

Abstract
Bovine lactoferricin (LfcinB) is a multi-functional peptide derived from proteolytic cleavage of bovine lactoferrin. LfcinB was found to antagonize the biological effects mediated by angiogenic growth factors such as vascular endothelial growth factor (VEGF) and fibroblast growth factor 2 (FGF-2) in endothelial cells. However, the effect of LfcinB on human articular cartilage remained unknown. Here, our findings demonstrate that LfcinB restored the proteoglycan loss promoted by catabolic factors (interleukin-1 β) IL-1β and FGF-2 in vitro and ex vivo. Mechanistically, LfcinB attenuated the effects of IL-1β and FGF-2 on the expression of cartilage-degrading enzymes (MMP-1, MMP-3, and MMP-13), destructive cytokines (IL-1β and IL-6), and inflammatory mediators (iNOS and TLR2). LfcinB induced protective cytokine expression (IL-4 and IL-10), and downregulated aggrecanase basal expression. LfcinB specifically activated ERK MAPK and Akt signaling pathways, which may account for its anti-inflammatory activity. We also revealed that LfcinB exerted similar protective effects on human synovial fibroblasts challenged by IL-1β, with minimal cytotoxicity. Collectively, our results suggest that LfcinB possesses potent anti-catabolic and anti-inflammatory bioactivities in human articular tissues, and may be utilized for the prevention and/or treatment of OA in the future.
Keywords: Lactoferricin, Cartilage, Synovium, Osteoarthritis
INTRODUCTION
Osteoarthritis (OA) is a debilitating disease characterized by self-perpetuating degradation of articular cartilage, osteophyte formation (Jeffery, 1975), subchondral bone remodeling (Kusakabe, 1977), and frequently at the clinical stage, synovitis and synovial angiogenesis (Haywood et al., 2003). Although its etiology remains poorly understood, numerous studies have identified a series of cellular molecular events which contribute to the onset or progression of OA, including enhanced expression of collagenases (Shlopov et al., 1997) and aggrecanases (Bondeson et al., 2008), and activation of pro-inflammatory cytokine-mediated pathways (Attur et al., 1998). In the osteoarthritic state, human articular chondrocytes fail to maintain the delicate metabolic balance, which leads to progressive extracelluar matrix (ECM) disruption, stressful cellular environment, and eventually, apoptosis of chondrocytes (Sharif et al., 2004). Inflamed synovium is also known to drive OA progression via synovial fibroblast/macrophage-based mechanisms (Abramson and Attur, 2009). Destructive cytokines and growth factors as well as fragments from degenerated cartilage matrix are present in excess in OA synovial fluid. These factors stimulate cell populations in the synovial lining, including synovial fibroblasts, to produce inflammatory mediators and proteolytic enzymes. Such paracrine/autocrine loops facilitate disease advancement, typically in an irreversible manner.
Members of the matrix metalloproteinase (MMP) family (MMP-1, MMP-3, and MMP-13) and members of a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) family (ADAMTS4, ADAMTS5) are recognized as essential participants in cartilage degradation. Both MMP-1 and MMP-13 are able to cleave the triple helix of fibrillar collagen (type I, II and III), thereby rendering the chains more susceptible to further degradation by other MMPs. Particularly, MMP-13 cleaves not only type-II collagen most efficiently among all collagenases (Mitchell et al., 1996), but also aggrecan, an essential component of cartilage proteoglycans (Fosang et al., 1996). MMP-3 (stromelysin) possesses a broad substrate spectrum, including aggrecan, type II collagen, and proMMP-1 (Kato et al., 1998). ADAMTS4 catalyzes the cleavage of core protein of aggrecan, versican and brevican, whereas ADAMTS5 specifically targets aggrecan core protein for degradation (Bondeson et al., 2008). Enhanced activity of ADAMTS4 and ADAMTS5 has been associated with OA (Malfait et al., 2002). Moreover, ablation of ADAMTS4 and ADAMTS5 conferred considerable protection on cartilage against proteoglycan degradation in several animal model-based studies (Glasson et al., 2005; Majumdar et al., 2007).
Inflammation-related cytokines have been implicated into the initiation and progression of OA. Chondrocytes upregulate the expression of cartilage-degrading enzymes and inflammatory mediators upon pro-inflammatory cytokine stimulation, which profoundly disturbs cartilage homeostasis. For example, IL-1β potently drives the production of destructive proteases and inhibits proteoglycan synthesis in articular chondrocytes (Daheshia and Yao, 2008; Pfander et al., 2004). By contrast, several anti-inflammatory cytokines have been proposed to mediate protective effects in articular joint. For instance, IL-10 has been demonstrate to downregulate IL-1β and TNF-α expression, as well as TNF-α-induced PGE2 release in OA synoviocytes (Alaaeddine et al., 1999). Overexpression of IL-10 significantly enhances collagen type II expression, and antagonizes TNF-α-mediated aggrecan repression in human articular chondrocytes(Li et al., 2011). Together with promising in vivo results, IL-10 appears to play a chondroprotective role in joints (De Rienzo et al., 2009).
Bovine lactoferricin (LfcinB) is a 25-residue multi-functional peptide derived from acidic hydrolysis of bovine lactoferrin (bLf), an ~80 kDa iron-binding glycoprotein. LfcinB has bactericidal, antifungal, antiparasitic, antitumor, antiviral and immunomodulatory activities (Gifford et al., 2005). LfcinB interacts with heparan sulfate, thus to prevent viral entry (Andersen et al., 2004). Furthermore, LfcinB inhibits FGF-2/vascular endothelial growth factor (VEGF165)-stimulated angiogenesis via competitive binding to heparin-like structures on endothelial plasma membrane (Mader et al., 2006). The multifunctional nature of LfcinB has rendered it an intriguing target in drug development and mechanistic research. Recently, we reported that LfcinB mediates anabolic and anti-catabolic effects in bovine intervertebral discs (Summers and Anderson, 1972). This finding incurred the question whether LfcinB also confers protection on human synovial joint.
In the present study, we aimed to characterize the bioactivities of LfcinB in human articular cartilage and synovium. Using primary chondrocytes, cartilage explants, and primary synovial fibroblasts, we determined whether LfcinB antagonizes IL-1β or FGF-2 in proteoglycan metabolism and regulation of multiple critical genes, including MMP family members, pro-inflammatory cytokines, anti-inflammatory cytokines, and inflammatory mediators.
MATERIALS AND METHODS
Reagents
Human recombinant FGF-2 was purchased from National Cancer Institute (Bethesda, MD). Human recombinant IL-1β was purchased from PeproTech (Rocky Hill, NJ). LfcinB was purchased from Biosynthesis (Lewisville, TX). Anti-MMP-13 antibody was purchased from Millipore (Billerica, MA). Anti-MMP-1 and anti-MMP-3 antibody were purchased from AbD Serotec (Raleigh, NC). Anti-ADAMTS4, anti-ADAMTS5, and anti-TLR2 antibody were purchased from Thermo Fisher Scientific (Rockford, IL). Anti-GAPDH antibody was purchased from Abcam (Cambridge, MA). Anti-phospho-ERK1/2 (Thr202/Tyr204), anti-phospho-Akt (Ser473), anti-ERK1/2, anti-Akt antibodies were obtained from Cell Signaling Technology (Danvers, MA).
Chondrocyte Isolation and Culture
Human femoral articular cartilage (age ranging from 40 to 75) was obtained through the Gift of Hope Organ and Tissue Donor Network (Elmhurst, IL) with prior approval by the local ethics committee and consent from families. Each specimen was graded for gross degenerative changes based on a modified 5-point scale of Collins (Muehleman et al., 1997). Osteoarthritic femoral cartilage was obtained from patients (age ranging from 40 to 70) through the Orthopedic Tissue and Implant Repository Study (Chicago, IL) with consent from the patients. Human tissues were handled according to the guidelines of the Human Investigation Committee of Rush University Medical Center. The cells were released by enzymatic digestion with pronase and collagenase as previously described (Im et al., 2003; Loeser et al., 2003). Alginate beads and monolayers were made for long-term and short-term culture, respectively. For monolayer culture, isolated chondrocytes were counted and plated onto 12-well plates at 8×105 cells/cm2 as previously described (Im et al., 2007; Im et al., 2003). Cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM)/F-12 (1:1) containing 10% fetal bovine serum and antibiotics (complete media) for 3 days before the treatments. Prior to treatments, chondrocytes were starved in serum-free DMEM/F-12 (1:1) for 24 hours. Media were replaced again with fresh serum-free DMEM/F-12 (1:1) 2 hours before stimulation. Cells were treated with LfcinB (50 and 100 μg/mL), in the presence or absence of FGF-2 (100 ng/mL) or IL-1β (10 ng/mL). Cells were harvested after 24 hours, and subjected to analytical procedures as described below. For alginate bead culture, grade 0 or 1 chondrocytes were suspended in 1.2% alginate at 2×106 cells/mL, and beads were formed in CaCl2 solution, as previously described (Gruber et al., 2006). Cells were cultured in DMEM/F-12 medium (1:1), supplemented with 1% mini-ITS+ premix (BD Biosciences, San Jose, CA) and 0.1% ascorbic acid, for 7 days prior to experiments. Cells were maintained for 7 days to allow for extracellular matrix assembly, before being treated with LfcinB (10 and 20 μg/mL), in the presence or absence of FGF-2 (50 ng/mL) or IL-1β (1 ng/mL). Triplicates were set up for each condition. Media were changed every other day over a 21-day period, before DNA and DMMB analyses.
Synovial Fibroblast Isolation and Culture
Human synovium was obtained together with articular cartilage through the Gift of Hope Organ and Tissue Donor Network (Elmhurst, IL). Each specimen was digested in 0.05% trypsin for 30 min, followed by collagenase digestion for 6-8 hours. Isolated synoviocytes underwent three consecutive passages before the experiments. The purity of synovial fibroblast preparation was confirmed by the fact that neither CD14 nor CD68 (macrophage markers) was expressed at detectable mRNA levels. Synovial fibroblasts were maintained as adherent cultures in DMEM supplemented with 10% FBS. Prior to stimulation, cells were starved in serum-free DMEM containing 0.2% lactalbumin hydrolysate (Sigma-Aldrich, St. Louis, MO) for 24 hours, and the media were replaced again before the addition of IL-1β (50 pg/mL) and LfcinB (50 and 100 μg/mL).
Reverse Transcription and Real-Time Polymerase Chain Reaction
Total RNA from normal and osteoarthritic articular chondrocytes was isolated using Trizol reagent (Invitrogen, Carlsbad, CA) following the instructions provided by the manufacturer. Reverse transcription (RT) was carried out with 1 μg total RNA using ThermoScript ™ RT-PCR system (Invitrogen, Carlsbad, CA) for first strand cDNA synthesis. For real-time PCR, cDNA was amplified using MyiQ Real-Time PCR Detection System (Bio-Rad, Hercules, CA). Relative gene expression was determined using the ΔΔCT method, as detailed by manufacturer guidelines (Bio-Rad, Hercules, CA). 18S rRNA was used as an internal control in the reaction for normalization. The standard deviations in samples represent at least five different donors from independent experiments. The primer sequences are summarized in Table 1.
Table 1. qPCR primer sequences.
| Gene | Primer Seq. (5′→3′) | NCBI Reference No. |
Annealing Tm |
|---|---|---|---|
| 18S rRNA | F: CGGCTACCACATCCAAGGAA R: GCTGGAATTACCGCGGCT |
NR_003286.2 | 60°C |
| MMP1 | F: AGTGACTGGGAAACCAGATGCTGA R: GCTCTTGGCAAATCTGGCGTGTAA |
NM_002421.3 | 60 °C |
| MMP3 | F: GCGTGGATGCCGCATATGAAGTTA R: AAACCTAGGGTGTGGATGCCTCTT |
NM_002422.3 | 60 °C |
| MMP13 | F:ACCCTGGAGCACTCATGTTTCCTA R:TGGCATCAAGGGATAAGGAAGGGT |
NM_002427.3 | 60 °C |
| ADAMTS4 | F: ACTGGGCTACTACTATGTGCTGGA R: CTTCTTCTTGGAGCCAATGATGCG |
NM_005099.4 | 60 °C |
| ADAMTS5 | F:CTGTGACGGCATCATTGGCTCAAA R:TTCAGGAATCCTCACCACGTCAGT |
NM_007038.3 | 60 °C |
| INOS | F: ATCACACGCCCACAGAGATCCA R:GCTTCAGGCTGTTGAGCCATGT |
NM_000625.4 | 55 °C |
| TLR2 | F: AAACGTTAACAATCCGGAGGCTGC R: TGTTGTGAAAGTAAACAAGGAACCAG |
NM_003264.3 | 60 °C |
| IL1B | F: ATGACCTGAGCACCTTCTTTCCCT R: GCATCGTGCACATAAGCCTCGTTA |
NM_000576.2 | 55 °C |
| IL6 | F: AAGCCAGAGCTGTGCAGATGAGTA R: TTCGTCAGCAGGCTGGCATTTGT |
NM_000600.3 | 60 °C |
| IL8 | F: TCTTGGCAGCCTTCCTGATTTCTG R: GGGTGGAAAGGTTTGGAGTATGTC |
NM_000584.3 | 60 °C |
| IL4 | F: GCCTCACATTGTCACTGCAAATCG R: AGGTGATATCGCACTTGTGTCCGT |
NM_000589.2 | 60 °C |
| IL10 | F: TTAAGGGTTACCTGGGTTGCCAAG R: AGTTCACATGCGCCTTGATGTCTG |
NM_000572.2 | 60 °C |
Dimethylethylene Blue (DMMB) Assay for Proteoglycan Accumulation and DNA Assay for Cell Numbers
On day 21 of alginate bead culture, the beads were collected and processed for proteoglycan assay using the DMMB binding method, as previously described (Gruber et al., 2006). Using PicoGreen (Molecular Probes, Carlsbad, CA), cell numbers were determined by quantification of total DNA in cell pellets, as previously described (Im et al., 2003). Proteoglycan accumulation per cell in the cell-associated matrix (CM) was quantified using DNA content for normalization (Im et al., 2003).
Immunoblotting
Chondrocytes were prepared using a modified lysis RIPA buffer (Muddasani et al., 2007). Total protein concentrations were determined by a bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL). An equal amount of protein was resolved by 10% SDS-polyacrylamide gels and transferred to nitrocellulose membrane for immunoblotting as described previously (Im et al., 2007). Immunoreactivity was visualized using the ECL system (Amersham Biosciences, Piscataway, NJ) and the Signal Visual Enhancer system (Pierce) to magnify the signal.
Cartilage Explant Culture
Full-thickness explants of 4 mm diameters were prepared. Explants then recovered in DMEM/F-12 (1:1, supplemented with 10% FBS) for 48 hours. Culture media were changed to DMEM/F-12 (1:1) supplemented with 1% mini-ITS+ premix (BD Biosciences, San Jose, CA) 48 hours before treatments started. The explants were treated with FGF-2 (100 ng/mL) or IL-1β (5 ng/mL), in the presence or absence of LfcinB (50 and 100 μg/mL).
Histology and Histomorphometry
After 11-day stimulation, explants were fixed in 4% paraformaldehyde, followed by paraffin embedding. Sections of 8 μm thickness were prepared. The sections were then deparaffinized, and stained with Safranin Orange dye to assess proteoglycan content in the ECM. Safranin-O-positive areas, defined as articular cartilage areas, were quantified using OsteoMeasure software (OsteoMetrics, Decatur, GA), in which the superficial zone and middle zone were included. Specimens from 3 donors were analyzed.
Cytotoxicity Assay
CellTiter 96 AQueous One Solution (Promega, Madison, WI) was used to determine the cell viability of synovial fibroblasts. Cells were plated on 96-well plates at 1×105 cells/well. Each condition was set up in quadruplet. Assays were performed according to the manufacturer’s instructions.
Statistical Analysis
Statistical significance was determined by one-way repeated measures ANOVA followed by Sidak post-hoc test, using the SPSS17 software (IBM Corporation, Somers, NY). P values lower than 0.05 were considered to be statistically significant in each test. Each value in the figures is presented as mean ± standard deviation.
RESULTS
LfcinB counteracts IL-1β or FGF-2-mediated suppression of proteoglycan accumulation in adult human articular chondrocytes
First we investigated the effect of LfcinB on proteoglycan accumulation in human articular chondrocytes. Cells cultured in 3-dimensional alginate beads that allow cells to maintain chondrocytic phenotype for a long period time were challenged by FGF-2 (50 ng/mL) or IL-1β (1 ng/mL), in the presence or absence of LfcinB (10 and 20 μg/mL) for 21 days. Then proteoglycan accumulation in cell-associated matrix was assessed by DMMB assay. Consistent with documented findings, FGF-2 and IL-β led to a dramatic decrease in proteoglycan accumulation by approximately 50% and 65%, respectively, compared with control (Figure 1; lanes 2 & 5). Co-administration of LfcinB dose-dependently rescued FGF-2- and IL-1β-suppressed proteoglycan accumulation (Figure 1; lanes 3, 4, & 6, 7), revealing the anti-catabolic activity of LfcinB in primary human articular chondrocytes. Assessments of DNA content suggest no statistical difference in cell proliferation between LfcinB-treated groups and the control group. In addition, our weekly cell viability assay results indicate that LfcinB did not elicit cytotoxicity in articular chondrocytes in our 21-day culture (data not shown).
Figure 1. LfcinB counteracts IL-1β/FGF-2-mediated effects on proteoglycan accumulation in human articular chondrocytes.
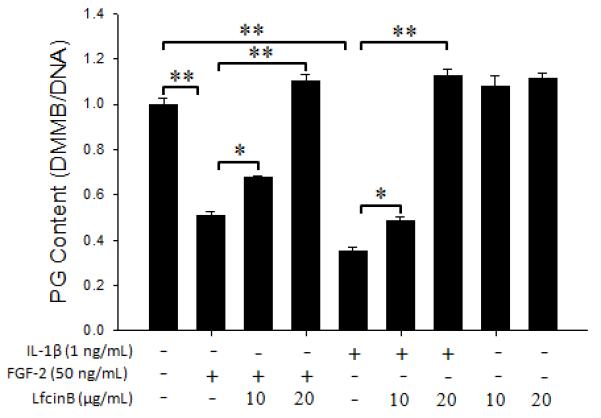
Chondrocytes were cultured in alginate beads for 7 days and subjected to different treatment conditions for 21 days, before dimethylmethylene blue (DMMB) assay for proteoglycan content. The gross proteoglycan amount in each group was normalized by DNA content determined by a PicoGreen method. *p<0.05; **p<0.01.
LfcinB suppresses cartilage-degrading enzyme production and iNOS expression in human articular chondrocytes
Although LfcinB effectively rescued proteoglycan loss by either IL-1β or FGF-2, we did not notice significant induction of proteoglycan accumulation when the cells were treated with LfcinB alone (Figure 1; lanes 8 & 9). Thus, we hypothesized that rescuing effects of LfcinB may be associated with inhibition of catabolism rather than extracellular matrix production. To test our hypothesis, normal chondrocytes cultured in monolayer were treated with LfcinB (50 and 100 μg/mL) for 24 hours. Cartilage-degrading enzyme production and expression levels were then analyzed by immunoblotting and real-time qPCR. We observed that LfcinB did not affect cell viability at either concentration (50 and 100 μg/mL, data not shown). LfcinB significantly downregulated MMP-1, MMP-3, and MMP-13 at both mRNA and protein levels in a concentration-dependent manner (Figure 2A; p<0.05). Similar inhibitory effects of LfcinB were also observed in aggrecanase expression (ADAMTS4, ADAMTS5) at both mRNA and protein levels (Figure 2B). In addition, LfcinB was able to repress inducible nitric oxide synthase (iNOS) mRNA expression (Figure 2C). In OA chondrocytes, LfcinB was also able to downregulate the target genes examined above (data not shown). Treatment with LfcinB had no significant influence on aggrecan expression regardless of the concentration used (ranging from 10~200 μg/mL) (data not shown).
Figure 2. LfcinB suppresses cartilage-degrading protease production and iNOS expression in human articular chondrocytes.

(A) Chondrocytes in monolayer were incubated with LfcinB (50 and 100 μg/mL) for 24 hours. Total RNA was extracted for quantification of MMP-1, MMP-3, and MMP-13 transcripts using qPCR. Conditioned media were collected for immunoblotting of MMP-1, MMP-3, and MMP-13. (B) Chondrocytes in monolayer were treated with LfcinB (50 and 100 μg/mL) for 24 hours. Total RNA was extracted for quantification of ADAMTS4 and ADAMTS5 mRNA levels. Conditioned media were collected for immunoblotting of ADAMTS4 and ADAMTS5. (C) Chondrocytes were treated with LfcinB (50 and 100 μg/mL) for 24 hours. Total RNA was extracted for quantification of iNOS transcripts. *p<0.05; **p<0.01.
LfcinB counteracts cytokine-induced cartilage-degrading enzymes in human articular chondrocytes
We further examined whether LfcinB could counteract specific protease expression in the presence of FGF-2 or IL-1β. Primary human articular chondrocytes in monolayer were stimulated with FGF-2 (100 ng/mL) or IL-1β (10 ng/mL), in the presence or absence of LfcinB (50 and 100 μg/mL). Both FGF-2 and IL-1β significantly augmented the expression of MMP-1, MMP-3, and MMP-13 at both mRNA (Figure 3A; lanes 2 & 5) and protein levels (Figure 3B; lanes 2 & 5), compared with control. Co-incubation with LfcinB significantly counteracted these catabolic effects, with the levels of cartilage-degrading enzymes returning to the control (untreated) levels at its higher concentration (100 μg/mL) (Figures 3A & 3B). These phenomena were also reproducibly observed using OA chondrocytes (data not shown). Together these results demonstrate the potent anti-catabolic activity of LfcinB in human articular chondrocytes.
Figure 3. LfcinB counteracts cytokine-induced cartilage-degrading enzymes in human articular chondrocytes.
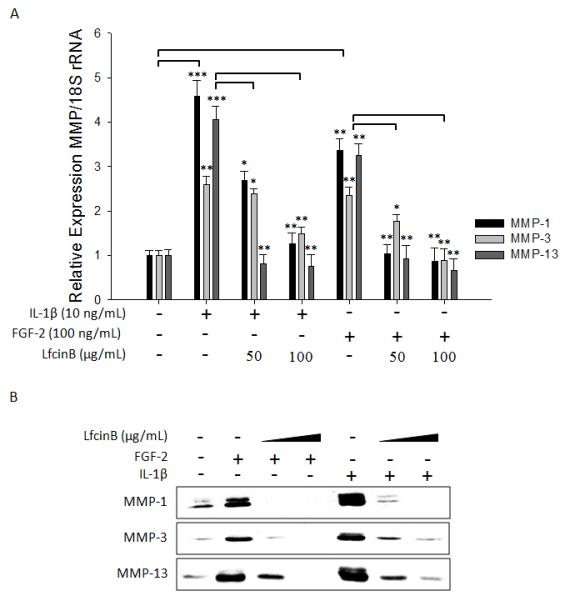
(A) Chondrocytes were treated with IL-1β (10 ng/mL) or FGF-2 (100 ng/mL), in the presence or absence of LfcinB (50 and 100 μg/mL), for 24 hours. Total RNA was then extracted for qPCR analyses of MMP-1, MMP-3, and MMP-13 transcripts. (B) In parallel, conditioned media were used for immunoblotting of secreted MMP-1, MMP-3, and MMP-13. *p<0.05; **p<0.01; ***p<0.001.
LfcinB represses pro-inflammatory cytokine and toll-like receptor 2 and induces anti-inflammatory cytokines in articular chondrocytes
In addition to upregulating destructive proteases, IL-1β is known to stimulate the expression of multiple pro-inflammatory cytokines, which perpetuate cartilage destruction and synovial inflammation via autocrine/paracrine loops (Goldring, 2000). Considering the remarkable potency of LfcinB in attenuating IL-1β-mediated effects, we hypothesized that LfcinB interferes with the inflammatory cytokine induction mediated by IL-1β. To test this notion, normal chondrocytes were stimulated by IL-1β (10 ng/mL), in the presence or absence of LfcinB (50 and 100 μg/mL) for 24 hours. Our qPCR analyses recapitulated previous findings that IL-1β is capable of inducing OA-associated pro-inflammatory cytokines, IL-1β and IL-6 (Goldring, 2000) (Figure 4A). Co-treatment with LfcinB, however, significantly inhibited the upregulation of IL-1β and IL-6 (Figure 4A), suggesting an anti-inflammatory role of LfcinB in human articular chondrocytes. Toll-like receptor (TLR)-mediated actions have been implicated in OA pathogenesis as an essential component of inflammatory response to inflammatory cytokines (Kim et al., 2006). Specifically, TLR2 activation leads to MMP induction and collagenolysis (Zhang et al., 2008), and TLR2 is upregulated upon IL-1β stimulation in human articular chondrocytes (Su et al., 2005). Treatment with LfcinB at both concentrations (50 and 100 μg/mL) markedly suppressed IL-1β-induced TLR2 expression at both mRNA and protein levels, suggesting its potent inhibitory effect on TLR signaling (Figure 4B; p<0.01).
Figure 4. LfcinB downregulates pro-inflammatory factors and induces anti-inflammatory cytokines in articular chondrocytes.

(A) Chondrocytes in monolayer were stimulated by IL-1β (10 ng/mL), in the presence or absence of LfcinB (50 and 100 μg/mL), for 24 hours. Total RNA was extracted for quantification of IL-1β and IL-6 transcripts using qPCR. (B) Chondrocytes were incubated with IL-1β (10 ng/mL), in the presence or absence of LfcinB (50 and 100 μg/mL) for 24 hours. TLR2 transcripts were quantified by qPCR. Immunoblotting was performed to assess TLR2 protein levels. (C) Chondrocytes were treated with LfcinB (50 and 100 μg/mL) for 24 hours, followed by qPCR analyses of IL-4 and IL-10 mRNA levels. *p<0.05; **p<0.01; ***p<0.001.
A set of anti-inflammatory cytokines, including IL-4 and IL-10, have been demonstrated to confer protection upon synovium and cartilage (Goldring, 2000; van der Kraan and van den Berg, 2000). To further characterize the anti-inflammatory property of LfcinB, we sought to determine whether it is able to stimulate anti-inflammatory cytokine expression. Normal chondrocytes in monolayer were treated with LfcinB for 24 hours. Our qPCR analyses revealed that LfcinB significantly induced anti-inflammatory cytokine IL-4 and IL-10 (Figure 4C). In addition, LfcinB-mediated effects on the pro-inflammatory mediators and anti-inflammatory cytokines examined above also held in OA chondrocytes (data not shown). Taken together, our results indicate that LfcinB exerts its anti-inflammatory actions via specific suppression of pro-inflammatory mediators and induction of anti-inflammatory cytokines.
Considering the anti-inflammatory activity of LfcinB, it was of interest to determine whether LfcinB directly regulates inflammatory signaling pathways, such as JNK MAPK and NFκB, in articular chondrocytes. Normal chondrocytes in monolayer were stimulated with LfcinB (50 μg/mL) for different durations (10, 30 and 60 min). Then we surveyed the activation status of each well-documented signaling pathway in chondrocytes by immunoblotting. Our results show that LfcinB rapidly and robustly activated ERK MAPK and Akt pathways (Figure 5), whereas JNK MAPK and NFκB pathways were not notably regulated by LfcinB within 2 hours after stimulation (data not shown). This finding suggests the possibility that LfcinB mediates anti-inflammatory effects via ERK and/or Akt pathways in articular chondrocytes.
Figure 5. LfcinB activates ERK MAPK and Akt pathways in articular chondrocytes.
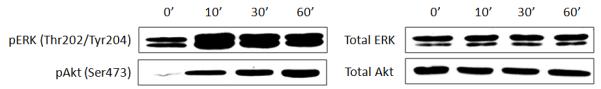
Cells in monolayer were incubated with LfcinB (50 μg/mL) for various durations (10, 30, and 60 min), before whole cell lysates were prepared. Immunoblotting was performed using anti-phospho-ERK and anti-phospho-Akt antibodies to reveal pathway activation. Total ERK and total Akt levels were used as loading controls.
LfcinB inhibits IL-1β- or FGF-2-mediated proteoglycan depletion in human articular cartilage ex vivo organ culture
Encouraged by our in vitro results from alginate beads and cell culture studies, we further tested if LfcinB-mediated anti-catabolic effects still hold in an ex vivo organ culture system. Adult human articular cartilage explants were incubated with FGF-2 (100 ng/mL) or IL-1β (5 ng/mL), in the presence or absence of LfcinB (50 and 100 μg/mL) for 11 days. Safranin-O Fast Green staining was chosen to assess alterations in gross proteoglycan content. Compared with control (Figure 6A; a and a’), IL-1β vastly depleted proteoglycan (Figure 6A; b’), and FGF-2 incurred proteoglycan loss to a lesser extent (Figure 6A; b). This observation is consistent with our previous reports (Im et al., 2008; Muddasani et al., 2007). Importantly, co-incubation with LfcinB restored the proteoglycan loss caused by FGF-2 or IL-1β in a dose-dependent manner (Figure 6A; c, d, c’ and d’). We also performed histomorphometrical analyses, in which areas with positive proteoglycan staining were quantified irrespective of staining intensity. Our data show that both IL-1β and FGF-2 decreased proteoglycan staining areas in cartilage explants, and LfcinB co-administration (50 and 100 μg/mL) effectively reversed such reduction (Figure 6B; p<0.01). These results further validated our in vitro results, and supported the anti-catabolic role of LfcinB in human articular cartilage.
Figure 6. LfcinB inhibits IL-1β/FGF-2-mediated proteoglycan depletion in articular cartilage ex vivo.
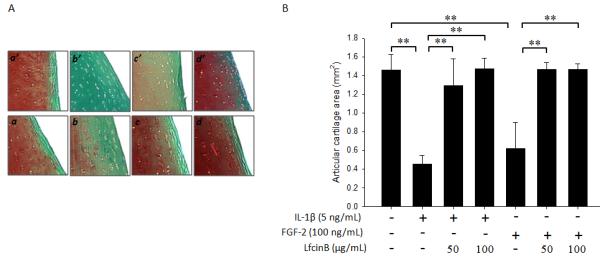
(A) Full-thickness cartilage explants with 4 mm diameters in serum-free media (plus mini-ITS™+ Premix) were treated with FGF-2 (100 ng/mL) or IL-1β (5 ng/mL), in the presence or absence of LfcinB (50 and 100 μg/mL), for 11 days. The explants were fixed in 4% paraformaldehyde and embedded in paraffin. Safranin O Fast Green staining was adopted to assess gross proteoglycan content in the extracellular matrix. (a & a’) Control; (b) FGF-2 (100 ng/mL); (c) FGF-2 plus LfcinB (50 μg/mL); (d) FGF-2 plus LfcinB (100 μg/mL); (b’) IL-1β (5 ng/mL); (c’) IL-1β plus LfcinB (50 μg/mL); (d’) IL-1β plus LfcinB (100 μg/mL). (B) Stained sections were analyzed for proteoglycan staining-positive areas using OsteoMeasure software. Superficial zone and middle zone were included into analyses, and cartilage areas were calculated based on sections from 3 different donors. **p<0.01.
LfcinB exerts anti-inflammatory effects in synovial fibroblasts
Synovial inflammation serves as a crucial component in OA pathogenesis. Considering the chondroprotective property of LfcinB, we then asked whether LfcinB also dampens the inflammatory responses in synovial fibroblasts. To address this question, human primary synovial fibroblasts were stimulated with IL-1β (50 pg/mL), in the presence or absence of LfcinB (50 and 100 μg/mL). The concentration of IL-1β was based on our initial experiments in which synovial cells responded with a greater sensitivity to IL-1β compared with articular chondrocytes. For example, IL-1β at the concentration of 10 ng/mL caused >100~200-fold induction of multiple inflammatory mediators (eg., IL-1β) in synovial cells (data not shown). Similar to what we observed in articular chondrocytes, stimulation of synovial cells with IL-1β potently upregulated the expression of MMP-1, MMP-3, and MMP-13 at mRNA and protein levels (Figure 7A) These inductions of cartilage degrading enzymes by IL-1β were effectively attenuated in the presence of LfcinB in a dose-dependent fashion (Figure 7A; p<0.01 at 100 μg/mL LfcinB). IL-1β also highly upregulated IL-1β, IL-8, TLR2, and iNOS expression in human synovial fibroblasts, and these responses were largely inhibited by LfcinB co-treatment (Figure 7B, 7C & 7D). To ensure that LfcinB-mediated downregulation of inflammatory mediators were not due to diminished cellularity, we performed cell viability assays in parallel. Incubation of synovial fibroblasts with LfcinB at various concentrations (0, 10, 50 and 100 μg/mL) for the experimental period (24 hours) showed no significant influence on cell viability (Figure 7E). Together these results demonstrate that, in addition to its protective effects on cartilage, LfcinB also acts as an anti-inflammatory molecule in human synovial fibroblasts.
Figure 7. LfcinB exerts anti-catabolic and anti-inflammatory effects in synovial fibroblasts.

(A) Synovial fibroblasts in monolayer were stimulated by IL-1β (50 pg/mL), in the presence or absence of LfcinB (50 and 100 μg/mL), for 24 hours. Then qPCR was performed to quantify MMP-1, MMP-3, and MMP-13 transcripts. In parallel, immunoblotting was carried out to assess the intracellular protein levels of MMP-1, MMP-3, and MMP-13. GADPH was used as loading controls. (B) Synovial fibroblasts were incubated with IL-1β (50 pg/mL), in the presence or absence of LfcinB (50 and 100 μg/mL), for 24 hours. The mRNA levels of IL-1β and IL-8 were measured by qPCR. (C) Synovial fibroblasts were treated with IL-1β (50 pg/mL), in the presence or absence of LfcinB (50 and 100 μg/mL), for 24 hours, followed by qPCR analyses of TLR2 transcripts. (D) Synovial fibroblasts were incubated with IL-1β (50 pg/mL), in the presence or absence of LfcinB (50 and 100 μg/mL), for 24 hours. The mRNA levels of iNOS were quantified by qPCR. (E) Synovial fibroblasts were plated on 96-well plates at 1×105 cells/well. Then cells were incubated with LfcinB (10, 50 and 100 μg/mL) for 24 hours. Each condition was set up in quadruplet. Assays were performed using CellTiter 96 AQueous One Solution. *p<0.05; **p<0.01.
DISCUSSION
The antibacterial, antiviral, antitumor, and immunomodulatory activities of LfcinB have been studied in a variety of tissues (Gifford et al., 2005; Mader et al., 2007; Zhang et al., 2007). Here, we for the first time characterize the biological effects of LfcinB in adult human articular cartilage and synovium. LfcinB significantly reversed the catabolic effects of FGF-2 and IL-1β by restoring proteoglycan loss in articular cartilage, antagonizing production of multiple matrix-degrading enzymes as well as inflammatory mediators while inducing anti-inflammatory cytokines in both articular cartilage and synovial fibroblasts. Collectively, our findings suggest that LfcinB elicits potent anti-catabolic and anti-inflammatory actions on human articular cartilage and synovium.
Our findings support the potent inhibitory action of LfcinB on FGF-2-mediated catabolic effects, including matrix-degrading enzyme expression, iNOS expression, and proteoglycan depletion. Interestingly, Mader and colleagues found that LfcinB inhibits FGF-2- and vascular endothelial growth factor (VEGF)-induced angiogenesis by competing for binding to heparan sulfate proteoglycans (HSPGs) on the surface of mice and human umbilical vein endothelial cells (Mader et al., 2006). In cartilage, these highly abundant, negatively charged HSPGs are found in the ECM and play an intricate role in growth factor signaling by forming complexes with the growth factor and its receptor. For example, HSPGs are required for binding of FGF-2 to FGF receptor (FGFR) to transduce the signal into the cell (Tumova et al., 2000).
More specifically, it has been shown that syndecan-4 facilitates binding of FGF-2 to FGF receptor (FGFR) to activate downstream signaling (Richardson et al., 1999; Volk et al., 1999). Therefore, we conjecture that LfcinB may compete with FGF-2 for HSPG binding on the chondrocyte cell surface, which serves as one possible mechanism for the LfcinB-mediated antagonism against FGF-2 signaling in human articular cartilage. Indeed, we have preliminary evidence that pre-incubation of LfcinB with exogenous heparin or heparan sulfate abolishes its signature transcriptional regulation in human articular chondrocyte as well as chondrocyte-like cells (disc cells) (unpublished data). Further studies are warranted to elucidate which HSPG member accounts for the counteractive effect of LfcinB on FGF-2 in chondrocytes.
Similarly, a study by Vallés et al. unveiled a critical role of heparan sulfate in IL-1 receptor (IL-1R) complex formation and subsequent signal transduction (Valles et al., 1999). More recently, Wang et al reported the involvement of syndecan in IL-1β signaling in chondrocyte-like nucleus pulposus cells (Wang et al., 2011). Thus, it is possible that LfcinB inhibits IL-1β signaling via disrupting its receptor complex assembly. Furthermore, LfcinB did not regulate the expression of IL-1R type I or type II (a decoy receptor) in our experimental system, suggesting LfcinB may not desensitize chondrocytes in response to IL-1β through beheading its signaling (data not shown).
The anti-catabolic and anti-inflammatory capacity of LfcinB in articular cartilage uncovered its potential in prevention and/or treatment of cartilage degeneration. The upregulation of matrix-degrading enzymes, such as MMP-13, ADAMTS4, and ADAMTS5 have been identified as a major contributor to joint degradation and OA (van den Berg, 2011). Hence the active participation of these proteases renders them targets of interest in OA treatment (De Rienzo et al., 2009; Li et al., 2011). Our data demonstrate that LfcinB can potently inhibit the expression of these key proteases. Our findings also revealed the capacity of LfcinB in repressing pro-inflammatory mediators IL-1β and IL-6 that account for cartilage erosion via autocrine/paracrine mechanisms. Simultaneously, LfcinB significantly induced anti-inflammatory cytokines (IL-4 and IL-10) that may dampen the inflammatory signaling pathways. Thus, LfcinB seems to confer protection on articular joint through coordinating anti-catabolic and anti-inflammatory processes to shut down detrimental paracrine/autocrine loops.
IL-1 induces TLR2 and TLR4 in chondrocytes (Kim et al., 2006; Su et al., 2005), and both TLRs are increased in OA cartilage in situ, which mediates innate immune response, thereby triggering inflammatory and immune attacks (Kim et al., 2006; Scanzello et al., 2008). Moreover, microbe-associated and endogenous TLR2 and TLR4 ligands [e.g., lipopolysaccharides (LPS), low molecular weight hyaluronan] stimulate catabolic responses in chondrocytes (Liu-Bryan and Terkeltaub, 2010; Schelbergen et al., 2011). In our study, LfcinB had no statistically significant influence on the basal expression of TLR4, suggesting the specific antagonistic action of LfcinB against TLR2 signaling (data not shown). TLR2 also directly contributes to inflammatory and oxidative stress-induced degenerative processes in chondrocytes by stimulating nitric oxide (Liu-Bryan et al., 2005). Our results show that LfcinB markedly attenuates IL-1β-induced expression of both iNOS and TLR2 in articular chondrocytes as well as synovial fibroblasts, suggesting LfcinB may also be involved in alleviation of oxidative stress in joint tissues.
Further studies are warranted to delineate the mechanisms whereby LfcinB exerts stimulatory and repressive effects on its target genes. Our data revealed that, although LfcinB does not directly impact on the inflammatory JNK MAPK or NFκB pathway, it may promote anti-inflammation via ERK and/or Akt signaling. In particular, LfcinB appears to trigger a non-canonical ERK pathway, because it does not modulate the activity of Elk-1, a classical target downstream of ERK (data not shown). This observation also explains why LfcinB represses MMP-13 expression instead of inducing it. Aside from defining the roles of ERK and Akt pathways in target gene regulation, elucidation of transcriptional mechanisms utilized by LfcinB is also of importance to our understanding of its mode of action.
To sum up, we have shown that LfcinB exerted anti-catabolic and anti-inflammatory effects against FGF-2 and IL-1β challenges in human articular cartilage and synovium. LfcinB seems to hold promise as a disease-modifying molecule in prevention and/or treatment of degenerative joint diseases. Additionally, considering its well-documented antimicrobial activities, LfcinB may also bring therapeutic benefits to septic arthritis. The present study warrants further investigation to uncover the cellular molecular mechanisms utilized by LfcinB in human articular chondrocytes, and also provides grounds for assessment of the potency of LfcinB in OA animal models.
ACKNOWLEDGMENTS
We would like to thank the tissue donors, Drs. Gabriella Cs-Szabo and Arkady Margulis, and the Gift of Hope Organ and Tissue Donor Network for adult human joint tissue samples. This work was supported by grants to HJI from NIH R01AR053220, the Arthritis Foundation, and the National Arthritis Research Foundation (HJI).
Contract grant sponsor: NIH, Arthritis Foundation, National Arthritis Research Foundation
Contract grant number: R01AR053220
ABBREVIATIONS
- OA
osteoarthritis
- LfcinB
bovine lactoferricin
- FGF-2
fibroblast growth factor 2
- IL-β
interleukin-1 β
- MMP
matrix metalloproteinase
- ADAMTS
a disintegrin and metalloproteinase with a thrombospondin type 1 motif
- TLR
Toll-like receptor
- iNOS
inducible nitric oxide synthase
- HSPG
heparan sulfate proteoglycan
- DMMB
dimethylmethylene blue
Footnotes
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
HJI, DC, and DY designed the experiments of this study. DY acquired the data. JS performed histomorphometry analyses. HJI, DC, GX, AJW and DY interpreted the data. HJI and DY drafted the manuscript. All authors read, edited, and approved the final manuscript.
REFERENCES
- Abramson SB, Attur M. Developments in the scientific understanding of osteoarthritis. Arthritis Res Ther. 2009;11(3):227. doi: 10.1186/ar2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaaeddine N, Di Battista JA, Pelletier JP, Kiansa K, Cloutier JM, Martel-Pelletier J. Inhibition of tumor necrosis factor alpha-induced prostaglandin E2 production by the antiinflammatory cytokines interleukin-4, interleukin-10, and interleukin-13 in osteoarthritic synovial fibroblasts: distinct targeting in the signaling pathways. Arthritis Rheum. 1999;42(4):710–718. doi: 10.1002/1529-0131(199904)42:4<710::AID-ANR14>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Andersen JH, Jenssen H, Sandvik K, Gutteberg TJ. Anti-HSV activity of lactoferrin and lactoferricin is dependent on the presence of heparan sulphate at the cell surface. J Med Virol. 2004;74(2):262–271. doi: 10.1002/jmv.20171. [DOI] [PubMed] [Google Scholar]
- Attur MG, Patel IR, Patel RN, Abramson SB, Amin AR. Autocrine production of IL-1 beta by human osteoarthritis-affected cartilage and differential regulation of endogenous nitric oxide, IL-6, prostaglandin E2, and IL-8. Proc Assoc Am Physicians. 1998;110(1):65–72. [PubMed] [Google Scholar]
- Bondeson J, Wainwright S, Hughes C, Caterson B. The regulation of the ADAMTS4 and ADAMTS5 aggrecanases in osteoarthritis: a review. Clin Exp Rheumatol. 2008;26(1):139–145. [PubMed] [Google Scholar]
- Daheshia M, Yao JQ. The interleukin 1beta pathway in the pathogenesis of osteoarthritis. J Rheumatol. 2008;35(12):2306–2312. doi: 10.3899/jrheum.080346. [DOI] [PubMed] [Google Scholar]
- De Rienzo F, Saxena P, Filomia F, Caselli G, Colace F, Stasi L, Giordani A, Menziani MC. Progress towards the identification of new aggrecanase inhibitors. Curr Med Chem. 2009;16(19):2395–2415. doi: 10.2174/092986709788682092. [DOI] [PubMed] [Google Scholar]
- Fosang AJ, Last K, Knauper V, Murphy G, Neame PJ. Degradation of cartilage aggrecan by collagenase-3 (MMP-13) FEBS Lett. 1996;380(1-2):17–20. doi: 10.1016/0014-5793(95)01539-6. [DOI] [PubMed] [Google Scholar]
- Gifford JL, Hunter HN, Vogel HJ. Lactoferricin: a lactoferrin-derived peptide with antimicrobial, antiviral, antitumor and immunological properties. Cell Mol Life Sci. 2005;62(22):2588–2598. doi: 10.1007/s00018-005-5373-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, Flannery CR, Peluso D, Kanki K, Yang Z, Majumdar MK, Morris EA. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434(7033):644–648. doi: 10.1038/nature03369. [DOI] [PubMed] [Google Scholar]
- Goldring MB. Osteoarthritis and cartilage: the role of cytokines. Curr Rheumatol Rep. 2000;2(6):459–465. doi: 10.1007/s11926-000-0021-y. [DOI] [PubMed] [Google Scholar]
- Gruber HE, Hoelscher GL, Leslie K, Ingram JA, Hanley EN., Jr. Three-dimensional culture of human disc cells within agarose or a collagen sponge: assessment of proteoglycan production. Biomaterials. 2006;27(3):371–376. doi: 10.1016/j.biomaterials.2005.06.032. [DOI] [PubMed] [Google Scholar]
- Haywood L, McWilliams DF, Pearson CI, Gill SE, Ganesan A, Wilson D, Walsh DA. Inflammation and angiogenesis in osteoarthritis. Arthritis Rheum. 2003;48(8):2173–2177. doi: 10.1002/art.11094. [DOI] [PubMed] [Google Scholar]
- Im HJ, Li X, Muddasani P, Kim GH, Davis F, Rangan J, Forsyth CB, Ellman M, Thonar EJ. Basic fibroblast growth factor accelerates matrix degradation via a neuro-endocrine pathway in human adult articular chondrocytes. J Cell Physiol. 2008;215(2):452–463. doi: 10.1002/jcp.21317. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Im HJ, Muddasani P, Natarajan V, Schmid TM, Block JA, Davis F, van Wijnen AJ, Loeser RF. Basic fibroblast growth factor stimulates matrix metalloproteinase-13 via the molecular cross-talk between the mitogen-activated protein kinases and protein kinase Cdelta pathways in human adult articular chondrocytes. J Biol Chem. 2007;282(15):11110–11121. doi: 10.1074/jbc.M609040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im HJ, Pacione C, Chubinskaya S, Van Wijnen AJ, Sun Y, Loeser RF. Inhibitory effects of insulin-like growth factor-1 and osteogenic protein-1 on fibronectin fragment- and interleukin-1beta-stimulated matrix metalloproteinase-13 expression in human chondrocytes. J Biol Chem. 2003;278(28):25386–25394. doi: 10.1074/jbc.M302048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery AK. Osteophytes and the osteoarthritic femoral head. J Bone Joint Surg Br. 1975;57(3):314–324. [PubMed] [Google Scholar]
- Kato M, Wang H, Kainulainen V, Fitzgerald ML, Ledbetter S, Ornitz DM, Bernfield M. Physiological degradation converts the soluble syndecan-1 ectodomain from an inhibitor to a potent activator of FGF-2. Nat Med. 1998;4(6):691–697. doi: 10.1038/nm0698-691. [DOI] [PubMed] [Google Scholar]
- Kim HA, Cho ML, Choi HY, Yoon CS, Jhun JY, Oh HJ, Kim HY. The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheum. 2006;54(7):2152–2163. doi: 10.1002/art.21951. [DOI] [PubMed] [Google Scholar]
- Kusakabe A. Subchondral cancellous bone in osteoarthrosis and rheumatoid arthritis of the femoral head. A quantitative histological study of trabecular remodelling. Arch Orthop Unfallchir. 1977;88(2):185–197. doi: 10.1007/BF00415099. [DOI] [PubMed] [Google Scholar]
- Li NG, Shi ZH, Tang YP, Wang ZJ, Song SL, Qian LH, Qian DW, Duan JA. New hope for the treatment of osteoarthritis through selective inhibition of MMP-13. Curr Med Chem. 2011;18(7):977–1001. doi: 10.2174/092986711794940905. [DOI] [PubMed] [Google Scholar]
- Liu-Bryan R, Pritzker K, Firestein GS, Terkeltaub R. TLR2 signaling in chondrocytes drives calcium pyrophosphate dihydrate and monosodium urate crystal-induced nitric oxide generation. J Immunol. 2005;174(8):5016–5023. doi: 10.4049/jimmunol.174.8.5016. [DOI] [PubMed] [Google Scholar]
- Liu-Bryan R, Terkeltaub R. Chondrocyte innate immune myeloid differentiation factor 88-dependent signaling drives procatabolic effects of the endogenous Toll-like receptor 2/Toll-like receptor 4 ligands low molecular weight hyaluronan and high mobility group box chromosomal protein 1 in mice. Arthritis Rheum. 2010;62(7):2004–2012. doi: 10.1002/art.27475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeser RF, Todd MD, Seely BL. Prolonged treatment of human osteoarthritic chondrocytes with insulin-like growth factor-I stimulates proteoglycan synthesis but not proteoglycan matrix accumulation in alginate cultures. J Rheumatol. 2003;30(7):1565–1570. [PubMed] [Google Scholar]
- Mader JS, Richardson A, Salsman J, Top D, de Antueno R, Duncan R, Hoskin DW. Bovine lactoferricin causes apoptosis in Jurkat T-leukemia cells by sequential permeabilization of the cell membrane and targeting of mitochondria. Exp Cell Res. 2007;313(12):2634–2650. doi: 10.1016/j.yexcr.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Mader JS, Smyth D, Marshall J, Hoskin DW. Bovine lactoferricin inhibits basic fibroblast growth factor- and vascular endothelial growth factor165-induced angiogenesis by competing for heparin-like binding sites on endothelial cells. Am J Pathol. 2006;169(5):1753–1766. doi: 10.2353/ajpath.2006.051229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar MK, Askew R, Schelling S, Stedman N, Blanchet T, Hopkins B, Morris EA, Glasson SS. Double-knockout of ADAMTS-4 and ADAMTS-5 in mice results in physiologically normal animals and prevents the progression of osteoarthritis. Arthritis Rheum. 2007;56(11):3670–3674. doi: 10.1002/art.23027. [DOI] [PubMed] [Google Scholar]
- Malfait AM, Liu RQ, Ijiri K, Komiya S, Tortorella MD. Inhibition of ADAM-TS4 and ADAM-TS5 prevents aggrecan degradation in osteoarthritic cartilage. J Biol Chem. 2002;277(25):22201–22208. doi: 10.1074/jbc.M200431200. [DOI] [PubMed] [Google Scholar]
- Mitchell PG, Magna HA, Reeves LM, Lopresti-Morrow LL, Yocum SA, Rosner PJ, Geoghegan KF, Hambor JE. Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J Clin Invest. 1996;97(3):761–768. doi: 10.1172/JCI118475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddasani P, Norman JC, Ellman M, van Wijnen AJ, Im HJ. Basic fibroblast growth factor activates the MAPK and NFkappaB pathways that converge on Elk-1 to control production of matrix metalloproteinase-13 by human adult articular chondrocytes. J Biol Chem. 2007;282(43):31409–31421. doi: 10.1074/jbc.M706508200. [DOI] [PubMed] [Google Scholar]
- Muehleman C, Bareither D, Huch K, Cole AA, Kuettner KE. Prevalence of degenerative morphological changes in the joints of the lower extremity. Osteoarthritis Cartilage. 1997;5(1):23–37. doi: 10.1016/s1063-4584(97)80029-5. [DOI] [PubMed] [Google Scholar]
- Pfander D, Heinz N, Rothe P, Carl HD, Swoboda B. Tenascin and aggrecan expression by articular chondrocytes is influenced by interleukin 1beta: a possible explanation for the changes in matrix synthesis during osteoarthritis. Ann Rheum Dis. 2004;63(3):240–244. doi: 10.1136/ard.2002.003749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson TP, Trinkaus-Randall V, Nugent MA. Regulation of basic fibroblast growth factor binding and activity by cell density and heparan sulfate. J Biol Chem. 1999;274(19):13534–13540. doi: 10.1074/jbc.274.19.13534. [DOI] [PubMed] [Google Scholar]
- Scanzello CR, Plaas A, Crow MK. Innate immune system activation in osteoarthritis: is osteoarthritis a chronic wound? Curr Opin Rheumatol. 2008;20(5):565–572. doi: 10.1097/BOR.0b013e32830aba34. [DOI] [PubMed] [Google Scholar]
- Schelbergen RF, Blom AB, van den Bosch MH, Sloetjes A, Abdollahi-Roodsaz S, Schreurs BW, Mort JS, Vogl T, Roth J, van den Berg WB, van Lent PL. Alarmins S100A8 and S100A9 elicit a catabolic effect in human osteoarthritic chondrocytes that is dependent on toll-like receptor 4. Arthritis Rheum. 2011 doi: 10.1002/art.33495. [DOI] [PubMed] [Google Scholar]
- Sharif M, Whitehouse A, Sharman P, Perry M, Adams M. Increased apoptosis in human osteoarthritic cartilage corresponds to reduced cell density and expression of caspase-3. Arthritis Rheum. 2004;50(2):507–515. doi: 10.1002/art.20020. [DOI] [PubMed] [Google Scholar]
- Shlopov BV, Lie WR, Mainardi CL, Cole AA, Chubinskaya S, Hasty KA. Osteoarthritic lesions: involvement of three different collagenases. Arthritis Rheum. 1997;40(11):2065–2074. doi: 10.1002/art.1780401120. [DOI] [PubMed] [Google Scholar]
- Su SL, Tsai CD, Lee CH, Salter DM, Lee HS. Expression and regulation of Toll-like receptor 2 by IL-1beta and fibronectin fragments in human articular chondrocytes. Osteoarthritis Cartilage. 2005;13(10):879–886. doi: 10.1016/j.joca.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Summers MD, Anderson DL. Granulosis virus deoxyribonucleic acid: a closed, double-stranded molecule. J Virol. 1972;9(4):710–713. doi: 10.1128/jvi.9.4.710-713.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumova S, Woods A, Couchman JR. Heparan sulfate proteoglycans on the cell surface: versatile coordinators of cellular functions. Int J Biochem Cell Biol. 2000;32(3):269–288. doi: 10.1016/s1357-2725(99)00116-8. [DOI] [PubMed] [Google Scholar]
- Valles S, Tsoi C, Huang WY, Wyllie D, Carlotti F, Askari JA, Humphries MJ, Dower SK, Qwarnstrom EE. Recruitment of a heparan sulfate subunit to the interleukin-1 receptor complex. Regulation by fibronectin attachment. J Biol Chem. 1999;274(29):20103–20109. doi: 10.1074/jbc.274.29.20103. [DOI] [PubMed] [Google Scholar]
- van den Berg WB. Osteoarthritis year 2010 in review: pathomechanisms. Osteoarthritis Cartilage. 2011;19(4):338–341. doi: 10.1016/j.joca.2011.01.022. [DOI] [PubMed] [Google Scholar]
- van der Kraan PM, van den Berg WB. Anabolic and destructive mediators in osteoarthritis. Curr Opin Clin Nutr Metab Care. 2000;3(3):205–211. doi: 10.1097/00075197-200005000-00007. [DOI] [PubMed] [Google Scholar]
- Volk R, Schwartz JJ, Li J, Rosenberg RD, Simons M. The role of syndecan cytoplasmic domain in basic fibroblast growth factor-dependent signal transduction. J Biol Chem. 1999;274(34):24417–24424. doi: 10.1074/jbc.274.34.24417. [DOI] [PubMed] [Google Scholar]
- Wang J, Markova D, Anderson DG, Zheng Z, Shapiro IM, Risbud MV. TNF-alpha and IL-1beta promote a disintegrin-like and metalloprotease with thrombospondin type I motif-5-mediated aggrecan degradation through syndecan-4 in intervertebral disc. J Biol Chem. 2011;286(46):39738–39749. doi: 10.1074/jbc.M111.264549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JX, Zhang SF, Wang TD, Guo XJ, Hu RL. Mammary gland expression of antibacterial peptide genes to inhibit bacterial pathogens causing mastitis. J Dairy Sci. 2007;90(11):5218–5225. doi: 10.3168/jds.2007-0301. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Hui W, Litherland GJ, Barter MJ, Davidson R, Darrah C, Donell ST, Clark IM, Cawston TE, Robinson JH, Rowan AD, Young DA. Differential Toll-like receptor-dependent collagenase expression in chondrocytes. Ann Rheum Dis. 2008;67(11):1633–1641. doi: 10.1136/ard.2007.079574. [DOI] [PubMed] [Google Scholar]