

Abstract
Myocardial ischaemia is associated with the generation of lipid peroxidation products such as HNE (4-hydroxy-trans-2-nonenal); however, the processes that predispose the ischaemic heart to toxicity by HNE and related species are not well understood. In the present study, we examined HNE metabolism in isolated aerobic and ischaemic rat hearts. In aerobic hearts, the reagent [3H]HNE was glutathiolated, oxidized to [3H]4-hydroxynonenoic acid, and reduced to [3H]1,4-dihydroxynonene. In ischaemic hearts, [3H]4-hydroxynonenoic acid formation was inhibited and higher levels of [3H]1,4-dihydroxynonene and [3H]GS-HNE (glutathione conjugate of HNE) were generated. Metabolism of [3H]HNE to [3H]4-hydroxynonenoic acid was restored upon reperfusion. Reperfused hearts were more efficient at metabolizing HNE than non-ischaemic hearts. Ischaemia increased the myocardial levels of endogenous HNE and 1,4-dihydroxynonene, but not 4-hydroxynonenoic acid. Isolated cardiac mitochondria metabolized [3H]HNE primarily to [3H]4-hydroxynonenoic acid and minimally to [3H]1,4-dihydroxynonene and [3H]GS-HNE. Moreover, [3H]4-hydroxynonenoic acid was extruded from mitochondria, whereas other [3H]HNE metabolites were retained in the matrix. Mitochondria isolated from ischaemic hearts were found to contain 2-fold higher levels of protein-bound HNE than the cytosol, as well as increased [3H]GS-HNE and [3H]1,4-dihydroxynonene, but not [3H]4-hydroxynonenoic acid. Mitochondrial HNE oxidation was inhibited at an NAD+/NADH ratio of 0.4 (equivalent to the ischaemic heart) and restored at an NAD+/NADH ratio of 8.6 (equivalent to the reperfused heart). These results suggest that HNE metabolism is inhibited during myocardial ischaemia owing to NAD+ depletion. This decrease in mitochondrial metabolism of lipid peroxidation products and the inability of the mitochondria to extrude HNE metabolites could contribute to myocardial ischaemia/reperfusion injury.
Keywords: aldehyde dehydrogenase, aldose reductase, glutathione transferase, 4-hydroxy-trans-2-nonenal (HNE), ischaemia, perfusion
INTRODUCTION
Myocardial ischaemia/reperfusion is accompanied by an increase in oxygen-derived free radicals [1,2]. These species are highly reactive. They stimulate, interrupt and modify many signalling events and, if unquenched, they induce tissue dysfunction or cell death. A causative role of ROS (reactive oxygen species) in myocardial ischaemia/reperfusion injury is supported by evidence showing that antioxidant treatment [1,2] or overexpression of antioxidant enzymes such as Mn-SOD (superoxide dismutase) [3], catalase [4], extracellular-SOD [5] or glutathione peroxidase [6] prevents ischaemia/reperfusion injury, and that partial deficiency of Mn-SOD [7] or the complete absence of glutathione peroxidase [8] or CuZn-SOD [9] renders the heart more sensitive to ischaemic injury. In addition, the generation of RNS (reactive nitrogen species), such as peroxynitrite, has also been linked to tissue injury associated with myocardial infarction [10]. Most free radicals are short-lived and their ability to alter cell signalling or incite tissue injury is paradoxically limited by their high reactivity. These radicals, however, generate a host of metastable secondary products derived from oxidized lipids. The generation of lipid peroxidation products in the heart is also associated with increased formation of RNS [11]. Products of lipid peroxidation generated from ROS or RNS could propagate and amplify cell injury even beyond that caused by free radicals themselves. In this role, products such as the α,β-unsaturated aldehydes are likely to be the most significant because they are highly reactive [12] and because they affect signal transduction [13], energy production [14], ion-channel function [15] and cell-death pathways [16]. Moreover, unsaturated aldehydes generated from lipid peroxidation form covalent adducts with proteins [12,17] and DNA [18], which could elicit acute stress responses or induce sustained tissue dysfunction.
Several studies show that unsaturated aldehydes such as HNE (4-hydroxy-trans-2-nonenal) are generated during myocardial ischaemia/reperfusion [19], and covalent adducts between HNE and myocardial proteins have been detected in the ischaemic heart [20]. Products of lipid peroxidation such as HNE are rapidly metabolized [21,22]; hence they are unlikely to linger in healthy and well-energized tissue. However, it remains unclear how unsaturated aldehydes, such as HNE, are cleared from the ischaemic heart. Our studies have shown that, in the aerobic heart, HNE is readily conjugated to glutathione (GS–HNE) or oxidized to HNA (4-hydroxynonenoic acid). In addition, HNE and its glutathione conjugate are both reduced by AR (aldose reductase) [21]. The overall competence of these pathways could, however, be significantly perturbed by ischaemia. Changes in tissue perfusion could prevent extrusion and washout of lipid peroxidation products and their metabolites, and inhibition of aerobic metabolism could affect the activity of the oxidoreductases involved in aldehyde metabolism. In addition, aldehyde-metabolizing enzymes may be modified or inactivated by ROS or their own substrates. As a result of these changes, HNE could accumulate in the heart and cause changes in cell signalling or function. Accordingly, in the present study we investigated how ischaemia affects HNE removal and show that ischaemia decreases the ability of the heart to oxidize HNE. We suggest that these events are caused by inhibition of mitochondrial oxidation of HNE via ALDH (aldehyde dehydrogenase) and that loss of ALDH activity is due to the ischaemia-induced decrease in the NAD+/NADH ratio.
EXPERIMENTAL
Materials
HNE was synthesized as described previously [21]. Anti-(cytochrome c oxidase subunit I) antibodies were purchased from Molecular Probes. Anti-ALDH2 antibodies were a gift from Dr Henry Weiner (Department of Biochemistry, Purdue University, West Lafayette, IN, U.S.A.). Polyclonal antibodies against recombinant human AR were raised and characterized as described previously [23].
Isolated perfused and ischaemic heart preparations
Animal studies were performed under protocols approved by the University of Louisville Animal Care Use Committee. Sprague-Dawley rats (200–260 g) were administered heparin (100 i.u./100 g), anaesthetized with sodium pentobarbital (60 mg/kg of body weight), and their hearts were excised, cannulated and perfused in the Langendorff retrograde mode as described previously [21]. Hearts were perfused at a constant perfusion pressure of 80 mmHg with KH (Krebs-Henseleit) buffer comprising 118 mM NaCl, 25 mM NaHCO3, 4.7 mM KCl, 1.25 mM KH2PO4, 3 mM CaCl2, 1.25 mM MgCl2, 0.5 mM EDTA and 10 mM glucose, and gassed with a 95% O2/5% CO2 gas mixture. The hearts were not paced. At the end of the experimental protocol, the hearts were immediately snap-frozen in liquid nitrogen and stored at −80 °C for biochemical analysis.
Isolation and subfractionation of rat heart mitochondria
Hearts were excised and rinsed in isolation buffer (pH 7.4), containing 220 mM mannitol, 70 mM sucrose and 5 mM Mops. The hearts were finely minced on ice with a razor blade and homogenized in a Glass Col homogenizer. Samples were centrifuged at 500 g for 10 min, and the supernatant was filtered through cheesecloth. The filtrate was centrifuged at 5000 g for 10 min to sediment the mitochondria. The mitochondrial pellet was washed in respiration buffer (pH 7.4) containing 120 mM KCl, 1 mM EGTA, 5 mM Mops and 5 mM KH2PO4, centrifuged at 5000 g for 10 min, and then resuspended in 1 ml of the respiration buffer. Cytosolic contamination of the mitochondrial fraction was 0.5% as measured by AR expression and lactate dehydrogenase activity. For subfractionation, the mitochondrial pellet was suspended and washed three times in equilibration buffer (pH 8.0), containing 20 mM Tris/HCl, 0.4% Chaps, 1 mM EDTA and 5 mM DTT (dithiothreitol). The mitochondria were centrifuged at 6000 g for 10 min and resuspended in 500 μl of the equilibration buffer containing 1:100 mammalian protease inhibitor cocktail (Pierce Biotechnology). Mitochondria were snap-frozen in liquid nitrogen and thawed three times. The mitochondria were then sonicated for 15 s and centrifuged for 30 min at 13000 g. The supernatant fraction was collected and designated the matrix fraction. To wash remaining matrix proteins from the membrane fraction, 2 ml of the equilibration buffer was added to the pellet and the mixture was sonicated for 15 s. This was repeated three times. After the final centrifugation step, the membrane fraction was resuspended in 300 μl of PBS containing 1% (v/v) Triton X-100.
Myocardial metabolism of HNE
For HNE metabolism, 50 nmol of [3H]HNE in 0.1 ml of oxygenated KH buffer was injected through a side port of the cannulated rat heart. Rat hearts were subjected to 10 or 70 min perfusion with KH buffer, 10 min perfusion followed by 30 min ischaemia (with [3H]HNE injection immediately prior to ischaemia), 10 min perfusion followed by 30 min ischaemia with 10 min reperfusion (with [3H]HNE injection either before ischaemia or immediately before reperfusion), or 10 min perfusion followed by 30 min ischaemia and 30 min reperfusion (with [3H]HNE injection after 30 min reperfusion). Perfusates were collected for 3, 6 and 10 min following HNE injection. At the end of each protocol, radioactivity in the perfusates and tissue extracts was measured, and radiolabelled metabolites of HNE were separated by HPLC and characterized by MS.
HPLC analysis and MS
The reagent [3H]HNE and its metabolites were synthesized as described previously [21]. Synthesized standards and metabolites of HNE were separated by reverse-phase HPLC as described previously [24]. The glutathione conjugates (GS–HNE and GS–DHN; DHN is 1,4-dihydroxynonene) eluted with a tR (retention time) of approx. 23 min, whereas DHN, HNA and HNE had a tR of 31, 36 and 43 min respectively. The purity and the chemical identity of the glutathione conjugates were established by HPLC and ESI–MS (electrospray ionization MS) as described previously [24]. The chemical identities of DHN and HNA were established by GC/MS as described previously [25]. To measure endogenous levels of HNE, DHN and HNA, D11-HNE and D11-HNE metabolites were prepared and used as internal standards as described for [3H]DHN and [3H]HNA [21]. No exogenous HNE was added in these experiments.
Measurement of myocardial NAD+/NADH ratios
Myocardial nucleotide ratios in perfused, ischaemic and reperfused hearts were measured using an NAD+/NADH quantification kit according to the manufacturer's instructions (BioVision).
In vitro HNE oxidation assay
Nucleotide-dependent HNE oxidation was measured in cardiac homogenates by HPLC using [3H]HNE as the substrate. Rat hearts were homogenized in 0.1 M potassium phosphate buffer (pH 7.4), containing 1 mM DTT, 1 mM EDTA and 1% protease inhibitor cocktail. The homogenate was sonicated on ice for 15 s and centrifuged at 14000 g for 15 min. The supernatant was then passed through a Sephadex G25 column equilibrated with nitrogen-saturated 0.1 M potassium phosphate buffer (pH 7.4). HNE oxidation reactions were carried out in 250 μl of potassium phosphate buffer containing 50 μg of myocardial protein, 1.1 mM total NAD(H) and 100 μM [3H]HNE for 30 min at 37 °C. To determine the dependence of HNE oxidation on aerobic- and ischaemia-representative nucleotide ratios, the NAD+/NADH ratios were measured in perfused, ischaemic and reperfused hearts. The [3H]HNE and [3H]HNA were separated by HPLC and their concentration was measured by scintillation counting.
Statistical analysis
Values are reported as means±S.E.M. An unpaired Student's t test was used to compare groups. For multiple group analysis, one-way ANOVA analysis was performed. P<0.05 was considered statistically significant.
RESULTS
Metabolism of exogenous HNE in the ischaemic heart
To examine how ischaemia affects HNE metabolism, we identified products of [3H]HNE generated in perfused and ischaemic rat hearts. As described previously [21], the major HNE metabolites generated in the perfused aerobic heart were the non-reduced and reduced glutathione conjugates of HNE (GS–HNE and GS–DHN; which together account for 22% of the metabolites recovered) and products of HNE oxidation (HNA; 39%) or reduction (DHN; 5%) (Figures 1A and 1D, and Table 1). Approx. 7% of the products remained unidentified, and 27% of the unmetabolized HNE was recovered in the perfusate. A similar profile of metabolites was observed in the ischaemic heart, although the relative distribution of the metabolites was altered. As shown in Figure 1(A), ESI–MS of peak I obtained from non-ischaemic hearts showed a strong peak at m/z 464 and another ion peak at m/z 446. These were assigned to MH+ and [MH-18]+ ions of GS–HNE. The 466 peak was assigned to the MH+ ion of GS–DHN. Peak I from ischaemic hearts, however, displayed only one prominent ion with m/z 466, indicating that in the ischaemic heart GS–HNE is completely reduced to GS–DHN (Figure 1B). Peak II displayed the tR on GC and an MS fragmentation pattern identical with that of DHN (Figure 1B). Peak III co-eluted with HNA. These results indicate that, in comparison with the aerobic heart, the ischaemic heart is less efficient in oxidizing HNE to HNA (39% compared with 16%), but it generates more DHN (increased from 5 to 25%) (Table 1). Similar changes (a decrease in HNA and an increase in DHN formation) were also observed in perfusates collected from hearts just after ischaemia (Figure 1C and Table 1). Collectively, these results suggest that ischaemia decreases the oxidative metabolism of HNE to HNA, while increasing the reduction of HNE to DHN and of GS–HNE to GS–DHN. Ischaemia also increases glutathione-linked metabolism of HNE in the whole heart.
Fig 1.

HPLC separation of HNE metabolites generated in the heart after a bolus injection of [3H]HNE. In each experiment, isolated adult rat hearts were equilibrated with aerobic buffer for 10 min and then 50 nmol of [3H]HNE in 0.1 ml of KH buffer was injected into the side port of the aortic cannula. The hearts were perfused with aerobic KH buffer (A), were subjected to 30 min of ischaemia (B), or were subjected to 30 min of ischaemia followed by 10 min of reperfusion (C). Peaks were assigned to GS–HNE/GS–DHN (peak I), DHN (peak II) and HNA (peak III), and unmetabolized HNE (peak IV). The identity of peak V is unknown. Inset (i) shows the ESI/mass spectrum of peak I. Ions with m/z 464 and 446 were assigned to MH+ and [MH-18]+ ions of GS–HNE. The peak with m/z 466.3 corresponds to the MH+ ion of GS–DHN. Inset (ii) shows the GC/mass spectrum of HPLC peak III after derivatization with BSTFA [bis-(trimethylsilyl)trifluoroacetamide]. The ion with a tR of 9.55 min, corresponding to the tR of HNA, displayed a fragmentation pattern (inset iii) similar to HNA. ESI–MS analysis of peak I from ischaemic heart tissue (B, inset iv) showed only a single peak with m/z 466.3 corresponding to GS–DHN. GC/MS analysis of peak II from the ischaemic heart showed an ion with a tR of 8.08 min (inset v) and a fragmentation pattern (inset vi) similar to reagent DHN. ESI–MS analysis of peak I obtained from perfusates obtained within the first 3 min of reperfusion showed a single ion (m/z=466.2) corresponding to GS–DHN (C, inset vii). Profiles are representative of 4–8 rats per group (see Table 1).
Table 1.
HNE metabolism in aerobic, ischaemic and post-ischaemic hearts
| Protocol | Sample | Peak I GS-conjugates | Peak II DHN | Peak III HNA | Peak IV HNE | Peak V | Others |
|---|---|---|---|---|---|---|---|
| Aerobic perfusion | Perfusate | 22±1 | 5±2 | 39±2 | 27±5 | 3±1 | 4±1 |
| Global ischaemia | Tissue extract | 39±3* | 25±2* | 16±3* | 4±2* | 10±3 | 6±3 |
| Ischaemia/reperfusion | Perfusate | 31±1* | 20±3* | 20±3* | 19±3† | 5±1 | 5±1 |
| Reperfusion I | Perfusate | 24±4† | 23±3* | 23±3* | 17±4† | 6±3 | 7±2 |
| Reperfusion II | Perfusate | 25±3† | 23±1* | 30±4† | 12±4* | 4±1 | 6±2 |
For aerobic perfusion experiments, [3H]HNE was injected 10 min after aerobic equilibration and metabolites were collected in the perfusate for the next 3 min. In the global ischaemia protocol, [3H]HNE was injected just before 30 min of ischaemia and metabolites were extracted from whole-heart tissue. For ischaemia/reperfusion, [3H]HNE was injected, the hearts were subjected to 30 min of ischaemia, and metabolites were collected within the first 3 min of reperfusion in the perfusate. In the reperfusion I protocol, [3H]HNE was injected immediately after 30 min of ischaemia, and the metabolites were collected in the next 3 min. In the reperfusion II protocol, HNE was injected after 30 min of ischaemia followed by 30 min of reperfusion, and metabolites were collected for the next 3 min. Values represent the percentage distribution of the total radioactivity recovered after HPLC and are expressed as the means±S.E.M. of four to eight hearts.
P<0.05 compared with aerobic perfusion;
P<0.05 compared with ischaemia.
To determine how reperfusion affects HNE metabolism, two series of experiments were performed. In the first series, hearts were subjected to ischaemia for 30 min and then injected with [3H]HNE at the beginning of reflow. In the second series, hearts were subjected to 30 min of ischaemia, and HNE was injected after 30 min of reperfusion. When the hearts were treated with HNE immediately upon reflow, the profile of HNE metabolites in the perfusate was similar to that obtained from the ischaemic myocardium, i.e. HNA formation was suppressed, DHN was increased and most of the glutathione conjugate of HNE was in the reduced form (Table 1, reperfusion I). Although there was no difference in the extent of glutathione conjugate formation, post-ischaemic hearts displayed diminished HNE oxidation. When the hearts were treated with [3H]HNE 30 min after reflow, oxidation of HNE was increased compared with non-reperfused hearts and reduction to DHN remained elevated (Table 1, reperfusion II). Overall, owing to the recovery of oxidative metabolism and the enhancement of reductive metabolism, the reperfused heart was more efficient at metabolizing HNE than the aerobic heart. These changes did not appear to be due to prolonged perfusion because 70 min of perfusion did not affect aerobic HNE metabolism (results not shown). These results demonstrate that ischaemia increases glutathione conjugation of HNE as well as the reduction of HNE and GS–HNE to DHN and GS–DHN respectively; however, no increase in glutathione transferase activity in the ischaemic heart was observed (results not shown). As reported previously [26,27], AR activity in the ischaemic heart was increased, as shown by greater conversion of GS–HNE into GS–DHN (Figure 1 and Table 1), but the increase in DHN formation was not prevented by AR inhibitors (results not shown). Hence, it appears that, in contrast with increases in other metabolic pathways, oxidation of HNE to HNA is suppressed by ischaemia, and the capacity of the heart to oxidize HNE recovers progressively during reperfusion.
Cardiac metabolism of endogenous HNE
The results obtained so far indicated that HNE is metabolized to multiple products and that the formation of HNA is inhibited and DHN is increased during ischaemia. To determine whether the metabolism of endogenously generated HNE is similarly affected, we measured the levels of endogenously generated HNE, HNA and DHN in hearts subjected to ischaemia and reperfusion using D11 analogues of these metabolites as internal standards. Low levels of HNE, HNA and DHN were detected in hearts perfused with aerobic buffer (Figure 2). In hearts subjected to 30 min of ischaemia, however, the concentration of HNE and DHN present in the heart increased 2–3-fold. No increase in HNA was observed (Figure 2E). Upon reperfusion, myocardial levels of the metabolites returned to their baseline values. Based on these observations, we conclude that ischaemia increases HNE formation in the heart; however, even though an increase in HNE was accompanied by an increase in DHN formation, HNA formation was not increased. These findings corroborate the observations with [3H]HNE and support the notion that HNE accumulates in the ischaemic heart in part due to lack of oxidation to HNA.
Fig 2.

Isolated rat hearts were perfused ex vivo for 40 min with aerobic buffer or were subjected to 30 min of ischaemia alone or were made ischaemic for 30 min followed by 5 min of reperfusion. After completion of the protocol, hearts were removed, homogenized in the presence of D11-HNE, D11-DHN and D11-HNA. The samples were derivatized with PFBHA [O-(2,3,4,5,6-pentafluorobenzyl)hydroxylamine] and BSTFA. (Ai) shows the gas chromatogram of HNE and D11-HNE in chemical ionization mode. (Aii) shows the separation of D11-DHN and D11-HNA in electron-impact ionization mode. DHN and HNA co-eluted with their D11 analogues (results not shown). To determine the relative metabolite levels, the abundance of the major ions was quantified by single ion monitoring. For quantification, ions in which C5H11/C5D11 fragments were intact [M-HF for HNE (m/z=403) and D11-HNE (m/z=414); M-CH3 for DHN (m/z=287) and D11-DHN (m/z=298); and M-CH3 for HNA (m/z=301) and D11-HNA (m/z=312)] were used. (B–D) show representative spectra illustrating the abundance of selected ions for quantification of HNE, DHN and HNA in hearts subjected to perfusion alone (P in B), ischaemia (I in C) or reperfusion after ischaemia (IR in D). (E) Group data showing myocardial abundance of HNA, DHN and HNE in perfused hearts and hearts subjected to ischaemia/reperfusion. Values are means±S.E.M. (n=four to six rats per group). *P<0.05 compared with hearts perfused with the aerobic buffer alone. P, perfused; I, ischaemia; IR, reperfusion after ischaemia.
HNE oxidation in the perfused heart
Previous studies suggest that oxidation of HNE to HNA could be catalysed by ALDH [21,22,25,28]. To delineate the role of this pathway, perfused hearts were treated with the ALDH inhibitor benomyl [29] immediately prior to injecting [3H]HNE. In aerobic hearts, treatment with benomyl inhibited HNA formation by 85% (Figure 3), suggesting that ALDH is the major route of HNE oxidation in the heart. Moreover, inhibition of ALDH led to a significant increase in DHN formation and slightly greater glutathione conjugation, indicating that ischaemic changes in HNE metabolism could be mimicked in the aerobic heart by inhibiting ALDH. This possibility strengthens the view that differences in the pattern of HNE metabolites generated in the aerobic and ischaemic hearts could be attributed to inhibition of ALDH.
Fig 3.

Following 10 min of KH buffer perfusion without or with the ALDH inhibitor, benomyl (20 μM), a bolus of [3H]HNE (50 nmol in 0.1 ml KH buffer) was injected into the aorta. The coronary perfusate was collected for 3 min and tritiated metabolites recovered in the perfusate were resolved by HPLC. Results are shown as the percentage of total HNE metabolites recovered in the perfusate. Values are means±S.E.M. (n=six rats per group). *P<0.05 compared with hearts perfused without benomyl.
Mitochondrial metabolism of HNE
Our previous studies show that there is little or no HNE-oxidizing activity in rat heart cytosol [21,30]. To determine whether mitochondria are the main sites of HNE oxidation, we examined HNE metabolism in isolated cardiac mitochondria. Intact mitochondria were isolated to high yield from naïve rat hearts (see the Experimental section). These mitochondria were intact and relatively free from cytosolic and myofibrillar components. To establish an appropriate dose of HNE that could be tolerated by mitochondria, we examined the effects of HNE on oxygen consumption. The standard P/O value of isolated mitochondria was 2.91±0.25, with an RCR (respiratory control ratio) of 5.22±0.76. Incubation with 10, 20 and 50 μM HNE decreased state 3 respiration. RCR values at 10, 20 and 50 μM HNE dropped to 4.92, 4.03 and 2.16 respectively, suggesting that respiratory complexes were inhibited at >20 μM HNE (results not shown). Hence, 15 μM HNE was used in all subsequent experiments.
For studying HNE metabolism, freshly isolated mitochondria were suspended in 1 ml of respiration buffer at a protein concentration of 2.5 mg/ml and incubated with 15 μM [3H]HNE for 30 min at 37 °C. At the end of the incubation, the mitochondria were separated from the medium by centrifugation, and the radioactivity retained in the mitochondria, as well as that in the medium, was measured. These experiments showed that 18±4% of the radioactivity (c.p.m. recovered) was in the mitochondrial pellet, whereas 75±5% of the radioactivity was in the incubation medium. HPLC separation of the metabolites retained inside the mitochondria revealed five peaks (Figure 4A). The tR of GS–HNE (representing 45±2% of the metabolites retained inside the mitochondria) was identical with the tR of HNE glutathione conjugates. ESI–MS analysis of this fraction showed a strong peak with an MH+ m/z of 464 and a [MH-18]+ product ion at 446 (Figure 4A, inset i), consistent with GS–HNE. No ions of m/z 466 or 480 were detected, indicating that GS–HNE was not converted into GS–DHN or GS–HNA in the mitochondria. The second peak of the mitochondrial extract, representing 22±2% of the radioactivity which was retained inside the pellet, eluted at a tR identical with DHN. Only traces of radioactivity were detected at the tR of HNA and HNE. A fifth HPLC peak in the mitochondrial extract, representing 31±2% of the metabolites inside the pellet, eluted at a tR of 54 min. The chemical identity of this peak remains unknown. Separation of radioactivity in the incubation medium by HPLC yielded two major peaks. The tR of the first major peak, representing 89±3% of the radioactivity recovered in the medium, was identical with HNA (Figure 4B). Fractions corresponding to this peak were pooled, silylated and subjected to GC/MS. The chromatograph showed a prominent peak with a tR of 9.5 min (Figure 4B, inset ii). The fragmentation pattern of this ion (Figure 4B, inset iii) was identical with that of HNA. The other prominent HPLC peak (peak IV) representing 6±2% of the radioactivity in the incubation medium had a tR similar to HNE and was assigned to unmetabolized HNE. Taken together, these results suggest that oxidation of HNE to HNA is the major route of HNE metabolism in cardiac mitochondria and that HNA is actively extruded from the mitochondria, whereas other HNE metabolites, i.e. GS–HNE and DHN, are retained in the matrix. To determine whether mitochondrial HNA was generated by ALDH, isolated mitochondria were incubated with the ALDH inhibitor, benomyl. After pre-incubating the mitochondria for 30 min at 37 °C with 20 μM benomyl, 15 nmol of [3H]HNE were added to the medium. After 30 min, the mitochondria were separated from the incubation medium, and the HNA generated in the medium was determined by HPLC. As shown in Figure 4(D), benomyl inhibited HNA formation by 82±2%, with a concomitant increase in unmetabolized HNE (Figure 4D, inset v), suggesting that ALDH is the main enzyme catalysing HNE oxidation in mitochondria. Western blot analysis of mitochondrial proteins confirmed the presence of ALDH2 in the mitochondrial matrix (Figure 4C, inset iv); little to no AR was detected in the mitochondria (results not shown). Measurement of the HNE metabolites in the pellets of control and benomyl-treated mitochondria showed nearly identical levels of GS–HNE, DHN and other compounds (Figures 4C and 4D).
Fig 4.

Isolated rat heart mitochondria were incubated with 15 μM [3H]HNE in 1.0 ml of respiration buffer for 30 min at 37 °C. Mitochondrial pellets (A and C) and the incubation medium (B and D) were separated by centrifugation, and radioactivity retained inside the mitochondrial pellet and extruded in the incubation medium was resolved by HPLC. Peak identification, as indicated, was based on the tR of GS–HNE (I), DHN (II), HNA (III) and HNE (IV). Inset (i) is the ESI/mass spectrum of the HPLC peak eluting at the tR identical with GS–HNE. The ions with m/z 464 and 446 were assigned to MH+ and [MH-18]+ ions of GS–HNE. The chemical identity of the major HPLC peak in (B) was established by GC/MS after derivatization with BSTFA. The metabolite eluted as a strong peak at 9.52 min (inset ii), similar to HNA. The fragmentation pattern (inset iii) of this peak was identical with HNA. To probe the role of ALDH in HNE oxidation, mitochondria were incubated with the ALDH inhibitor, benomyl (20 μM), for 30 min at 37 °C. The mitochondria were then incubated with 15 μM [3H]HNE, and metabolites in the pellet (C) and in the medium (D) were separated by HPLC. Expression, as shown by Western blotting (10 μg of protein/well), of ALDH2 in whole mitochondria (WM), the mitochondrial matrix (Mat) and the mitochondrial membrane (Mem) fractions of the rat heart, is shown in inset (iv). Cytochrome c oxidase subunit 1 (COX IV-1) was used as a mitochondrial membrane marker. The percentage change in HNE metabolite distribution in control and benomyl-treated mitochondria is shown in inset (v). Values are means±S.E.M. (n=four to six individual experiments). *P<0.05 compared with the control.
These results suggest that, in contrast with the intact heart in which ALDH inhibition by benomyl or ischaemia led to an increase in metabolism via other pathways, inhibition of HNE oxidation in the mitochondria is not compensated by an increase in other metabolic pathways, but instead results in a net decrease in HNE metabolism.
To examine mitochondrial HNE metabolism during ischaemia, we determined the mitochondrial and cytosolic distribution of radiolabelled metabolites of [3H]HNE in ischaemic hearts. For this, [3H]HNE was injected into the side port of the cannulated heart, and the heart was subjected to 30 min of global ischaemia. After ischaemia, the hearts were homogenized in the presence of 2 μM cyclosporin to prevent spurious MPT (mitochondrial permeability transition) during homogenization. Mitochondrial and cytosolic fractions were separated by centrifugation and the mitochondrial pellet was extensively washed to remove any adherent radioactivity. Measurements of radioactivity in these samples showed 11±1% retention in the mitochondrial pellet and 85±2% in the cytosol (Figure 5). Less than 2% of the radioactivity was recovered in the mitochondrial pellet in hearts homogenized in the absence of cyclosporin. HPLC separation of the radioactivity recovered from the cytosolic fraction showed that 41±2% of the radioactivity eluted as glutathione conjugates, 31±2% as DHN, 20±3% as HNA and 8±1% as peak V (Figure 5A, inset i). HPLC separation of the radioactivity retained inside the mitochondria showed that the glutathione conjugates of HNE accounted for 26±1% of the radioactivity recovered from the mitochondrial extract, whereas 48±5% radioactivity was DHN, and 16±1% eluted as peak V (Figure 5B, inset ii); 10±2% of the radioactivity retained inside the mitochondria was bound to the proteins, whereas only 3±1% of [3H]HNE recovered in the cytosol was protein-bound (Figure 5, inset iii). No HNA was recovered in the mitochondrial pellet, indicating that, although mitochondria are the major site of HNE oxidation, HNA is extruded from the mitochondria into the cytosol, whereas other metabolites of HNE, such as GS–HNE and DHN, are retained in the mitochondrial matrix.
Fig 5.

Isolated rat hearts were equilibrated with aerobic buffer for 10 min, and 50 nmol of [3H]HNE in 0.1 ml of KH buffer was injected into the side port of the aortic cannula, followed by 30 min of ischaemia. After the ischaemic protocol, the mitochondria were isolated as described in the Experimental section in the presence of 2 μM cyclosporin A. Peaks were assigned to GS-HNE/GS-DHN (peak I), DHN (peak II), HNA (peak III) and unmetabolized HNE (peak IV). The identity of peak V is unknown. HNE metabolites from the cytosol (A, inset i) and mitochondria (B, inset ii) were quantified and are shown as the percentage of total recovered HNE metabolites. Inset (iii) shows the protein-bound [3H]HNE in cytosolic and mitochondrial fractions. Values represent the means±S.E.M. (n=four rat hearts).
Mechanism of the ischaemic decrease in HNE oxidation
Our results so far suggested that HNE is oxidized to HNA and that this process is inhibited during ischaemia. To understand the mechanism of this inhibition, we studied the pyridine nucleotide dependence of HNE metabolism in the mitochondria and determined whether changes in pyridine nucleotides may be responsible for ischaemic inhibition of HNE metabolism. Mitochondrial lysates were incubated with 15 μM [3H]HNE in the presence of NAD+ or NADP+. After the incubation, proteins were precipitated and metabolites in the supernatant were separated by HPLC. As shown in Figure 6(A), NAD+ supported oxidation of HNE to HNA; however, no HNA was found in supernatants of mitochondrial lysates incubated with NADP+ (Figure 6B). No additional metabolites were detected. These results suggest that NAD+-mediated oxidation is the major route of HNE metabolism in cardiac mitochondria.
Fig 6.
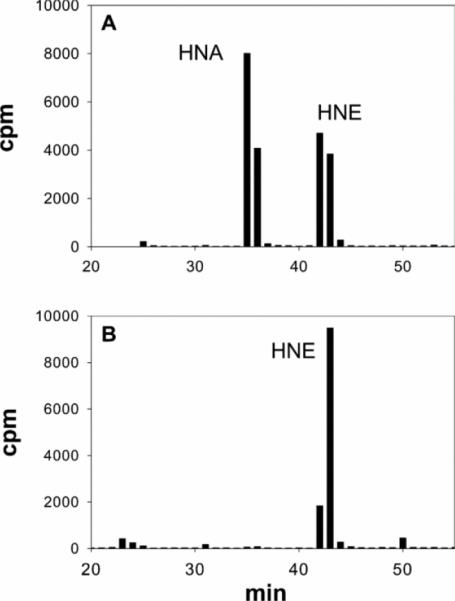
HPLC profiles obtained from mitochondrial lysates incubated with [3H]HNE in the presence of the indicated pyridine nucleotides. Isolated rat heart mitochondria were solubilized in respiration buffer, containing 1% Nonidet P40, and 2 mg of mitochondrial protein was incubated with 15 μM [3H]HNE in the presence of (A) 1.0 mM NAD+ or (B) 2 mM NADP+ for 30 min at 37 °C. Protein was then precipitated with trichloroacetic acid and metabolites in the supernatant were separated by HPLC and quantified by scintillation counting.
To determine whether inhibition of HNE oxidation during ischaemia could be due to changes in NAD+, we measured the NAD+/NADH ratio in perfused, ischaemic and ischaemia/reperfused hearts. As shown in Figure 7(A), hearts subjected to 40 min of aerobic perfusion had tissue NAD+/NADH ratios greater than 10; however, when the hearts were subjected to 10 min of perfusion followed by 30 min of ischaemia, the NAD+/NADH ratio decreased to 0.38. Reperfusion of the ischaemic myocardium for 5 min resulted in a significant rebound of the nucleotide ratio to 2.8, and 60 min of reperfusion caused the ratio to increase further to 8.6. These observations indicate that ischaemia and reperfusion cause dramatic changes in the redox state of NAD(H).
Fig 7.
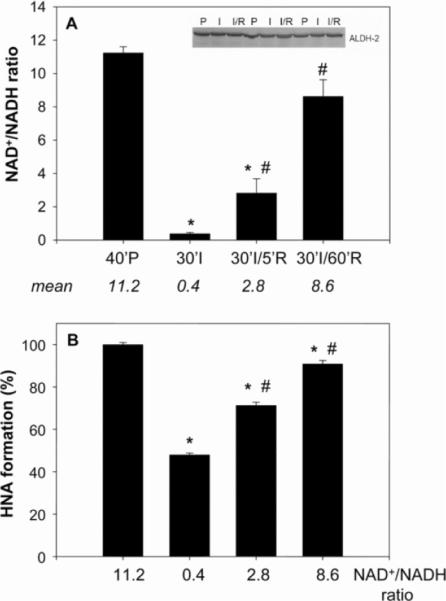
(A) Hearts were subjected to aerobic perfusion for 40 min, global ischaemia for 30 min, or global ischaemia for 30 min followed by 5 or 60 min reperfusion, and their NAD+ and NADH contents were measured. Mean values of the ratio are shown below the histograms. The inset shows Western blots from perfused (P), ischaemic (I) and reperfused (I/R) heart lysates developed using an anti-ALDH2 antibody. Values are means±S.E.M. (n=three individual rat hearts). *P<0.05 compared with the 40 min perfusion control (40′P); #P<0.05 compared with the 30 min ischaemia sample (30′I). (B) Myocardial lysates were incubated with the indicated ratio of NAD+/NADH and [3H]HNE. The [3H]HNA generated was separated by HPLC and quantified by scintillation counting. Values are means±S.E.M. (n=three hearts per group). *P<0.01 compared with 11.2 NAD+/NADH ratio; #P<0.01 compared with 0.4 NAD+/NADH ratio.
To determine whether changes in the NAD+/NADH ratio during ischaemia and reperfusion could affect HNE metabolism, oxidation of HNE was measured in myocardial homogenates in vitro by HPLC using [3H]HNE as a substrate and the NAD+/NADH ratio measured in the perfused, ischaemic and reperfused myocardium. As shown in Figure 7(B), incubation of myocardial protein with nucleotides comparable in ratio with that found in the ischaemic heart resulted in a 52.3±0.7% decrease in HNE oxidation compared with HNE oxidation at the nucleotide ratio measured in aerobic heart (=11.2). Using nucleotide ratios similar to those in hearts subjected to 30 min of ischaemia followed by 5 min reperfusion caused a 27.7±0.6% decrease in HNE oxidation, and using nucleotide ratios measured 60 min after reperfusion restored HNE oxidation to near control levels, showing only 9.2±1.7% inhibition. The level of inhibition of oxidative HNE metabolism that we found in the whole heart during ischaemia ex vivo was similar to that found in these in vitro measurements (in both measurements, HNE oxidation was inhibited by ~50%), suggesting that NAD+ loss during myocardial ischaemia is sufficient to cause a decrease in whole-heart HNE oxidation. In addition to changes in pyridine nucleotides, modification, inactivation or degradation of ALDH could be an additional reason for inhibition of HNE oxidation in the ischaemic heart. To test whether changes in HNE metabolism are due to changes in mitochondrial ALDH2 protein, Western blot analysis was performed. The levels of ALDH2 protein in aerobic, ischaemic and reperfused hearts were similar (Figure 7A, inset), indicating that the loss of HNE oxidation in the ischaemic heart could not be due to degradation of ALDH2, and that recovery of HNE oxidation upon reperfusion was not due to increased de novo synthesis of the enzyme. To test whether the enzyme is inactivated during ischaemia, mitochondria were isolated from the perfused and ischaemic heart, suspended in normoxic respiration buffer, and tested for their ability to oxidize HNE to HNA. As shown in Figure 8, mitochondria from perfused and ischaemic hearts showed similar abilities to oxidize HNE. These results indicate that ALDH2 itself is not inactivated by ischaemia and that mitochondria isolated under normoxic conditions from the ischaemic heart are just as efficient in oxidizing HNE to HNA as those from aerobic hearts.
Fig 8.
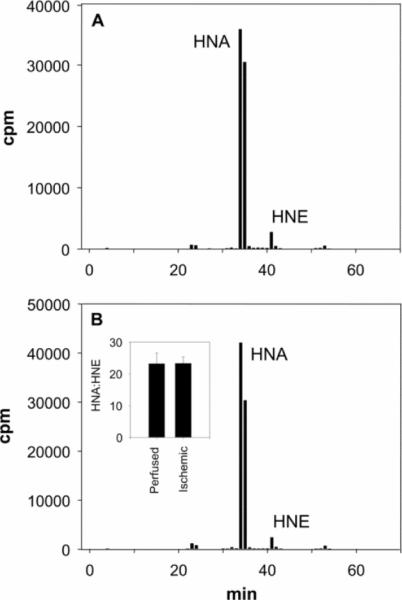
HPLC profiles of mitochondria isolated from (A) perfused and (B) ischaemic hearts. After isolation, mitochondria were resuspended in aerobic respiration buffer (2.5 mg/ml) and exposed to 15 mM [3H]HNE for 30 min at 37 °C. After incubation, mitochondria were sedimented by centrifugation, and metabolites in the supernatant were separated by HPLC and quantified by scintillation counting. Inset (i) shows HNA/HNE ratios (means±S.E.M.) of perfused and ischaemic rat hearts (n=3).
DISCUSSION
The results of the present study show that ischaemia profoundly affects myocardial metabolism of HNE leading to a decrease in HNE oxidation and a corresponding increase in reduction. Our results suggest that the inhibition of HNE oxidation during ischaemia could be attributed in part to metabolic failure imposed by ischaemic changes in NAD+/NADH ratios in the heart which prevent ALDH-catalysed HNE oxidation. We found that oxidation via ALDH is the major route of mitochondrial HNE metabolism and that, when this pathway fails during ischaemia, the mitochondria are unable to extrude glutathione conjugates and to up-regulate compensatory pathways of reductive metabolism, leading to a greater accumulation of HNE-modified protein in the mitochondria than in the cytosol. These findings provide new insight into mechanisms that elevate the sensitivity of the ischaemic heart to products of lipid peroxidation and those that render the mitochondria more susceptible to oxidative stress during myocardial ischaemia and reperfusion injury.
Several previous studies have reported that myocardial ischaemia/reperfusion elevates tissue levels of aldehydes derived from lipid peroxidation [19,31] and results in the appearance of proteins covalently adducted to these aldehydes [20]. To understand how ischaemia affects metabolism of aldehydes that are generated during oxidative stress, we examined how the model lipid peroxidation product HNE is metabolized in the cytosol and in mitochondria in the ischaemic and reperfused heart. In agreement with previous work [21,22,25], we found that, in the aerobic heart, HNE is readily conjugated to glutathione, oxidized to HNA or reduced to DHN. Oxidation to HNA appears to be quantitatively more important, because it accounts for 40% of the total HNE consumed ([21] and Figure 1). In contrast, in the ischaemic heart, the conversion of HNE into HNA was significantly inhibited (to 40% of its pre-ischaemic value) and was reversible after prolonged reperfusion. Similar to exogenous HNE, endogenous HNE generated during myocardial ischaemia was readily metabolized; however, the relative abundance of endogenously generated free HNE and its metabolites was distinctly different from the ischaemic hearts exposed to exogenous HNE. This could be due to multiple reasons. Unlike HNE delivered exogenously to the heart (at relatively high concentrations), the amount of HNE generated endogenously during myocardial ischaemia may be relatively low and at or below the Km of aldehyde-metabolizing enzymes, resulting in the accumulation of free, unmetabolized HNE and relatively low levels of metabolites. Alternatively, the abundance and availability of aldehyde-metabolizing enzymes may differ for HNE generated within the heart and that delivered exogenously as a bolus. Because of such potential differences, we studied the metabolites generated from both exogenous and endogenous HNE. Nonetheless, similar to exogenous HNE-treated ischaemic hearts, HNE oxidation was found not to be a major pathway for the metabolism of HNE generated endogenously during myocardial ischaemia/reperfusion. Based on these results, we suggest that ischaemic inhibition of HNE oxidation may be due to inhibition of mitochondrial ALDH. This is supported by our observation that the ALDH inhibitor benomyl inhibited HNA formation in the perfused heart and in isolated mitochondria and that ALDH is localized to the mitochondrial matrix (Figure 4). Taken together with our previous results showing that there is little to no HNE oxidizing activity in myocardial cytosol [21], it appears that mitochondria are the major sites of cardiac HNE oxidation, and it is this oxidation, catalysed by ALDH, which is inhibited during ischaemia.
The observation that ischaemia inhibits ALDH is consistent with our measurements showing that ischaemia decreases the NAD+/NADH ratio. Ischaemia for 30 min decreased the ratio from 11.2 to 0.38 (Figure 7). These measurements are in close agreement with previous work showing that the ratio decreases from 9.5 to 0.8 within 10 min of ischaemia [32]. The ischaemic decline in the NAD+/NADH ratio is reflected in the dramatic increase in the lactate/pyruvate ratio [33], indicating that, owing to lack of oxygen, the ischaemic heart is unable to regenerate NAD+ from NADH. This decrease in NAD+ is likely to be further exacerbated upon reperfusion, since reflow induces some mitochondria to undergo permeability transition. This results in the release of NAD+ to the cytosol, where it is degraded by glycohydrolase [34]. Indeed, our measurements indicate that reperfused myocardium contains 42% less NAD+ than the pre-ischaemic tissue (results not shown). Hence, although the NAD+/NADH ratio is restored upon reperfusion, the total amount of NAD+ is less than in pre-ischaemic tissue. However, for HNE metabolism to recover, the restoration of the NAD+/NADH ratio, rather than the absolute NAD+ concentration, appears to be more important. We suggest that the ischaemic deficiency in HNE oxidation is a reflection of severe anaerobic NAD+ depletion, which prevents HNE oxidation by ALDH. This decrease in HNE oxidation in the ischaemic heart is likely to promote myocardial accumulation and toxicity of HNE and related aldehydes generated by lipid peroxidation.
Although ischaemic deficits in HNE oxidation are likely to promote HNE accumulation both in the cytosol and mitochondria, oxidative changes are likely to be more severe in mitochondria. Cardiac mitochondria are rich in unsaturated fatty acids [35], and they contain high levels of haem, non-haem iron and semiquinone, all of which can catalyse free-radical formation [36]. Such radicals have been detected in the ischaemic heart, and mitochondria are believed to be the major site of free-radical generation during ischaemia/reperfusion [37], making mitochondria more vulnerable to oxidative injury. The present study reveals two additional sources of mitochondrial sensitivity to oxidative injury. First, in comparison with the cytosol, mitochondria depend more upon oxidation to remove HNE. In isolated mitochondria, approx. 70% of HNE was oxidized to HNA, whereas in the cytosol, particularly during ischaemia, most of HNE metabolism appears to be linked to glutathione conjugation or reduction to DHN. Moreover, even though inhibition of ALDH resulted in an increase in HNE metabolism via other pathways in the cytosol, this was not the case with the mitochondria. Thus during ischaemia, when unfavourable nucleotide ratios prevent oxidation, mitochondria are unable to up-regulate other pathways of metabolism and accumulate more HNE adducted proteins than the cytosol. Secondly, mitochondria were unable to extrude glutathiolated or reduced HNE. We found that, when incubated with HNE, mitochondria were unable to extrude GS–HNE, which was retained in the matrix. In additional experiments, we have found that mitochondria are also unable to extrude oxidized glutathione (B. G. Hill, A. Bhatnagar and S. Srivastava unpublished work). In most eukaryotic cells, the efflux of oxidized glutathione and glutathione conjugates across the cell membrane is mediated by several transporters including the multidrug resistant protein, P-glycoprotein, GSSG transporter and RLIP76 [38–40]. Apparently, cardiac mitochondria lack such transporters, and they are unable to remove these conjugates from their matrix. Although GSSG can be reduced by mitochondrial glutathione reductase, the accumulation of glutathione conjugates of xenobiotics or lipid peroxidation products in the mitochondria may be, for many reasons, problematic. For instance, even though conjugation removes α,β unsaturation, glutathione conjugates of 2-trans-butenal, 2-trans-hexenal and 2-trans,6-cis-nonadienal induce DNA damage [41], and the glutathione conjugate of acrolein stimulates radical formation [42] and causes nephrotoxicity [43]. Hence, glutathione conjugates generated in the mitochondria could cause local, mitochondrial-specific injury. Moreover, glutathiolation of unsaturated aldehydes is potentially reversible [12]; therefore spontaneous reversal of glutathione conjugates could deliver free aldehydes back to the mitochondrial matrix.
Mitochondrial injury is a pivotal event in the evolution of ischaemic injury from reversible dysfunction to irreversible cell death [44]. Mitochondrial swelling is an early event in ischaemic injury [45] and mitochondria are the major source of ROS production during reperfusion [46]. Mitochondria are also dominant integrators, checkpoints and amplifiers of cell-death pathways [47]. In hearts subjected to ischaemia/reperfusion, predominantly mitochondrial proteins undergo HNE modification [48], indicating that cardiac mitochondria are likely to be highly vulnerable to HNE generated during ischaemia/reperfusion. This is consistent with our observation that mitochondria isolated from ischaemic hearts contained 2-fold higher levels of protein-bound radioactivity than the ischaemic heart cytosol. Given in vitro data showing that exposure to HNE induces mitochondrial injury [14,49] and inhibits mitochondrial respiration [50], we speculate that accumulation of HNE in the ischaemic heart mitochondria, due to a decrease in ALDH-mediated oxidation and the inability of the mitochondria to extrude HNE metabolites, may be a significant factor in mitochondrial injury during myocardial ischaemia/reperfusion.
In summary, these studies on HNE metabolism provide new concepts regarding the metabolism of lipid peroxidation products in the ischaemic heart. Reductive metabolism of reactive aldehydes and conjugation to glutathione are the primary metabolic pathways that are active in the cytosol in the ischaemic heart; the contribution of these pathways to aldehyde detoxification in the cytosol is increased during ischaemia. In contrast, mitochondria rely primarily on oxidative metabolism for the inactivation of cytotoxic aldehydes generated during oxidative stress, and this pathway is inhibited during ischaemia. The redox state of mitochondria appears to be the primary modulator of the metabolism of aldehydes, such as HNE, during ischaemia; the decline in the NAD+/NADH ratio during ischaemia results in inhibition of ALDH and decreases mitochondrial HNE metabolism. Furthermore, mitochondria are unable to extrude glutathione-conjugated aldehydes and products of reduction. Collectively, this could leave myocardial mitochondria particularly vulnerable to oxidative damage during episodes of ischaemia/reperfusion.
ACKNOWLEDGEMENTS
We thank Dan Riggs, David Hoetker and Denny Clark for technical assistance.
FUNDING This work was supported in part by the National Institutes of Health [grant numbers HL65618, HL55477, HL59378, ES 011594, ES11860]; and the American Heart Association [grant number 0415165B] (predoctoral fellowship to B. G. H.).
Abbreviations used
- ALDH
aldehyde dehydrogenase
- AR
aldose reductase
- BSTFA
bis-(trimethylsilyl)trifluoroacetamide
- DHN
1,4-dihydroxynonene
- DTT
dithiothreitol
- ESI–MS
electrospray ionization MS
- GS–DHN
glutathione conjugate of DHN
- HNA
4-hydroxynonenoic acid
- HNE
4-hydroxy-trans-2-nonenal
- GS–HNE
glutathione conjugate of HNE
- KH buffer
Krebs–Henseleit buffer
- RCR
respiratory control ratio
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- tR
retention time
REFERENCES
- 1.Downey JM. Free radicals and their involvement during long-term myocardial ischemia and reperfusion. Annu. Rev. Physiol. 1990;52:487–504. doi: 10.1146/annurev.ph.52.030190.002415. [DOI] [PubMed] [Google Scholar]
- 2.Kloner RA, Jennings RB. Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 2. Circulation. 2001;104:3158–3167. doi: 10.1161/hc5001.100039. [DOI] [PubMed] [Google Scholar]
- 3.Chen Z, Siu B, Ho YS, Vincent R, Chua CC, Hamdy RC, Chua BH. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J. Mol. Cell. Cardiol. 1998;30:2281–2289. doi: 10.1006/jmcc.1998.0789. [DOI] [PubMed] [Google Scholar]
- 4.Li G, Chen Y, Saari JT, Kang YJ. Catalase-overexpressing transgenic mouse heart is resistant to ischemia-reperfusion injury. Am. J. Physiol. 1997;273:H1090–H1095. doi: 10.1152/ajpheart.1997.273.3.H1090. [DOI] [PubMed] [Google Scholar]
- 5.Chen EP, Bittner HB, Davis RD, Folz RJ, Van Trigt P. Extracellular superoxide dismutase transgene overexpression preserves postischemic myocardial function in isolated murine hearts. Circulation. 1996;94:II412–II417. [PubMed] [Google Scholar]
- 6.Yoshida T, Watanabe M, Engelman DT, Engelman RM, Schley JA, Maulik N, Ho YS, Oberley TD, Das DK. Transgenic mice overexpressing glutathione peroxidase are resistant to myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 1996;28:1759–1767. doi: 10.1006/jmcc.1996.0165. [DOI] [PubMed] [Google Scholar]
- 7.Asimakis GK, Lick S, Patterson C. Postischemic recovery of contractile function is impaired in SOD2+/− but not SOD1+/− mouse hearts. Circulation. 2002;105:981–986. doi: 10.1161/hc0802.104502. [DOI] [PubMed] [Google Scholar]
- 8.Yoshida T, Maulik N, Engelman RM, Ho YS, Magnenat JL, Rousou JA, Flack JE, III, Deaton D, Das DK. Glutathione peroxidase knockout mice are susceptible to myocardial ischemia reperfusion injury. Circulation. 1997;96:II20. [PubMed] [Google Scholar]
- 9.Yoshida T, Maulik N, Engelman RM, Ho YS, Das DK. Targeted disruption of the mouse Sod I gene makes the hearts vulnerable to ischemic reperfusion injury. Circ. Res. 2000;86:264–269. doi: 10.1161/01.res.86.3.264. [DOI] [PubMed] [Google Scholar]
- 10.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang P, Xu X, Hu X, van Deel ED, Zhu G, Chen Y. Inducible nitric oxide synthase deficiency protects the heart from systolic overload-induced ventricular hypertrophy and congestive heart failure. Circ. Res. 2007;100:1089–1098. doi: 10.1161/01.RES.0000264081.78659.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biol. Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 13.Usatyuk PV, Natarajan V. Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced actin remodeling and barrier function in endothelial cells. J. Biol. Chem. 2004;279:11789–11797. doi: 10.1074/jbc.M311184200. [DOI] [PubMed] [Google Scholar]
- 14.Echtay KS, Esteves TC, Pakay JL, Jekabsons MB, Lambert AJ, Portero-Otin M, Pamplona R, Vidal-Puig AJ, Wang S, Roebuck SJ, Brand MD. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003;22:4103–4110. doi: 10.1093/emboj/cdg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhatnagar A. Electrophysiological effects of 4-hydroxynonenal, an aldehydic product of lipid peroxidation, on isolated rat ventricular myocytes. Circ. Res. 1995;76:293–304. doi: 10.1161/01.res.76.2.293. [DOI] [PubMed] [Google Scholar]
- 16.Yang Y, Sharma A, Sharma R, Patrick B, Singhal SS, Zimniak P, Awasthi S, Awasthi YC. Cells preconditioned with mild, transient UVA irradiation acquire resistance to oxidative stress and UVA-induced apoptosis: role of 4-hydroxynonenal in UVA-mediated signaling for apoptosis. J. Biol. Chem. 2003;278:41380–41388. doi: 10.1074/jbc.M305766200. [DOI] [PubMed] [Google Scholar]
- 17.Uchida K, Szweda LI, Chae HZ, Stadtman ER. Immunochemical detection of 4-hydroxynonenal protein adducts in oxidized hepatocytes. Proc. Natl. Acad. Sci. U.S.A. 1993;90:8742–8746. doi: 10.1073/pnas.90.18.8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartsch H, Nair J. Accumulation of lipid peroxidation-derived DNA lesions: potential lead markers for chemoprevention of inflammation-driven malignancies. Mutat. Res. 2005;591:34–44. doi: 10.1016/j.mrfmmm.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 19.Blasig IE, Grune T, Schonheit K, Rohde E, Jakstadt M, Haseloff RF, Siems WG. 4-Hydroxynonenal, a novel indicator of lipid peroxidation for reperfusion injury of the myocardium. Am. J. Physiol. 1995;269:H14–H22. doi: 10.1152/ajpheart.1995.269.1.H14. [DOI] [PubMed] [Google Scholar]
- 20.Eaton P, Li JM, Hearse DJ, Shattock MJ. Formation of 4-hydroxy-2-nonenal-modified proteins in ischemic rat heart. Am. J. Physiol. 1999;276:H935–H943. doi: 10.1152/ajpheart.1999.276.3.H935. [DOI] [PubMed] [Google Scholar]
- 21.Srivastava S, Chandra A, Wang LF, Seifert WE, Jr, DaGue BB, Ansari NH, Srivastava SK, Bhatnagar A. Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. J. Biol. Chem. 1998;273:10893–10900. doi: 10.1074/jbc.273.18.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srivastava S, Conklin DJ, Liu SQ, Prakash N, Boor PJ, Srivastava SK, Bhatnagar A. Identification of biochemical pathways for the metabolism of oxidized low-density lipoprotein derived aldehyde-4-hydroxy trans-2-nonenal in vascular smooth muscle cells. Atherosclerosis. 2001;158:339–350. doi: 10.1016/s0021-9150(01)00454-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shinmura K, Bolli R, Liu SQ, Tang XL, Kodani E, Xuan YT, Srivastava S, Bhatnagar A. Aldose reductase is an obligatory mediator of the late phase of ischemic preconditioning. Circ. Res. 2002;91:240–246. doi: 10.1161/01.res.0000029970.97247.57. [DOI] [PubMed] [Google Scholar]
- 24.Ramana KV, Bhatnagar A, Srivastava S, Yadav UC, Awasthi S, Awasthi YC, Srivastava SK. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE): role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. J. Biol. Chem. 2006;281:17652–17660. doi: 10.1074/jbc.M600270200. [DOI] [PubMed] [Google Scholar]
- 25.Srivastava S, Dixit BL, Cai J, Sharma S, Hurst HE, Bhatnagar A, Srivastava SK. Metabolism of lipid peroxidation product, 4-hydroxynonenal (HNE) in rat erythrocytes: role of aldose reductase. Free Radical Biol. Med. 2000;29:642–651. doi: 10.1016/s0891-5849(00)00351-8. [DOI] [PubMed] [Google Scholar]
- 26.Kaiserova K, Srivastava S, Hoetker JD, Awe SO, Tang XL, Cai J, Bhatnagar A. Redox activation of aldose reductase in the ischemic heart. J. Biol. Chem. 2006;281:15110–15120. doi: 10.1074/jbc.M600837200. [DOI] [PubMed] [Google Scholar]
- 27.Kaiserova K, Tang XL, Srivastava S, Bhatnagar A. Role of nitric oxide in regulating aldose reductase activation in the ischemic heart. J. Biol. Chem. 2008;283:9101–9112. doi: 10.1074/jbc.M709671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luckey SW, Petersen DR. Metabolism of 4-hydroxynonenal by rat Kupffer cells. Arch. Biochem. Biophys. 2001;389:77–83. doi: 10.1006/abbi.2001.2307. [DOI] [PubMed] [Google Scholar]
- 29.Staub RE, Quistad GB, Casida JE. Mechanism for benomyl action as a mitochondrial aldehyde dehydrogenase inhibitor in mice. Chem. Res. Toxicol. 1998;11:535–543. doi: 10.1021/tx980002l. [DOI] [PubMed] [Google Scholar]
- 30.Srivastava S, Chandra A, Ansari NH, Srivastava SK, Bhatnagar A. Identification of cardiac oxidoreductase(s) involved in the metabolism of the lipid peroxidation-derived aldehyde-4-hydroxynonenal. Biochem. J. 1998;329:469–475. doi: 10.1042/bj3290469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maulik N, Engelman DT, Watanabe M, Engelman RM, Rousou JA, Flack JE, III, Deaton DW, Gorbunov NV, Elsayed NM, Kagan VE, Das DK. Nitric oxide/carbon monoxide. A molecular switch for myocardial preservation during ischemia. Circulation. 1996;94:II398–II406. [PubMed] [Google Scholar]
- 32.Ferrari R, Cargnoni A, Bernocchi P, Pasini E, Curello S, Ceconi C, Ruigrok TJ. Metabolic adaptation during a sequence of no-flow and low-flow ischemia. A possible trigger for hibernation. Circulation. 1996;94:2587–2596. doi: 10.1161/01.cir.94.10.2587. [DOI] [PubMed] [Google Scholar]
- 33.Vuorinen K, Ylitalo K, Peuhkurinen K, Raatikainen P, Ala-Rami A, Hassinen IE. Mechanisms of ischemic preconditioning in rat myocardium. Roles of adenosine, cellular energy state, and mitochondrial F1F0-ATPase. Circulation. 1995;91:2810–2818. doi: 10.1161/01.cir.91.11.2810. [DOI] [PubMed] [Google Scholar]
- 34.Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD(+) and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 35.Gross RW. Identification of plasmalogen as the major phospholipid constituent of cardiac sarcoplasmic reticulum. Biochemistry. 1985;24:1662–1668. doi: 10.1021/bi00328a014. [DOI] [PubMed] [Google Scholar]
- 36.Zweier JL, Kuppusamy P, Williams R, Rayburn BK, Smith D, Weisfeldt ML, Flaherty JT. Measurement and characterization of postischemic free radical generation in the isolated perfused heart. J. Biol. Chem. 1989;264:18890–18895. [PubMed] [Google Scholar]
- 37.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc. Natl. Acad. Sci. U.S.A. 1987;84:1404–1407. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gerk PM, Vore M. Regulation of expression of the multidrug resistance-associated protein 2 (MRP2) and its role in drug disposition. J. Pharmacol. Exp. Ther. 2002;302:407–415. doi: 10.1124/jpet.102.035014. [DOI] [PubMed] [Google Scholar]
- 39.Hoffmann U, Kroemer HK. The ABC transporters MDR1 and MRP2: multiple functions in disposition of xenobiotics and drug resistance. Drug Metab. Rev. 2004;36:669–701. doi: 10.1081/dmr-200033473. [DOI] [PubMed] [Google Scholar]
- 40.Sharma R, Singhal SS, Cheng J, Yang Y, Sharma A, Zimniak P, Awasthi S, Awasthi YC. RLIP76 is the major ATP-dependent transporter of glutathione-conjugates and doxorubicin in human erythrocytes. Arch. Biochem. Biophys. 2001;391:171–179. doi: 10.1006/abbi.2001.2395. [DOI] [PubMed] [Google Scholar]
- 41.Dittberner U, Eisenbrand G, Zankl H. Genotoxic effects of the α, β-unsaturated aldehydes 2-trans-butenal, 2-trans-hexenal and 2-trans, 6-cis-nonadienal. Mutat. Res. 1995;335:259–265. doi: 10.1016/0165-1161(95)00029-1. [DOI] [PubMed] [Google Scholar]
- 42.Adams JD, Jr, Klaidman LK. Acrolein-induced oxygen radical formation. Free Radical Biol. Med. 1993;15:187–193. doi: 10.1016/0891-5849(93)90058-3. [DOI] [PubMed] [Google Scholar]
- 43.Horvath JJ, Witmer CM, Witz G. Nephrotoxicity of the 1:1 acrolein-glutathione adduct in the rat. Toxicol. Appl. Pharmacol. 1992;117:200–207. doi: 10.1016/0041-008x(92)90238-n. [DOI] [PubMed] [Google Scholar]
- 44.Jennings RB, Ganote CE. Mitochondrial structure and function in acute myocardial ischemic injury. Circ. Res. 1976;38:I80–I91. [PubMed] [Google Scholar]
- 45.Greve G, Rotevatn S, Svendby K, Grong K. Early morphologic changes in cat heart muscle cells after acute coronary artery occlusion. Am. J. Pathol. 1990;136:273–283. [PMC free article] [PubMed] [Google Scholar]
- 46.Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, Tritto I, Cirillo P, Condorelli M, Chiariello M, Flaherty JT. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J. Biol. Chem. 1993;268:18532–18541. [PubMed] [Google Scholar]
- 47.Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ. Res. 2003;93:292–301. doi: 10.1161/01.RES.0000087542.26971.D4. [DOI] [PubMed] [Google Scholar]
- 48.Lucas DT, Szweda LI. Cardiac reperfusion injury: aging, lipid peroxidation, and mitochondrial dysfunction. Proc. Natl. Acad. Sci. U.S.A. 1998;95:510–514. doi: 10.1073/pnas.95.2.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kristal BS, Park BK, Yu BP. 4-Hydroxyhexenal is a potent inducer of the mitochondrial permeability transition. J. Biol. Chem. 1996;271:6033–6038. doi: 10.1074/jbc.271.11.6033. [DOI] [PubMed] [Google Scholar]
- 50.Picklo MJ, Amarnath V, McIntyre JO, Graham DG, Montine TJ. 4-Hydroxy-2(E)-nonenal inhibits CNS mitochondrial respiration at multiple sites. J. Neurochem. 1999;72:1617–1624. doi: 10.1046/j.1471-4159.1999.721617.x. [DOI] [PubMed] [Google Scholar]