

Abstract
Purpose
The Ewing Sarcoma Family of Tumors (ESFTs) comprises a group of aggressive, malignant bone and soft tissue tumors that predominantly affect children and young adults. These tumors frequently share expression of the EWS-FLI-1 translocation, which is central to tumor survival but not present in healthy cells. In this study, we examined EWS-FLI-1 antigens for their capacity to induce immunity against a range of ESFT types.
Design
Computer prediction analysis of peptide binding, HLA-A2.1 stabilization assays, and induction of Cytotoxic T-Lymphocytes (CTL) in immunized HLA-A2.1 transgenic mice were used to assess the immunogenicity of native and modified peptides derived from the fusion region of EWS-FLI-1 type 1. CTL-killing of multiple ESFT family members in vitro, and control of established xenografts in vivo, was assessed. We also examined whether these peptides could induce human CTLs in vitro.
Results
EWS-FLI-1 type 1 peptides were unable to stabilize cell surface HLA-A2.1 and induced weak CTL activity against Ewing Sarcoma cells. In contrast, peptides with modified anchor residues induced potent CTL killing of Ewing Sarcoma cells presenting endogenous (native) peptides. The adoptive transfer of CTL specific for the modified peptide YLNPSVDSV resulted in enhanced survival of mice with established Ewing Sarcoma xenografts. YLNPSVDSV-specific CTL displayed potent killing of multiple ESFT types in vitro: Ewing Sarcoma, pPNET, Askin’s Tumor, and Biphenotypic Sarcoma. Stimulation of human Peripheral Blood Mononuclear Cells with YLNPSVDSV peptide resulted in potent CTL-killing.
Conclusions
These data show that YLNPSVDSV peptide is a promising antigen for ESFT immunotherapy and warrants further clinical development.
Keywords: Ewing Sarcoma Family of Tumors, Ewing Sarcoma, pPNET, Askin’s Tumor, Biphenotypic sarcoma, EWS-FLI-1, Immunotherapy, vaccine, cancer, HLA-A2.1, HLA-A*0201
INTRODUCTION
Immunotherapy is a promising approach for the treatment of established neoplastic disease. Many tumors share the expression of tumor-specific antigens not found in healthy cells, raising the possibility of invoking immune responses that selectively target diseased cells. Ewing Sarcoma, and the historically named Primitive Neuroectodermal Tumor (pPNET), Biphenotypic Sarcoma and Askin’s tumor, all share recurrent translocations that involve fusion of the EWS gene (22q-12) to genes encoding ETS transcription factors, of which, fusion to the FLI-1 gene occurs in about 90% of cases (1). For this reason, these tumors are now more broadly known as the Ewing Sarcoma Family of Tumors (ESFTs).
ESFTs are among the most aggressive tumors encountered in children and young adults, both morphologically and clinically. They require equally aggressive therapy, including surgery or radiation of the primary tumor site, and intensive multi-agent chemotherapy (2). Presently, ~70% of Ewing Sarcoma patients with localized disease (3), and just 20–30% of patients with metastatic disease (4), survive for 5 years after initial diagnosis; additional therapeutic options are badly needed.
Tumor recurrence is a serious problem following ESFT therapy, and 30–40% of Ewing Sarcoma patients with localized tumors develop recurrent disease (5). Such patients have a 5-year survival rate of just 13% (6). The addition of adjunctive, immune-activating, therapies delivered during the first remission has the potential to reduce recurrence rates (7), both through the capacity of the immune system to mop up residual disease, and through the stimulation of immune “memory”. The junction region where the EWS and FLI-1 genes fuse, also known as the breakpoint, confers a region of novelty in the EWS-FLI-1 protein, and therefore provides a means to target selectively the immune system to EWS-FLI-1+ tumor cells. At least 11 subtypes of the EWS-FLI-1 gene exist in which exons 7, 9, or 10 from EWS are combined with exons 4, 5, 6, 7, or 8 from FLI-1 (8). Although the breakpoint sequences of each subtype are different, EWS-FLI-1 Type 1 fusions (EWS exon 7 fused to FLI-1 exon 6) are present in approximately 57% of cases (9). Thus, immunotherapies targeting the EWS-FLI-1 Type 1 protein may provide clinical benefit to approximately half of all ESFT patients.
In this study we investigated the capacity of peptides derived from EWS-FLI-1 Type 1, presented on a common HLA-type, HLA-A2.1, to activate CTL capable of recognizing and killing ESFT carrying the EWS-FLI-1 Type 1 translocation. Anticipating that native peptides would be poorly immunogenic (10), we also studied the immune-stimulating properties of native peptides modified at key binding residues in order to improve the binding affinity for HLA-A2.1. We show that EWS-FLI-1 Type 1-derived peptides have a poor capacity to activate CTL, resulting in weak CTL killing of ESFT cells. The modification of key binding residues, in contrast, results in potent CTL activation, strong CTL killing across a range of ESFT types and increased survival of mice with established Ewing Sarcoma xenografts.
MATERIALS AND METHODS
Mice
HLA-A2/Kb transgenic mice were bred in our facility with the kind consent of Dr. L. Sherman (The Scripps Research Institute, La Jolla, CA) (11). SCID/beige mice were purchased from Taconic (Germantown, NY, USA). Experiments were conducted under an animal protocol approved by the Animal Care and Use Committee at Beth Israel Deaconess Medical Center
Antibodies and reagents
Anti-HLA-A2.1 mAb (BB7.2), and anti-CD8 (53–6.7) were purchased from BD biosciences. Anti-CD40 (3/23) was from Serotec (Oxford, UK). PE-HLA-A2.1/YLNPSVDSV, and PE-HLA-A2.1/GILGFVFTL MHC Pentamers were from ProImmune (Oxford, UK). All peptides were synthesized by Thinkpeptides (ProImmune Ltd). Modified peptide sequences were compared against a human protein sequence database (http://web.expasy.org/blast/) performed at the Swiss Institute of Bioinformatics using the BLAST network service and the NCBI BLAST 2 software; no homology to human proteins was observed.
Cell lines
TC-71 and TC-32 cells were a kind gift from Dr. Aykut Üren, Georgetown University Medical Center, Washington, D.C (12). Rh1 (EW8) cells were a kind gift from Dr. Peter Houghton, Nationwide Children's Hospital, Columbus, Ohio (13). TC-174 and TTC-547 were a kind gift from Prof. Timothy Triche, University of Southern California, Los Angeles, CA (14). T2, SK-PN-DW and SK-ES-1 were from the ATCC, initially authenticated by STR profiling, and passaged for less than 6 months following resuscitation. A673 and EWS502 were provided by S.L., and WE-68, SK-N-MC, STA-ET-1, and TC-83 by F.V.V. RD-ES was a kind gift from Dr. Lee Helman, National Cancer Institute, Bethesda, MD.
Screening of ESFT cell lines
HLA-A2.1 expression was determined by FACS. Cells with low or undetectable HLA-A2.1 expression were cultured for 48 hours in the presence of 300U/ml rhIFN-γ (R&D Systems) to increase surface MHC expression, and re-analysed. The presence of the EWS-FLI-1 type 1 (EWS exon 7 fused to FLI-1 exon 6) or EWS-FLI-1 type 2 (EWS exon 7 fused to FLI-1 exon 5) translocations was determined by RT-PCR as described (15).
Target peptide prediction
The online “SYFPEITHI” database (http://www.syfpeithi.de/) was used to predict HLA-A2.1:peptide binding affinities, and the “BIMAS” database (http://www-bimas.cit.nih.gov/molbio/hla_bind/) to predict HLA-A2.1: peptide dissociation half-times. Binding to HLA-A2.1 was assessed using the REVEAL MHC-Peptide Binding Assay service of Proimmune Ltd.
T2 assay for peptide stabilization of cell-surface HLA-A2.1
Peptide stabilization of cell-surface MHC molecules was assessed using human T2 cells (TAP-, HLA-A2.1+) as described (23).
CTL killing assays
HLA-A2.1/Kb mice were immunized i.d. (100µl; both flanks) with 200µg test peptides in conjunction with CASAC (Combined Adjuvant for Synergistic Activation of Cellular Immunity) - a potent adjuvant for the activation of CD8 T-cells (16) comprising 50µg CpG1826 (Invitrogen), 25µg anti-CD40, and 100ng rmIFN-γ (R&D Systems) emulsified into the Sigma Adjuvant System (Sigma). After 8 days splenocytes and inguinal lymph nodes cells were harvested and restimulated in vitro for 5 days with 1µg/ml peptide. CD8 cells were negatively selected using MACS beads (Miltenyi biotech), and cultured with CFSE-labeled human ESFT targets. After 5 hours, cell death was assessed by 7-AAD co-staining of CFSE-labeled target cells using a FACSCanto II Analyzer as described (17). An irrelevant peptide, GILGFVFTL (Influenza A matrix peptide 58–66), was used to determine non-specific CTL killing, which was subtracted from all subsequent test data.
In vitro generation of Human CTL
Characterized, cryopreserved human PBMC were purchased from Cellular Technology Ltd (Cleveland, OH, USA). HLA-A2+ donors were selected based on known recall reactivity to Influenza A peptides. PBMC were cultured for 4 weeks to generate CTL according to an established protocol (18). To assess CTL activity, PBMC were cultured with CFSE-labeled TC-71 cells as above at a ratio of 20:1.
Creation of TC-71-Luc
A lentiviral vector encoding both green fluorescent protein and firefly luciferase was used for the transduction of TC-71 cells. The vector was generated using a third generation packaging system (19), co-transfecting 293T cells (System Biosciences) with the lentiviral expression plasmid pGreenFire1-CMV (System Biosciences) and packaging plasmids pMDLg/pRRE, pRSV.Rev, and pMD.G (Addgene) using Lipofectamine 2000 (Invitrogen). A 1:1 mix of 293T supernatant and fresh medium containing Polybrene (Sigma; final concentration of 8 µg/ml) was added to TC-71 cells at ~25% confluence; plates were spun at 1,000g for 60 min at 32°C to enhance transduction efficiency (20). The virus-containing media were then replaced with fresh media and the cells were cultured for 48 hours before assessment of GFP expression by FACS.
TC-71-Luc xenograft treatment model
SCID/beige were inoculated i.v. with 2×106 TC-71-Luc cells. Five and 12 days later, PBS (no T-cell control) or 3×106 CD8 T-cells derived, cultured, and negatively selected as described in CTL killing assays above were adoptively transferred i.v. Tumor growth was monitored by bioluminescence imaging. Blind assessments of animal health were performed.
Bioluminescence Imaging
The Xenogen IVIS-50™ Bioluminescence Imaging System was used to assess tumor growth. SCID/beige mice were anesthetized with isoflurane and injected i.p. with 150 mg/kg of D-luciferin (Caliper LifeSciences) in PBS. Ten minutes after injection, and under continued isoflurane inhalation, the mice were imaged for a period of 5 minutes. Using Living Image® software (Xenogen), the luminescence signal was represented as a heat map superimposed over a grayscale photograph of the animal.
MRI Imaging
Mouse tumor images were acquired on an ASPECT Model M2 1T tabletop MRI scanner (ASPECT Magnet Technologies Ltd., Netanya, Israel). VivoQuant (inviCRO, Boston MA) software was used to visualize DICOM datasets.
Statistical analysis
Statistical comparisons of mean values were performed using unpaired Student’s t-test. Statistical comparisons of survival curves were performed using the logrank test with the null hypothesis that treatments did not change survival. P < 0.05 (*) was considered significant. P < 0.005 (**) and P < 0.001 (***) are indicated.
RESULTS
Identification of EWS-FLI-1 Type 1 target peptides
The affinity with which peptides bind to MHC Class-I is critical for the induction of CTL activation. Therefore, we used the SYFPEITHI and BIMAS databases to predict the binding affinity of native peptide sequences that span the breakpoint region of EWS-FLI-1 type 1 to HLA-A2.1. For each peptide that spans the fusion region between EWS and FLI-1, a relatively low affinity score was predicted (Table 1). Of these, highest SYFPEITHI score, 15, was predicted for QQNPSYDSV.
Table 1.
Binding efficiencies of native 9-mer peptides spanning the fusion region in EWS-FLI-1 Type 1
| Amino-Acids | Peptide sequence | SYFPEITHI | BIMAS |
|---|---|---|---|
| 264–272 | SSSYGQQNP | 3 | 0.000 |
| 265–273 | SSYGQQNPS | 6 | 0.002 |
| 266–274 | SYGQQNPSY | 5 | 0.000 |
| 267–275 | YGQQNPSYD | 2 | 0.002 |
| 268–276 | GQQNPSYDS | 3 | 0.008 |
| 269–277 | QQNPSYDSV | 15 | 5.183 |
| 270–278 | QNPSYDSVR | 1 | 0.000 |
N – The fusion point between EWS and FLI-1 is marked by the creation of a codon for an asparagine residue not found in either of the parent genes.
The low predicted binding scores for these peptides suggested that these sequences would not bind with the affinity required to induce CTL activation, and would therefore not be suitable for use in targeting CTL responses against cells carrying the EWS-FLI-1 Type 1 translocation. However, appropriately modified tumor peptides can often prime the immune response to act against native tumor peptides incapable of inducing an immune response on their own (21). We therefore substituted key anchor residues in 3 native peptides with the most promising SYFPEITHI affinity scores: SYGQQNPSY (score = 5), SSYGQQNPS (score = 6), and QQNPSYDSV (score =15), according to previously described HLA-A2.1-binding principles (22). As expected, the predicted binding affinities of peptides modified in this manner increased substantially both SYFPEITHI and BIMAS scores following the substitution of residues at positions 1, 2, 6, and/or 9 with Tyrosine (Y), Leucine (L), Isoleucine (I) and/or Valine (V: see Table 2).
Table 2.
Native junction peptide modifications and predicted binding affinity
| Peptide I.D. | Peptide sequence | SYFPEITHI | BIMAS |
|---|---|---|---|
| 1 | SYGQQNPSY | 5 | 0.000 |
| 2 | SLGQQNPSY | 15 | 0.075 |
| 3 | SYGQQNPSL | 15 | 0.003 |
| 4 | SIGQQNPSL | 23 | 2.937 |
| 5 | SIGQQNPSV | 23 | 9.563 |
| 6 | SLGQQNPSV | 25 | 69.552 |
| 7 | SLGQQNPSL | 25 | 21.362 |
| 8 | SLGQQVPSL | 29 | 49.134 |
| 9 | SSYGQQNPS | 6 | 0.002 |
| 10 | SLYGQQNPS | 16 | 0.238 |
| 11 | SIYGQQNPV | 24 | 30.603 |
| 12 | SLYGQQNPL | 26 | 68.360 |
| 13 | YLYGQQNPL | 26 | 314.455 |
| 14 | QQNPSYDSV | 15 | 5.1.83 |
| 15 | QQNPSVDSV | 19 | 7.947 |
| 16 | QQNPSLDSV | 19 | 7.947 |
| 17 | QINPSYDSV | 23 | 7.029 |
| 18 | QLNPSYDSV | 25 | 51.121 |
| 19 | QLNPSYDSL | 25 | 15.701 |
| 20 | YLNPSYDSV | 27 | 235.155 |
| 21 | QINPSVDSV | 27 | 10.778 |
| 22 | OLNPSYASV | 27 | 104.328 |
| 23 | QLNPSVDSV | 29 | 78.385 |
| 24 | YLNPSVDSV | 31 | 360.571 |
Residues underlined represent modifications from the native sequence.
Because computer models of peptide:MHC binding affinity have only 60–80% predictive accuracy (23), we measured empirically the ability of native and modified peptides to bind to HLA-A2.1. Binding to HLA-A2.1 was detected using the REVEAL MHC-Peptide Binding Assay; an antibody staining assay based on the conformational changes that only occur when peptide, MHC class I, and β2m form a complex. As shown in Figure 1 (A–C), most of the modified peptides, but not all, displayed a stronger binding capacity than the corresponding native peptides from which they were derived. Together, these data show that the HLA-A2.1-binding of native EWS-FLI-1 Type 1 junction peptides can be improved through the modification of anchor residues.
Figure 1. Binding, peptide-mediated MHC stabilization, and CTL induction of native and modified peptides.
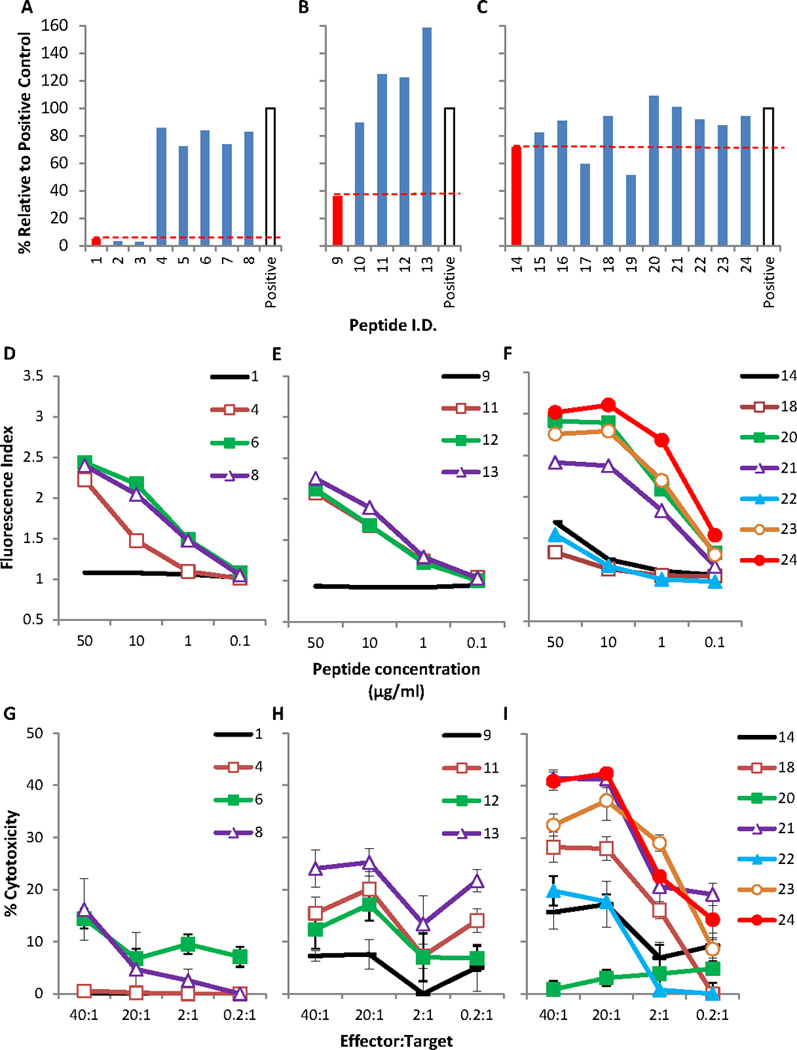
(A–C) Three native peptides (red bars) and 21 modified peptides (blue bars) were analyzed for binding to HLA-A2.1 using the REVEAL MHC-Peptide Binding Assay. Peptide sequences are shown in Table 2: (A) Native peptide SYGQQNPSY (#1) and its modified derivatives, (B) Native peptide SSYGQQNPS (#9) and its modified derivatives, (C) Native peptide QQNPSYDSV (#14) and its modified derivatives. Each peptide was given a score reported quantitatively as a percentage of the signal generated by the test peptide versus a positive control peptide (GILGFVFTL, Flu. open bars). (D–F) The ability of native peptides (D) SYGQQNPSY (#1), (E) SSYGQQNPS (#9), (F) QQNPSYDSV (#14) and their modified derivatives to stabilize HLA-A2.1 molecules. The Fluorescence Index is the median fluorescence intensity (MFI) with the indicated concentration of test peptide divided by the MFI in the absence of peptide. (G–I) CTL induction in HLA-A2/Kb transgenic mice by native peptides (G) SYGQQNPSY (#1), (H) SSYGQQNPS (#9), (I) QQNPSYDSV (#14) and their modified derivatives was assessed through the killing of TC-71 Ewing’s Sarcoma target cells. Graphs show a representative experiment from multiple independent experiments with similar results. Error bars represent SD.
Peptide modification increases stabilization of HLA-A2.1 molecules on the cell surface
We also sought to determine whether modified peptides would show an improved capacity to stabilize MHC molecules on the live cell surface, as the ability of peptide/MHC complexes to remain on the cell surface is critical to the initiation of T-cell activation. We selected modified peptides with a predicted SYFPEITHI score greater than 21 and improved binding to HLA-A2.1 for further study. Human T2 cells express HLA-A2.1 but lack Transporters associated with Antigen Processing (TAP); consequently their cell surface MHC molecules are empty. The association of exogenously added peptides with the empty HLA-A2.1 molecules stabilizes them and results in increased levels of cell surface HLA-A2.1 molecules, recognizable by monoclonal antibodies and flow cytometry. Unlike the native peptide sequences SYGQQNPSY (#1) and SSYGQQNPS (#9) from which they were derived, each of the modified peptides (#4, #6, #8, and #11, #12, and #13 respectively) showed improved stabilization of cell surface MHC (Fig. 1D–E). The native sequence QQNPSYDSV (#14) was found to stabilize cell surface MHC molecules at a high concentration (50µg/ml), but not at low concentrations <10µg/ml (Fig. 1F). Two of the modified QQNPSYDSV peptides (#18 and #22) did not show improved MHC stabilization when compared to the native sequence QQNPSYDSV (#14). The remaining four peptides (#20, #21, #23, and #24) revealed an excellent capacity for MHC stabilization, even at a 50-fold reduction in peptide concentration (1µg/ml). One peptide in particular, YLNPSVDSV (#24), was found to stabilize MHC to a similar extent as 50µg/ml of the native peptide QQNPSYDSV (#14) at a 500-fold lower peptide concentration (0.1µg/ml). These data show that modifying native EWS-FLI-1 Type 1 junction peptides can result in better retention of peptide/MHC complexes on the cell surface.
CTL raised against modified peptides display improved killing of Ewing Sarcoma cells
A critical question when modifying tumor peptides to induce CTL activation is whether or not CTL raised against the modified peptide will recognize and kill tumor cells expressing the native peptide. We conducted CTL assays using purified CD8 T-cells derived from HLA-A2.1/Kb mice immunized with native or modified peptides. We used an unmodified human Ewing sarcoma cell line: TC-71, as a tumor target, which we confirmed as HLA-A2.1+ by flow cytometry (data not shown). The EWS-FLI-1 Type 1 translocation was also confirmed by RT-PCR using TaqMan probes (data not shown). As shown in Figure 1G, the native peptide SYGQQNPSY (#1) was not able to induce a detectable CTL response, and the CTL responses induced by the three modified SYGQQNPSY peptides (#4, #6, and #8) were weak (≤15% killing). The native peptide SSYGQQNPS (#9) also induced weak CTL activity (<10%: Fig. 1.H), although each of the three modified SSYGQQNPS peptides (#11, #12, and #13) induced an appreciable increase in CTL activity, with up to ~25% killing. Interestingly, these 3 peptides all induced strong CD8 T-cell proliferation during in vitro restimulation (data not shown). However this did not appear to correlate with a high level of target cell killing. Of the native peptides, QQNPSYDSV (#14) induced the strongest CTL response with ~15–18% killing (Fig. 1.I). Surprisingly, its modified derivative, YLNPSYDSV (#20), did not induce CTL killing despite displaying an increased capacity to stabilize cell-surface MHC (see Fig 1.F). The strongest CTL activity was displayed by modified peptides QINPSVDSV (#21) and YLNPSVDSV (#24) which both displayed over 40% killing. Peptide YLNPSVDSV (#24) was found to be considerably more immunogenic than native peptide QQNPSYDSV (#14), as evidenced by the consistently increased size of draining lymph nodes following vaccination (Supplementary Fig. S1).
Collectively, these data show that the CTL-inducing capacity of native EWS-FLI-1 Type 1 junction peptides presented on HLA-A2.1 is low, but modification of key MHC-binding anchor residues can result in the activation of CTL clones capable of recognizing and killing unmodified tumor cells expressing native EWS-FLI-1 Type 1 junction peptides. Due to its consistent ability to induce strong immune activation, peptide YLNPSVDSV (#24) was selected for further studies.
Adoptive immunotherapy with EWS-FLI-1 Type 1 –targeting CTL results in enhanced survival of tumor-bearing mice
The TC-71 Ewing sarcoma cell line is known to be tumorigenic in SCID/beige mice (12), and has been used for preclinical in vivo studies for Ewing Sarcoma (12, 24). Following the stable transduction of the firefly luciferase gene into TC-71 cells (TC-71-Luc), TC-71-Luc cells were transferred i.v. into SCID/beige mice. On days 5 and 12 post-tumor challenge, we adoptively transferred CD8 T-cells derived from HLA-A2.1/Kb mice immunized with YLNPSVDSV (#24) or irrelevant control peptide (Flu: GILGFVFTL). Bioluminescence imaging of the mice 21 days post-tumor challenge indicated fewer mice with detectable tumors in the YLNPSVDSV-treated group (Fig. 2A). Treatment of the xenografts with T-cells specific for YLNPSVDSV significantly increased mouse survival time when compared with untreated mice or mice receiving T-cells recognizing an irrelevant peptide (Flu: GILGFVFTL) (Fig. 2B, P < 0.05). Ovaries and kidneys/adrenals were a strong metastatic site for these tumors (Fig. 2C). Figure 2D depicts a paraspinal suprarenal tumor imaged in a mouse recipient of EWS-FLI-1 type 1-targeting T-cells. These data are consistent with a recent study (25) in which Rag2-/-;γc-/- mice, hosts for TC-71 Ewing Sarcoma xenografts. We did not see a correlation between treatment group and tumor location. The results indicate that the adoptive transfer of YLNPSVDSV-specific CTL, but not irrelevant control CTL, into mice with established Ewing Sarcoma xenografts results in enhanced survival.
Figure 2. Adoptive transfer of T-cells into mice with established xenograft tumors.
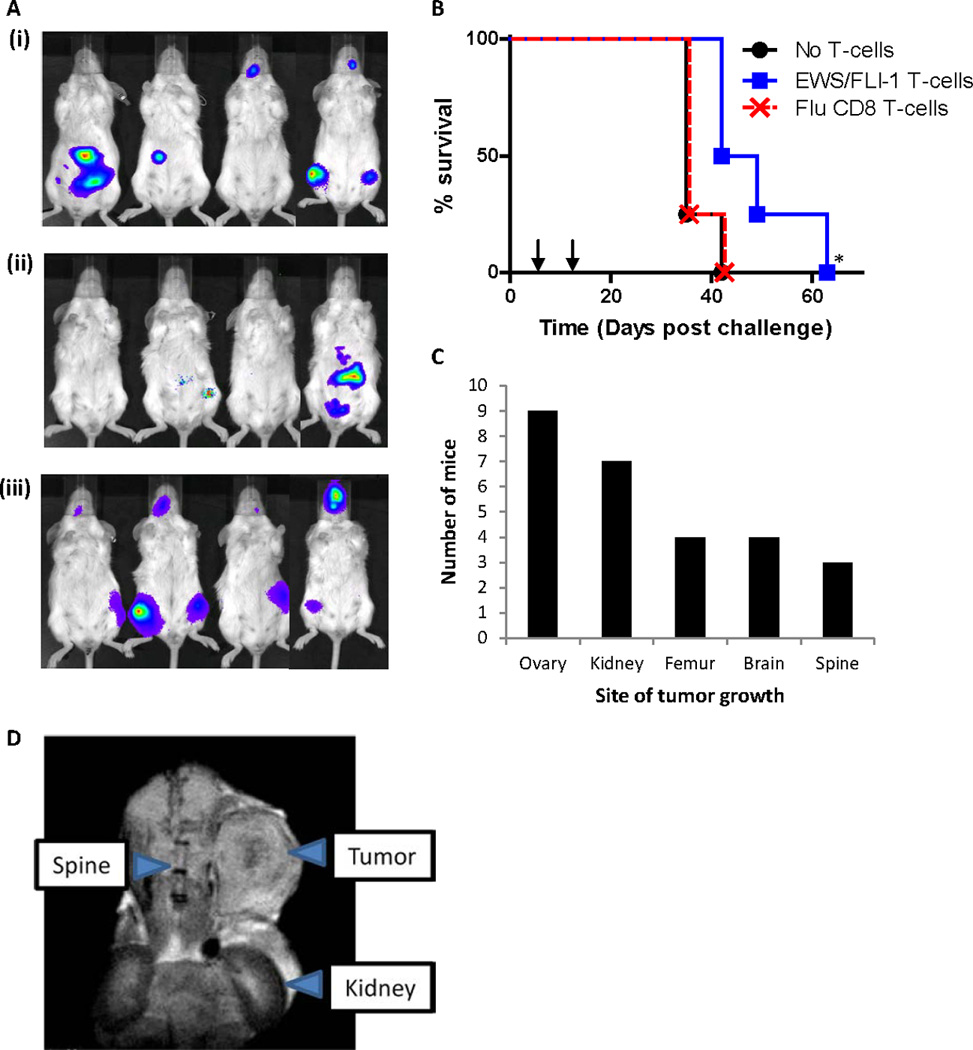
SCID/beige mice were challenged with viable TC-71-Luc cells i.v. and treated on days 5 and 12 with CD8 T-cells from immunized HLA-A2/Kb mice (n=4). (A) Bioluminescence imaging of mice 21 days post tumor challenge with: (i) PBS alone (No T-cells), (ii) EWS-FLI-1 (YLNPSVDSV-specific) T-cells, and (iii) Flu (GILGFVFTL-specific) T-cells. (B) Mouse survival post tumor challenge. Treatments are represented by arrows. *indicates significant increase (p<0.05) in survival of mice recipient of EWS-FLI-1 (YLNPSVDSV-specific) T-cells over both control groups. (C) Tumor frequency in autopsied mice. (D) MRI imaging of abdominal tumor mass in a mouse recipient of EWS-FLI-1 (YLNPSVDSV-specific) T-cells.
EWS-FLI-1 Type 1 –targeting CTL kill multiple members of the Ewing Sarcoma Family of Tumors
The ESFTs are grouped together because they share expression of fusion genes like the EWS-FLI-1 translocation. We therefore asked whether multiple ESFT members would be susceptible to EWS-FLI-1- targeted CTL killing. An array of human ESFT tumor lines were screened to identify lines that are both HLA-A2.1+ and carry the EWS-FLI-1 Type 1 translocation. As shown in Table 3, 11 suitable lines were identified spread across multiple different EWS-FLI-1+ ESFT tumor types. We identified a further 3 EWS-FLI-1 Type 1+ cell lines of unknown tumor specificity (“ES Cell lines”). Three negative control lines were also identified: RD-ES (EWS-FLI-1 Type 1-, HLA-A2.1-), SK-N-MC (EWS-FLI-1 Type 1+, HLA-A2.1-), and SK-ES-1 (EWS-FLI-1 Type 1-, HLA-A2.1+). We conducted CTL assays using purified CD8 T-cells derived from HLA-A2.1/Kb mice immunized with native (QQNPSYDSV #14) or modified (YLNPSVDSV #24) peptides. As shown in Figure 3A, killing by CTL specific for EWS-FLI-1 Type 1 (above 10%, and after subtraction of non-specific CTL killing) was displayed against 8 out of the 11 target lines, including at least one example of killing in each of the ESFT tumor types tested. Furthermore, YLNPSVDSV-specific CTL (#24) showed significantly improved killing against 7 out of these 8 lines when compared with QQNPSYDSV-specific CTL (#14). CTL killing directed against the negative control lines (RD-ES, SK-N-MC, and SK-ES-1), in which EWS-FLI-1 Type 1-peptides are not presented on HLA-A2.1 molecules, was not significantly above background, as expected. These data show that CTL which target the EWS-FLI-1 Type 1 translocation display potent killing of multiple EWS-FLI-1 Type 1+ expressing cells in an HLA-A2.1 dependent manner. These data also underline the improved cytotoxic capacity of CTL raised using a modified (YLNPSVDSV #24) EWS-FLI-1 Type 1 peptide in comparison to CTL raised using the native (QQNPSYDSV #14) peptide.
Table 3.
Summary of ESFT cell lines studied.
| Cell line | ESFT type | EWS-FLI-1 type |
HLA-A2.1 | IFN-γ | Description | Ref |
|---|---|---|---|---|---|---|
| TC-71 | ES | 1 | ++ | +++ | Recurrent tumor of the humerus derived from a 22-year old male |
(38) |
| WE-68 | ES | 1 | +++ | Primary tumor derived from the fibula of a 19-year old female |
(39) | |
|
A673> (RMS1598) |
Unknown | 1 | ++ | +++ | Unknown subtype: Derived from the muscle of a 15-year old female. Initially believed to be a rhabdomyosarcoma. |
(40) |
| EWS502 | Unknown | 1 | + | +++ | Unknown subtype | (41) |
| Rh1 (EW8) | Unknown | 1 | +++ | Unknown subtype: Initially believed to be an alveolar rhabdomyosarcoma. |
(13) | |
| TC-32 | pPNET | 1 | +++ | Primary tumor derived from the pelvis of a 17-year old female |
(38 | |
| SK-PN-DW | pPNET | 1 | + | +++ | Primary tumor derived from the presacrum of a 17-year old male |
(42) |
| STA-ET.1 | pPNET | 1 | ++ | +++ | Recurrent tumor of the humerus derived from a 13-year old female |
(43) |
| TTC-547 | BPT | 1 | − | +++ | Primary tumor derived from the retroperitoneum of a 13-year old female |
(14) |
| TC-174 | BPT | 1 | +++ | Primary tumor originating in the soft tissue of the thigh of a 9-year old female |
(14) | |
| TC-83 | AT | 1 | +++ | Derived from the chest wall of a 13-year old female |
(44) | |
| RD-ES | ES | 2 | − | − | Derived from the humerus of a 19-year old male. |
(45) |
| SK-N-MC | pPNET/AT | 1 | − | − | Derived from the supraorbital metastasis of 14-year old female |
(46) |
| SK-ES-1 | pPNET | 2 | + | +++ | Isolated from an 18-year old male | (47) |
ES: Ewing’s Sarcoma, Unknown: Tumor type undefined, pPNET: peripheral Primitive NeuroEctodermal Tumor, AT: Askin’s Tumor, and BPT: Biphenotypic sarcoma. Percent HLA-A2.1 expression as determined by FACS: Not detected: (−), 1–40: (+), 41–89: (++), 90–100: (+++)). IFN-γ: HLA-A2.1 expression following 48 hours in the presence of 300U/ml rhIFN-γ.
Figure 3. Peptide-mediated CTL killing of multiple ESFT targets.
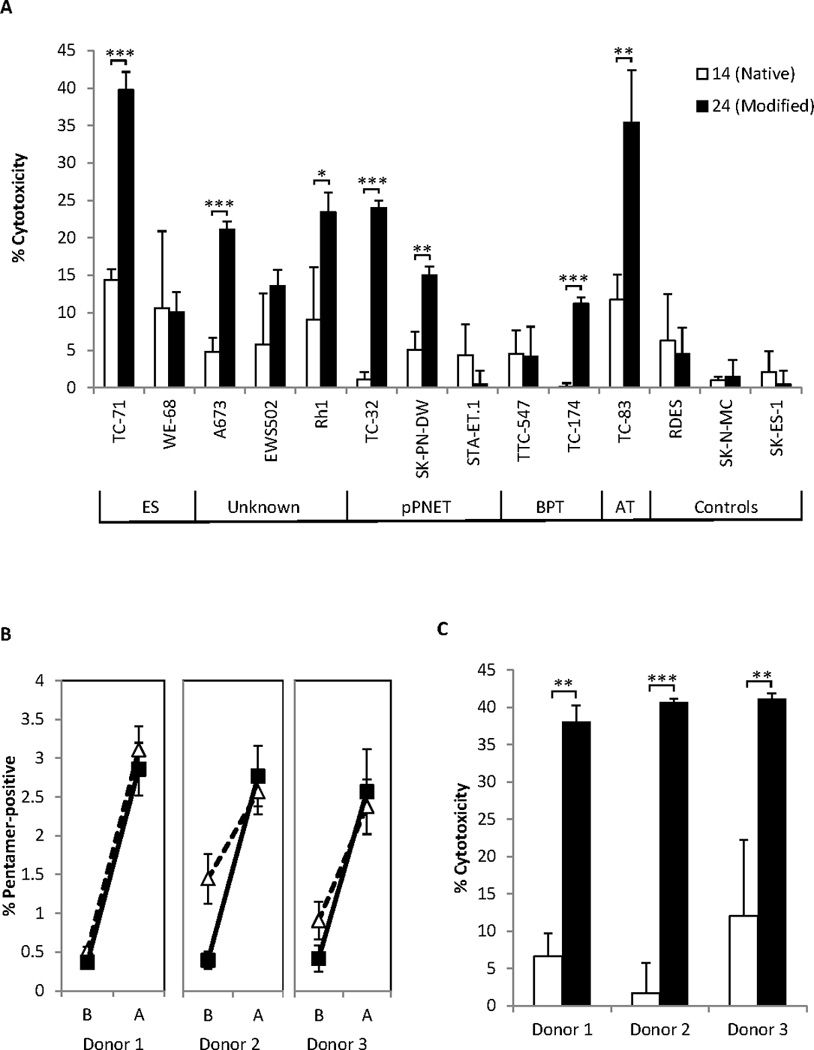
(A) CTL were induced in HLA-A2/Kb mice by immunization with native peptide QQNPSYDSV (#14) or its modified derivative YLNPSVDSV (#24). The capacity of CTL to kill 11 EWS-FLI-1 Type 1+, HLA-A2.1+ ESFT targets (described in Table 3) was subsequently assessed. Graphs show combined data from 3 independent experiments with similar results. Error bars represent SEM. (B–C) Healthy human PBMC from 3 different HLA-A2.1+ donors were stimulated with native- QQNPSYDSV (#14) or modified - YLNPSVDSV (#24) peptide for 4 weeks to generate CTL. (B) Expansion of human CTL lines. Pentamer-positive staining as a percentage of total CD8 cells. Closed squares = YLNPSVDSV, open triangles = GILGFVFTL, B = Before culture, A = After culture. Graphs show combined data from 3 independent experiments. Error bars represent SEM. (C) EWS-FLI-1 Type 1-targeted cytotoxic activity using TC-71 cells as targets. White bars = QQNPSYDSV (#14), Black bars = YLNPSVDSV (#24). Graphs show combined data from 2 independent experiments. Error bars represent SD.
Stimulation of PBMCs from healthy human donors with modified EWS-FLI-1 Type 1 peptide induces T-cell expansion and potent EWS-FLI-1 Type 1-targeted CTL Activity
Since our results suggested that YLNPSVDSV (#24) peptide could induce EWS-FLI-1-directed CTLs in the HLA-A2.1/Kb mouse, we expected this peptide to be an epitope for human CTL. In the absence of available blood from HLA-A2.1+ patients known to carry the EWS-FLI-1 Type 1 translocation, we attempted to generate CTL from the PBMCs of healthy donors.
Pre-characterized PBMC from 3 different HLA-A2.1+ donors with known recall responses Influenza A peptides were stimulated with native (QQNPSYDSV #14), modified (YLNPSVDSV #24), or positive control (GILGFVFTL; Flu) peptide. The degree of peptide-specific CD8 T-cell clonal expansion in the modified- and control- peptide populations was assessed through CD8 and MHC pentamer co-staining. Due to the low binding affinity of the native peptide to HLA-A2.1 we were unable to make HLA-A2.1/QQNPSYDSV pentamer.
As shown in Fig. 3B, the baseline level of YLNPSVDSV-specific CD8 T-cells in unstimulated PBMC was low (~0.4%) compared with GILGFVFTL peptide (~0.5–1.4%); this is expected, due to the prior exposure of these patients to Influenza A. After 4 weeks of peptide stimulation, however, the level of CD8 T-cells specific for YLNPSVDSV peptide expanded to between 2.57% and 2.86%. We further examined the cytotoxic activity of these CTLs against TC-71 cells. As shown in Figure 3C, for each of the 3 donors tested YLNPSVDSV-specific CTL displayed substantially improved killing of TC-71 Ewing sarcoma cells compared to QQNPSYDSV-specific CTL. We also saw killing of TC-71 cells using YLNPSVDSV-specific CTL derived from a further 2 healthy donors, although comparative native-peptide specific CTL assays were not performed due to lack of expansion of these cells in culture (data not shown). These results identify CD8 T-cells capable of recognizing a modified (YLNPSVDSV #24) peptide from the junction region of the EWS-FLI-1 Type 1 translocation in the peripheral blood of healthy donors, and show that they can be readily activated by peptide, to kill efficiently cells bearing the EWS-FLI-1 Type 1 translocation.
DISCUSSION
The EWS-FLI-1 oncogene is well recognized as a highly promising therapeutic target for ESFT (26). The complete lack of EWS-FLI-1 expression in healthy cells, coupled with the absolute requirement of diseased cells to maintain expression of this gene (27, 28), have led to the development of several EWS-FLI-1-targeting therapies in recent years. While many of these studies have concentrated on disrupting the function of this oncogene (29, 30), studies are also being conducted which attempt to activate the immune system specifically to target cells carrying EWS-FLI-1.
In a pilot clinical trial conducted by Dagher and colleagues (31), EWS-FLI-1-targeted immunotherapy was investigated using native junction peptides. In this study, patient peripheral blood leukocytes (PBL) were pulsed with long (15–18aa) peptides spanning the breakpoint region for both EWS-FLI-1 Type 1 and Type 2, irradiated, and injected intravenously into patients, who were then put on IL-2 therapy. No patients showed any evidence of peptide-specific cytokine release, and all patients suffered toxicity as a result of IL-2 therapy. However, this study was primarily a safety study incorporating immune compromised patients with bulky, rapidly progressing tumors. Importantly, no evidence of peptide-induced toxicity was found.
A subsequent study by the same group (7) demonstrated improved 5-year survival rates in patients following the transfer of autologous T-cells, harvested pre-chemotherapy and reintroduced post-chemotherapy, in combination with long breakpoint peptide-pulsed dendritic cell vaccines. However, survival did not correlate with immune responses to peptide vaccines, which were poor in comparison to responses induced by a commercially available Flu vaccine delivered around the same time as the dendritic cell/peptide vaccines. Thus, improved survival may have been attributed to immune reconstitution rather than EWS-FLI-1-targeted immunotherapy. Subsequently, the authors highlighted the need for the discovery of more immunogenic EWS-FLI-1 antigens and more potent vaccine-delivery systems.
Since Caucasians have the highest incidence of Ewing sarcoma (32), and HLA-A2.1 is present in approximately 40% of Caucasians (33), we focused our study on HLA-A2.1-restricted presentation of EWS-FLI-1 Type 1 peptides. Our results suggest that native peptides from the breakpoint region of EWS-FLI-1 Type 1 bind with weak affinity to HLA-A2.1, resulting in poor stability of peptide/MHC complexes on the cell surface. Despite co-immunizing HLA-A2/Kb mice with native peptides in combination with a potent CTL-inducing adjuvant (CASAC) (16, 34), strong CTL activity could not be induced. In contrast, modified peptides, in which we substituted key binding residues of the native peptides, displayed improved HLA-A2.1-binding affinity, an enhanced capacity to stabilize cell-surface HLA-A2.1 molecules, and induction of potent CTLs targeting EWS-FLI-1 Type 1. Importantly, CTL raised using modified peptides were able to recognize and kill tumor cells endogenously expressing native EWS-FLI-1 Type 1 peptides.
CTL induced with modified peptide (YLNPSVDSV) and recognizing EWS-FLI-1 Type 1 were adoptively transferred into immune compromised mice bearing established TC-71 xenografts. This produced enhanced survival, but not a cure. In the absence of host regulatory T-cell (T-reg) activity in our mice, it is likely that the numbers of transferred T-cells were simply insufficient to control the aggressive growth of TC-71 cells. Although not investigated in the present study, this model might be an attractive one with which to examine the combined influence of conventional cytoreductive therapies, such as chemotherapy, radiation, and surgery to reduce significantly the tumor load, with subsequent immunotherapy to prevent tumor recurrence.
YLNPSVDSV-specific CTL were found to be capable of killing multiple ESFT tumor types. In 7 out of the 8 target lines sensitive to CTL-killing, these CTL significantly improved killing when compared with CTL raised against the native peptide QQNPSYDSV (#14). Interestingly, we did not see convincing CTL killing against 3 cell lines: WE-68, STA-ET.1 and TTC-547, despite the pre-incubation of these cells with 300U/ml rhIFN-γ to increase surface MHC expression. This raises the possibility that these tumor lines have a defect in the processing or presentation of endogenous EWS-FLI-1 peptides on their cell surface. Alternatively, it has been demonstrated that some ESFT lines down-regulate caspase-8 in culture, rendering them resistant to Fas-mediated killing (35).
The successful activation of CD8 T-cells from healthy human PBMC using the modified YLNPSVDSV peptide indicates that CD8 T-cell clones capable of recognizing this sequence exist and are not depleted during selection in the thymus. Furthermore, YLNPSVDSV-specific CTL can be readily activated by peptide to result in the potent killing of cells bearing the EWS-FLI-1 Type 1 translocation. Subsequent experiments need to be carried out to ensure that YLNPSVDSV-specific T-cells present in the blood of patients with EWS-FLI-1 Type 1+ HLA-A2.1+ are similarly responsive to stimulation with YLNPSVDSV. For peptide-based ESFT vaccines to reach their full potential, a library of peptides known to activate EWS-FLI-1-targeting CTL across multiple HLA-restrictions may need to be created. In addition, immunogenic MHC Class II-restricted peptides spanning the EWS-FLI-1 breakpoint region (10) merit further investigation for their capacity to induce tumor-specific helper T-cell responses, as ultimately a multi-pronged approach is likely to yield the most effective antitumor immunity.
Due to the aggressive growth rates of pediatric sarcomas, the propensity of advanced-stage tumors to down-regulate MHC expression (36), and the importance of MHC class I expression for patient survival (37), both the timing and partnering of immunotherapies with cytoreductive therapies for ESFT warrant further investigation.
Supplementary Material
Translational Relevance.
EWS-FLI-1 is a novel translocation expressed by the large majority of Ewing Sarcoma Family of Tumors (ESFTs). This translocation is known to be critical for the persistence of disease, and is absent from normal, healthy, cells. EWS-FLI-1 is therefore a highly desirable target for immunotherapy. To date two pilot clinical trials have attempted to induce EWS-FLI-1-targeted immune responses in the clinic. Although both studies reported modest, yet encouraging, increases in survival expectancy, evidence for the induction of EWS-FLI-1-specific immune recall responses was weak or absent in most patients; a correlation with an EWS-FLI-1 – specific immune response could not be demonstrated. In this manuscript we show that epitopes derived from the novel fusion region in EWS-FLI-1 are incapable of inducing HLA-A2.1-restricted Cytotoxic T-lymphocytes (CTL) that recognize and kill ESFT. However, CTL that recognize a modified EWS-FLI-1 peptide, YLNPSVDSV, potently kill several different ESFT tumor types. This peptide may significantly improve clinical outcomes for ESFT patients undergoing immunotherapy.
ACKNOWLEDGEMENTS
We thank Linda Sherman for permission to use HLA-A2/Kb transgenic mice. We also thank Aykut Üren, Georgetown University Medical Center, Washington, D.C; Peter Houghton, Nationwide Children's Hospital, Columbus, Ohio; Timothy Triche, University of Southern California, Los Angeles, CA; and Lee Helman, National Cancer Institute, Bethesda, MD, for their generous contribution of rare ESFT cell lines.
GRANT SUPPORT
This work was supported by a grant from the Children’s Cancer Research Fund to JWW.
Abbreviations
- ESFT
Ewing Sarcoma Family of Tumors
- pPNET
peripheral Primitive NeuroEctodermal Tumor
- CTL
Cytotoxic T-Lymphocyte
- MHC
Major Histocompatability Complex
- PBMC
Peripheral Blood Mononuclear Cell
Footnotes
This paper is dedicated to Sam Ward, Ewing’s victim
The authors disclose no potential conflicts of interest
REFERENCES
- 1.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–165. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- 2.Lessnick SL, Dei Tos AP, Sorensen PH, Dileo P, Baker LH, Ferrari S, et al. Small round cell sarcomas. Seminars in oncology. 2009;36:338–346. doi: 10.1053/j.seminoncol.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. The New England journal of medicine. 2003;348:694–701. doi: 10.1056/NEJMoa020890. [DOI] [PubMed] [Google Scholar]
- 4.Subbiah V, Anderson P, Lazar AJ, Burdett E, Raymond K, Ludwig JA. Ewing's sarcoma: standard and experimental treatment options. Current treatment options in oncology. 2009;10:126–140. doi: 10.1007/s11864-009-0104-6. [DOI] [PubMed] [Google Scholar]
- 5.Leavey PJ, Mascarenhas L, Marina N, Chen Z, Krailo M, Miser J, et al. Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children's Oncology Group. Pediatric blood & cancer. 2008;51:334–338. doi: 10.1002/pbc.21618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bacci G, Ferrari S, Longhi A, Donati D, De Paolis M, Forni C, et al. Therapy and survival after recurrence of Ewing's tumors: the Rizzoli experience in 195 patients treated with adjuvant and neoadjuvant chemotherapy from 1979 to 1997. Ann Oncol. 2003;14:1654–1659. doi: 10.1093/annonc/mdg457. [DOI] [PubMed] [Google Scholar]
- 7.Mackall CL, Rhee EH, Read EJ, Khuu HM, Leitman SF, Bernstein D, et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:4850–4858. doi: 10.1158/1078-0432.CCR-07-4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlott T, Nagel H, Ruschenburg I, Reimer S, Droese M. Reverse transcriptase polymerase chain reaction for detecting Ewing's sarcoma in archival fine needle aspiration biopsies. Acta Cytol. 1997;41:795–801. doi: 10.1159/000332706. [DOI] [PubMed] [Google Scholar]
- 9.Zucman J, Melot T, Desmaze C, Ghysdael J, Plougastel B, Peter M, et al. Combinatorial generation of variable fusion proteins in the Ewing family of tumours. The EMBO journal. 1993;12:4481–4487. doi: 10.1002/j.1460-2075.1993.tb06137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyer-Wentrup F, Richter G, Burdach S. Identification of an immunogenic EWS-FLI1-derived HLA-DR-restricted T helper cell epitope. Pediatr Hematol Oncol. 2005;22:297–308. doi: 10.1080/08880010590935194. [DOI] [PubMed] [Google Scholar]
- 11.Vitiello A, Marchesini D, Furze J, Sherman LA, Chesnut RW. Analysis of the HLA-restricted influenza-specific cytotoxic T lymphocyte response in transgenic mice carrying a chimeric human-mouse class I major histocompatibility complex. The Journal of experimental medicine. 1991;173:1007–1015. doi: 10.1084/jem.173.4.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uren A, Merchant MS, Sun CJ, Vitolo MI, Sun Y, Tsokos M, et al. Beta-platelet-derived growth factor receptor mediates motility and growth of Ewing's sarcoma cells. Oncogene. 2003;22:2334–2342. doi: 10.1038/sj.onc.1206330. [DOI] [PubMed] [Google Scholar]
- 13.Smith MA, Morton CL, Phelps D, Girtman K, Neale G, Houghton PJ. SK-NEP-1 and Rh1 are Ewing family tumor lines. Pediatric blood & cancer. 2008;50:703–706. doi: 10.1002/pbc.21099. [DOI] [PubMed] [Google Scholar]
- 14.Sorensen PH, Shimada H, Liu XF, Lim JF, Thomas G, Triche TJ. Biphenotypic sarcomas with myogenic and neural differentiation express the Ewing's sarcoma EWS/FLI1 fusion gene. Cancer research. 1995;55:1385–1392. [PubMed] [Google Scholar]
- 15.Lewis TB, Coffin CM, Bernard PS. Differentiating Ewing's sarcoma from other round blue cell tumors using a RT-PCR translocation panel on formalin-fixed paraffin-embedded tissues. Mod Pathol. 2007;20:397–404. doi: 10.1038/modpathol.3800755. [DOI] [PubMed] [Google Scholar]
- 16.Wells JW, Cowled CJ, Farzaneh F, Noble A. Combined triggering of dendritic cell receptors results in synergistic activation and potent cytotoxic immunity. J Immunol. 2008;181:3422–3431. doi: 10.4049/jimmunol.181.5.3422. [DOI] [PubMed] [Google Scholar]
- 17.Kim GG, Donnenberg VS, Donnenberg AD, Gooding W, Whiteside TL. A novel multiparametric flow cytometry-based cytotoxicity assay simultaneously immunophenotypes effector cells: comparisons to a 4 h 51Cr-release assay. J Immunol Methods. 2007;325:51–66. doi: 10.1016/j.jim.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gritzapis AD, Voutsas IF, Lekka E, Tsavaris N, Missitzis I, Sotiropoulou P, et al. Identification of a novel immunogenic HLA-A*0201-binding epitope of HER-2/neu with potent antitumor properties. J Immunol. 2008;181:146–154. doi: 10.4049/jimmunol.181.1.146. [DOI] [PubMed] [Google Scholar]
- 19.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bunnell BA, Muul LM, Donahue RE, Blaese RM, Morgan RA. High-efficiency retroviral-mediated gene transfer into human and nonhuman primate peripheral blood lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:7739–7743. doi: 10.1073/pnas.92.17.7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maslak PG, Dao T, Gomez M, Chanel S, Packin J, Korontsvit T, et al. A pilot vaccination trial of synthetic analog peptides derived from the BCR-ABL breakpoints in CML patients with minimal disease. Leukemia. 2008;22:1613–1616. doi: 10.1038/leu.2008.7. [DOI] [PubMed] [Google Scholar]
- 22.Ruppert J, Sidney J, Celis E, Kubo RT, Grey HM, Sette A. Prominent role of secondary anchor residues in peptide binding to HLA-A2.1 molecules. Cell. 1993;74:929–937. doi: 10.1016/0092-8674(93)90472-3. [DOI] [PubMed] [Google Scholar]
- 23.Pinilla-Ibarz J, Korontsvit T, Zakhaleva V, Roberts W, Scheinberg DA. Synthetic peptide analogs derived from bcr/abl fusion proteins and the induction of heteroclitic human T-cell responses. Haematologica. 2005;90:1324–1332. [PubMed] [Google Scholar]
- 24.Manara MC, Landuzzi L, Nanni P, Nicoletti G, Zambelli D, Lollini PL, et al. Preclinical in vivo study of new insulin-like growth factor-I receptor--specific inhibitor in Ewing's sarcoma. Clin Cancer Res. 2007;13:1322–1330. doi: 10.1158/1078-0432.CCR-06-1518. [DOI] [PubMed] [Google Scholar]
- 25.Nanni P, Nicoletti G, Landuzzi L, Croci S, Murgo A, Palladini A, et al. High metastatic efficiency of human sarcoma cells in Rag2/gammac double knockout mice provides a powerful test system for antimetastatic targeted therapy. Eur J Cancer. 2010;46:659–668. doi: 10.1016/j.ejca.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 26.Uren A, Toretsky JA. Ewing's sarcoma oncoprotein EWS-FLI1: the perfect target without a therapeutic agent. Future Oncol. 2005;1:521–528. doi: 10.2217/14796694.1.4.521. [DOI] [PubMed] [Google Scholar]
- 27.Toomey EC, Schiffman JD, Lessnick SL. Recent advances in the molecular pathogenesis of Ewing's sarcoma. Oncogene. 2010;29:4504–4516. doi: 10.1038/onc.2010.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erkizan HV, Uversky VN, Toretsky JA. Oncogenic partnerships: EWS-FLI1 protein interactions initiate key pathways of Ewing's sarcoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:4077–4083. doi: 10.1158/1078-0432.CCR-09-2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grohar PJ, Woldemichael GM, Griffin LB, Mendoza A, Chen QR, Yeung C, et al. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. Journal of the National Cancer Institute. 2011;103:962–978. doi: 10.1093/jnci/djr156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erkizan HV, Scher LJ, Gamble SE, Barber-Rotenberg JS, Sajwan KP, Uren A, et al. Novel peptide binds EWS-FLI1 and reduces the oncogenic potential in Ewing tumors. Cell cycle (Georgetown, Tex. 2011;10:3397–3408. doi: 10.4161/cc.10.19.17734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dagher R, Long LM, Read EJ, Leitman SF, Carter CS, Tsokos M, et al. Pilot trial of tumor-specific peptide vaccination and continuous infusion interleukin-2 in patients with recurrent Ewing sarcoma and alveolar rhabdomyosarcoma: an inter-institute NIH study. Medical and pediatric oncology. 2002;38:158–164. doi: 10.1002/mpo.1303. [DOI] [PubMed] [Google Scholar]
- 32.Jawad MU, Cheung MC, Min ES, Schneiderbauer MM, Koniaris LG, Scully SP. Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: an analysis of 1631 cases from the SEER database, 1973–2005. Cancer. 2009;115:3526–3536. doi: 10.1002/cncr.24388. [DOI] [PubMed] [Google Scholar]
- 33.Nijman HW, Houbiers JG, Vierboom MP, van der Burg SH, Drijfhout JW, D'Amaro J, et al. Identification of peptide sequences that potentially trigger HLA-A2.1-restricted cytotoxic T lymphocytes. European journal of immunology. 1993;23:1215–1219. doi: 10.1002/eji.1830230603. [DOI] [PubMed] [Google Scholar]
- 34.Wells JW, Choy K, Lloyd CM, Noble A. Suppression of allergic airway inflammation and IgE responses by a class I restricted allergen peptide vaccine. Mucosal immunology. 2009;2:54–62. doi: 10.1038/mi.2008.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Hooge AS, Berghuis D, Santos SJ, Mooiman E, Romeo S, Kummer JA, et al. Expression of cellular FLICE inhibitory protein, caspase-8, and protease inhibitor-9 in Ewing sarcoma and implications for susceptibility to cytotoxic pathways. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:206–214. doi: 10.1158/1078-0432.CCR-06-1457. [DOI] [PubMed] [Google Scholar]
- 36.Berghuis D, de Hooge AS, Santos SJ, Horst D, Wiertz EJ, van Eggermond MC, et al. Reduced human leukocyte antigen expression in advanced-stage Ewing sarcoma: implications for immune recognition. J Pathol. 2009;218:222–231. doi: 10.1002/path.2537. [DOI] [PubMed] [Google Scholar]
- 37.Yabe H, Tsukahara T, Kawaguchi S, Wada T, Torigoe T, Sato N, et al. Prognostic significance of HLA class I expression in Ewing's sarcoma family of tumors. J Surg Oncol. 2011;103:380–385. doi: 10.1002/jso.21829. [DOI] [PubMed] [Google Scholar]
- 38.Whang-Peng J, Triche TJ, Knutsen T, Miser J, Kao-Shan S, Tsai S, et al. Cytogenetic characterization of selected small round cell tumors of childhood. Cancer Genet Cytogenet. 1986;21:185–208. doi: 10.1016/0165-4608(86)90001-4. [DOI] [PubMed] [Google Scholar]
- 39.van Valen F, Jurgens H, Winkelmann W, Keck E. Beta-adrenergic agonist- and prostaglandin-mediated regulation of cAMP levels in Ewing's sarcoma cells in culture. Biochem Biophys Res Commun. 1987;146:685–691. doi: 10.1016/0006-291x(87)90583-3. [DOI] [PubMed] [Google Scholar]
- 40.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, et al. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. Journal of the National Cancer Institute. 1973;51:1417–1423. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- 41.Lessnick SL, Dacwag CS, Golub TR. The Ewing's sarcoma oncoprotein EWS/FLI induces a p53-dependent growth arrest in primary human fibroblasts. Cancer Cell. 2002;1:393–401. doi: 10.1016/s1535-6108(02)00056-9. [DOI] [PubMed] [Google Scholar]
- 42.Askin FB, Rosai J, Sibley RK, Dehner LP, McAlister WH. Malignant small cell tumor of the thoracopulmonary region in childhood: a distinctive clinicopathologic entity of uncertain histogenesis. Cancer. 1979;43:2438–2451. doi: 10.1002/1097-0142(197906)43:6<2438::aid-cncr2820430640>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 43.Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M. MIC2 is a specific marker for Ewing's sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewing's sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer. 1991;67:1886–1893. doi: 10.1002/1097-0142(19910401)67:7<1886::aid-cncr2820670712>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 44.van Valen F. Ewing's Sarcoma Family of Tumors. In: John RW, Masters BP, editors. Human Cell Culture. Dordrecht: Kluwer Academic Publishers; 1999. pp. 55–85. [Google Scholar]
- 45.Sano K, Nakamura H, Mabuchi S, Tanaka T, Nakagawara A, Takai Y. Expression of the smg p25A (a ras p21-like GTP-binding protein) gene in human neuroblastoma cell lines and tumor tissues. Cancer research. 1990;50:7242–7245. [PubMed] [Google Scholar]
- 46.Biedler JL, Helson L, Spengler BA. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer research. 1973;33:2643–2652. [PubMed] [Google Scholar]
- 47.Bloom ET. Further definition by cytotoxicity tests of cell surface antigens of human sarcomas in culture. Cancer research. 1972;32:960–967. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.