

Abstract
We are the first to report allosterism during styrene oxidation by recombinant CYP2E1 and human liver microsomes. At low styrene concentrations, oxidation is inefficient because of weak binding to CYP2E1 (Ks = 830 μM). A second styrene molecule then binds CYP2E1 with higher affinity (Kss = 110 μM) and significantly improves oxidation to achieve a kcat of 6.3 nmol · min−1 · nmol CYP2E1−1. The transition between these metabolic cycles coincides with reported styrene concentrations in blood from exposed workers; thus, this CYP2E1 mechanism may be relevant in vivo. Scaled modeling of the in vitro-positive allosteric mechanism for styrene metabolism to its in vivo clearance led to significant deviations from the traditional model based on Michaelis-Menten kinetics. Low styrene levels were notably much less toxic than generally assumed. We interrogated the allosteric mechanism using the CYP2E1-specific inhibitor and drug 4-methylpyrazole, which we have shown binds two CYP2E1 sites. From the current studies, styrene was a positive allosteric effector on 4-methylpyrazole binding, based on a 10-fold increase in 4-methylpyrazole binding affinity from Ki 0.51 to Ksi 0.043 μM. The inhibitor was a negative allosteric effector on styrene oxidation, because kcat decreased 6-fold to 0.98 nmol · min−1 · nmol CYP2E1−1. Consequently, mixtures of styrene and other molecules can induce allosteric effects on binding and metabolism by CYP2E1 and thus mitigate the efficiency of their metabolism and corresponding effects on human health. Taken together, our elucidation of mechanisms for these allosteric reactions provides a powerful tool for further investigating the complexities of CYP2E1 metabolism of drugs and pollutants.
Introduction
Styrene is a monocyclic compound that is used primarily in the industrial manufacture of synthetic rubber. Styrene is classified as a class 2B carcinogen because of limited evidence for animal and human carcinogenicity (Bond, 1989; Barale, 1991; National Toxicology Program, 2008) and was included on the 2011 Agency for Toxic Substances and Disease Registry Substance Priority List for Superfund Sites. Bioactivation of styrene involves oxidation to mainly styrene oxide and to a lesser extent 4-vinylphenol by cytochromes P450 (P450s) (Fig. 1) (Bond, 1989). Styrene oxide is a genotoxic metabolite that undergoes cleavage to the less toxic metabolite 1,2-phenylethanediol (styrene glycol). This degradative pathway occurs very slowly in water but is rapidly catalyzed by epoxide hydrolase. CYP2E1 is primarily responsible for the initial critical step in styrene activation to a reactive metabolite (Mendrala et al., 1993; Nakajima et al., 1994; Kim et al., 1997; Wenker et al., 2001), and thus, an understanding of the contribution of CYP2E1 to the metabolic clearance of styrene is necessary for adequately estimating risk associated with exposure to this pollutant.
Fig. 1.

Metabolic pathways for styrene. EH, epoxide hydrolase.
Numerous groups used human liver microsomes (HLM) to measure the efficiency of in vitro styrene oxidation as a model for metabolic clearance of styrene. Kinetic profiles for steady-state reactions demonstrated distinct high- and low-affinity enzyme activities. The high-affinity activity was attributed to CYP2E1 on the basis of correlative activity assays (Kim et al., 1997; Wenker et al., 2001) and inhibitor phenotyping (Koop, 1990; Wenker et al., 2001). Kinetic data were analyzed by the Michaelis-Menten equation to obtain Vmax and Km parameters used for modeling metabolic clearance (Vmax/Km) of the pollutant (Mendrala et al., 1993; Nakajima et al., 1994; Wenker et al., 2001). This strategy demonstrated that a high-affinity (CYP2E1) activity was approximately 100-fold more efficient than a low-affinity activity, indicating that the metabolic efficiency of CYP2E1 dominates styrene metabolism (Wenker et al., 2001).
Nevertheless, the design of those studies may have compromised an adequate analysis of styrene metabolism and parameterization of styrene metabolism by CYP2E1. First, liver microsomes reflect the inherent diversity of enzyme activities, which may hinder studying the role of an individual P450, e.g., CYP2E1. Recombinant enzymes avoid this issue and thus provide an important complement to microsomal studies. Second, initial rate determinations at a relatively low number of substrate concentrations may fail to reveal the true kinetic profile, including deviations from a traditional hyperbolic relationship between initial rates and substrate concentrations. Third, use of Lineweaver-Burk plots to determine kinetic parameters and analyze kinetic mechanisms is not a reliable approach. The linearization of data does not equally weigh data points and masks deviations in the trend of the data predicted by the Michaelis-Menten mechanism. These shortcomings may generate inaccurate parameters that describe concentration-dependent changes in styrene metabolism by CYP2E1.
Many metabolic reactions catalyzed by CYP2E1 result in hyperbolic kinetic profiles that can be explained by the simple single-binding-site Michaelis-Menten kinetic mechanism. However, there are exceptions suggesting CYP2E1 metabolism can be more complex than generally assumed (Miller, 2008). Some aromatic and heterocyclic compounds inhibit CYPE1 activity through two-site binding (Hargreaves et al., 1994; Collom et al., 2008), and the metabolism of several CYP2E1 substrates results in nonhyperbolic kinetic profiles, suggesting the involvement of multiple binding sites (Koop, 1986; Venkatakrishnan et al., 1998; Spatzenegger et al., 2003; Harrelson et al., 2008). A limitation of these studies has been the absence of a mechanism to parameterize and thus explain the observed metabolic kinetics for CYP2E1. We were the first to validate an allosteric mechanism for CYP2E1 oxidation of a substrate, in this case the maker substrate 4-nitrophenol (Collom et al., 2008). Binding data and inhibition kinetics demonstrated substrate 4-nitrophenol and inhibitor 4-methylpyrazole bound to catalytic and allosteric effector sites and affected CYP2E1 activity. The prevalence of this two-site binding mechanism for CYP2E1 remains largely unexplored. Styrene shares many of the structural and functional attributes of molecules shown to bind CYP2E1 and induce allosteric effects (Hargreaves et al., 1994; Collom et al., 2008; Harrelson et al., 2008); consequently, we hypothesized that styrene metabolism involving a two-site binding allosteric mechanism contributes to its metabolic clearance.
Herein, we tested this hypothesis through in vitro studies using recombinant CYP2E1 and HLM pooled from 150 donors as a model for metabolism by the average human liver (Crespi et al., 2009). Kinetic profiles for steady-state metabolism of styrene indicated positive (homotypic) cooperativity. A two-site binding catalytic mechanism explained these data by involving an increase in affinity for a second styrene molecule binding after the first binding event. We interrogated the mechanism using the CYP2E1-specific drug 4-methylpyrazole (marketed as fomepizole), which associates with two binding sites (Collom et al., 2008). This inhibitor provided an excellent reporter to validate both styrene-binding sites and assess heterotypic allosteric interactions between styrene and 4-methylpyrazole. Inhibition kinetic profiles for 4-methylpyrazole were fit globally to the most probable mechanism determined from 16 different possibilities. Finally, we assessed the potential impact of positive cooperativity on metabolic clearance of styrene and its relevance to typical styrene levels in the blood of exposed workers.
Materials and Methods
Reagents.
All chemicals used in this study were American Chemical Society grade or higher. The following chemicals were purchased from Sigma-Aldrich (St. Louis, MO): styrene (substrate), 8,9-styrene oxide (product), styrene glycol (product), benzyl alcohol (internal standard), and 4-methylpyrazole (inhibitor). Pooled HLM (HLM 150) and human recombinant CYP2E1 (Supersomes) were purchased from BD Biosciences (San Jose, CA).
Steady-State Styrene Oxidation Studies.
We first examined the steady-state metabolism of styrene by CYP2E1 to its major metabolite, styrene oxide. Either 50 nM recombinant CYP2E1 or 0.5 mg/ml protein for HLM 150 was incubated with 15 concentrations of styrene between 3.0 μM and 1.0 mM in 50 mM potassium phosphate buffer, pH 7.4, at 37°C. On the basis of solubility, all styrene stocks were prepared in methanol. Reactions contained a final concentration of 1% methanol, which has been reported to have little inhibitory effect (Nakajima et al., 1994; Collom et al., 2008). Reactions were initiated by the addition of 1.0 mM NADPH and, after 30 min, were quenched by barium sulfate precipitation. Specifically, equal volumes of saturated barium hydroxide solution and 10% zinc sulfate solution containing the internal standard (15 μM benzyl alcohol) were added to each reaction. Although styrene is mainly metabolized to styrene oxide (Bond, 1989), the basic conditions of the quench process led to complete conversion of styrene oxide to styrene glycol. The glycol was then used as a measure of the total oxidative metabolism. The quenched mixture was centrifuged, and the resulting supernatant was analyzed by high-performance liquid chromatography (HPLC) to quantify the styrene glycol content. Under these experimental conditions, product formation was linear with respect to time.
HPLC Analysis of Styrene Oxidation under Steady-State Conditions.
For the quantitation of styrene glycol, we developed an in-house HPLC method based on one reported previously (Nakajima et al., 1994). Samples containing styrene (substrate), styrene glycol (product), and internal standard (benzyl alcohol) were injected onto a Waters Breeze HPLC system (Waters, Milford, MA) and resolved using a 4.6 × 150 mm Zorbax Eclipse 5-μm XDB-C18 column (Agilent Technologies, Santa Clara, CA) heated to 45°C using a gradient method involving H2O and acetonitrile at a flow rate of 1.2 ml/min. Elution of the compounds was monitored at a wavelength of 200 nm. Peak areas for the compound of interest were normalized to the internal standard and then quantitated relative to known standards. The detection limit of styrene glycol was 400 nM under these conditions. Standard solutions of glycol were prepared fresh every 2 weeks because of slow degradation of prepared stocks over time.
Analysis of Kinetic Profiles for Styrene Oxidation under Steady-State Conditions.
Analysis of the kinetic data was performed initially by an iterative least-squares nonlinear regression analysis using GraphPad Prism version 4.0 (GraphPad Software Inc., San Diego, CA). The data were fit to the Hill equation, as has been reported for previously reported cases of positive cooperative mechanisms. Next, to obtain parameters for the equilibrium constants and reaction rate, the data were analyzed globally using DynaFit version 3.28 (BioKin, Ltd., Watertown, MA) (Kuzmic, 1996) for model discrimination analysis to identify the most probable mechanism and corresponding parameters for styrene metabolism.
For this comparative analysis, data were fit to the Michaelis-Menten (single-binding-site) and two-site binding mechanisms (Fig. 2) in which the binary [enzyme-substrate (ES)] complex, the ternary [enzyme-substrate-substrate (ESS)] complex, or both complexes are active. The binding events presented in the model are ordered with respect to stoichiometry, but occupancy of each binding site was not distinguishable. The DynaFit script file used for the model discrimination analysis is included as Supplemental Fig. S1.
Fig. 2.

Possible reaction mechanisms for CYP2E1 activity toward styrene. E, CYP2E1; S, styrene; P, product (styrene oxide).
Inhibition of Styrene Oxidation by CYP2E1 in HLM Using 4-Methylpyrazole.
We measured steady-state metabolism of styrene by HLM in the presence of 4-methylpyrazole. Experimental conditions were identical to the uninhibited reactions, except all reactions were performed in the presence of 40 μM 4-methylpyrazole. 4-Methylpyrazole stocks were prepared in methanol. All reactions contained a final concentration of 1% methanol. The reactions were initiated, quenched, and analyzed using the same conditions as those used for the uninhibited experiments.
Inhibition Kinetics for 4-Methylpyrazole toward Styrene Oxidation by Recombinant CYP2E1.
Kinetic inhibition experiments for 4-methylpyrazole studies involved 50 nM recombinant CYP2E1 incubated with 0, 10, 50, 100, 225, 350, 500, 750, and 1000 μM styrene in 50 mM potassium phosphate, pH 7.4, at 37°C. Reactions were performed in the presence of four concentrations of 4-methylpyrazole (0.1, 0.5, 2, and 40 μM) and were subsequently performed, quenched, and analyzed by HPLC as described for the steady-state reactions. Each experiment included three independent replicates.
The most probable mechanism of inhibition was identified from the resulting kinetic profiles with 4-methylpyrazole using DynaFit 3.28 (BioKin, Ltd.) (Kuzmic, 1996). Inhibition data were fit to 16 different mechanisms whereby 4-methylpyrazole bound to one or two sites on CYP2E1 as generalized in Fig. 3. Given the observed positive cooperativity during styrene oxidation, all of the mechanisms included two substrate binding sites leading to two active complexes (ES and ESS). In addition, we included the possibility of an allosteric effect of 4-methylpyrazole on styrene metabolism, hence, kcat1 and kcat2. Table 1 summarizes the possible inhibition mechanisms, which were compared in this analysis.
Fig. 3.

Possible inhibition mechanisms for CYP2E1 styrene activity by 4-methylpyrazole. E, CYP2E1; S, styrene; P, product (styrene oxide); I, 4-methylpyrazole.
TABLE 1.
Possible models for inhibition of CYP2E1
| Model | Complexes Formed | Binding Allostery? |
Parameters and Consequencesa | |
|---|---|---|---|---|
| Substrate (on Inhibitor) | Inhibitor (on Substrate) | |||
| Competitive | ES, ESS, EI | No | No | Ks, Kss, Ki, kcat |
| Uncompetitive | ES, ESS, ESI | No | No | Ks, Kss, Ksi, kcat1, kcat2? |
| Mixed | ||||
| Allosteric both | ES, ESS, EI, ESI | Yes | Yes | Ks, Kss, Ki, Ksi, Kis, kcat1, kcat2? |
| Allosteric Substrate | ES, ESS, EI, ESI | Yes | No | Ks, Kss, Ki, Ksi, Kis = Ks, kcat1, kcat2? |
| Allosteric inhibitor | ES, ESS, EI, ESI | No | Yes | Ks, Kss, Ki = Ksi, Kis, kcat1, kcat2? |
| Noncompetitive | ES, ESS, EI, ESI | No | No | Ks, Kss, Ki = Ksi, Kis = Ks, kcat1, kcat2? |
EI, enzyme-inhibitor.
In all models except competitive, ESI may be active or not. Both possibilities were included in discriminations.
These mechanisms are similar to the traditional mechanisms for inhibition; thus, the same nomenclature was used to describe the models. For the competitive inhibition model, inhibitor can only bind to the free enzyme; thus, there is no allostery between substrate and enzyme. Likewise, in the uncompetitive inhibition model, the inhibitor binds the ES complex exclusively, and thus, there is no allostery. For the mixed allosteric mechanisms, the inhibitor binds both the free enzyme and ES complex with different inhibition constants. In the first mixed allosteric model, substrate and inhibitor both have an allosteric effect on each other. In the second mixed allosteric model, only the substrate acts allosterically on the inhibitor (Ki ≠ Ksi), but the inhibitor does not act allosterically on the substrate (Kis = Ks). Alternatively, the mixed allosteric inhibitor model describes the possibility where the inhibitor acts allosterically on the substrate (Kis ≠ Ks), but the substrate does not act allosterically on the inhibitor (Ki = Ksi). In the noncompetitive model, the inhibition constants for inhibitor binding free enzyme and the ES complex are equivalent, and no allostery is observed. In all models except for competitive inhibition, the enzyme-substrate-inhibitor (ESI) complex could be either active or not. If active, the kcat for product formation from the ESI complex could either be equal to or different from the kcat for the uninhibited reaction. All of these possibilities were included in the discrimination.
Similar to the convention used previously, binding events are ordered with respect to stoichiometry to determine possible allosteric effects if substrate or inhibitor is bound first, but occupancy of each individual binding site is not distinguishable through this analysis. Parameters for the uninhibited styrene oxidation were used from the previous studies and held constant, whereas the Ki, Kis, Ksi, and kcat2 (where applicable) were allowed to vary. The script file used for the analysis is given in Supplemental Fig. S2.
Modeling Styrene Metabolic Clearance.
A readily accessible and powerful approach to modeling metabolic clearance of drugs and pollutants uses kinetic parameters obtained through in vitro studies (Houston, 1994; Houston and Kenworthy, 2000). As described previously (Houston and Kenworthy, 2000), we fit our data to mechanistic equations describing the relationship between styrene concentration and its clearance (rate/[styrene]). To predict the metabolic clearance of styrene due to the Michaelis-Menten mechanism, a plot was generated using eq. 1 and kinetic parameters derived from fitting the microsomal kinetic data studies. To predict the metabolic clearance of styrene due to the positive allosteric (sigmoidal) mechanism, a plot was generated using eq. 2 and kinetic parameters derived from fitting the microsomal kinetic data to the Hill equation. Equation 1 is as follows:
where ν is the initial rate of the reaction, Vmax is the maximal rate of the reaction, and Km is the Michaelis constant. Equation 2 is as follows:
where ν is the initial rate of the reaction, Vmax is the maximal rate of the reaction, n is the Hill coefficient, and S50 the concentration of substrate at half-maximal rate for the reaction.
Results
Steady-State Styrene Metabolism by Recombinant CYP2E1 Reveals Nonhyperbolic Kinetics.
Oxidation of styrene by recombinant CYP2E1 led to a significantly nonhyperbolic kinetic profile (Fig. 4). A comparative analysis between fitting the data to the Michaelis-Menten equation and the Hill equation demonstrated strong preference for the Hill equation [>99% probability on the basis of Akaike's information criterion (AIC)]. This analysis yielded a Hill coefficient of 1.6, indicating significant positive cooperativity (Table 2). Because the Hill equation does not provide mechanistic insight about CYP2E1 catalysis, we did a comparative analysis of the steady-state data fit to either the Michaelis-Menten (single-site) mechanism or multiple two-site cooperative mechanisms in which CYP2E1 possesses catalytic and allosteric sites (Fig. 2). The most probable mechanism based on AIC was a two-site (allosteric) mechanism in which ES and ESS form, and both result in catalytically active complexes (Supplemental Table S1). The first binding event is very weak, whereas the second binding event leading to formation of the ESS complex is significantly stronger. These different catalytic cycles operating at low and high styrene concentrations result in the observed positive cooperativity in the kinetic profile. The parameters obtained using this model are summarized in Table 2.
Fig. 4.

Steady-state oxidation of styrene by recombinant CYP2E1. Solid line represents allosteric mechanism. Dashed line represents Michaelis-Menten mechanism. For reactions, 50 nM recombinant CYP2E1 Supersomes, styrene (varied from 14–1000 μM), and 1 mM NADPH were incubated at 37°C and pH 7.4. The reported values represent the average from nine individual experiments, including the mean ± S.D.
TABLE 2.
Parameters for steady-state metabolism of styrene by CYP2E1
The nonsymmetrical 95% confidence intervals for parameters are shown in parentheses.
| Enzyme System | Parameters for Hill Equation |
Parameters for Allosteric Modela |
||||
|---|---|---|---|---|---|---|
| Vmaxb | Hill Slope (n) | S50 | Vmaxb | Ks | Kss | |
| μM | μM | |||||
| Recombinant CYP2E1 | 6.7 (5.9–7.4) | 1.5 (1.2–1.9) | 270 (210–330) | 6.3 (5.9–6.9) | 830 (500–3700) | 110 (16–350) |
| HLM 150c | 0.79 (0.65–0.92) | 1.6 (1.2–2.0) | 110 (80–140) | 0.74 (0.69–0.84)c | 375 (210–3500) | 39 (2.8–140) |
See Fig. 2. For HLM 150, data were fit from 0 to 350 μM styrene.
Units are nmol · min−1 · nmol−1 for recombinant CYP2E1 or nmol · min−1 · mg protein−1 for HLM 150.
Ninety percent confidence intervals were used for analysis of the HLM 150 discrimination.
Biphasic Kinetics for Steady-State Styrene Metabolism by HLM 150.
Steady-state metabolism of styrene by human liver microsomal fractions yielded high-affinity and low-affinity contributions to the kinetic profile (Fig. 5, A and B). The high-affinity portion of the kinetic profile (up to 350 μM styrene), corresponding to the contribution of CYP2E1, was also nonhyperbolic, like the kinetic profile for recombinant enzyme. A comparative analysis of the HLM data demonstrated that the Hill equation was again strongly favored (>95% probability based on AIC) over the Michaelis-Menten equation (Fig. 5, B). A similar analysis using DynaFit software (BioKin, Ltd.) resulted in a strong preference for one of two positive cooperativity models. The first model, which was the preferred model with recombinant enzyme, described weak initial binding improving the binding of a second molecule of styrene with both the ES and ESS complexes being active. The second model was one in which the ESS complex was the only active complex, and there was essentially equivalent affinity for the free enzyme or ESS complex. Neither model had closed intervals, so the confidence intervals had to be lowered to 90%. At this level of confidence, the first model had closed intervals, but the second model had open intervals, so the second model was excluded (Supplemental Table S2). The parameters and corresponding confidence intervals for the preferred model are given in Table 2.
Fig. 5.

Steady-state oxidation of styrene by HLM 150. A, for reactions, 0.50 mg/ml protein, styrene (varied from 5–1000 μM), and 1 mM NADPH were incubated at 37°C and pH 7.4 in the absence (squares) or presence (triangles) of 40 μM 4-methylpyrazole. The reported values represent the average from six individual experiments, including the mean ± S.D. B, representation of data obtained at lower styrene concentrations (up to 350 μM) in the absence of 4-methylpyrazole, which were fit to allosteric and Michaelis-Menten mechanisms using DynaFit software (BioKin, Ltd.). The solid line represents the allosteric mechanism, and the dashed line represents the Michaelis-Menten mechanism.
The introduction of the CYP2E1-specific inhibitor 4-methylpyrazole to the HLM 150 reaction significantly altered the kinetic profile (Fig. 5, A). The overall plot became more monophasic and hyperbolic, and hence, there was no sigmoidicity. In fact, the high-affinity CYP2E1 portion of the profile was essentially eliminated. There was also a 4-fold decrease in maximal rate of turnover.
Two-Site Inhibition of Styrene Oxidation by 4-Methylpyrazole.
We probed the catalytic and allosteric sites occupied by styrene during CYP2E1 metabolism by determining the mechanism for 4-methylpyrazole inhibition of the reaction using recombinant CYP2E1. To avoid ambiguity from the significant contribution of multiple P450s, the experiment was not performed with HLM. Of 16 possible mechanisms, generalized in Fig. 3 and described in Table 1, there was significant statistical support for the mixed allosteric substrate model with an active ESI complex having a different kcat from the uninhibited reaction (Supplemental Table S3). All other models had relatively higher AIC values, i.e., ΔAICc, ≫2, and thus are less likely to explain the data (Burnham and Anderson, 2002). A plot of the data along with the statistically preferred fit from this mechanism is shown in Fig. 6.
Fig. 6.

Steady-state oxidation of styrene by recombinant CYP2E1 in the presence of 4-methylpyrazole. For reactions, 25 nM CYP2E1, styrene (varied from 14–1000 μM), and 1 mM NADPH were incubated at 37°C and pH 7.4. The reported values reflect the average of four experimental replicates, including the mean ± S.D. The concentrations of 4-methylpyrazole added to the reaction were 0, 0.1, 0.5, 4, and 20 μM, and corresponding kinetic profiles were indicated by increasing darkness in symbol coloring from white to black. Data were fit to the mixed allosteric substrate model (Fig. 3; Table 1) with ESI active.
In the preferred mechanism, there was a positive allosteric effect on binding and a negative one on catalysis. 4-Methylpyrazole bound with high affinity toward CYP2E1 (Ki 0.51 = μM; 95% confidence interval, 0.13–5.7). Nevertheless, the presence of bound styrene improved inhibitor binding 10-fold (Ksi = 0.043 μM; 95% confidence interval, 0.022–0.069). 4-Methylpyrazole had no allosteric effect on substrate binding (Kis = Ks). The affinity of 4-methylpyrazole for these sites was much higher than that observed for the substrate, whereby Ks and Kss were 830 and 110 μM, respectively. In addition to competing with styrene for binding sites, 4-methylpyrazole was a negative allosteric effector on catalysis, causing a 6-fold decrease in kcat from 6.3 (Table 2) to 0.98 nmol · min−1 · nmol CYP2E1−1 (95% confidence interval, 0.59–1.5). The observed values for the equilibrium constants are not consistent with the thermodynamic relationship that Ki/Kis must equal Ks/Ksi, because Ks and Ki are apparent constants. The values reflect the contributions of all possible ES and enzyme-inhibitor complexes, respectively, even though only one proceeds in the next step of the metabolic pathway, such as catalysis. Overall, the kinetic profile exhibited a loss in sigmoidicity and a decrease in maximal turnover (Fig. 3).
Impact of Positive Cooperativity on Metabolic Clearance of Styrene.
We used kinetic parameters from fits of the microsomal kinetic data to either the Michaelis-Menten or allosteric mechanisms to assess the effects of alternative mechanisms on styrene metabolic clearance. The fit of the high-affinity activity to the Michaelis-Menten equation yielded Vmax and Km values equal to 1.2 nmol · min−1 · mg protein−1 (95% confidence interval, 1.0–1.4) and 250 μM (95% confidence interval, 160–340), respectively. These values were used to simulate a clearance curve using eq. 1 (dashed line in Fig. 7). For the allosteric mechanism, values from the fit of the data to the Hill equation (Table 2) were used to plot a curve based on eq. 2 (solid line in Fig. 7). The resulting log plot demonstrates little difference in predicted metabolic clearance of styrene at high concentrations between the mechanisms. However, the plots begin a significant deviation at approximately 70 μM styrene, reaching a 10-fold difference by 0.1 μM styrene in predicted metabolic clearance between the different Michaelis-Menten and allosteric mechanisms.
Fig. 7.

Styrene clearance involving either Michaelis-Menten (dashed line) or positively cooperative (solid line) kinetics. GraphPad Prism software (GraphPad Software, Inc.) was used to simulate clearance curves for styrene on the basis of microsomal kinetic parameters for the respective mechanisms and equations published by Houston and Kenworthy (2000).
Discussion
The consequences of exposure to drugs and pollutants are often assessed through the use of in vitro systems to estimate in vivo metabolic clearance. Herein, we show that in vitro CYP2E1 metabolism of styrene into genotoxic styrene oxide does not conform to the traditional Michaelis-Menten mechanism. Rather, kinetic profiles generated by recombinant CYP2E1 and HLM indicated positive cooperativity during styrene metabolism. A two-site allosteric mechanism for CYP2E1 effectively explained the shift from an inefficient catalytic complex to a more efficient one as a function of styrene concentration. Modeling styrene metabolic clearance demonstrated significant differences between predictions by the traditional Michaelis-Menten and the observed positively cooperative mechanisms. Moreover, this transition in styrene metabolism occurred at low concentrations that coincide with styrene levels reported in blood from workers exposed to the pollutant. Consequently, these findings may have profound implications on predicting the toxicological effects resulting from styrene exposure.
Unlike our findings, others have reported styrene kinetic profiles by HLM that were consistent with the Michaelis-Menten kinetics. Several reasons may explain these different observations. First, we used steady-state studies with recombinant CYP2E1 to establish the kinetic profile and metabolic mechanism and then replicated those results using the more complex microsomal system. Second, we measured initial rates at a wide range of styrene concentrations. This design led to a high resolution kinetic profile that enabled a clear distinction between a hyperbolic curve predicted by the Michaelis-Menten mechanism and the positive cooperative one observed in this study. Third, we statistically assessed the most probable mechanism to explain the untransformed (nonlinearized) kinetic data. This approach is not possible through a reciprocal Lineweaver-Burk plot of the data, which unevenly weighs data points and masks non-Michaelis-Menten behavior. For comparison to previous studies (Mendrala et al., 1993; Nakajima et al., 1994; Wenker et al., 2001), we recast data from our microsomal reactions as a Lineweaver-Burk plot shown in Fig. 8. High-affinity activity (Fig. 8, black squares) attributed to CYP2E1 fits well to the linear regression predicted by a derivation of the Michaelis-Menten equation, even though a two-site allosteric mechanism best explains the reaction kinetics. Taken together, current studies by our group provide a more thorough and accurate investigation of styrene metabolism by CYP2E1.
Fig. 8.
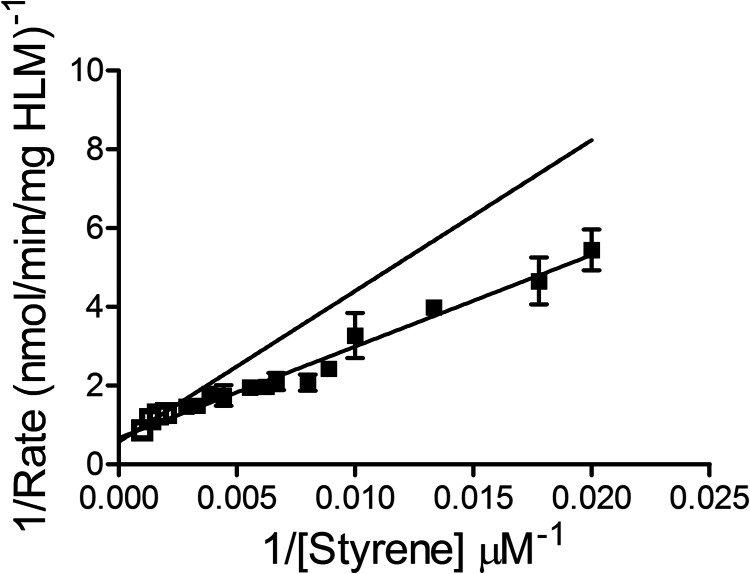
Lineweaver-Burk (double reciprocal) plot of steady-state oxidation of styrene by HLM 150. The two phases of the kinetic profile were fit to individual linear regressions using GraphPad Prism (GraphPad Sofware, Inc.). For high-affinity CYP2E1 activity (black squares), reflected rates were obtained at styrene concentrations up to 350 M as used in model discrimination studies (see Fig. 5), whereas the lower affinity enzyme activity was attributed to rates at higher substrate concentrations (unfilled squares).
Kinetic profiles for several CYP2E1 substrates have demonstrated positive cooperativity. CYP2E1 hydroxylation of m-xylene yielded a hyperbolic Michaelis-Menten kinetic profile for 2,4-dimethylphenol but a positively cooperative one for m-methylbenzylalcohol (Hill coefficient 1.4) (Harrelson et al., 2008). Formation of m-methylbenzylalcohol involved an orientation like that required for the styrene reaction, suggesting that this binding mode may favor multiple binding events. The oxidation of 7-ethoxycoumarin yielded a similar Hill coefficient of 1.6 (Spatzenegger et al., 2003), and there was a deviation of the kinetic profile for phenacetin from linearity at low substrate concentrations, although the authors did not investigate the possibility of cooperativity (Venkatakrishnan et al., 1998). When characterized, these significant Hill coefficients were similar in magnitude to that observed for the styrene reaction and were strikingly comparable to results shown for other more traditionally recognized allosteric P450s CYP2C9 and CYP3A4 (reviewed in Niwa et al., 2008). Nonetheless, analysis of kinetic profiles by the Hill equation does not provide a mechanistic perspective for interpreting a basis for allosteric reactions or providing a foundation for predicting their occurrence.
We addressed this knowledge gap by demonstrating that a two-site allosteric mechanism explained positive cooperativity for in vitro styrene metabolism (Fig. 9). At low concentrations, styrene bound CYP2E1 weakly and underwent oxidation to styrene oxide. Product release liberated free enzyme to enter another catalytic cycle and reform the CYP2E1-styrene complex. An increase in styrene concentration created a new catalytic cycle in which a second styrene molecule bound the CYP2E1-styrene complex. The resulting ternary complex was catalytically active. Styrene underwent oxidation and release to regenerate the CYP2E1-styrene complex. Depending on the styrene concentration, this complex either oxidized the bound styrene to the oxide or bound another styrene molecule. On the basis of our studies, styrene bound free enzyme approximately 10-fold more poorly than the CYP2E1-styrene complex. Consequently, CYP2E1 metabolism of styrene shifted from a low affinity, less efficient catalytic cycle to a higher affinity, more efficient catalytic cycle, resulting in the observed positive cooperativity in the kinetic profile.
Fig. 9.

Stylized depiction of the allosteric mechanism for CYP2E1 metabolic reactions. In the case of styrene, substrate binds very poorly to CYP2E1 to form an active binary complex, which releases product and regenerates the free enzyme. At higher styrene concentrations, a second substrate molecule binds the ES complex with higher affinity than the first binding event. This ternary complex also generates product and regenerates the ES complex to bind another substrate molecule for a subsequent round of catalysis. Substrate concentration then regulates the respective metabolic cycles for CYP2E1. Preference for these catalytic cycles is further modulated by variations in allosteric effects dependent on the structure of bound effector molecules.
The allosteric mechanism for in vitro styrene metabolism may occur under in vivo conditions, according to biomonitoring studies for industrial workers (Löf et al., 1986; Somorovská et al., 1999; Prieto et al., 2002). Typical styrene levels in blood of low to medium exposed workers, e.g., maintenance workers and sprayers, ranged from 24 to 188 ng/ml (0.24–1.9 μM). Heavily exposed manual lamination workers led to styrene blood levels as high as 2100 ng/ml (20 μM). Actual peak styrene levels would be more than those reported from studies given rapid distribution and metabolism of styrene (Bond, 1989). Blood was typically taken after work shifts sometimes hours after exposure. For example, a 2-h difference between the time of exposure to that for sampling would lead to a 6-fold decrease in styrene blood levels from an initial peak value because of the half-life of styrene (41 min) (Wigaeus et al., 1983). In addition, styrene blood levels in workers could range from 144 to 12,600 ng/ml (1.2–120 μM) at the time of exposure. Taken together, styrene blood levels in workers occur over a range comparable to changes in the efficiency of CYP2E1 metabolism, and thus, it is conceivable that positive cooperativity affects in vivo toxicological effects resulting from styrene exposure.
Current in vivo studies on styrene toxicity do not include a sufficient range of exposure levels to provide unequivocal evidence for this allosteric mechanism. Consequently, we showed that modeling styrene metabolic clearance on the basis of traditional Michaelis-Menten kinetics versus a positively allosteric mechanism yielded significantly different predictions at low to moderate styrene levels as reported for industrial workers. This deviation in styrene clearance arose from the contribution of the less efficient CYP2E1 catalytic cycle at low styrene concentrations. The traditional clearance model did not take into account those kinetics and thus overpredicted styrene metabolic clearance rates. Differences between models predictions began at approximately 70 μM and continued to increase as styrene concentration decreased. By 0.1 μM styrene, the traditional clearance model predicted a 10-fold higher rate than that for the positive allosteric mechanism. The toxicological impact of the clearance model involving positive allostery could be significant. Metabolism results in the generation of genotoxic and potentially carcinogenic styrene oxide, and thus, these changes in in vivo clearance values may be linked to the carcinogenic potential of styrene in exposed individuals. For styrene, positive cooperativity led to less efficient metabolism, and hence activation of styrene, at lower exposure levels such that the toxic potential would be less than that predicted by more traditional models. Present toxicological models for styrene do not incorporate positive allostery for CYP2E1; thus, its contribution to the metabolic activation and clearance of styrene may confound ongoing efforts to establish the carcinogenic potential of styrene.
The inclusion of inhibition studies with the drug 4-methylpyrazole (marketed as fomepizole) yielded two significant findings. Inhibition studies demonstrated that 4-methylpyrazole bound to the same two sites as styrene and thus further validated the proposed two-site allosteric mechanism for styrene metabolism. Moreover, bound 4-methylpyrazole and styrene interacted with each other directly or through protein structure causing heterotypic interactions that were reflected in parameters for binding and catalysis (in the latter case for styrene). Styrene had a positive allosteric effect on 4-methylpyrazole binding (Ki ≫ Kis). In structural terms, this effect may reflect the contribution of bound styrene to the hydrophobicity of the active site, which strongly favors 4-methylpyrazole binding. In addition, simultaneous binding of 4-methylpyrazole and styrene to CYP2E1 yielded a catalytically active complex. Unlike the second bound styrene molecules, 4-methylpyrazole had a negative allosteric effect on catalysis, a 6-fold decrease in maximal turnover compared with the uninhibited reaction. The binding mode for inhibitor may block product release and thus decrease overall substrate turnover. Elucidation of these complex interactions was made possible through our proposed allosteric mechanism. In this case, we explained heterotypic interactions between the pollutant styrene and drug 4-methylpyrazole on styrene metabolism. If the two-site allosteric mechanism for CYP2E1 is more universal, it may provide a critical mechanistic perspective to understanding other complex compound-compound interactions involving styrene.
Concluding Remarks.
Predicting the consequences of exposure to drugs and pollutants requires the appropriate mechanistic models to parameterize in vitro kinetic data accurately and estimate their in vivo metabolic clearance correctly (Houston and Kenworthy, 2000; Atkins, 2004; Guengerich, 2008). We are the first to report the significance of a two-site binding allosteric mechanism in CYP2E1 metabolism of styrene, a common pollutant and potential carcinogen. The observed positive allostery profoundly affected scaling of in vitro kinetics to predict metabolic activation and clearance of styrene. Low styrene levels were notably much less toxic than assumed by the traditional model based on Michaelis-Menten kinetics. Other compounds can further modulate the kinetics for styrene metabolism and hence affect the consequences for exposure. Taken together, our elucidation of mechanisms for these reactions provides a powerful tool for further investigating the complexities of CYP2E1 metabolism of drugs and pollutants.
Supplementary Material
Acknowledgments
We thank Drew R. Jones for technical assistance with developing the high-performance liquid chromatographic method used to analyze styrene metabolism.
This work was supported in part by the National Institutes of Health National Center for Research Resources [Grant P20-RR-16460] (IDeA Networks of Biomedical Research Excellence Program; to J.H.H. and G.P.M.); the Summer Undergraduate Research Fellowship sponsored by the Biochemistry and Molecular Biology Department at the University of Arkansas for Medical Sciences (to J.H.H. and G.P.M.); a bridging grant from the University of Arkansas for Medical Sciences (to G.P.M.); and the Arkansas Tobacco Settlement Proceeds Act of 2000 (to G.B).
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
- P450
- cytochrome P450
- HLM
- human liver microsomes
- HPLC
- high-performance liquid chromatography
- ES
- enzyme-substrate
- ESS
- enzyme-substrate-substrate
- ESI
- enzyme-substrate-inhibitor
- AIC
- Akaike's information criterion.
Authorship Contributions
Participated in research design: Hartman, Boysen, and Miller.
Conducted experiments: Hartman.
Contributed new reagents or analytic tools: Boysen, and Miller.
Performed data analysis: Hartman and Miller.
Wrote or contributed to the writing of the manuscript: Hartman, Boysen, and Miller.
References
- Atkins WM. (2004) Implications of the allosteric kinetics of cytochrome P450s. Drug Discov Today 9:478–484 [DOI] [PubMed] [Google Scholar]
- Barale R. (1991) The genetic toxicology of styrene and styrene oxide. Mutat Res 257:107–126 [DOI] [PubMed] [Google Scholar]
- Bond JA. (1989) Review of the toxicology of styrene. Crit Rev Toxicol 19:227–249 [DOI] [PubMed] [Google Scholar]
- Burnham KP, Anderson DR. (2002) Model Selection and Multimodel Inference: A Practical Information-Theoretic Approach, Springer, New York [Google Scholar]
- Collom SL, Laddusaw RM, Burch AM, Kuzmic P, Perry MD, Jr, Miller GP. (2008) CYP2E1 substrate inhibition. Mechanistic interpretation through an effector site for monocyclic compounds. J Biol Chem 283:3487–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi CL, Francis E, Patten C. (2009) Application Note #465: Optimizing Donor Number for Consistent Pooled Human Liver Microsomes, BD Biosciences - Discovery Labware, Billerica, MA [Google Scholar]
- Guengerich FP. (2008) Cytochrome p450 and chemical toxicology. Chem Res Toxicol 21:70–83 [DOI] [PubMed] [Google Scholar]
- Hargreaves MB, Jones BC, Smith DA, Gescher A. (1994) Inhibition of p-nitrophenol hydroxylase in rat liver microsomes by small aromatic and heterocyclic molecules. Drug Metab Dispos 22:806–810 [PubMed] [Google Scholar]
- Harrelson JP, Atkins WM, Nelson SD. (2008) Multiple-ligand binding in CYP2A6: probing mechanisms of cytochrome P450 cooperativity by assessing substrate dynamics. Biochemistry 47:2978–2988 [DOI] [PubMed] [Google Scholar]
- Houston JB. (1994) Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol 47:1469–1479 [DOI] [PubMed] [Google Scholar]
- Houston JB, Kenworthy KE. (2000) In vitro-in vivo scaling of CYP kinetic data not consistent with the classical Michaelis-Menten model. Drug Metab Dispos 28:246–254 [PubMed] [Google Scholar]
- Kim H, Wang RS, Elovaara E, Raunio H, Pelkonen O, Aoyama T, Vainio H, Nakajima T. (1997) Cytochrome P450 isozymes responsible for the metabolism of toluene and styrene in human liver microsomes. Xenobiotica 27:657–665 [DOI] [PubMed] [Google Scholar]
- Koop DR. (1986) Hydroxylation of p-nitrophenol by rabbit ethanol-inducible cytochrome P-450 isozyme 3a. Mol Pharmacol 29:399–404 [PubMed] [Google Scholar]
- Koop DR. (1990) Inhibition of ethanol-inducible cytochrome P450IIE1 by 3-amino-1,2,4-triazole. Chem Res Toxicol 3:377–383 [DOI] [PubMed] [Google Scholar]
- Kuzmic P. (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal Biochem 237:260–273 [DOI] [PubMed] [Google Scholar]
- Löf A, Lundgren E, Nydahl EM, Nordqvist MB. (1986) Biological monitoring of styrene metabolites in blood. Scand J Work Environ Health 12:70–74 [DOI] [PubMed] [Google Scholar]
- Mendrala AL, Langvardt PW, Nitschke KD, Quast JF, Nolan RJ. (1993) In vitro kinetics of styrene and styrene oxide metabolism in rat, mouse, and human. Arch Toxicol 67:18–27 [DOI] [PubMed] [Google Scholar]
- Miller GP. (2008) Advances in the interpretation and prediction of CYP2E1 metabolism from a biochemical perspective. Expert Opin Drug Metab Toxicol 4:1053–1064 [DOI] [PubMed] [Google Scholar]
- Nakajima T, Elovaara E, Gonzalez FJ, Gelboin HV, Raunio H, Pelkonen O, Vainio H, Aoyama T. (1994) Styrene metabolism by cDNA-expressed human hepatic and pulmonary cytochromes P450. Chem Res Toxicol 7:891–896 [DOI] [PubMed] [Google Scholar]
- National Toxicology Program (2008) Final report on carcinogens background document for styrene, in Report on Carcinogens Background Document for Styrene, pp i–398 Department of Health and Human Services, Research Triangle Park, NC: [PubMed] [Google Scholar]
- Niwa T, Murayama N, Yamazaki H. (2008) Heterotropic cooperativity in oxidation mediated by cytochrome p450. Curr Drug Metab 9:453–462 [DOI] [PubMed] [Google Scholar]
- Prieto MJ, Marhuenda D, Cardona A. (2002) Analysis of styrene and its metabolites in blood and urine of workers exposed to both styrene and acetone. J Anal Toxicol 26:23–28 [DOI] [PubMed] [Google Scholar]
- Somorovská M, Jahnová E, Tulinská J, Zámecníková M, Sarmanová J, Terenová A, Vodicková L, Lísková A, Vallová B, Soucek P, et al. (1999) Biomonitoring of occupational exposure to styrene in a plastics lamination plant. Mutat Res 428:255–269 [DOI] [PubMed] [Google Scholar]
- Spatzenegger M, Liu H, Wang Q, Debarber A, Koop DR, Halpert JR. (2003) Analysis of differential substrate selectivities of CYP2B6 and CYP2E1 by site-directed mutagenesis and molecular modeling. J Pharmacol Exp Ther 304:477–487 [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan K, von Moltke LL, Greenblatt DJ. (1998) Human cytochromes P450 mediating phenacetin O-deethylation in vitro: validation of the high affinity component as an index of CYP1A2 activity. J Pharm Sci 87:1502–1507 [DOI] [PubMed] [Google Scholar]
- Wenker MA, Kezić S, Monster AC, de Wolff FAA. (2001) Metabolic capacity and interindividual variation in toxicokinetics of styrene in volunteers. Hum Exp Toxicol 20:221–228 [DOI] [PubMed] [Google Scholar]
- Wigaeus E, Löf A, Bjurström R, Nordqvist MB. (1983) Exposure to styrene. Uptake, distribution, metabolism and elimination in man. Scand J Work Environ Health 9:479–488 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.