

Abstract
The cAMP receptor protein (CRP) of Escherichia coli undergoes a conformational change in response to cAMP binding that allows it to bind specific DNA sequences. Using an in vivo screening method following the simultaneous randomization of the codons at positions 127 and 128 (two C-helix residues of the protein interacting with cAMP), we have isolated a series of novel constitutively active CRP variants. Sequence analysis showed that this group of variants commonly possesses leucine or methionine at position 127 with a β-branched amino acid at position 128. One specific variant, T127L/S128I CRP, showed extremely high cAMP-independent DNA binding affinity comparable with that of cAMP-bound wild-type CRP. Further biochemical analysis of this variant and others revealed that Leu127 and Ile128 have different roles in stabilizing the active conformation of CRP in the absence of cAMP. Leu127 contributes to an improved leucine zipper at the dimer interface, leading to an altered intersubunit interaction in the C-helix region. In contrast, Ile128 stabilizes the proper position of the β4/β5 loop by functionally communicating with Leu61. By analogy, the results suggest two direct local effects of cAMP binding in the course of activating wild-type CRP: (i) C-helix repositioning through direct interaction with Thr127 and Ser128 and (ii) the concomitant reorientation of the β4/β5 loop. Finally, we also report that elevated expression of T127L/S128I CRP markedly perturbed E. coli growth even in the absence of cAMP, which suggests why comparably active variants have not been described previously.
The Escherichia coli cAMP receptor protein (CRP2; also known as the catabolite activator protein) activates transcription at >100 promoters (1–5). CRP was originally identified as a protein factor responsible for catabolite repression in E. coli (6, 7) and since then has been a paradigm for bacterial transcription activators. CRP is a homodimer, each subunit of which is composed of two distinct domains connected by a hinge region: an N-terminal effector-binding domain and a C-terminal DNA-binding domain (see Fig. 1A) (8). In the presence of its effector cAMP, CRP becomes competent to bind its target DNA and to recruit RNA polymerase to activate transcription (2, 4).
FIGURE 1.

A, shown is the structure of active cAMP-bound E. coli CRP (Protein Data Bank code 1CGP). CRP is composed of a homodimer with an interface along the long C-helices. The bound cAMP molecules and residues Thr127, Ser128, and Leu61 are visible in the center of this figure and are labeled in one monomer. The C-helices, D-helices, hinge regions, and β4/β5 loops are indicated by arrows. The F-helices are responsible for DNA binding. The ARs involved in the interaction with RNA polymerase (AR1, AR2, and AR3) are also indicated. B, the relative C-helix positions are different between active CRP (black) and inactive CooA (gray) when one of the two C-helices in each protein is aligned (right). The effector-free inactive CooA structure is from the Protein Data Bank (code 1FT9). This view is from effector-binding domains toward DNA-binding domains. Both figures were generated using Swiss-PdbViewer Version 3.7.
A number of methods have been used to demonstrate that this DNA binding ability is cAMP-dependent and accompanied by a conformational change. (i) Although CRP is protease-resistant, it becomes protease-sensitive in the presence of micromolar cAMP. Most common cleavage sites are located in the D-helix, and the protease data suggest that the N- and C-terminal domains of CRP communicate through the hinge (see Fig. 1A) (5). (ii) Both NMR data (9) and protein footprinting experiments (10) suggest a change of secondary structure around residues 127–139 (hinge region) and an increase in the solvent exposure of the DNA-binding F-helix upon cAMP binding. (iii) Constitutively active CRP variants have often been found to be altered in the proximity of the hinge region, particularly at Gly141 and Ala144 (11–13). Although evidence suggesting the importance of the hinge region in CRP activation is substantial, it is unlikely that cAMP directly affects this region because the bound cAMP and hinge are separated by >10 Å (8). We therefore still do not know the locally critical event(s) that trigger global conformational changes in CRP upon cAMP binding.
In this regard, the repositioning of the long C-helices at the dimer interface upon cAMP binding is attractive as such a trigger because such a repositioning would necessarily change the environment of the hinge region. There are three lines of evidence in support of this model. First, the C-helices assume a coiled-coil interaction, which usually utilizes hydrophobic residues at the crucial a- and d-positions in the heptad repeat. As Passner et al. (14) pointed out, because each cAMP molecule interacts extensively with both C-helices, the C-helices would have a different orientation with respect to each other in the presence and absence of cAMP. Interestingly, Thr127, a non-optimal leucine zipper residue, occupies an important d-position in the coiled-coil structure and interacts with cAMP, as does the adjacent Ser128. More specifically, Thr127 forms a hydrogen bond with N-6 of the bound cAMP, and Ser128 forms a hydrogen bond with the cAMP bound to the other subunit (8). For this reason, CRP residues Thr127 and Ser128 have been the focus of considerable study, and their alteration has been reported to perturb a variety of properties such as substrate discrimination (15), cooperativity (16), protein structure (17), and CRP activity (15, 18). The T127L/S128A CRP variant has significant activity even in the absence of cAMP (19), and the cAMP-bound structure of the protein shows a modest C-helix bend at the altered positions relative to the wild-type (WT) cAMP-bound CRP structure (17). Given these results, it is quite possible that the initial effect of cAMP binding is the repositioning of the C-helices through these residues.
Second, studies of CooA, a member of the CRP family of transcription activators, provide additional insight concerning the local change in CRP structure induced by cAMP binding. The structure of inactive CooA has been determined, and the structural comparison of cAMP-bound CRP with effector-free CooA (20) reveals a modest repositioning of these two C-helices with respect to each other (see Fig. 1B). Although not an ideal comparison, this is consistent with the C-helix repositioning hypothesis as the trigger for the activation of each protein. The importance of the C-helix region as a signal pathway in CooA was further tested genetically. The CooA region homologous to CRP residues 125–130 on the C-helix was genetically randomized, and the resulting library of mutants was screened for those that activated CooA in the absence of the effector CO. Effector-independent CooA variants were readily found and uniformly possessed a consensus leucine zipper motif, consistent with the hypothesis (21). In another line of study, a novel CooA variant that is able to respond to the non-natural effector imidazole has been found, and the molecular basis for this is most easily explained by invoking a similar, but locally different, C-helix repositioning (22).
Last, two structures of PrfA, a CRP homolog that regulates virulence in Listeria monocytogenes, have been recently reported (23): that of WT PrfA and that of G145S PrfA, a hyperactive variant. Although the mutated site is not in the C-helices, the comparison of the two structures, which correlate with differences in activity, revealed a noticeable straightening of the C-helices in G145S PrfA (23). Such correlation between C-helix repositioning and DNA-binding activity further substantiates the possibility of C-helix repositioning in the activation of this family of proteins.
The goal of this study was to test the hypothesis that C-helix repositioning is a critical local event triggering CRP activation upon cAMP binding. For this purpose, we completely randomized the codons at Thr127 and Ser128 and screened for highly active CRP variants in the absence of cAMP, anticipating that certain mutations would mimic the effect of cAMP. By isolating a series of highly cAMP-independent CRP variants altered at these positions and characterizing these variants, we provide genetic evidence in support of the hypothesis. Furthermore, in the course of this study, we recognized that another local event must occur in addition to C-helix repositioning for complete CRP activation: movement of the β4/β5 loop. This loop is indicated in Fig. 1A, and the base of the loop comes into contact with the bound cAMP (14). We propose that cAMP repositions the β4/β5 loop through its hydrophobic interactions with loop residues, including Leu61.
EXPERIMENTAL PROCEDURES
Strains, Plasmids, and Recombinant DNA Methodology
The bacterial strains, plasmids, and promoter fragments used in this study are listed in Table 1. Standard methods for the isolation and manipulation of DNA fragments (24) were used throughout the study. Synthetic oligonucleotides used for sequencing, PCR amplification, and mutant construction were purchased from Integrated DNA Technologies, Inc. (Coralville, IA). Bacterial strains carrying different plasmids were propagated in LC medium (1% Tryptone, 0.5% yeast extract, and 0.5% NaCl) with 15 μg/ml tetracycline, 30 μg/ml kanamycin, 25 μg/ml chloramphenicol, or 50 μg/ml ampicillin as appropriate.
TABLE 1.
Bacterial strains, plasmids, and promoters used in this work
| Brief description | Source/Ref. | |
|---|---|---|
| Bacterial strains | ||
| HB101 | ||
| M182 | Δ(lacIPOZY) × 74galK galU strA | Ref. 46 |
| UQ3259 | F− his StrR relA1 ilv∷Tn10 cya (SA2755) | S. Adhya |
| UQ3260 | F− his StrR relA1 crp∷cam (SA2777) | S. Adhya |
| UQ3261 | F− his StrR relA1 ilv∷Tn10 cya and crp∷cam | This work |
| UQ3588 | HB101 with ilv∷Tn10 cya and crp∷cam recA56 TnR | This work |
| UQ3740 | M182 carrying λ prophage with CC(−41.5)∷lacZ fusion (RLG4649) | Ref. 26 |
| UQ3741 | M182 carrying λ prophage with CC(−61.5)∷lacZ fusion (RLG4650) | Ref. 27 |
| UQ3809 | UQ3740 with ilv∷Tn10 cya and crp∷cam | This work |
| UQ3811 | UQ3741 with ilv∷Tn10 cya and crp∷cam | This work |
|
| ||
| Plasmids | ||
| pEXT20 | Ref. 32 | |
| pHY26-1 | pEXT20 plasmid bearing E. coli crp allele (His6-tagged) | This work |
|
| ||
| Promoters | ||
| pSR | pBR322 derivative containing λ OOP transcription terminator | Ref. 47 |
| pSR/CC(−41.5) | Class II derivative of melR promoter with consensus CRP-binding site centered at −41.5 | Ref. 26 |
| pSR/CC(−61.5) | Class I derivative of melR promoter with consensus CRP-binding site centered at −61.5 | Ref. 27 |
Cloning of crp and Generation of Mutants
E. coli crp was cloned into EcoRI-HindIII-digested pEXT20 after PCR amplification of the chromosomal DNA from DH5α using EcoRI-containing 5′- and HindIII-containing 3′-primers designed as described previously (21). Six histidine codons were subsequently added between the last codon and the stop codon of the crp gene for easy purification of the corresponding protein. Site-directed mutagenesis involved PCR amplification of crp-containing pEXT20 with primers designed to incorporate the desired nucleotide changes as described (25). The method used for codon randomization was essentially identical to that used for site-directed mutagenesis except that the primers contained randomized codons at the desired positions. For the screening of constitutively active CRP variants, we used strain UQ3811 (see Table 1 for details). The assay monitors the ability of the CRP variants to cause β-galactosidase accumulation in colonies on agar plates. Based on colony color, variants could be classified as active, weakly active, and inactive. The crp genes for selected variants were sequenced to determine the causative residue changes.
Measurement of in Vivo β-Galactosidase Activity
Each cya crp strain harboring either a Class I- or II-type reporter system was constructed by introducing cya and crp mutant alleles sequentially into either strain UQ3740 (RLG4649) (26) or UQ3741 (RLG4650) (27) using P1 transduction (see Table 1). For the in vivo β-galactosidase assay, cells were grown overnight at 37 °C in LC medium containing 50 μg/ml ampicillin to full growth. The next day, cells were diluted to A600 nm = 0.1 in fresh LC medium containing 50 μg/ml ampicillin and different levels of isopropyl β-d-thiogalactopyranoside (IPTG) and grown at 37 °C and 220 rpm. Cells at A600 nm = 1–1.5 were used for the measurement of β-galactosidase activity by a standard method (28).
Measurement of Growth Rates in Strains Containing WT CRP and Selected Variants
WT, T127L/S128A, and T127L/S128I CRP-containing cells in the UQ3588 background (see Table 1) were grown overnight at 37 °C in LC medium containing 50 μg/ml ampicillin to full growth. The next day, each strain was diluted to A600 nm = 0.1 in fresh LC medium containing 50 μg/ml ampicillin, and different levels of IPTG were added with or without 2 mm cAMP. Each strain was grown using 10-ml culture volumes in 125- or 250-ml Erlenmeyer flasks at 37 °C and 220 rpm, and A600 nm was monitored every hour.
Purification of CRP Proteins
For biochemical analysis, each His-tagged CRP protein was purified as follows. Cultures of UQ3811 cells transformed with plasmids encoding appropriate CRP variants were grown aerobically at 220 rpm in 2× LC medium at 37 °C to A600 nm = 0.5, at which point, the synthesis of CRP was induced by the addition of 1 mm IPTG. Cultures were grown for another 5 h before harvesting and storing at −80 °C. Cell pellets were resuspended in 50 mm Tris-HCl (pH 8.0) and 500 mm KCl, and broken using a French pressure system. The lysates were cleared by centrifugation at 10,000 × g for 10 min, and the supernatants were loaded onto a nickel-nitrilotriacetic acid column (Novagen). After washing with 50 mm imidazole, the CRP proteins were eluted with 250 mm imidazole, 50 mm Tris-HCl (pH 8.0), and 500 mm KCl; precipitated with 42.5% ammonium sulfate; and stored at −80 °C until uses. All protein preparations were >95% homogeneous.
Measurement of the in Vitro DNA-binding Activity of CRP Proteins
In vitro DNA binding assays were performed using the fluorescence polarization technique with a Beacon 2000 fluorescence polarization detector (Pan-vera Corp., Madison, WI). A fluorescent DNA probe was generated in which a 26-bp target DNA containing CCpmelR (5′-GTAAATGTGATGTACATCACATGGAT-3′) (29) was labeled with Texas Red on one end of the duplex. Binding assays were performed in 50 mm Tris-HCl (pH 8.0), 50 mm KCl, and 1 mm EDTA. The probe was used at a concentration of 5 nm in the presence of 6.4 μm salmon sperm DNA. Dissociation constants (Kd) were calculated by fitting the binding data to an equation as described previously (30).
In Vitro Transcription Assays
Multiple-round in vitro transcription assays were performed as described by Savery et al. (27). Reactions (25 μl-final volume) were performed with 5 nm RNA polymerase, 200 nm WT or T127L/S128I CRP, either 0.1 or 1 mM cAMP, 25 ng of super-coiled plasmid templates, 200 μm ATP, 200 μm CTP, 200 μm GTP, 10 μm UTP, and 5 μCi of [α-32P]UTP in buffer containing 40 mm Tris acetate (pH 7.9), 100 mm KCl, 10 mm MgCl2, 1 mm dithiothreitol, and 100 μg/ml bovine serum albumin. For DNA templates, we used pSR/CC(−41.5) (26) and pSR/CC(−61.5) (27), which contain the λ OOP Rho factor-independent transcription terminator and which both produce a transcript of 123 nucleotides. The 108-nucleotide RNA I transcript served as a control (31). The pSR/CC(−41.5) and pSR/CC(−61.5) plasmids were prepared using a Qiagen miniprep kit and further purified by phenol extraction. Reactions were started by the addition of RNA polymerase, and after 15 min at room temperature, products were loaded onto denaturing 7% (w/v) polyacrylamide gels and analyzed using a Typhoon Trio Variable Mode Imager (Amersham Biosciences).
RESULTS
Genetic Selection for Constitutively Active Residue 127 and 128 CRP Variants
As noted in the Introduction, it was our working hypothesis that CRP, like CooA, employs repositioning of the C-helices upon effector binding as a major element of signal transduction within the protein. Given the importance of C-helix residues Thr127 and Ser128 for CRP activation (15, 19) and of the d-position of Thr127 in the coiled-coil heptad repeat, we predicted that some substitutions at these positions might create a C-helix repositioning without cAMP that is similar to that caused by cAMP in WT CRP. If the hypothesis were correct, we predicted that such cAMP-independent variants would be found to contain residues more conducive to the formation of a consensus leucine zipper, as seen in the mutational analysis of CooA (21).
The codons at residues 127 and 128 were simultaneously randomized in a library of crp-expressing plasmids, and the resultant plasmid pool was transformed into UQ3811 (Table 1), a cya crp E. coli reporter strain that contains the lacZ gene under the control of the Class I CC(−61.5) promoter (27). Expression of the mutated crp gene in the pEXT20 vector can be regulated by IPTG (32). Approximately 4000 colonies were screened at either 0 or 100 μm IPTG for those with activity in the absence of cAMP, as evidenced by their blue color.
Without IPTG added, 2.3% of the colonies were light blue above a white background. Under the assay conditions used, no colony was dark blue. The crp regions of the light blue colonies were then sequenced, revealing that all the variants (eight of eight) contained exclusively a T127L/S128I substitution, although with different codons (Table 2). This result suggests that T127L/S128I CRP is a particularly active cAMP-independent variant.
TABLE 2. Screening results for constitutively active CRP variants randomly altered at Th127 and Ser128.
The codons at positions 127 and 128 of CRP were simultaneously randomized, and the the pool of randomized clones (crp gene in pEXT20) was transformed into strain UQ3811 and screened on LC agar plates containing 50 μg/ml ampicillin, 20 μg/ml 5-bromo-4-chloro-3-indolyl-β -D-galactopyranoside (X-gal), and 0 or 100 μM IPTG.
| Screened without IPTG |
Screened with 100 μM IPTG |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CRP | Position 127 |
Position 128 |
CRP | Position 127 |
Position 128 |
CRP | Position 127 |
Position 128 |
||||||
| Codon | Amino acid | Codon | Amino acid | Codon | Amino acid | Codon | Amino acid | Codon | Amino acid | Codon | Amino acid | |||
| LB-1a | TTA | Leu | ATA | Ile | DB-1'b | ATA | Ile | ATT | Ile | LB-1'c | ATG | Met | ACA | Thr |
| LB-2 | CTG | Leu | ATT | Ile | DB-2' | ATG | Met | GTA | Val | LB-2' | TTG | Leu | CAA | Gln |
| LB-3 | CTG | Leu | ATA | Ile | DB-3' | CTT | Leu | GTG | Val | LB-3' | TTG | Leu | CTA | Leu |
| LB-4 | CTG | Leu | ATT | Ile | DB-4' | TTG | Leu | GTC | Val | LB-4' | ACA | Thr | ATA | Ile |
| LB-5 | TTG | Leu | ATC | Ile | DB-5' | CTA | Leu | GTG | Val | LB-5' | CTG | Leu | ACT | Thr |
| LB-6 | CTG | Leu | ATA | Ile | DB-6' | CTT | Leu | GTG | Val | LB-6' | CTC | Leu | ATG | Met |
| LB-7 | TTA | Leu | ATT | Ile | DB-7' | ATG | Met | GTA | Val | LB-7' | CTA | Leu | ACT | Thr |
| LB-8 | TTA | Leu | ATA | Ile | DB-8' | CTA | Leu | AAG | Lys | LB-8' | ATG | Met | ATG | Met |
Light blue (2.3% of the total population).
Dark blue (3.4% of the total population).
Light blue (8.5% of the total population).
With 100 μm IPTG added, 3.4% of the total colonies turned dark blue, and 8.5% turned light blue. Sequence analysis showed that most dark blue colonies had Leu or Met at position 127 (seven of eight) (Table 2). At position 128, β-branched amino acids such as Ile and Val were found (seven of eight), resulting in three T127L/S128V and one T127M/S128V independent CRP variants. In the variants with light blue color, a similar Leu or Met preference at position 127 was seen (seven of eight), whereas β-branched chain amino acids were less frequent at position 128 (four of eight). Surprisingly, T127L/S128I CRP, which was consistently found in the 0 μm IPTG screen, was not found with 100 μm IPTG. We believe that large amounts of a highly active CRP variant such as T127L/S128I CRP make E. coli cells sick, as will be detailed below. These randomization results suggest that Leu or Met at position 127 and a β-branched amino acid at position 128 can effectively shift the equilibrium of the protein into a highly active form even without the effector cAMP and that the specific mutants detected depend on IPTG (and therefore CRP expression) levels.
An Improved Leucine Zipper Motif Centered at Position 127 Stabilizes the Active Conformation of CRP
To test systematically whether an improved leucine zipper motif at residues 127 and 128 stabilizes the active conformation of CRP, we created several CRP variants (T127L/S128(L/M/V) and T127(I/M/V)/S128I) that are similar to T127L/S128I CRP. We also created T127L/S128A CRP because it was the most active cAMP-independent variant characterized previously (16, 17, 19).
When we measured the in vivoβ-galactosidase activities of these CRP variants at either a Class I or II promoter, we noticed that the in vivo activities were highly dependent upon protein expression and easily saturated at higher protein levels: with 100 μm IPTG, all the tested CRP variants, including T127L/S128A, showed significant in vivo activities, which were not distinguishable from one another (data not shown). To obtain meaningful activity values, we therefore analyzed activity only in the absence of IPTG (Table 3). As a group, all the CRP variants showed higher activities without cAMP than did WT CRP at both Class I and II promoters, although the pattern was more apparent at the Class II promoter (Table 3). It is also worth noting that, under conditions of low protein expression, all the new CRP variants were better than the well studied T127L/S128A CRP variant in terms of cAMP-independent activity. Among the variants, T127L/S128I CRP showed the highest cAMP-independent activity, confirming the validity of the screen. The effector-independent activity of T127L/S128I CRP was comparable with that of WT CRP in the presence of cAMP. However, in the presence of cAMP, most CRP variants (except T127L/S128I and T127M/S128I) showed poorer activities than did WT CRP. This might mean that either (i) those variants are not saturated by the externally supplied 2 mM cAMP or (ii) fully cAMP-bound forms of these variants are functionally different from that of WT CRP. In vitro DNA binding measurements were necessary to differentiate between these two possibilities and to rule out differential protein accumulation as another variable.
TABLE 3. In vivo β-galactosidase activities and in vitro DNA-binding activities of CRP proteins mutated at residues 127 and 128.
The in vivo activity was measured in the background of UQ3811/UQ3809, a cya crp Class I/II E. coli reporter strain that contains the lacZ gene under the control of the CC(−61.5) or CC(−41.5) promoter. The in vivo activity value indicates the mean of multiple measurements (expressed as Miller units) and shows a variability of <20%. For in vivo assays, the cells were grown aerobically at 37 °C and 220 rpm in LC medium without IPTG. cAMP (2 mM) was externally added to the medium to determine the cAMP-dependent in vivo activity of each variant. The Kd values of the CRP proteins for DNA binding determined by the fluorescence polarization method were highly reproducible, with a range of variability of 30%. All proteins are His6-tagged at their C termini. NT, not tested.
| CRP | β-Galactosidase activity |
DNA-binding activity, Kd |
|||||
|---|---|---|---|---|---|---|---|
| Class I (CC(−61.5)) |
Class II (CC(−41.5)) |
||||||
| −cAMP | +cAMP | −cAMP | +cAMP | −cAMP | +0.1 mM cAMP | +1 mM cAMP | |
| Miller units | Miller units | nM | |||||
| Vector | 34 | 36 | 54 | 53 | |||
| Wild-type | 36 | 139 | 57 | 398 | >5000 | 10 | 12 |
| T127L/S128I | 182 | 186 | 435 | 502 | 35 | 14 | 16 |
| T127M/S128I | 110 | 158 | 335 | 497 | 40 | 15 | 18 |
| T127I/S128I | 54 | 76 | 133 | 211 | 186 | 18 | 28 |
| T127L/S128V | 55 | 82 | 134 | 205 | 444 | 21 | 25 |
| T127L/S128M | 40 | 43 | 83 | 98 | 1429 | 276 | 255 |
| T127V/S128I | 42 | 42 | 89 | 81 | 1482 | 37 | 42 |
| T127L/S128L | 38 | 40 | 71 | 70 | 3083 | 68 | 76 |
| T127L/S128A | 37 | 39 | 60 | 62 | ~4000 | 57 | 56 |
| T127L/S128L/L61V | NT | NT | NT | NT | 954 | 47 | 37 |
| T127L/ S128I/L61V | NT | NT | NT | NT | 434 | 100 | 104 |
We therefore purified each protein and examined its ability to bind a 26-mer CRP consensus DNA sequence in the presence and absence of cAMP. With this system, WT CRP bound DNA with Kd values of 10 and 12 nm in the presence of 0.1 and 1 mm cAMP, respectively, whereas it did not show any measurable DNA binding affinity without cAMP (Fig. 2A and Table 3). On the other hand, T127L/S128I CRP readily bound DNA in the absence of cAMP: its DNA binding affinity was only ~4-fold poorer than that of cAMP-bound WT CRP (Fig. 2B and Table 3). The other CRP variants all displayed better cAMP-independent DNA binding affinities than did WT CRP and even T127L/S128A CRP. Because the common property of these variants is a better leucine zipper motif at position 127, we suggest that a stronger intersubunit interaction in the region at residues 127 and 128 stabilizes the active conformation of CRP. However, the strong preference for Ile or Val at position 128 among the most active variants (Table 2) does not follow the hypothesis because residue 128 occupies an e-position, which is not an important site for a leucine zipper motif. The basis for the pattern at position 128 is detailed below.
FIGURE 2. In vitro DNA-binding activities of WT and T127L/S128I CRP proteins.
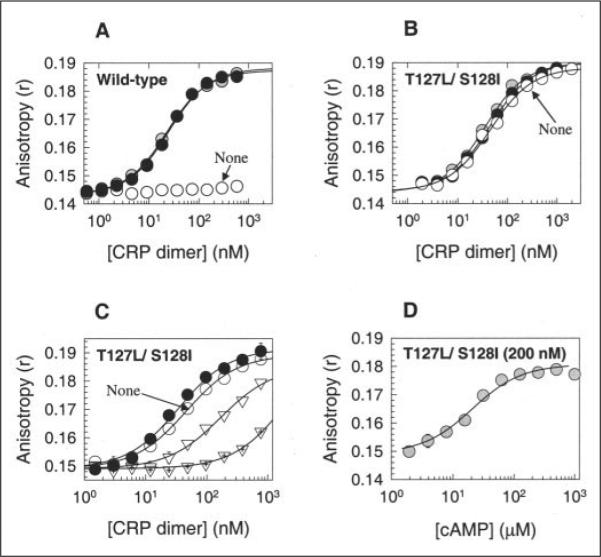
A and B, for WT CRP and T127L/S128I CRP, respectively, the activities were measured by the fluorescence polarization method. Open circles, no cAMP; shaded circles, 0.1 mM cAMP; closed circles, 1 mM cAMP. Solid lines show the best fit of the data to an equation described by Lundblad et al. (30). C, cGMP caused the loss of DNA-binding activity in T127L/S128I CRP, and cAMP restored the activity. Open circles, no cAMP; inverted open triangles, 0.1 mM cGMP; crossed inverted open triangles, 1 mM cGMP; closed circles, 1 mM cGMP + 1 mM cAMP. D, T127L/S128I CRP (200 nM) showed better affinity for cAMP than for cGMP. The DNA-binding activity of the protein was measured with various cAMP levels in the presence of 1 mM cGMP. Both WT and T127L/S128I CRP proteins were His6-tagged at their C termini.
All the residue 127 and 128 CRP variants displayed a higher affinity for DNA in response to cAMP addition (Table 3). The DNA binding affinity of most variants with cAMP was comparable with that of WT CRP. This suggests that these variants continue to bind cAMP and that the substitutions do not dramatically perturb functional action of CRP by cAMP. However, the T127L/S128M, T127L/S128L, and T127L/ S128A CRP variants were notably poor in terms of DNA affinity even with cAMP (Table 3). In the case of T127L/S128A CRP, it has been reported that the binding affinity of the protein for cAMP is not severely perturbed (17). Although we cannot completely rule out incomplete cAMP binding to the other two variants, this seems unlikely because the affinities of these proteins with 0.1 mm cAMP were not further increased by adding cAMP up to 1 mm. Rather, we imagine that this result can be explained in one of two ways. In the first, the cAMP binding might fail to shift the equilibrium fully to the active form, so the population of protein has a lower apparent DNA affinity. In the second, which is not exclusive of the first, there is an equilibrium shift, but the “active species” is not precisely the same as that of WT CRP, such that the DNA-binding domains are mispositioned, resulting in lower DNA affinity.
The striking cAMP independence of T127L/S128I CRP for high affinity DNA binding caused us to examine the variant further. We were concerned that this particular variant might bind cAMP poorly, so most of its DNA-binding activity in the presence of cAMP might be from that of the cAMP-free form. We therefore examined how cGMP affects the DNA-binding activity of this protein. As shown in Fig. 2C, T127L/S128I CRP showed a significant decrease in DNA affinity at 0.1 mm cGMP (Kd = 165 nm) and a very dramatic decrease at 1 mm cGMP (Kd = 2006 nm). This suggests not only poor affinity of this variant for cGMP, but also a clear negative effect on DNA binding caused by cGMP binding. Structural modeling suggests that, if cGMP binds in the anti-form, the NH2 moiety linked to C-2 of cGMP can sterically perturb part of the protein (Val49, Ser62, and Leu64), although the importance of these residues for CRP function has not been well defined. When 1 mm cAMP was then added to this protein already premixed with 1 mm cGMP, the affinity for DNA returned to the level seen with only 1 mm cAMP (Fig. 2C). This demonstrates that cAMP efficiently replaces the bound cGMP in T127L/S128I CRP and that the cAMP-bound form is at least as active as the cAMP-free form. To assess the relative affinity of the protein for cAMP and cGMP, we carried out a cAMP titration in a reaction mixture containing 1 mm cGMP and measured DNA binding (Fig. 2D). The result shows that T127L/S128I CRP had >55-fold higher affinity for cAMP than for cGMP. In summary, T127L/S128I CRP binds to cAMP efficiently, but it closely mimics the active conformation of cAMP-bound WT CRP regardless of the presence or absence of the effector. Finally, the negative response of this CRP variant to cGMP is different from that of hinge region CRP* variants such as G141Q and A144T, which have been reported to show positive responses to cGMP (12, 33). However, it is rather similar to the cases of the L148R and D53H CRP variants, the affinities of which for lac or gal DNA sequence in the absence of effector are stronger than in the presence of cGMP (18).
Genetic Evidence Suggesting Functional Communication between Ile128 and Leu61 in the T127L/S128I CRP Variant
The residues at position 127 that cause the cAMP-independent effects clearly follow the expected rule for a d-position residue in a leucine zipper. However, the effects of position 128 shown in Table 3 require a different explanation because this e-position residue should have minor importance in a leucine zipper, other than possible interhelical salt bridges. We imagined three general possibilities for the differential activity of different substitutions at position 128 in the Leu127 background: destabilization of the inactive form, stabilization of the active form by a direct effect on C-helix repositioning, and stabilization of the active form by a direct effect on another portion of the protein. We then assumed that Ile makes an exceptionally good interaction with some partner to provide one of these effects. Because we do not know the inactive structure of CRP, a direct test of the first possibility was impossible, but we tested the other two.
In the second possibility, we noted that residue 128 is an e-position in the leucine zipper motif and that residues at this position often interact with the residues at the ǵ-position, which in this case would be Arg123. Although e–ǵ interactions are normally ionic, one might imagine that a hydrophobic interaction between Ile128 and Arg123 might further stabilize the C-helices in a position similar to that found in cAMP-bound WT CRP, albeit by a molecularly distinct mechanism. By this hypothesis, one would predict that optimal residues at position 128 would depend on the residue at position 123. To test this, we simultaneously randomized positions 123 and 128 in a background with Leu127 and demanded variants with high activity in the absence of cAMP. We again screened without IPTG. A variety of relatively small and hydrophilic residues were found at position 123 (Ser, Asp, Thr, Lys, and Asn) and the position 128 residues were almost exclusively Ile and Val. Although we do not understand the value of hydrophilicity at position 123, the presence of small residues at position 123, which should make residue 123-residue 128 interactions impossible, and the lack of any apparent “matching” of residues at these two positions seem to disprove the starting hypothesis of a residue 123-residue 128 interaction. These results also cast doubt on the first hypothesis (destabilization of the inactive form) because it seems very unlikely that Ile and Val are the only residues at position 128 that significantly destabilize the inactive form without perturbing the active state.
The third possibility is the stabilization of the active form by a direct effect on another portion of the protein. Specifically, cAMP binding might cause the repositioning of the β4/β5 loop, which is part of the effector-binding domain and which makes a number of contacts with the DNA-binding domain in the active form (8), thus stabilizing this form. By this model, one would suppose that Ile or Val at position 128 is particularly adept at making a necessary interaction in the pocket normally filled by cAMP to help stimulate this repositioning. In a test of this model, we therefore considered Leu61 to be a possible candidate for functional communication with residue 128 for two reasons. First, Leu61 lies at the base of the β4/β5 loop and might contact a large residue at position 128 in the absence of cAMP based on the structural modeling using Swiss-PdbViewer Version 3.7 (available at www.expasy.org/ spdbv). Second, Lee et al. (34) have shown that cAMP binding perturbs the methyl proton resonance of this residue, demonstrating their proximity. We therefore randomized position 61 in the T127L/S128L CRP background, which was relatively inactive by itself (Table 3), and screened for the most active variants in the absence of cAMP. All four sequenced variants with this property had Val61 and had ~3-fold higher cAMP-independent affinity for DNA than did the T127L/S128L CRP variant itself (Table 3). By the model, this result means that Leu128 interacts better with Val61 than with Leu61 to stabilize the active form. To rule out the possibility that Val61 is better than Leu61 independent of the specific 128 residue, Val61 was introduced into the T127L/S128I CRP background. The T127L/S128I/L61V CRP variant displayed 10-fold poorer cAMP-independent affinity for DNA in the absence of cAMP than did T127L/S128I CRP alone (Table 3), showing that the effect of the residue at position 61 depends on that at position 128. These results strongly suggest a residue-specific communication between the two regions in the active CRP* form. This supports the model in which Ile or Val at position 128 stabilizes the active form of CRP through repositioning the β4/β5 loop, a result normally achieved by the bound cAMP.
Effect of cAMP on the Positioning of Activating Regions in T127L/S128I CRP
The activating regions (ARs) are the surfaces of CRP and related proteins that interact with different subunits of RNA polymerase to activate transcription (2). As a consequence, for CRP to function in vivo, it must not only properly position its F-helices, but also these ARs. Although the effect of cAMP on the conformational change for DNA binding of CRP has been extensively studied, the issue of cAMP affecting AR positioning has not been addressed because it is technically difficult. In an in vitro transcription experiment, AR positioning can be probed only when the DNA-binding domains have been properly positioned, and for WT CRP, this requires cAMP. However, the cAMP-independent DNA binding of T127L/S128I CRP avoids this problem and allowed us the opportunity to test in vitro if cAMP directly affects AR positioning. The fact that T127L/S128I CRP displays substantial activity in vivo without cAMP shows that AR positioning is at least acceptable in this variant, and therefore, cAMP is not an absolute requirement for transcriptional activation by CRP. However, in vivo transcription data reflect many unknown factors such as the accumulation level of a protein and the availability of the effector, making such data hard to interpret.
To quantitatively investigate the effect of cAMP binding on the AR surfaces of T127L/S128I CRP, we carried out an in vitro transcription assay with T127L/S128I CRP and with WT CRP as the control. T127L/ S128I CRP was used at 200 nm, which should saturate the template DNA in this analysis. Thus, under these conditions, any effect of cAMP on the in vitro transcription of the protein would result from AR positioning. Both Class I (CC(−61.5)) and Class II (CC(−41.5)) promoters were examined because different AR surfaces are used at each of these. With WT CRP, transcription from both promoters was stimulated by 0.1 mm cAMP (Fig. 3), and the activity at the Class II promoter was about two times better than that at the Class I promoter, consistent with a previous report (35). There was a slight but reproducible improvement of activity for WT CRP when the cAMP level was raised from 0.1 to 1 mm that we do not understand (note that the in vitro DNA-binding activity of WT CRP was slightly better with 0.1 mm cAMP), but otherwise, the results were as expected. The transcriptional activities of cAMP-free T127L/S128I CRP were almost identical to those of cAMP-bound WT CRP at both Class I and II promoters (Fig. 3). The result confirms the in vivo data showing that cAMP-free T127L/S128I CRP is normally active for transcription as well as DNA binding. In other words, the AR surfaces of T127L/S128I CRP in the absence of cAMP are positioned similarly to those of WT CRP in the presence of cAMP. This might suggest that, although cAMP binding to WT CRP certainly causes a change in AR positions concomitant with the reorientation of the DNA-binding F-helices, there is no reason to believe that it changes the AR surfaces independent of the repositioning of the DNA-binding domain.
FIGURE 3. Multiple-round in vitro transcription assays of WT and T127L/S128I CRP proteins.
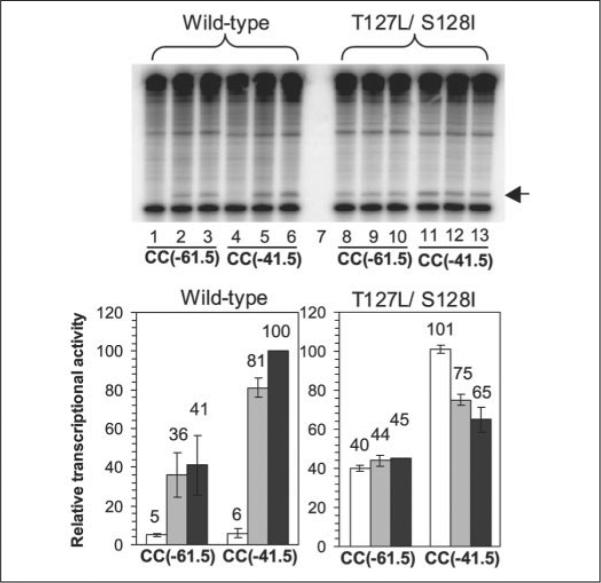
Measurements were carried out for both Class I (CC(−61.5)) and Class II (CC(−41.5)) promoters under three conditions (no cAMP, 0.1 mM cAMP, and 1 mM cAMP). The gel is of radiolabeled transcripts from the assays. Lanes 1, 4, 8, and 11, no cAMP; lanes 2, 5, 9, and 12, 0.1 mM cAMP; lanes 3, 6, 10, and 13, 1 mM cAMP. Lane 7 is empty. The bars in the histograms represent the average of two independent assays. White bars, no cAMP; light gray bars, 0.1 mM cAMP; dark gray bars, 1 mM cAMP. The data in the histograms are presented as a percentage of activation by WT CRP at the CC(−41.5) promoter in the presence of 1 mM cAMP. The experiment was performed with supercoiled templates (26, 27) in the presence of RNA polymerase containing σ70. CRP-dependent transcripts are indicated by the arrow and are located just above the control transcripts from the RNA I promoter. Both proteins were His6-tagged at their C termini and used at a concentration of 200 nM.
Unexpectedly, the addition of cAMP to T127L/S128I CRP caused a reproducible decrease in in vitro transcriptional activity at the Class II promoter by 25–50%, but not at the Class I promoter (Fig. 3). Because Class I promoters use only AR1 and Class II promoters use AR1, AR2, and AR3, this result suggests that cAMP does not affect AR1, but can inappropriately rearrange AR2 or AR3 in T127L/S128I CRP. The alteration of two cAMP contact residues might lead to altered AR2/AR3 positioning in the presence of cAMP.
A High Level of CRP Activity Is Lethal to E. coli
Although not described above, we initially screened a library of CRP-expressing clones randomized at positions 127 and 128 for constitutively active CRP variants using either strain UQ3261 or UQ3588 (see Table 1 for details). In these strains, a modest level of IPTG is an absolute requirement to turn on the natural lac assay system. When a screening was done with 200 μm IPTG, many constitutive mutants were found, but some of them proved to be genetically unstable during isolation, implying that a deleterious growth property might be associated with that constitutive activity. We assume that the same reason explains our failure to find T127L/S128I CRP in the screen at 100 μm IPTG in strain UQ3811 (Table 2). The observation that elevated levels of CRP activity might deleteriously affect growth has been made before (11, 13, 36), but not fully analyzed.
We then deliberately tested whether elevated expression of highly active CRP variants causes poor E. coli growth. We grew strains lacking the wild-type crp and cya genes (UQ3588 and cya crp E. coli HB101), but expressing WT, T127L/S128A, or T127L/S128I CRP from a plasmid under IPTG regulation, aerobically in LC medium in the absence of cAMP. As evident in Fig. 4A, the strain with T127L/S128I CRP showed modest growth impairment at 100 μm IPTG and strong impairment at 1 mm IPTG. Although the strain containing WT CRP showed almost identical growth rates independent of IPTG concentration (Fig. 4B), the strain with T127L/S128A CRP showed a slight improvement in growth at higher IPTG levels (Fig. 4C). WT and variant CRP proteins appear to accumulate similarly based on the fact that we obtained a very similar purification yield of these proteins when expressed at 1 mm IPTG. This suggests that it is CRP activity (not the CRP protein level) that is causative for the growth perturbation. Finally, given the weak constitutive activity of T127L/S128A CRP, this result suggests that some CRP activity is important for optimal growth and that this variant requires modest levels of expression to provide that necessary activity to compensate for the growth impairment of the parental cya crp strain.
FIGURE 4. Effects of T127L/S128I, T127L/S128A, and WT CRP proteins on the growth of host E. coli strains.

Overnight cultures of the UQ3588 host strain (Table 1) expressing different CRP proteins (C-terminally His-tagged) under IPTG-dependent regulation were diluted in LC medium (see “Experimental Procedures”) supplemented with 50 μg/ml ampicillin, 0–1 mM IPTG, and 0 cAMP (A–C) or 2 mM cAMP (D–F). Open symbols, no IPTG; lightgraysymbols,0.01mM IPTG;darkgraysymbols,0.1mM IPTG;blacksymbols,1mM IPTG. Each strain was grown with shaking at 37 °C and 220 rpm and sampled every hour for the measurement of A600 nm.
Externally added cAMP (2 mm) did not cause any difference in the growth pattern of either T127L/S128I or T127L/S128A CRP (Fig. 4, D and F). Rather surprisingly, we also failed to see a dramatic growth perturbation in the strain with WT CRP in the presence of cAMP (Fig. 4E). One might expect that high expression of WT CRP in the presence of cAMP would cause severe growth perturbation, as did T127L/S128I CRP even without cAMP, unless the exogenously provided cAMP is not reaching a sufficiently high concentration within the cells to activate a large population of WT CRP. To test whether endogenous cAMP can reach a level so that overexpressed WT CRP is detrimental, we transformed the same WT CRP-containing plasmid into the parental wild-type E. coli HB101 (cya+) strain and tested the effect of elevated CRP levels on the growth. Severe growth perturbation occurred at 1 mm IPTG in the wild-type HB101 background (data not shown), consistent with the hypothesis that high CRP activity is detrimental to E. coli. This indicates that only modest internal cAMP levels result from a 2 mm exogenous addition, at least in the HB101 background. Taken together, these results explain why effector-independent CRP variants as active as those reported here have not been previously described.
DISCUSSION
C-helix Repositioning and Proper Positioning of the β/β5 Loop Are Two Local Triggering Signals for the Conformational Change in CRP in the Course of Activation by cAMP
We have shown here that cAMP-free T127L/S128I CRP closely resembles cAMP-bound WT CRP in terms of DNA affinity and transcriptional activity. By genetic selection and biochemical analysis, we were able to dissect the effects of the substitutions into two mechanistically different parts: (i) a stronger intersubunit interaction because of Leu127, the optimal leucine zipper residue at the d-position in the heptad repeat, and (ii) proper β4/β5 loop positioning because of the ability of Ile128 to functionally communicate with (at least) Leu61. These two local signals are depicted in Fig. 5A in a structural model for T127L/S128I CRP based on the structure of cAMP-bound WT CRP (Protein Data Bank code 1CGP). For Ile128, a conformer has been arbitrarily chosen to show that contact between residues 61 and 128 is plausible, and for Leu127, a leucine zipper conformation is depicted.
FIGURE 5. Structural model for high constitutive activity of T127L/S128I CRP.

The hypothesized local structure of T127L/S128I CRP (A) was derived from the structure of active cAMP-bound WT CRP (B) (Protein Data Bank code 1CGP). In this view along a portion of the C-helices, the DNA-binding domains are in the distance. The T127L/S128I CRP structure is not meant to represent the actual one, but rather to indicate a reasonable set of Leu127 and Ile128 conformers that might explain its high constitutive activity. In each figure, two subunits are differently colored. In A, the side chains of Leu61, Leu127, and Ile128 residues are shown. In B, the side chains of Leu61, Thr127, and Ser128 residues and cAMP molecules are shown, and the known interactions (adopted from Ref. 14) are also indicated. The structure was generated using Swiss-PdbViewer Version 3.7.
We propose that the Leu127 and Ile128 substitutions induce similar local conformational changes to cAMP binding in WT CRP (Fig. 5B). Under this assumption, the results support two direct local changes by cAMP in the course of the activation of WT CRP, which is consistent with a previous structure-based prediction about CRP activation (14). First, cAMP causes C-helix repositioning, thereby resulting in a new conformation that allows a stronger intersubunit interaction at the region of positions 127 and 128 of the C-helices. We hypothesize that cAMP draws Thr127 and Ser128 into such a new position and that both residues are required to stabilize an active C-helix conformation (Fig. 5B). We further hypothesize that the new conformation of the C-helices has a direct effect on the hinge region at the C-proximal end and stabilizes the hinge region of the protein in a way to properly position the DNA-binding domains. The general notion of hinge region stabilization in activation has been proposed previously (11, 12), although not in the context of C-helix repositioning. Second, we believe that cAMP repositions the β4/β5 loop of WT CRP. Consistent with this view is the observation that the methyl proton resonance of Leu61 is changed by cAMP binding to CRP when studied by NMR (34). In a structural view (Fig. 5B), it appears that cAMP directly fills the cavity between the C-helix and β4/β5 loop through hydrophobic interactions. As suggested previously (37), these hydrophobic interactions involve Leu61, but are not limited to this residue. The properly positioned β4/β5 loop would stabilize the hinge region in the active form, as proposed by Passner et al. (14).
When we performed a similar screening for constitutively active variants of CooA, variants were found with improved leucine zippers (21). These had substantial activity without the effector CO, but also showed a significant increase in activity to approximately the wild-type level upon CO addition. This was interpreted to mean that the subunit reorientation of the C-helices in CooA is normally limited by other interactions within the effector domain, such as Pro2 axial ligation to the heme (21). Alternatively, the existence of an additional signal transduction route has been suggested in CooA (21). Our results with CRP suggest that movement of the β4/β5 loop might be part of the activation pathway of CooA as well.
Two different PrfA structures (WT PrfA and G145S PrfA) that differ in both C-helix conformations and DNA binding affinities are known. Consistent with our proposed view of CRP activation, a significant structural difference has also been found in the relative positions of the β4/β5 loops between the two PrfA proteins (23). It remains to be determined if these changes reflect the normal response of PrfA to effector binding because the very existence of such an effector is in doubt, but the correlation between the C-helix repositioning and DNA affinity is very suggestive. Therefore, the results of the present study, together with CooA and PrfA data, predict that the other members of this large protein family might use the same signaling pathways, although they will certainly use different specific effector recognition mechanisms to trigger these pathways.
What Are the Roles of Thr127 and Ser128 in WT CRP?
Thr127 and Ser128 of WT CRP are C-helix residues that make specific contacts with cAMP based on the structures of active CRP (8, 14). One might have assumed that the identity of these residues would be crucial for both cAMP binding and CRP activation. However, numerous studies from different research groups have shown that these residues play a minor role in cAMP binding (15, 38–41). Our result that a variety of CRP variants altered at positions 127 and 128 continue to bind cAMP is also consistent with the conclusion. It is also interesting to note that, in a recent structure-based sequence comparison of a wide array of cAMP-binding proteins (42), the residues homologous to Thr127 and Ser128 are not conserved at all. Our data further suggest that the wild-type residues Thr127 and Ser128 are not unique in the stabilization of the active conformation in response to cAMP binding, as evidenced by the fact that T127L/S128I, T127M/S128I, T127I/S128I CRP variants have normal DNA binding affinities in the presence of cAMP (Table 3). Finally, Lee et al. (15) claimed that hydrophobic amino acid substitutions at either position 127 or 128 broaden the specificity of the CRP effector to include cGMP, at least when assayed in vivo. This is, however, not a general property of changes at these positions because T127L/S128I CRP still clearly discriminates for cAMP over cGMP (Fig. 2, C and D). What then are the roles of these residues in WT CRP?
Our current view is that Thr127 and Ser128 allow an optimal regulation of CRP activity by cAMP because of two properties. (i) In the absence of cAMP, they introduce a disruption in an otherwise strong leucine zipper interface along the C-helices, thereby reducing the level of CRP activity without the effector. (ii) By a hydrogen-bonding network with cAMP, they effectively allow a new C-helix conformation that favors the positioning of the DNA-binding domains to interact with DNA. Quite possibly, Thr127 and Ser128 are the best combination to perform these two dissimilar functions. It is worth noting that sequence alignment of closely related CRP homologs (either putative or confirmed) showed that a Thr/Ser or Thr/Thr combination at these positions is typical (data not shown).
Among the non-CRP members of the extended CRP/FNR family of proteins, analogous residues are not as conserved, but some similarity in roles might be suggested. In CooA, a Cys residue occupies the position analogous to Thr127, and its replacement by residues that are preferable for a leucine zipper results in some degree of CO-independent activity (21). Indeed, even FNR, the other well studied member of the protein family, has a leucine zipper disruption at the same position: in this case, Asp. Although FNR seems to have fundamental differences in its response to regulatory signals, changing this Asp to Ala causes FNR to be effector-independent (43). In each case, a residue that is not optimal for a leucine zipper allows two different possible C-helix conformations: one that favors the positioning of the DNA-binding domains to interact with DNA and another that does not. In the absence of effector, the equilibrium between these conformations is shifted toward the inactive form, which in turn allows effector binding to have a biologically significant effect by shifting the equilibrium the other way. It is perhaps instructive that both CRP and CooA normally have small residues at these positions, which might be particularly appropriate for allowing the choice in conformations. In conclusion, there is a common disruption in the coiled-coil C-helices among this extended family of proteins that is consistent with the proposed C-helix repositioning as a common activation mechanism.
Comparison of T127L/S128I CRP with Known cAMP-independent CRP Variants
Most of the CRP variants described in this study are substantially more active than is T127L/S128A CRP, which was the most active cAMP-independent CRP variant reported previously (16, 17, 19). This result shows the advantage of structure-informed mutagenesis combined with an in vivo screen selection scheme as long as the lethal effects of high CRP activity are circumvented. In the past, moderately active cAMP-independent CRP variants have been found in other regions of the protein as well (11–13, 36, 44). One of the most common regions where these have been found is the D-helix, and they have been typically explained by suggesting that these substitutions affect interactions between the C- and D-helices. We agree with this view, but with the following two modifications. (i) The effects of these substitutions must be interpreted with respect to the repositioning of the C-helices as well, and this view well explains the earlier findings by Garges and Adhya (45) that a T127A mutation can suppress the cAMP-independent activity of a D-helix CRP* variant, G141S. (ii) Such substitutions certainly shift the equilibrium toward the active form, but might do so either by destabilizing the inactive form or, as typically interpreted, stabilizing the active form. Obviously, we require a structure of the inactive form for more progress to be made.
Acknowledgments
We thank Wilma Ross and Rick Gourse for numerous technical suggestions and assistance with the in vitro transcription assays. We also thank Rick Gourse for helpful critical comments on the manuscript and Jose Serate and Jacob Sonntag for technical assistance.
Footnotes
The work was supported by the College of Agricultural and Life Sciences of the University of Wisconsin at Madison and by National Institutes of Health Grant GM53228 (to G. P. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: CRP, cAMP receptor protein; WT, wild-type; IPTG, isopropyl β-D-thiogalactopyranoside; ARs, activating regions.
REFERENCES
- 1.Kolb A, Busby S, Buc H, Garges S, Adhya S. Annu. Rev. Biochem. 1993;62:749–795. doi: 10.1146/annurev.bi.62.070193.003533. [DOI] [PubMed] [Google Scholar]
- 2.Busby S, Ebright RH. J. Mol. Biol. 1999;293:199–213. doi: 10.1006/jmbi.1999.3161. [DOI] [PubMed] [Google Scholar]
- 3.Brown CT, Callan CG., Jr. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2404–2409. doi: 10.1073/pnas.0308628100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawson CL, Swigon D, Murakami KS, Darst SA, Berman HM, Ebright RH. Curr. Opin. Struct. Biol. 2004;14:10–20. doi: 10.1016/j.sbi.2004.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harman JG. Biochim. Biophys. Acta. 2001;1547:1–17. doi: 10.1016/s0167-4838(01)00187-x. [DOI] [PubMed] [Google Scholar]
- 6.Zubay G, Schwartz D, Beckwith J. Proc. Natl. Acad. Sci. U. S. A. 1970;66:104–110. doi: 10.1073/pnas.66.1.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Emmer M, deCrombrugghe B, Pastan I, Perlman R. Proc. Natl. Acad. Sci. U. S. A. 1970;66:480–487. doi: 10.1073/pnas.66.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schultz SC, Shields GC, Steitz TA. Science. 1991;253:1001–1007. doi: 10.1126/science.1653449. [DOI] [PubMed] [Google Scholar]
- 9.Won HS, Yamazaki T, Lee TW, Yoon MK, Park SH, Kyogoku Y, Lee B-J. Biochemistry. 2000;39:13953–13962. doi: 10.1021/bi000012x. [DOI] [PubMed] [Google Scholar]
- 10.Baichoo N, Heyduk T. Protein Sci. 1999;8:518–528. doi: 10.1110/ps.8.3.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Adhya S, Garges S. Proc. Natl. Acad. Sci. U. S. A. 1992;89:9700–9704. doi: 10.1073/pnas.89.20.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garges S, Adhya S. Cell. 1985;41:745–751. doi: 10.1016/s0092-8674(85)80055-6. [DOI] [PubMed] [Google Scholar]
- 13.Harman JG, McKenney K, Peterkofsky A. J. Biol. Chem. 1986;261:16332–16338. [PubMed] [Google Scholar]
- 14.Passner JM, Schultz SC, Steitz TA. J. Mol. Biol. 2000;304:847–859. doi: 10.1006/jmbi.2000.4231. [DOI] [PubMed] [Google Scholar]
- 15.Lee EJ, Glasgow J, Leu SF, Belduz AO, Harman JG. Nucleic Acids Res. 1994;22:2894–2901. doi: 10.1093/nar/22.15.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi Y, Wang S, Krueger S, Schwarz FP. J. Biol. Chem. 1999;274:6946–6956. doi: 10.1074/jbc.274.11.6946. [DOI] [PubMed] [Google Scholar]
- 17.Chu SY, Tordova M, Gilliland GL, Gorshkova I, Shi Y, Wang S, Schwarz FP. J. Biol. Chem. 2001;276:11230–11236. doi: 10.1074/jbc.M010428200. [DOI] [PubMed] [Google Scholar]
- 18.Dai J, Lin SH, Kemmis C, Chin AJ, Lee JC. Biochemistry. 2004;43:8901–8910. doi: 10.1021/bi0499359. [DOI] [PubMed] [Google Scholar]
- 19.Moore JL, Gorshkoya II, Brown JW, McKenney KH, Schwarz FP. J. Biol. Chem. 1996;271:21273–21278. doi: 10.1074/jbc.271.35.21273. [DOI] [PubMed] [Google Scholar]
- 20.Lanzilotta WN, Schuller DJ, Thorsteinsson MV, Kerby RL, Roberts GP, Poulos TL. Nat. Struct. Biol. 2000;7:876–880. doi: 10.1038/82820. [DOI] [PubMed] [Google Scholar]
- 21.Kerby RL, Youn H, Thorsteinsson MV, Roberts GP. J. Mol. Biol. 2003;325:809–823. doi: 10.1016/s0022-2836(02)01203-2. [DOI] [PubMed] [Google Scholar]
- 22.Youn H, Kerby RL, Roberts GP. J. Biol. Chem. 2004;279:45744–45752. doi: 10.1074/jbc.M404336200. [DOI] [PubMed] [Google Scholar]
- 23.Eiting M, Hageluken G, Schubert WD, Heinz DW. Mol. Microbiol. 2005;56:433–446. doi: 10.1111/j.1365-2958.2005.04561.x. [DOI] [PubMed] [Google Scholar]
- 24.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 25.Chiang LW, Kovari I, Howe MM. PCR Methods Applications. 1993;2:210–217. doi: 10.1101/gr.2.3.210. [DOI] [PubMed] [Google Scholar]
- 26.Savery NJ, Lloyd GS, Kainz M, Gaal T, Ross W, Ebright RH, Gourse RL, Busby SJ. EMBO J. 1998;17:3439–3447. doi: 10.1093/emboj/17.12.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savery NJ, Lloyd GS, Busby SJ, Thomas MS, Ebright RH, Gourse RL. J. Bacteriol. 2002;184:2273–2280. doi: 10.1128/JB.184.8.2273-2280.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller J. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 29.Gaston K, Bell A, Kolb A, Buc H, Busby S. Cell. 1990;62:733–743. doi: 10.1016/0092-8674(90)90118-x. [DOI] [PubMed] [Google Scholar]
- 30.Lundblad JR, Laurance M, Goodman RH. Mol. Endocrinol. 1996;10:607–612. doi: 10.1210/mend.10.6.8776720. [DOI] [PubMed] [Google Scholar]
- 31.Tomizawa J, Itoh T. Proc. Natl. Acad. Sci. U. S. A. 1981;78:6096–6100. doi: 10.1073/pnas.78.10.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dykxhoorn DM, St. Pierre R, Linn T. Gene (Amst.) 1996;177:133–136. doi: 10.1016/0378-1119(96)00289-2. [DOI] [PubMed] [Google Scholar]
- 33.Cheng X, Lee JC. J. Biol. Chem. 1994;269:30781–30784. [PubMed] [Google Scholar]
- 34.Lee B-J, Aiba H, Kyogoku Y. Biochemistry. 1991;30:9047–9054. doi: 10.1021/bi00101a020. [DOI] [PubMed] [Google Scholar]
- 35.Rhodius VA, Busby SJ. J. Mol. Biol. 2000;299:311–324. doi: 10.1006/jmbi.2000.3737. [DOI] [PubMed] [Google Scholar]
- 36.Aiba H, Nakamura T, Mitani H, Mori H. EMBO J. 1985;4:3329–3332. doi: 10.1002/j.1460-2075.1985.tb04084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomlinson SR, Tutar Y, Harman JG. Biochemistry. 2003;42:3759–3765. doi: 10.1021/bi027126o. [DOI] [PubMed] [Google Scholar]
- 38.Polit A, Bonarek P, Kepys B, Kedracka-Krok S, Gorecki A, Wasylewski Z. J. Biol. Chem. 2003;278:43020–43026. doi: 10.1074/jbc.M306398200. [DOI] [PubMed] [Google Scholar]
- 39.Leu SF, Baker CH, Lee EJ, Harman JG. Biochemistry. 1999;38:6222–6230. doi: 10.1021/bi982938z. [DOI] [PubMed] [Google Scholar]
- 40.Lin SH, Lee JC. Biochemistry. 2002;41:11857–11867. doi: 10.1021/bi026099z. [DOI] [PubMed] [Google Scholar]
- 41.Shi Y, Wang S, Schwarz FP. Biochemistry. 2000;39:7300–7308. doi: 10.1021/bi000225m. [DOI] [PubMed] [Google Scholar]
- 42.Berman HM, Ten Eyck LF, Goodsell DS, Haste NM, Korney A, Taylor SS. Proc. Natl. Acad. Sci. U. S. A. 2005;102:45–50. doi: 10.1073/pnas.0408579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moore LJ, Kiley PJ. J. Biol. Chem. 2001;276:45744–45750. doi: 10.1074/jbc.M106569200. [DOI] [PubMed] [Google Scholar]
- 44.Ryu S, Kim J, Adhya S, Garges S. Proc. Natl. Acad. Sci. U. S. A. 1993;90:75–79. doi: 10.1073/pnas.90.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garges S, Adhya S. J. Bacteriol. 1988;170:1417–1422. doi: 10.1128/jb.170.4.1417-1422.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Casadaban MJ, Cohen SN. J. Mol. Biol. 1980;138:179–207. doi: 10.1016/0022-2836(80)90283-1. [DOI] [PubMed] [Google Scholar]
- 47.Kolb A, Kotlarz D, Kusano S, Ishihama A. Nucleic Acids Res. 1995;23:819–826. doi: 10.1093/nar/23.5.819. [DOI] [PMC free article] [PubMed] [Google Scholar]