

Abstract
A key step in the regulation of networks that control gene expression is the sequence-specific binding of transcription factors to their DNA recognition sites. A more complete understanding of these DNA–protein interactions will permit a more comprehensive and quantitative mapping of the regulatory pathways within cells, as well as a deeper understanding of the potential functions of individual genes regulated by newly identified DNA-binding sites. Here we describe a DNA microarray-based method to characterize sequence-specific DNA recognition by zinc-finger proteins. A phage display library, prepared by randomizing critical amino acid residues in the second of three fingers of the mouse Zif268 domain, provided a rich source of zinc-finger proteins with variant DNA-binding specificities. Microarrays containing all possible 3-bp binding sites for the variable zinc fingers permitted the quantitation of the binding site preferences of the entire library, pools of zinc fingers corresponding to different rounds of selection from this library, as well as individual Zif268 variants that were isolated from the library by using specific DNA sequences. The results demonstrate the feasibility of using DNA microarrays for genome-wide identification of putative transcription factor-binding sites.
An understanding of the sequence specificity of DNA–protein interactions has resulted from studies of the effects of mutations in the DNA-binding sites and the amino acid residues implicated in sequence-specific binding. The zinc-finger transcription factors are among the best understood families in terms of sequence-specific DNA binding. Rational zinc-finger design by using structure-based (1) and database-guided (2) approaches has permitted some progress in revealing certain rules that govern these discriminating contacts (3–6). In addition, phage display has emerged as a powerful tool to select for zinc fingers that recognize given target DNA sites (5, 7–9). Although this technology has permitted millions of protein variants to be sampled simultaneously, the effects of individual mutants have had to be measured one at a time by using nitrocellulose-binding assays (10), gel mobility-shift analysis (11), ELISA (12), Southwestern blotting (13), or reporter constructs (14). Because these methods are generally too laborious to be used for the analysis of a large number of DNA–protein interactions, it has not been possible to gather data on vast collections of variant DNA–protein pairings. Although in vitro selections (15) and “binding-site signatures” (4) have permitted the sampling of multiple DNA-binding sites for a given DNA-binding protein, these in vitro selections provide only a partial view of binding-site specificity, because only the tightest binding interactions are selected, whereas information about suboptimal interactions is lost in the experimental process. It is possible that these lower-affinity DNA sites are functionally significant in transcriptional regulation of gene expression. Therefore, we have taken advantage of DNA microarray technologies, which have revolutionized mRNA expression analysis (16), in developing a highly parallel method for studying the sequence specificity of DNA–protein interactions (17).
We describe the use of DNA microarrays to study DNA recognition by the zinc finger, because this domain is one of the most common structural motifs found in eukaryotic transcription factors (18). An important member of the Cys2His2 class of this family of proteins is the mouse transcription factor Zif268. Zif268 serves as a valuable model system for studying zinc finger–DNA recognition, because crystallographic data of the Zif268 DNA–protein complex are available (18, 19). In a simple model of the DNA–protein interactions, three fingers in the DNA-binding domain bind as independent modules to three tandem 3-bp subsites (Fig. 1A). This modularity has been exploited in studies aimed at unraveling the rules governing the interactions between zinc-finger residues and the DNA bases they contact (3–5).
Figure 1.

Design of microarrays for binding experiments. (A) Model depicting interactions between the Zif268 phage display library and the DNA used in phage selections. The three zinc fingers of Zif268 (F1, F2, and F3) are aligned to show contacts to the nucleotides of the DNA-binding site as inferred from the crystal structure of Zif268 and biochemical experiments. The zinc-finger amino acid positions are numbered relative to the first helical residue (position 1). The randomized positions in the α helix of the second finger are circled. DNA base pairs marked N were fixed as given sequences and used to select sequence-specific zinc-finger phage from the library. (B) Design of the DNA sequences spotted on the microarrays used in the microarray-binding experiments. F1, F2, and F3 refer to fingers 1, 2, and 3 of Zif268 variants, and the boxes indicate the three corresponding 3-bp binding sites for the three fingers. DNA base pairs marked N were systematically varied to explore the 64 different 3-bp binding sites for finger 2. The diagram shows attachment of the DNA to a glass slide via an amino linker. (C) Entire microarray, showing all nine replicates, bound by wild-type Zif268 phage. The fluorescence intensities of the spots are shown in false color, corresponding to the DNA-binding affinities of the protein for the different DNA sequences. The Cy3-labeled alignment oligonucleotide was spotted above and below each column, as well as to the right and left of each row, along the perimeter of the nine replicates. In addition, four spots of DNA containing the wild-type Zif268-binding site were spotted at higher concentrations in each of the four corners (row 1, columns 1–4; rows 1–4, column 10; row 8, columns 7–10; rows 5–8, column 1) of each replicate, as a positive control for wild-type phage binding to the microarrays in preliminary experiments. (D) Amino acid sequences of the variant α-helical regions in finger 2 of the Zif268 variants used in this study. The randomized positions are marked with an X. The three primary recognition positions are highlighted. The names of the clones are listed to the Left of these sequences. The first variant listed is wild-type Zif268.
Materials and Methods
Synthesis of DNA Microarrays.
Cy3-labeled oligonucleotide was spotted for alignment purposes. The set of 64 oligonucleotides, synthesized to represent all possible 3-nt central-finger sites for Zif268 zinc fingers, was combined with a 5′ amino-tagged universal primer in a 2:1 molar ratio in a Sequenase (United States Biochemical) reaction. The completed extension reactions were exchanged into 150 mM K2 HPO4, pH 9.0, by using CentriSpin-10 spin columns (Princeton Separations, Adelphia, NJ). Exact methods and oligonucleotides are published in the Supplemental Methods on the PNAS web site, www.pnas.org.
Glass slides (Gold Seal, Gold Seal Products, Portsmouth, NH) were cleaned for 0.5–2 h in 2 M nitric acid. After rinsing in distilled water, the slides were soaked in distilled water for 5–15 min and then washed once with acetone. The slides were silanized by immersing them for 15 min in a solution of 1% aminopropyl-methyl-diethoxysilane (Fluka) dissolved in 95% acetone. After washing the slides twice in acetone, they were baked for 30 min at 75°C. The surface of the slides was then activated by placing them in a solution of 0.5% 1,4-diphenylene-diisothiocyanate (PDC) (Fluka) dissolved in a solution consisting of 40 ml of pyridine and 360 ml of anhydrous N,N-dimethylformamide for 2–4 h. The slides were then washed twice with methanol, twice with acetone, and stored in a dessicator until use. A custom-built arraying robot equipped with piezoelectric printheads was used to print the microarrays. After printing, the microarrays were incubated overnight at room temperature, then for 1 h at 37°C in a humidity chamber containing 300 mM K2 HPO4, pH 9.0. The rest of the PDC surface was inactivated by a 10-min incubation in 1% ammonium hydroxide/0.1% SDS/200 mM NaCl. After washing in 4 × SSC, the slides were neutralized in 6 × standard saline phosphate/EDTA (0.18 M NaCl/10 mM phosphate, pH 7.4/1 mM EDTA)/0.01% Triton X-100, washed twice in 4 × SSC, then washed in 2 × SSC and spun dry in a clinical centrifuge. Slides were stored in a closed box at room temperature until use.
Phage ELISAs.
To determine apparent dissociation constants (K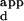 s), phage ELISAs were carried out at least in triplicate, essentially as described (4), with some modifications. Exact methods and oligonucleotides are described in the Supplemental Methods. Because these measurements provide apparent, not actual, Kds, all final observed K
s), phage ELISAs were carried out at least in triplicate, essentially as described (4), with some modifications. Exact methods and oligonucleotides are described in the Supplemental Methods. Because these measurements provide apparent, not actual, Kds, all final observed K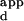 values were scaled by the same constant so that the K
values were scaled by the same constant so that the K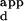 for wild-type Zif268 with the sequence containing the 3-bp finger 2 binding-site TGG was equal to 3.0 nM.
for wild-type Zif268 with the sequence containing the 3-bp finger 2 binding-site TGG was equal to 3.0 nM.
Microarray Protein Binding.
For production of Zif phage, overnight bacterial cultures of TG1 (or JM109) cells, each producing a particular zinc-finger phage or pool of phages, were grown at 30°C in 2 × TY medium containing 50 mM zinc acetate and 15 mg/ml tetracycline (2 × TY/Zn/Tet). Culture supernatants containing phage were diluted 2-fold by addition of PBS/Zn containing 4% (wt/vol) nonfat dried milk, 2% (vol/vol) Tween 20, and 100 mg/ml salmon testes DNA (Sigma). The slides were blocked with 2% milk in PBS/Zn for 1 h, then washed once with PBS/Zn/0.1% Tween 20, then once with PBS/Zn/0.01% Triton X-100. The diluted phage solutions were then added to the slides, and binding was allowed to proceed for 1 h. The slides were then washed five times with PBS/Zn/1% Tween 20, and then three times with PBS/Zn/0.01% Triton X-100. Mouse anti-(M13) antibody (Amersham Pharmacia) was diluted in PBS/Zn containing 2% milk, preincubated for at least 1 h, and added to the slide. After incubation for 1 h at room temperature, the slides were washed three times with PBS/Zn/0.05% Tween 20, and three times with PBS/Zn/0.01% Triton X-100. R-phycoerythrin-conjugated goat anti-(mouse IgG) (Sigma) was diluted in PBS/Zn containing 2% milk, preincubated for at least 1 h, and added to the slides. After incubation for 1 h at room temperature, the slides were washed three times with PBS/Zn/0.05% Tween 20, three times with PBS/Zn/0.01% Triton X-100, and once with PBS/Zn, and then scanned.
Microarray Data Analysis.
Microarrays were scanned essentially as described (16). The signal intensities of each of the spots in the scanned images were quantified by using imagene Version 3.0 software (BioDiscovery, Los Angeles, CA). Subsequent analyses were performed with perl scripts written by M.L.B. After background subtraction, the relative signal intensity of each of the spots within a replicate was calculated as a fraction of the highest signal intensity for a spot containing one of the 64 different 37-bp sequences. To normalize for possible variability in the DNA concentrations of the different DNA samples that were spotted onto the microarrays, each of the average relative signal intensities from zinc-finger phage binding was divided by each of the respective average relative signal intensities from SybrGreen I staining (see Supplemental Methods for details).
Results
We used DNA microarrays to examine the spectrum of binding-site specificities of a collection of Zif268 mutants selected from a phage display library of the second finger. Quantitative measurements of more than 750 DNA–protein interactions were gathered from 10 different microarray-binding assays by using wild-type Zif268, four mutants, and seven pools of mutants (Fig. 2 A–E and 3 A and B). Double-stranded DNAs containing the wild-type binding sites for fingers 1 and 3 and all possible 3-bp binding sites for finger 2 of wild-type Zif268 were created by primer extension on unique oligonucleotides by using a universal primer (Fig. 1B). Nine replicates of each of these 64 different sequences were printed onto glass slides. Phage displaying the three Zif268 zinc fingers were harvested directly from bacterial cultures and bound to the microarrays. The bound zinc-finger phage were labeled fluorescently by using a primary antibody against a phage coat protein and an R-phycoerythrin-conjugated secondary antibody (Fig. 1C). After DNA concentration normalization, each of the fluorescence intensities was expressed as a fraction of the fluorescence intensity of the DNA sequence, with the highest average intensity for the particular Zif268 mutant being examined.
Figure 2.

Wild-type and variant Zif268 zinc-finger phage bound to microarrays. One of nine replicates on each microarray slide is shown for each of the binding experiments described. Spots with high relative signal intensities for each of the Zif268 variants labeled in descending numerical order according to decreasing K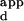 values, and the sequences corresponding to each of these numbered spots are listed between the microarray images and the sequence logos. The fluorescence intensities of the spots are shown in false color, corresponding to the DNA-binding affinities. The color bars were calibrated from the K
values, and the sequences corresponding to each of these numbered spots are listed between the microarray images and the sequence logos. The fluorescence intensities of the spots are shown in false color, corresponding to the DNA-binding affinities. The color bars were calibrated from the K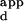 s, as determined by using ELISA. Sequence logos depicting the DNA-binding site preferences of the variant zinc fingers are shown to the Right of the microarrays. The numbers along the base of the sequence logo indicate the 5′, middle, and 3′ nucleotides of the 3-bp DNA-binding site for finger 2. The values along the y axis indicate the number of bits of information at each position of the 3-bp DNA-binding site. The height of each nucleotide at each position of the 3-bp DNA-binding site is determined by multiplying the relative DNA-binding affinity of the nucleotide by the total information at that position, so that a taller printed nucleotide is more beneficial for tight binding than a shorter one. The nucleotides are sorted so that the nucleotide most beneficial for tight binding is on top. (A) Wild type; (B) RGPD; (C) REDV; (D) LRHN; (E) KASN. Single-letter abbreviations for the amino acid residues are as follows: A, Ala; D, Asp; E, Glu; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; R, Arg; S, Ser; T, Thr; V, Val.
s, as determined by using ELISA. Sequence logos depicting the DNA-binding site preferences of the variant zinc fingers are shown to the Right of the microarrays. The numbers along the base of the sequence logo indicate the 5′, middle, and 3′ nucleotides of the 3-bp DNA-binding site for finger 2. The values along the y axis indicate the number of bits of information at each position of the 3-bp DNA-binding site. The height of each nucleotide at each position of the 3-bp DNA-binding site is determined by multiplying the relative DNA-binding affinity of the nucleotide by the total information at that position, so that a taller printed nucleotide is more beneficial for tight binding than a shorter one. The nucleotides are sorted so that the nucleotide most beneficial for tight binding is on top. (A) Wild type; (B) RGPD; (C) REDV; (D) LRHN; (E) KASN. Single-letter abbreviations for the amino acid residues are as follows: A, Ala; D, Asp; E, Glu; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; R, Arg; S, Ser; T, Thr; V, Val.
Figure 3.

Evolution of sequence-specific DNA-binding zinc fingers from selections of the phage display library. Phage pools isolated from different rounds of selections analyzed by using DNA microarrays. One of nine replicates on each microarray slide is shown for each of the binding experiments described. Spots with high relative signal intensities in each of the rounds are labeled to indicate the bound DNA sequence. (A) Rounds 2–4 of the selection by using the middle triplet GCG. Round 1 (not shown) did not have any outstanding spots. Round 2 shows binding to the wild-type Zif268 DNA-binding site, which is spotted at a high concentration on the periphery of the array (see supplemental data). (B) Rounds 1–3 of the selection using the middle triplet TCC. Round 1 did not have any outstanding spots. (C) Portions of the sequences present at the GAC and TCC spots on the microarrays. The 9-bp binding sites for variant zinc-finger phage are underlined, and the 3-bp binding sites for finger 2 are boldfaced.
As a validation of this protocol, wild-type Zif268 phage were bound to a microarray (Figs. 1C and 2A). The dynamic range of relative fluorescence intensities spanned two orders of magnitude and corresponded to a dynamic range of apparent binding constants (K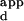 s) that spanned at least three orders of magnitude. To evaluate the relationship between the normalized fluorescence intensities and the DNA-binding affinities of the zinc fingers, the binding affinities of wild-type Zif268 phage for a set of DNA sequences were determined by performing zinc-finger phage ELISA (20) at a series of DNA concentrations. The relative fluorescence intensities were found to correlate well with a hyperbolic function of the K
s) that spanned at least three orders of magnitude. To evaluate the relationship between the normalized fluorescence intensities and the DNA-binding affinities of the zinc fingers, the binding affinities of wild-type Zif268 phage for a set of DNA sequences were determined by performing zinc-finger phage ELISA (20) at a series of DNA concentrations. The relative fluorescence intensities were found to correlate well with a hyperbolic function of the K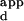 s, based on fractional occupancy (see supplemental data for details) (Fig. 4). Therefore, for each variant Zif phage, a calibration curve was constructed by determining the K
s, based on fractional occupancy (see supplemental data for details) (Fig. 4). Therefore, for each variant Zif phage, a calibration curve was constructed by determining the K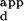 s of a few representative sequences that spanned the range of relative fluorescence intensities on the microarrays spotted with all different 3-bp binding sites for finger 2. These calibration curves were used to interpolate the K
s of a few representative sequences that spanned the range of relative fluorescence intensities on the microarrays spotted with all different 3-bp binding sites for finger 2. These calibration curves were used to interpolate the K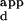 s for the remaining sequences on the microarrays (Table 2A, which is published as supplemental data on the PNAS web site). These binding-site preferences were then used to construct sequence logo representations (21) of each variant's binding-site profile (Fig. 2 A–E). Each of the input sequences for logo construction was weighted according to the inverse of its K
s for the remaining sequences on the microarrays (Table 2A, which is published as supplemental data on the PNAS web site). These binding-site preferences were then used to construct sequence logo representations (21) of each variant's binding-site profile (Fig. 2 A–E). Each of the input sequences for logo construction was weighted according to the inverse of its K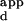 , so that the sequences with the highest binding affinities had the greatest contribution in creation of the sequence logo (see supplemental data for details).
, so that the sequences with the highest binding affinities had the greatest contribution in creation of the sequence logo (see supplemental data for details).
Figure 4.

Relationship between relative fluorescence intensity and DNA-binding affinity. (A) SybrGreen I-stained microarray; (B) low laser-power scan of wild-type Zif268 bound to a microarray; (C) high laser-power scan of wild-type Zif268 bound to a microarray. Red pixels indicate saturated signal intensity. (D) Plot showing the relationship between relative signal intensity and K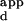 . Error bars indicate 1 SD of the SybrGreen I normalized binding data. These sequences were chosen because they span a range of relative fluorescence intensities. For this evaluation, a separate set of microarrays was spotted with these DNA sequences. Because some of these sequences contain mutations in the binding sites for fingers 1 and 3 of Zif268, they were not printed on the microarrays containing all different 3-bp binding sites for finger 2.
. Error bars indicate 1 SD of the SybrGreen I normalized binding data. These sequences were chosen because they span a range of relative fluorescence intensities. For this evaluation, a separate set of microarrays was spotted with these DNA sequences. Because some of these sequences contain mutations in the binding sites for fingers 1 and 3 of Zif268, they were not printed on the microarrays containing all different 3-bp binding sites for finger 2.
Identifying small differences in the DNA sequence specificity of distinct transcription factors is highly desirable, because a single nucleotide difference in a DNA-binding site can dictate which transcription factor binds at that site. To determine whether our approach could be used to distinguish proteins with very similar binding-site preferences, we used two related Zif268 mutants in microarray-binding experiments (Fig. 2 B and C; Table 2B of supplemental data). The mutant RGPD was selected from the phage display library by using the DNA sequences GCG and GCT, whereas the mutant REDV was selected by using GCG and GTG. The microarray-binding results not only verify binding to these respective sequences but also indicate that RGPD binds fairly well to CCG and to GCT (although RGPD was not recovered from a selection by using CCG). These results demonstrate that microarray-binding experiments can be used to distinguish the DNA-binding site preferences of transcription factors, even those with very similar DNA-binding specificities.
We next used this microarray approach to determine the binding-site preferences of zinc fingers with poorly characterized sequence specificity. The mutants LRHN and KASN had been isolated repeatedly after independent sets of in vitro selections by using many different 3-bp binding sites for the second finger (ACT, AAA, TTT, CCT, CTT, TTC, AGT, CGA, CAT, AGA, AGC, and AAT). Although LRHN was isolated in all of these selections, microarray-binding experiments revealed that this variant is highly specific for the DNA sequence containing the 3-bp binding site TAT (Table 2C of supplemental data). Moreover, the LRHN–TAT complex is almost as tight as the wild-type Zif268–DNA complex in these experiments. Meanwhile, microarray-binding experiments showed KASN to be fairly nonspecific, with binding to a number of DNA sequences consistent with a central 3-bp consensus (A/C/T)NT (Table 2D of supplemental data). These results indicate that microarray-binding experiments are highly sensitive in determining the DNA-binding site preferences of DNA-binding proteins, as the K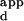 of the tightest KASN–DNA complex is over 80 times weaker than the interaction of wild-type Zif268 with its optimal binding site.
of the tightest KASN–DNA complex is over 80 times weaker than the interaction of wild-type Zif268 with its optimal binding site.
We calculated three types of DNA-binding specificity scores as metrics of how specific a protein is for its most tightly binding DNA site, as well as of the general spectrum of affinities that the protein has for variant DNA sites (Table 1). These analyses show that the Zif268 variants perform very differently in site-specific DNA recognition. Such binding specificity scores will allow us to determine which transcription factors are likely to bind at only a few specific sequences versus those which are likely to bind more uniformly throughout the genome.
Table 1.
DNA-binding specificity scores
| Variant | Single substitution |
K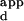 range range |
Overall preference |
|---|---|---|---|
| REDV | 510 | 1,100 | 920 |
| LRHN | 40 | 89 | 74 |
| RGPD | 38 | 75 | 65 |
| Wild type | 31 | 78 | 66 |
| KASN | 2.8 | 5.1 | 3.2 |
A mean K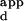 was calculated for the set of sequences consisting of all possible single base-pair substitutions in the central 3 bp of the Zif268 DNA-binding site of the sequence with the highest binding affinity. The single substitution score is defined as this mean K
was calculated for the set of sequences consisting of all possible single base-pair substitutions in the central 3 bp of the Zif268 DNA-binding site of the sequence with the highest binding affinity. The single substitution score is defined as this mean K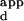 , divided by the K
, divided by the K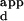 of the sequence with the highest binding affinity. The K
of the sequence with the highest binding affinity. The K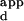 range is defined as the K
range is defined as the K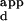 of the sequence with the lowest DNA-binding affinity, divided by the K
of the sequence with the lowest DNA-binding affinity, divided by the K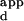 of the sequence with the highest DNA-binding affinity. The K
of the sequence with the highest DNA-binding affinity. The K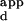 range serves as a measure of the specificity of a variant for the sequence with the highest binding affinity versus the sequence it binds most weakly. The overall preference of a variant is defined as the mean K
range serves as a measure of the specificity of a variant for the sequence with the highest binding affinity versus the sequence it binds most weakly. The overall preference of a variant is defined as the mean K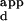 of the set of all the sequences on the microarray except for the one with the highest binding affinity, divided by the K
of the set of all the sequences on the microarray except for the one with the highest binding affinity, divided by the K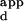 of the sequence with the highest binding affinity.
of the sequence with the highest binding affinity.
In addition, we used these microarrays to test whether the zinc-finger library's bias against binding DNA triplets with 5′ A or C could have been deduced before selection experiments. Therefore, we applied the entire library of zinc-finger clones to a microarray and analyzed binding of the entire population to the 64 different triplets. In general, triplets with a 5′ T were bound significantly better than their counterparts with a 5′ G, which in turn were bound significantly better than their counterparts with a 5′ A or C (T>G>A/C) (Fig. 5). The reason for this is that the 5′ nucleotide of the middle triplet is potentially specified by a combination of contacts from fingers 2 and 3 (see base pair 5 in Fig. 1A), but only the residue from finger 2 is randomized in the library (see Supplemental Discussion, which is published on the PNAS web site). The library is therefore biased toward binding sequences that have the specificity of wild-type Zif268 for the 5′ nucleotide of the middle triplet, i.e., T or, to a lesser degree, G. The use of microarrays to determine the DNA sequence preferences of entire libraries of DNA-binding domains will be extremely useful in guiding in vitro selections and for determining which of a set of libraries is best suited for selection by using a particular DNA sequence.
Figure 5.

Binding of the entire zinc-finger phage display library to a microarray indicates that DNA triplets with a 5′ T or G are bound preferentially over triplets with a 5′ A or C. The data are plotted to analyze binding as a function of the 5′ nucleotide. The average relative fluorescence intensity of all 64 different triplet-binding sites was normalized to 1; therefore, a value less than 1 indicates the particular sequence is bound less than average, and a value greater than 1 indicates the particular sequence is bound greater than average.
To see whether the evolution of sequence-specific phage from library selections could be followed, pools of library members eluted from different panning rounds of the GCG selection were bound to microarrays (Fig. 3A). Specifically bound sequences could be detected starting with round 2 (Supplemental Discussion). A significant change in the DNA-binding preference of the population occurred during the third round of selection, from T(A/T)G in round 2 to G(C/T)G in round 3. There was very little change in the DNA-binding specificity of the population during the fourth round of selection. These microarray-binding data are a useful aid to carrying out successful phage selections and can be used to guide the improvement of selection conditions or to determine the endpoint of a selection.
As a further demonstration of the utility of this approach for tracking the evolution of binding-site specificity during phage selections before sequencing of the selected phage, rounds 1–3 of the TCC selection were applied to microarrays, because this selection appeared to fail to produce zinc fingers specific for TCC (Fig. 3B). Sequencing analysis of clones obtained from this selection revealed that the experiment had not yielded zinc fingers specific for TCC but had instead produced zinc-finger clones that were also selected by the triplet GAC. Without the use of sequence information for the selected zinc fingers, microarray-binding experiments of phage pools from rounds 1–3 indicate that the selection was driven by zinc-finger phage that bind to the sequence present at the GAC spot on the microarrays. These phage were selected because the sequence GCGGACGCA is the complement of TGCGTCCGC, which is contained within the sequence of the DNA used in the TCC selection. GCGGACGCA is present on the complementary strand of the DNA sequence at the TCC spot on the microarrays, offset by 1 bp from the intended register of the 3-bp binding sites (Fig. 3C). The microarray results also indicate that GCGGACGCG (the top strand of the GAC spot) is bound more tightly by the phage pool eluted from round 3 of the TCC selection than are either GCGTCCGCG or GCGGACGCA, which are present on the top and bottom strands, respectively, of the DNA at the TCC spot on the microarrays.
Discussion
Our microarray-based method encourages comprehensive and directly comparable measures that in the past would have been prohibitive because of laborious experimental procedures. Because dozens of microarray-binding experiments could be performed in parallel in a single day, this technology provides significant cost and time advantages over conventional methods such as gel mobility-shift assays and nitrocellulose-binding assays, which can take months or even years to measure the effects of mutations for a large set of variant DNA–protein interactions. The materials and instrumentation used in our microarray experiments are commercially available and widely used in laboratories using DNA microarrays for mRNA expression analysis. In addition, the antibodies used for detection of the bound proteins are universal, in that they can be used regardless of what DNA-binding domain is displayed on the phage (see supplemental data). These experiments are not limited to zinc-finger proteins, as other structural classes of DNA-binding domains have been displayed on the surface of phage, including homeodomains (22), helix–turn–helix motifs (23), β sheets (24), leucine zippers (25), and steroid receptors (26). Furthermore, epitope-tagged DNA-binding proteins or whole transcription factors could be used instead of displaying the proteins on the surface of phage.
These microarray-binding experiments are highly scalable and thus could readily be adapted both for the combinatorial analysis of longer binding sites and for whole-genome analyses of transcription factor-binding sites. A full set of sequences spanning all possible 8-bp binding sites would consist of roughly 65,000 spots, which could fit onto a single microscope slide (see Supplemental Discussion). As for genomic microarrays, preliminary experiments indicate that the upper bound for DNA fragment lengths suitable for protein-binding experiments is at least 1 kb (see Fig. 7, which is published as supplemental data on the PNAS web site). Therefore, a microarray spotted with ≈12,000 sequences spanning ≈12 Mb of the entire Saccharomyces cerevisiae genome (27) could be used for the characterization of the sequence specificity of S. cerevisiae transcription factors as well as for the identification of genes putatively regulated by these proteins. Preliminary data indicate that DNA-binding domains of S. cerevisiae transcription factors, displayed on phage, are capable of specific binding to DNA microarrays (Fig. 6). Furthermore, such binding experiments using microarrays spotted with DNA corresponding to S. cerevisiae intergenic and coding regions have shown that this approach can be used to characterize the sequence specificity of transcription factors (M.L.B., P. N. Estep, X.H., and G.M.C., unpublished work). Likewise, a microarray consisting of roughly 30,000 spots of 1-kb sequence, enriched for the portions of the 120-Mb Drosophila melanogaster genome likely to contain regulatory elements, could be used to characterize the DNA-binding specificities of over 670 D. melanogaster transcription factors, at least 135 of which are zinc-finger proteins (28). Extrapolating these gene ratios to estimate the number of transcription factors and zinc-finger proteins in the human genome (29), there are thousands of transcription factors, approximately 1,000 of which are zinc-finger proteins (see supplemental data for details), which could be characterized by using these methodologies.
Figure 6.

Specific binding of S. cerevisiae transcription factors Rpn4 and Zap1 to microarrays. The image on the Left is a portion of a microarray bound by Rpn4; the image on the Right is a portion of a microarray bound by Zap1. The large white spots along three sides of the microarray images are alignment spots that indicate the positions of each row and column. The first two columns of experimental spots, indicated by “+” and “−” and labeled “Zap1,” correspond to those spots containing 37-bp long positive and 39-bp long negative control sequences for binding by Zap1. The next two columns of experimental spots, indicated by “+” and “−” and labeled “Rpn4,” correspond to those spots containing positive and negative control sequences (both 33 bp long) for binding by Rpn4.
Such microarray-binding experiments would produce datasets that would be useful not only for predicting functions for previously uncharacterized transcription factors but also for elucidating regulatory networks. Additionally, the effects of different concentrations of cofactors as well as the effects of alternate cofactors or binding conditions could be measured. This technology will also be immediately useful in engineering designer zinc-finger DNA-binding domains for the control of gene expression in biotechnology applications ranging from functional genomics to gene therapy. Finally, as more and more of these experiments are performed, the vast datasets produced could yield the necessary data required to determine what rules exist governing DNA recognition by sequence-specific transcription factors.
Supplementary Material
Acknowledgments
We thank Mark Isalan, Felix Lam, and Martin Steffen for technical assistance. We also thank Aaron Klug of the Medical Research Council and Pete Estep, John Aach, Jason Hughes, Robi Mitra, and other members of the Church Lab for helpful discussions and critical reading of the manuscript. This work was supported by a grant from the Office of Naval Research (N00014–99-1–0783).
Abbreviation
-
K
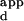
apparent dissociation constant
References
- 1.Elrod-Erickson M, Pabo C. J Biol Chem. 1999;274:19281–19285. doi: 10.1074/jbc.274.27.19281. [DOI] [PubMed] [Google Scholar]
- 2.Desjarlais J R, Berg J M. Proteins. 1992;12:101–104. doi: 10.1002/prot.340120202. [DOI] [PubMed] [Google Scholar]
- 3.Desjarlais J R, Berg J M. Proc Natl Acad Sci USA. 1992;89:7345–7349. doi: 10.1073/pnas.89.16.7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choo Y, Klug A. Proc Natl Acad Sci USA. 1994;91:11168–11172. doi: 10.1073/pnas.91.23.11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choo Y, Klug A. Proc Natl Acad Sci USA. 1994;91:11163–11167. doi: 10.1073/pnas.91.23.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choo Y, Klug A. Curr Opin Struct Biol. 1997;7:117–125. doi: 10.1016/s0959-440x(97)80015-2. [DOI] [PubMed] [Google Scholar]
- 7.Rebar E J, Pabo C O. Science. 1994;263:671–673. doi: 10.1126/science.8303274. [DOI] [PubMed] [Google Scholar]
- 8.Jamieson A, Kim S, Wells J. Biochemistry. 1994;33:5689–5695. doi: 10.1021/bi00185a004. [DOI] [PubMed] [Google Scholar]
- 9.Wu H, Yang W, Barbas C F., III Proc Natl Acad Sci USA. 1995;92:344–348. doi: 10.1073/pnas.92.2.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woodbury C P, von Hippel P H. Biochemistry. 1983;22:4730–4737. doi: 10.1021/bi00289a018. [DOI] [PubMed] [Google Scholar]
- 11.Garner M M, Revzin A. Nucleic Acids Res. 1981;9:3047–3060. doi: 10.1093/nar/9.13.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choo Y, Klug A. Nucleic Acids Res. 1993;21:3341–3346. doi: 10.1093/nar/21.15.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowen B, Steinberg J, Laemmli U K, Weintraub H. Nucleic Acids Res. 1980;8:1–20. doi: 10.1093/nar/8.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanes S D, Brent R. Science. 1991;251:426–430. doi: 10.1126/science.1671176. [DOI] [PubMed] [Google Scholar]
- 15.Oliphant A, Brandl C, Struhl K. Mol Cell Biol. 1989;9:2944–2949. doi: 10.1128/mcb.9.7.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schena M, Shalon D, Davis R W, Brown P O. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 17.Bulyk M L, Gentalen E, Lockhart D J, Church G M. Nat Biotechnol. 1999;17:573–577. doi: 10.1038/9878. [DOI] [PubMed] [Google Scholar]
- 18.Pavletich N P, Pabo C O. Science. 1991;252:809–817. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- 19.Elrod-Erickson M, Rould M, Nekludova L, Pabo C. Structure (Cambridge, UK) 1996;4:1171–1180. doi: 10.1016/s0969-2126(96)00125-6. [DOI] [PubMed] [Google Scholar]
- 20.Griffiths A, Williams S, Hartley O, Tomlinson I, Waterhouse P, Crosby W, Kontermann R, Jones P, Low N, Allison T, et al. EMBO J. 1994;13:3245–3260. doi: 10.1002/j.1460-2075.1994.tb06626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider T D, Stormo G D, Gold L. J Mol Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- 22.Connolly J, Augustine J, Francklyn C. Nucleic Acids Res. 1999;27:1182–1189. doi: 10.1093/nar/27.4.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.d'Alencon E, Ehrlich S. J Bacteriol. 2000;182:2973–2977. doi: 10.1128/jb.182.10.2973-2977.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruan B, Hoskins J, Wang L, Bryan P. Protein Sci. 1998;7:2345–2353. doi: 10.1002/pro.5560071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crameri R, Suter M. Gene. 1993;137:69–75. doi: 10.1016/0378-1119(93)90253-y. [DOI] [PubMed] [Google Scholar]
- 26.Chusacultanachai S, Glenn K A, Rodriguez A O, Read E K, Gardner J F, Katzenellenbogen B S, Shapiro D J. J Biol Chem. 1999;274:23591–23598. doi: 10.1074/jbc.274.33.23591. [DOI] [PubMed] [Google Scholar]
- 27.Goffeau A, Barrell B, Bussey H, Davis R, Dujon B, Feldmann H, Galibert F, Hoheisel J, Jacq C, Johnston M, et al. Science. 1996;546:563–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 28.Adams M, Celniker S, Holt R, Evans C, Gocayne J, Amanatides P, Scherer S, Li P, Hoskins R, Galle R, et al. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 29.Pennisi E. Science. 2000;288:1146–1147. doi: 10.1126/science.288.5469.1146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}