue factor (normally an integral membrane protein) restores full biological activity to this recombinant fragment when it is bound to Ni-NTA-liposomes [21]. During the course of those studies we discovered that even in the absence of tissue factor, liposomes containing DOGS-NTA-Ni elicit substantial procoagulant activity. We now show that Ni-NTA-liposomes are novel and potent activators of the contact pathway, enhancing the four initial reactions: autoactivation of FXII and PK, and reciprocal activation of FXII and PK by kallikrein and FXIIa, respectively. Activation of this pathway drives coagulation via cleavage of FXI, ultimately resulting in thrombin generation and fibrin formation. It could be especially advantageous to incorporate a potent activator of the contact pathway onto the same liposome surface that also supports the downstream (membrane-dependent) clotting reactions.
The degree of PK autoactivation by Ni-NTA-liposomes was comparable to that with dextran sulfate in the presence of its cofactor, HK. No autoactivation of PK was observed in the absence of HK, consistent with the role of this cofactor in anchoring PK to a surface. Likewise, the Kd derived for the interaction of PK with immobilized Ni2+ was several orders of magnitude higher than that observed for FXI and FXII and no binding of PK was observed to immobilized Cu2+. In contrast to the results obtained in the FXII activation studies, the NTA moiety itself was capable of stimulating PK autoactivation and FXIIa-mediated PK activation, with immobilized Ni2+ only slightly increasing these rates. This suggests that the NTA group, which is anionic in nature, is sufficiently charged to support binding of PK via HK, thereby resulting in autocatalysis or cleavage by FXIIa. Alternatively, zinc ions that are already complexed to HK may interact with the NTA moiety on the lipid surface to support binding and activation of PK. Despite these observations with PK in the activity assays, the NTA group alone was not sufficient to induce clotting in plasma. The enhanced rate of PK activation observed with Ni-NTA-liposomes in the presence of HK most likely reflects binding of this cofactor, as plasma depletion studies and BIAcore experiments indicate that HK binds strongly to immobilized Ni2+.
Bacterial infection is often accompanied by activation of the contact pathway in the host organism. Bacterial strains from E. coli and S. typhimurium display fibrous proteins, commonly referred to as curli, on their surfaces [24-26]. These strains bind FXII, PK and HK, and support activation of FXII and HK under certain conditions [25]. Addition of these curlieated strains to plasma, in vitro, stimulates release of bradykinin [25]. It is feasible that metal ions on the surface of pathogenic organisms could be involved in activation of the contact pathway. Indeed, there are many examples of pathogens using immobilized metal ions in their virulent attacks. Many strains of pathogenic bacteria utilize Cu, Zn superoxide dismutase to protect themselves from oxidative damage by the host [27,28]. These proteins are expressed either in the periplasmic space or the outer membrane of these pathogenic organisms [29,30] and have unique N-terminal extensions that are rich in histidine residues and bind divalent metal ions, including Ni2+, with high affinity. Efficient colonization of the gram-negative pathogenic bacterium Helicobacter pylori requires urease and hydrogenase enzymes, both of which utilize Ni2+ as a cofactor [31]. Canatoxin, a highly toxic variant form of urease from jack bean, specifically binds both Ni2+ and Zn2+ [32]. Studies indicate that this toxin possesses activities distinct from the enzyme’s capacity to hydrolyze urea, including the ability to induce platelet activation and aggregation [33,34]. The question of whether Ni2+ bound on these proteins is accessible on their surface and capable of activating the contact pathway remains to be determined.
Exposure to particulate matter from air pollution is linked to arterial and venous thrombosis and is a risk factor for cardiovascular disease and stroke [35,36]. A recent report demonstrated that particulate matter can support activation of FXII in vitro and that traffic-related ultra-fine particles induce contact pathway-mediated thrombin generation in vivo [37]. The mechanisms underlying these effects require definition, but it is interesting to speculate that the immobilization of transition metals on particulate matter, in a manner similar to that defined here for lipids, may stimulate FXIIa-mediated coagulation. Indeed, transition metals such as Ni2+ can be found at high concentrations in particulate matter [38-40], and its presence has been found to correlate positively with cardiovascular and respiratory mortality [41,42]. Water-soluble metals such as Zn2+ and Ni2+ are capable of translocating to extrapulmonary organs, thereby providing a basis for them to elicit biological effects throughout the systemic circulation [43].
We have shown that immobilization of metal ions on liposomes induces potent activation of the contact pathway and drives FXII-mediated coagulation. These results indicate that when blood contacts immobilized transition metals such as Ni2+ and Cu2+, perhaps during pathogenic infection or when individuals are exposed to a high degree of pollution, a procoagulant response may occur as a result of activation of the contact pathway. Several studies have now documented a positive association between Ni2+ content of particulate matter and onset of thromboembolic disease (reviewed by [42]). The addition of Ni2+ to unleaded fuel and its presence as a catalyst in catalytic converters means it is abundant in areas of increased traffic pollution. This highlights the need to study the effects of immobilization of this metal on particulate matter in relation to activation of the contact system. Further work is necessary to define the exact nature of this interaction in terms of the concentrations of Ni2+ that are achieved in the circulation and the threshold at which it can induce contact activation and drive thrombin formation.
Acknowledgements
This work was supported by Grant R01 HL47014 from the National Heart, Lung and Blood Institute (NHLBI) and Grant 06-2328 from the Roy J. Carver Charitable Trust. E.K.W. was supported in part by Training Grant T32 GM007283 from the National Institutes of Health (NIH). N.J.M. was supported by the British Heart Foundation (PG/07/122 and FS/11/02/28579).
Footnotes
Disclosure of Interests The authors state they have no conflict of interest.
References
- 1.Revak SD, Cochrane CG, Griffin JH. The binding and cleavage characteristics of human Hageman factor during contact activation. A comparison of normal plasma with plasmas deficient in factor XI, prekallikrein, or high molecular weight kininogen. J Clin Invest. 1977;59:1167–75. doi: 10.1172/JCI108741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griffin JH. Role of surface in surface-dependent activation of Hageman factor (blood coagulation factor XII) Proc Natl Acad Sci U S A. 1978;75:1998–2002. doi: 10.1073/pnas.75.4.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tans G, Rosing J, Griffin JH. Sulfatide-dependent autoactivation of human blood coagulation Factor XII (Hageman Factor) J Biol Chem. 1983;258:8215–22. [PubMed] [Google Scholar]
- 4.Rojkjaer R, Schousboe I. The surface-dependent autoactivation mechanism of factor XII. Eur J Biochem. 1997;243:160–6. doi: 10.1111/j.1432-1033.1997.0160a.x. [DOI] [PubMed] [Google Scholar]
- 5.Schousboe I. Inositolphospholipid-accelerated activation of prekallikrein by activated factor XII and its inhibition by beta 2-glycoprotein I. Eur J Biochem. 1988;176:629–36. doi: 10.1111/j.1432-1033.1988.tb14323.x. [DOI] [PubMed] [Google Scholar]
- 6.Tankersley DL, Finlayson JS. Kinetics of activation and autoactivation of human factor XII. Biochemistry. 1984;23:273–9. doi: 10.1021/bi00297a016. [DOI] [PubMed] [Google Scholar]
- 7.Tans G, Rosing J, Berrettini M, Lammle B, Griffin JH. Autoactivation of human plasma prekallikrein. J Biol Chem. 1987;262:11308–14. [PubMed] [Google Scholar]
- 8.Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, Song Y, Tzima E, Kennerknecht E, Niepmann M, von Bruehl ML, Sedding D, Massberg S, Gunther A, Engelmann B, Preissner KT. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci U S A. 2007;104:6388–93. doi: 10.1073/pnas.0608647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Meijden PE, Munnix IC, Auger JM, Govers-Riemslag JW, Cosemans JM, Kuijpers MJ, Spronk HM, Watson SP, Renne T, Heemskerk JW. Dual role of collagen in factor XII-dependent thrombus formation. Blood. 2009;114:881–90. doi: 10.1182/blood-2008-07-171066. [DOI] [PubMed] [Google Scholar]
- 10.Smith SA, Mutch NJ, Baskar D, Rohloff P, Docampo R, Morrissey JH. Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci U S A. 2006;103:903–8. doi: 10.1073/pnas.0507195103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, Schmidbauer S, Gahl WA, Morrissey JH, Renne T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139:1143–56. doi: 10.1016/j.cell.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Motta G, Rojkjaer R, Hasan AA, Cines DB, Schmaier AH. High molecular weight kininogen regulates prekallikrein assembly and activation on endothelial cells: a novel mechanism for contact activation. Blood. 1998;91:516–28. [PubMed] [Google Scholar]
- 13.Shore JD, Day DE, Bock PE, Olson ST. Acceleration of surface-dependent autocatalytic activation of blood coagulation factor XII by divalent metal ions. Biochemistry. 1987;26:2250–8. doi: 10.1021/bi00382a027. [DOI] [PubMed] [Google Scholar]
- 14.Shimada T, Kato H, Iwanaga S. Accelerating effect of zinc ions on the surface-mediated activation of factor XII and prekallikrein. J Biochem. 1987;102:913–21. doi: 10.1093/oxfordjournals.jbchem.a122132. [DOI] [PubMed] [Google Scholar]
- 15.Greengard JS, Griffin JH. Receptors for high molecular weight kininogen on stimulated washed human platelets. Biochemistry. 1984;23:6863–9. doi: 10.1021/bi00321a090. [DOI] [PubMed] [Google Scholar]
- 16.Bernardo MM, Day DE, Halvorson HR, Olson ST, Shore JD. Surface-independent acceleration of factor XII activation by zinc ions. II. Direct binding and fluorescence studies. J Biol Chem. 1993;268:12477–83. [PubMed] [Google Scholar]
- 17.Herwald H, Morgelin M, Svensson HG, Sjobring U. Zinc-dependent conformational changes in domain D5 of high molecular mass kininogen modulate contact activation. Eur J Biochem. 2001;268:396–404. doi: 10.1046/j.1432-1033.2001.01888.x. [DOI] [PubMed] [Google Scholar]
- 18.Bock PE, Srinivasan KR, Shore JD. Activation of intrinsic blood coagulation by ellagic acid: insoluble ellagic acid-metal ion complexes are the activating species. Biochemistry. 1981;20:7258–66. doi: 10.1021/bi00528a032. [DOI] [PubMed] [Google Scholar]
- 19.Neuenschwander PF, Bianco-Fisher E, Rezaie AR, Morrissey JH. Phosphatidylethanolamine augments factor VIIa-tissue factor activity: enhancement of sensitivity to phosphatidylserine. Biochemistry. 1995;34:13988–93. doi: 10.1021/bi00043a004. [DOI] [PubMed] [Google Scholar]
- 20.Smith SA, Morrissey JH. Rapid and efficient incorporation of tissue factor into liposomes. J Thromb Haemost. 2004;2:1155–62. doi: 10.1111/j.1538-7836.2004.00772.x. [DOI] [PubMed] [Google Scholar]
- 21.Waters EK, Morrissey JH. Restoring full biological activity to the isolated ectodomain of an integral membrane protein. Biochemistry. 2006;45:3769–74. doi: 10.1021/bi052600m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mutch NJ, Myles T, Leung LL, Morrissey JH. Polyphosphate binds with high affinity to exosite II of thrombin. J Thromb Haemost. 2010;8:548–55. doi: 10.1111/j.1538-7836.2009.03723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darst SA. A new twist on protein crystallization. Proc Natl Acad Sci U S A. 1998;95:7848–9. doi: 10.1073/pnas.95.14.7848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herwald H, Morgelin M, Bjorck L. Contact activation by pathogenic bacteria: a virulence mechanism contributing to the pathophysiology of sepsis. Scand J Infect Dis. 2003;35:604–7. doi: 10.1080/00365540310016268. [DOI] [PubMed] [Google Scholar]
- 25.Herwald H, Morgelin M, Olsen A, Rhen M, Dahlback B, Muller-Esterl W, Bjorck L. Activation of the contact-phase system on bacterial surfaces--a clue to serious complications in infectious diseases. Nat Med. 1998;4:298–302. doi: 10.1038/nm0398-298. [DOI] [PubMed] [Google Scholar]
- 26.Colman RW. The contact system: a proinflammatory pathway with antithrombotic activity. Nat Med. 1998;4:277–8. doi: 10.1038/nm0398-277. [DOI] [PubMed] [Google Scholar]
- 27.Battistoni A. Role of prokaryotic Cu,Zn superoxide dismutase in pathogenesis. Biochem Soc Trans. 2003;31:1326–9. doi: 10.1042/bst0311326. [DOI] [PubMed] [Google Scholar]
- 28.Krishnakumar R, Craig M, Imlay JA, Slauch JM. Differences in enzymatic properties allow SodCI but not SodCII to contribute to virulence in Salmonella enterica serovar Typhimurium strain 14028. J Bacteriol. 2004;186:5230–8. doi: 10.1128/JB.186.16.5230-5238.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D’Orazio M, Folcarelli S, Mariani F, Colizzi V, Rotilio G, Battistoni A. Lipid modification of the Cu,Zn superoxide dismutase from Mycobacterium tuberculosis. Biochem J. 2001;359:17–22. doi: 10.1042/0264-6021:3590017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinman HM. Bacteriocuprein superoxide dismutase of Photobacterium leiognathi. Isolation and sequence of the gene and evidence for a precursor form. J Biol Chem. 1987;262:1882–7. [PubMed] [Google Scholar]
- 31.Mobley HLT. Urease. In: Mobley H, Mendz G, Hazell S, editors. Helicobacter pylori: Physiology and Genetics. ASM Press; Washington, D.C.: 2001. pp. 179–91. [PubMed] [Google Scholar]
- 32.Follmer C, Barcellos GB, Zingali RB, Machado OL, Alves EW, Barja-Fidalgo C, Guimaraes JA, Carlini CR. Canatoxin, a toxic protein from jack beans (Canavalia ensiformis), is a variant form of urease (EC 3.5.1.5): biological effects of urease independent of its ureolytic activity. Biochem J. 2001;360:217–24. doi: 10.1042/0264-6021:3600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Follmer C, Real-Guerra R, Wasserman GE, Olivera-Severo D, Carlini CR. Jackbean, soybean and Bacillus pasteurii ureases: biological effects unrelated to ureolytic activity. Eur J Biochem. 2004;271:1357–63. doi: 10.1111/j.1432-1033.2004.04046.x. [DOI] [PubMed] [Google Scholar]
- 34.Carlini CR, Guimaraes JA, Ribeiro JM. Platelet release reaction and aggregation induced by canatoxin, a convulsant protein: evidence for the involvement of the platelet lipoxygenase pathway. Br J Pharmacol. 1985;84:551–60. doi: 10.1111/j.1476-5381.1985.tb12940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brook RD, Rajagopalan S, Pope CA, 3rd, Brook JR, Bhatnagar A, Diez-Roux AV, Holguin F, Hong Y, Luepker RV, Mittleman MA, Peters A, Siscovick D, Smith SC, Jr., Whitsel L, Kaufman JD, American Heart Association Council on Epidemiology Prevention. Council on the Kidney in Cardiovascular Disease. Council on Nutrition. Physical Activity and Metabolism Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331–78. doi: 10.1161/CIR.0b013e3181dbece1. [DOI] [PubMed] [Google Scholar]
- 36.Mateen FJ, Brook RD. Air pollution as an emerging global risk factor for stroke. JAMA. 2011;305:1240–1. doi: 10.1001/jama.2011.352. [DOI] [PubMed] [Google Scholar]
- 37.Kilinc E, van Oerle R, Borissoff JI, Oschatz C, Gerlofs-Nijland ME, Janssen NA, Cassee FR, Sandstrom T, Renne T, Ten Cate H, Spronk HM. Factor XII activation is essential to sustain the procoagulant effects of particulate matter. J Thromb Haemost. 2011 doi: 10.1111/j.1538-7836.2011.04280.x. [DOI] [PubMed] [Google Scholar]
- 38.Ripanucci G, Grana M, Vicentini L, Magrini A, Bergamaschi A. Dust in the underground railway tunnels of an Italian town. J Occup Environ Hyg. 2006;3:16–25. doi: 10.1080/15459620500444004. [DOI] [PubMed] [Google Scholar]
- 39.Seaton A, Cherrie J, Dennekamp M, Donaldson K, Hurley JF, Tran CL. The London Underground: dust and hazards to health. Occup Environ Med. 2005;62:355–62. doi: 10.1136/oem.2004.014332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moreno T, Querol X, Alastuey A, de la Rosa J, Sanchez de la Campa AM, Minguillon M, Pandolfi M, Gonzalez-Castanedo Y, Monfort E, Gibbons W. Variations in vanadium, nickel and lanthanoid element concentrations in urban air. Sci Total Environ. 2010;408:4569–79. doi: 10.1016/j.scitotenv.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 41.Cao J, Xu H, Xu Q, Chen B, Kan H. Fine particulate matter constituents and cardiopulmonary mortality in a heavily polluted Chinese city. Environ Health Perspect. 2012;120:373–8. doi: 10.1289/ehp.1103671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Z, Chau PY, Lai HK, Wong CM. A review of effects of particulate matter-associated nickel and vanadium species on cardiovascular and respiratory systems. Int J Environ Health Res. 2009;19:175–85. doi: 10.1080/09603120802460392. [DOI] [PubMed] [Google Scholar]
- 43.Wallenborn JG, McGee JK, Schladweiler MC, Ledbetter AD, Kodavanti UP. Systemic translocation of particulate matter-associated metals following a single intratracheal instillation in rats. Toxicol Sci. 2007;98:231–9. doi: 10.1093/toxsci/kfm088. [DOI] [PubMed] [Google Scholar]