

Abstract
Inhibition of cell proliferation by fenoterol and fenoterol derivatives in 1321N1 astrocytoma cells is consistent with β2-adrenergic receptor (β2-AR) stimulation. However, the events that result in fenoterol-mediated control of cell proliferation in other cell types are not clear. Here, we compare the effect of the β2-AR agonists (R,R′)-fenoterol (Fen) and (R,R′)-4-methoxy-1-naphthylfenoterol (MNF) on signaling and cell proliferation in HepG2 hepatocarcinoma cells by using Western blotting and [3H]thymidine incorporation assays. Despite the expression of β2-AR, no cAMP accumulation was observed when cells were stimulated with isoproterenol or Fen, although the treatment elicited both mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt activation. Unexpectedly, isoproterenol and Fen promoted HepG2 cell growth, but MNF reduced proliferation together with increased apoptosis. The mitogenic responses of Fen were attenuated by 3-(isopropylamino)-1-[(7-methyl-4-indanyl)oxy]butan-2-ol (ICI 118,551), a β2-AR antagonist, whereas those of MNF were unaffected. Because of the coexpression of β2-AR and cannabinoid receptors (CBRs) and their impact on HepG2 cell proliferation, these Gαi/Gαo-linked receptors may be implicated in MNF signaling. Cell treatment with (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-napthalenylmethanone (WIN 55,212-2), a synthetic agonist of CB1R and CB2R, led to growth inhibition, whereas inverse agonists of these receptors blocked MNF mitogenic responses without affecting Fen signaling. MNF responses were sensitive to pertussis toxin. The β2-AR-deficient U87MG cells were refractory to Fen, but responsive to the antiproliferative actions of MNF and WIN 55,212-2. The data indicate that the presence of the naphthyl moiety in MNF results in functional coupling to the CBR pathway, providing one of the first examples of a dually acting β2-AR-CBR ligand.
Introduction
(R,R′)-fenoterol (Fen) is a potent and selective agonist of the β2-adrenergic receptor (β2-AR), with an EC50cAMP value of 0.3 nM for the stimulation of cAMP accumulation in human embryonic kidney cells expressing human β2-AR (Jozwiak et al., 2010). We had reported the synthesis and characterization of a number of Fen analogs and stereoisomers with a range of β2-AR selectivity and potency (Jozwiak et al., 2007, 2010). One of these analogs, (R,R′)-4-methoxy-1-naphthylfenoterol (MNF), has a β2-AR/β1-AR selectivity of 573 with an EC50cAMP value of 3.90 nM.
β2-ARs associate with heterotrimeric G proteins (e.g., GS, Gi), ion channels, and cytosolic scaffold proteins, including β-arrestin, to initiate various signaling pathways and modulate the activity of intracellular effectors such as adenylyl cyclase and mitogen-activated protein kinases (Audet and Bouvier, 2008; Kahsai et al., 2011). The difference in the G protein and β-arrestin signaling by β2-AR agonists has been attributed to interaction with ligand-specific GPCR conformations and functional selectivity, which is based on the assumption that the β2-AR exists in an inactive (R) state and one or more ligand-specific active conformations (R*n) (Seifert and Dove, 2009). The basis for the ligand-specific differences in pharmacological outcome lies in the interplay between the molecular structure of the agonist (Kahsai et al., 2011) and the cellular environment of the receptor. In the first instance, we have shown that the Gs/Gi selectivity of Fen is a function of molecular structure and stereochemistry, because Fen preferentially activated GS signaling in a cardiomyocyte contractility model, whereas (S,R′)-fenoterol and MNF activated both GS and Gi proteins (Woo et al., 2009; Jozwiak et al., 2010).
We have also demonstrated that β2-AR agonists such as Fen and isoproterenol exert antiproliferative effects in the human-derived 1321N1 astrocytoma cell line specifically through the cAMP-dependent pathway (Toll et al., 2011), whereas Yuan et al. (2010) reported that isoproterenol dose-dependently induced the growth of the human-derived HepG2 hepatocellular carcinoma cell line. This cell type-specific divergence on cell proliferation by β2-AR agonists raises questions about whether there is cross-talk between β2-AR and other receptor-linked signaling cascades. A link between β2-AR and other GPCRs, including α2-AR, bradykinin, oxytocin, and cannabinoid receptors (CBRs), has been described previously (Uberti et al., 2005; Haack et al., 2010; Hudson et al., 2010; Wrzal et al., 2012). Of significance, CBRs have been reported to modulate β2-AR activity (Gardiner et al., 2005). The two known cannabinoid receptors, CB1R and CB2R, are coupled to Gαi/o proteins and consequently inhibit adenylyl cyclase activity upon agonist binding (Pertwee, 2006). It is interesting that the endogenous endocannabinoid anandamide induces cell death in hepatic stellate cells (Yang et al., 2010) and promotes necrosis of HepG2 cells via activation of CB1R and CB2R (Wu et al., 2010). Treatment with selective pharmacological CB2R agonists promotes neural progenitor cell proliferation and survival both in vitro and in vivo (Palazuelos et al., 2006). However, despite the fact that β2-AR and CBRs are coexpressed and can affect similar cellular processes in a variety of tissues, no studies have examined the possible role that CBRs may have in influencing cell fate decision mediated by β2-agonists.
The current study was designed to investigate the effect of the molecular structure and stereochemistry of β2-AR agonists on [3H]thymidine incorporation in HepG2 cells and compare these results to similar studies conducted with the 1321N1 and human-derived U87MG glioblastoma cell lines. Here, we report that proliferative responses that occur with Fen and isoproterenol were opposite to those induced by MNF, even though the latter fenoterol analog is also a full and potent β2-AR agonist. Unexpectedly, we show that the CBR pathway is intimately involved in the cell type-dependent control of cell proliferation by MNF. These data suggest an additional level of molecular regulation between fenoterol analogs.
Materials and Methods
Materials.
(R,R′)-, (R,S′)-, (S,R′)-, and (S,S′)-fenoterol and the fenoterol analogs (R,R′)-ethylfenoterol, (R,R′)-4′-aminofenoterol, (R,R′)-1-naphthylfenoterol, and (R,R′)- and (R,S′)-4-methoxy-1-naphthylfenoterol were synthesized as described previously (Jozwiak et al., 2007, 2010). [3H]thymidine (70–90 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). Eagle's minimum essential medium, trypsin solution, phosphate-buffered saline (PBS), fetal bovine serum (FBS), and 100× solutions of sodium pyruvate (100 mM), l-glutamine (200 mM), and penicillin/streptomycin (a mixture of 10,000 units/ml penicillin and 10,000 μg/ml streptomycin) were obtained from Quality Biological (Gaithersburg, MD). (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-napthalenylmethanone (WIN 55,212-2), 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-(1-piperidyl)pyrazole-3-carboxamide (AM251), and 1-[2-(morpholin-4-yl)ethyl]-2-methyl-3-(4-methoxybenzoyl)-6-iodoindole (AM630) were purchased from Cayman Chemical (Ann Arbor, MI). 3-(Isopropylamino)-1-[(7-methyl-4-indanyl)oxy]butan-2-ol (ICI 118,551) hydrochloride, (R)-isoproterenol, pertussis toxin, 3-isobutyl-1-methylxanthine (IBMX), and probenecid were obtained from Sigma-Aldrich (St. Louis, MO). The primary antibodies for β2-AR were obtained from Enzo Life Sciences, Inc. (Farmingdale, NY) and Abcam Inc. (Cambridge, MA). Rabbit antiphospho-Akt (Ser-473), phospho-ERK1/2, total Akt, and total ERK2 were from Cell Signaling Technology (Danvers, MA), and anti-β-actin was purchased from Abcam Inc. Rat anti-multidrug resistance protein (MRP) 4 and goat anti-MRP5 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Maintenance and Treatment of Cell Lines.
Human HepG2 hepatocarcinoma cells and human U87MG glioma cells (American Type Culture Collection, Manassas, VA) were maintained in Eagle's minimum essential medium supplemented with 1% l-glutamine, 1% sodium pyruvate, 1% penicillin/streptomycin, and 10% Hyclone FBS (Thermo Fisher Scientific, Waltham, MA). The human 1321N1 astrocytoma cells (European Collection of Cell Cultures, Porton Down, Wiltshire, UK and Sigma-Aldrich) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS and penicillin/streptomycin. All cell lines were cultured at 37°C in 5% CO2, and the medium was replaced every 2 to 3 days. The passage numbers of the cell lines used ranged from 15 to 30.
Unless otherwise indicated, cells at 70 to 80% confluence were depleted of serum for 3 h, followed by the addition of ICI 118551, AM251, AM630 or WIN 55,212-2 for 1 h before treatment with vehicle, Fen, MNF, and other fenoterol derivatives at the indicated concentrations.
[3H]Thymidine Incorporation Assay.
Cells were seeded in 12-well plates at approximately 50,000 cells/well and incubated at 37°C. After 24 h, the wells were rinsed with PBS and replaced with serum-free medium containing the appropriate concentration of the test compounds. After another 24-h incubation at 37°C, 1 μCi of [3H]thymidine was added to each well and incubated at 37°C for 16 h. [3H]thymidine incorporation into DNA was monitored after the cells were washed twice with PBS and then lysed in 600 μl of 0.1 N NaOH for 30 min with shaking. The lysate was then mixed with 3 ml of liquid scintillation cocktail (Beckman Coulter, Fullerton, CA), and radioactivity was measured by liquid scintillation counting using a Beckman Coulter LS6000IC scintillation counter. Data are shown as cpm incorporated compared with the control cells.
cAMP Accumulation.
HepG2 cells were seeded in 96-well plates and grown to confluence. Cells were rinsed in Krebs-HEPES buffer, pH 7.4, preincubated for 20 min with the buffer supplemented with 50 μM IBMX, a phosphodiesterase inhibitor, and then 1 μM isoproterenol, Fen, or MNF was added for an additional 10 min. In some experiments, the MRP inhibitor probenecid (2.5 mM) was included in the preincubation buffer. The levels of cAMP accumulated in cells were determined and normalized to the amount of protein per well as described previously (Toll et al., 2011).
RNA Interference.
HepG2 and 1321N1 cells were plated at a density of 3 × 105 cells/well in six-well cell culture plates and incubated for 48 h. Transfection of the cells was carried out in Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA) with 50 nM each of a combination of three siRNAs (Applied Biosystems, Foster City, CA and Santa Cruz Biotechnology, Inc.) targeted against human β2-AR or a nonsilencing siRNA control (Santa Cruz Biotechnology, Inc.). Forty-eight hours later, cell lysates were prepared and immunoblotted for β2-AR by Western blotting.
RNA Extraction, cDNA Synthesis, and Reverse Transcription-PCR Analysis.
Total RNA was isolated from HepG2, 1321N1, and U87MG cells by using the RNeasy Mini kit (QIAGEN, Valencia, CA). The RNA preparation included a DNase digestion step. RNA concentration and quality was measured by using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE). To obtain cDNA, 1 μg of total RNA was reverse-transcribed by using the Promega reverse transcription kit (Promega, Madison, WI). PCRs were performed to determine the expression of CB1R, CB2R, and β2-AR mRNAs by using glyceraldehyde-3-phosphate dehydrogenase as an internal control. The PCR primers and conditions are found in Supplemental Table 1.
Cell Cycle Analysis.
Cell cycle distributions were performed by flow cytometry on propidium iodide (PI)-stained nuclei prepared by the NIM technique (Kopp and Wersto, 1992). DNA histograms of at least 10,000 cells acquired on a BD FACScanto II (BD Biosciences, San Jose, CA) were deconvoluted by using the Multicycle program (Phoenix Flow Systems, San Diego, CA) for estimates of the percentage of cells in the G0/1, S, and G2+M phases of the cell cycle. Debris and doublets were removed from the analysis by software algorithms.
Apoptosis Assay.
The degree of apoptosis induced by drug treatment was assayed by flow cytometry using the Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit (Invitrogen) following the standard manufacturer's protocol. In brief, HepG2 cells (5 × 105) were grown on 100-mm dishes for 24 h followed by treatment with vehicle, Fen, or MNF, all in serum-free medium. Cells were subsequently harvested after 24-h incubation, washed in cold PBS, and resuspended in 100 μl of 1× annexin-binding buffer to maintain a density of ∼1 × 106 cells/ml. Five microliters of Alexa Fluor 488 Annexin V and 1 μl of 100 μg/ml propidium iodide were added to the cell suspensions. Cells were then incubated at room temperature for 15 min, and 400 μl of 1× annexin-binding buffer was added followed by gentle mixing. Stained cells were analyzed on a BD FACSCanto II flow cytometer.
Western Blotting.
Cells were lysed with radioimmunoprecipitation assay buffer containing EGTA and EDTA (Boston BioProducts, Ashland, MA). The lysis buffer was mixed with a protease inhibitor cocktail (Sigma-Aldrich). Protein concentrations were measured by using the bicinchoninic acid reagent (Thermo Fisher Scientific). Proteins (20 μg/well) were separated on 4 to 12% precast gels (Invitrogen) using SDS-polyacrylamide gel electrophoresis under reducing conditions and electrophoretically transferred onto polyvinylidene fluoride membrane (Invitrogen). Western blots were performed according to standard methods, and the antibodies were used at a dilution recommended by the manufacturer. The visualization of immunoreactive bands was performed by using the ECL Plus Western Blotting Detection System (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK), and their quantification was done by volume densitometry using ImageJ software (National Institutes of Health, Bethesda, MD) and normalization to β-actin.
Statistical Analysis.
Results were expressed as relative to the control value. Experiments were performed in at least two to three different culture preparations, and two to three dishes for each experimental condition were plated in each preparation. Results are expressed as means ± S.E. Statistical comparisons between groups were made by t test. Analyses were performed by using SigmaPlot software (Systat Software, Inc. San Jose, CA), Prism 4 (GraphPad Software, Inc., La Jolla, CA), and Excel (Microsoft, Redmond, WA), with p values ≤0.05 considered significant.
Results
Expression of β2-AR in the HepG2 and 1321N1 Cell Lines.
The mRNA and protein levels of the β2-AR were determined in HepG2 hepatocarcinoma cells and 1321N1 astrocytoma cells. Semiquantitative PCR analysis indicated higher expression of β2-AR mRNA in 1321N1 cells compared with HepG2 cells (Supplemental Fig. 1A). Accordingly, 1321N1 cells expressed more β2-AR protein than HepG2 cells, when three commercial antibodies were tested by using a Western blot technique (Supplemental Fig. 1B). As indicated in Fig. 1A, the knockdown of β2-AR expression by siRNA-based approach led to ∼70% reduction in the level of β2-AR protein, validating the specificity of these antibodies. U87MG cells were found previously to be devoid of β2-AR (Toll et al., 2011).
Fig. 1.

Responses of HepG2 cells to β-agonist stimulation. A, HepG2 and 1321N1 cells were transfected with the negative control siRNA (−) or β2-AR siRNA (+) for 48 h. Cell lysates were immunoblotted with a specific anti-β2-AR antibody (ab40834), using Hsp90 as a loading control. B, increase in cAMP accumulation in HepG2 cells was observed with forskolin (10 μM), but not with 1 μM of either isoproterenol (Iso), Fen, or MNF. Data shown are from a single experiment conducted in quadruplicate. Error bars indicate mean ± S.D. C, serum-starved HepG2 cells were incubated in the presence of Iso (1 μM) or Fen (1 μM) for 5, 10, and 30 min. Cell lysates were immunoblotted with antibodies against phosphorylated Akt (p-Akt; Ser473) and total Akt, as well as phosphorylated ERK1/2 (p-ERK1/2) and total ERK2. The experiments shown in B and C were repeated twice with comparable results. The positions of molecular mass markers (in kilodaltons) are shown to the left of the immunoblots.
Effect of β-AR Agonists on cAMP Accumulation and Phosphorylation of Akt and ERK1/2 in HepG2 Cells.
Isoproterenol, Fen, or MNF at 1.0 μM did not elicit an increase in cAMP production in HepG2 cells, whereas cell treatment with the adenylyl cyclase activator forskolin induced significant accumulation of cAMP (Fig. 1B). Because of the ability of cyclic nucleotides to move across the cell membrane via the drug efflux pump MRP4 (ABCC4) and MRP5 (ABCC5) (Wielinga et al., 2003; Cheng et al., 2010), we investigated whether the active export of cAMP accounted for the apparent lack of effect of isoproterenol and fenoterol compounds on intracellular cAMP accumulation. HepG2 cells were pretreated with the MRP inhibitor probenecid (Copsel et al., 2011), followed by agonist stimulation. The presence of probenecid did not produce an increase in the intracellular cAMP levels under conditions where phosphodiesterase-mediated cAMP hydrolysis was inhibited by IBMX and the cells stimulated either with β2-AR agonists or forskolin (data not shown). Moreover, Western blotting of total cell extracts revealed that the MRP4/5 protein expression levels were below the limit of detection (data not shown). These data indicate that the export of cAMP plays a little role, if any, in the apparent lack of effect of β2-AR agonists on cAMP accumulation in HepG2 cells. Rather, this behavior may indicate uncoupling between agonist-stimulated β2-AR and Gαs protein. Alternatively, because of the low number of β2-ARs in HepG2 cells, agonist-stimulated adenylyl cyclase activity may be below detectable levels.
Previous studies have demonstrated that β2-AR can signal to the mitogen-activated protein kinases ERK1 and ERK2 (Ahn et al., 1999; Fan et al., 2001) independently of a functional adenylyl cyclase coupling (Agarwal and Glasel, 1999). The effect of isoproterenol and Fen on Akt and ERK1/2 activation was assessed by imunoblotting using selective antibodies to phosphorylated peptides that correspond to the active forms of Akt and ERK1/2. We observed that treatment of HepG2 cells with these β-agonists induced a time-dependent increase in Akt and ERK activation (Fig. 1C), which was blocked by ICI 118551, a β2-AR inhibitor (data not shown). Stimulation with MNF had no effect on the activation of Akt and ERK (data not shown). These results indicate that different types of agonist-stimulated β2-AR signaling events can occur, probably through the coupling to different Gα proteins and/or other G protein-independent pathways (Shenoy et al., 2006).
The Effects of Isoproterenol and Fen Analogs on the Proliferation of HepG2 Cells.
The effect of isoproterenol, Fen, and selected fenoterol analogs on cell proliferation was determined in HepG2 cells. Both isoproterenol and Fen produced a significant increase in cell proliferation, as assessed by [3H]thymidine incorporation, with EC50 values of 0.40 ± 0.08 and 1.17 ± 0.37 μM, respectively (Table 1; Fig. 2A). Yuan et al. (2010) reported similar potency of isoproterenol toward HepG2 cell proliferation, where saturation was reached at 10 μM of the β-AR agonist. The EC50 value reported here for isoproterenol was 400-fold lower than that reported in U118 cells (Toll et al., 2011). The lower potency of isoproterenol in HepG2 cells raised the concern that this cell model may not contain spare β2-AR receptors that would increase the efficacy of isoproterenol as an agonist (Nicolas et al., 1991).
TABLE 1.
Structures, percentage change in thymidine incorporation, and IC50/EC50 of fenoterol and analogs that were used for this study
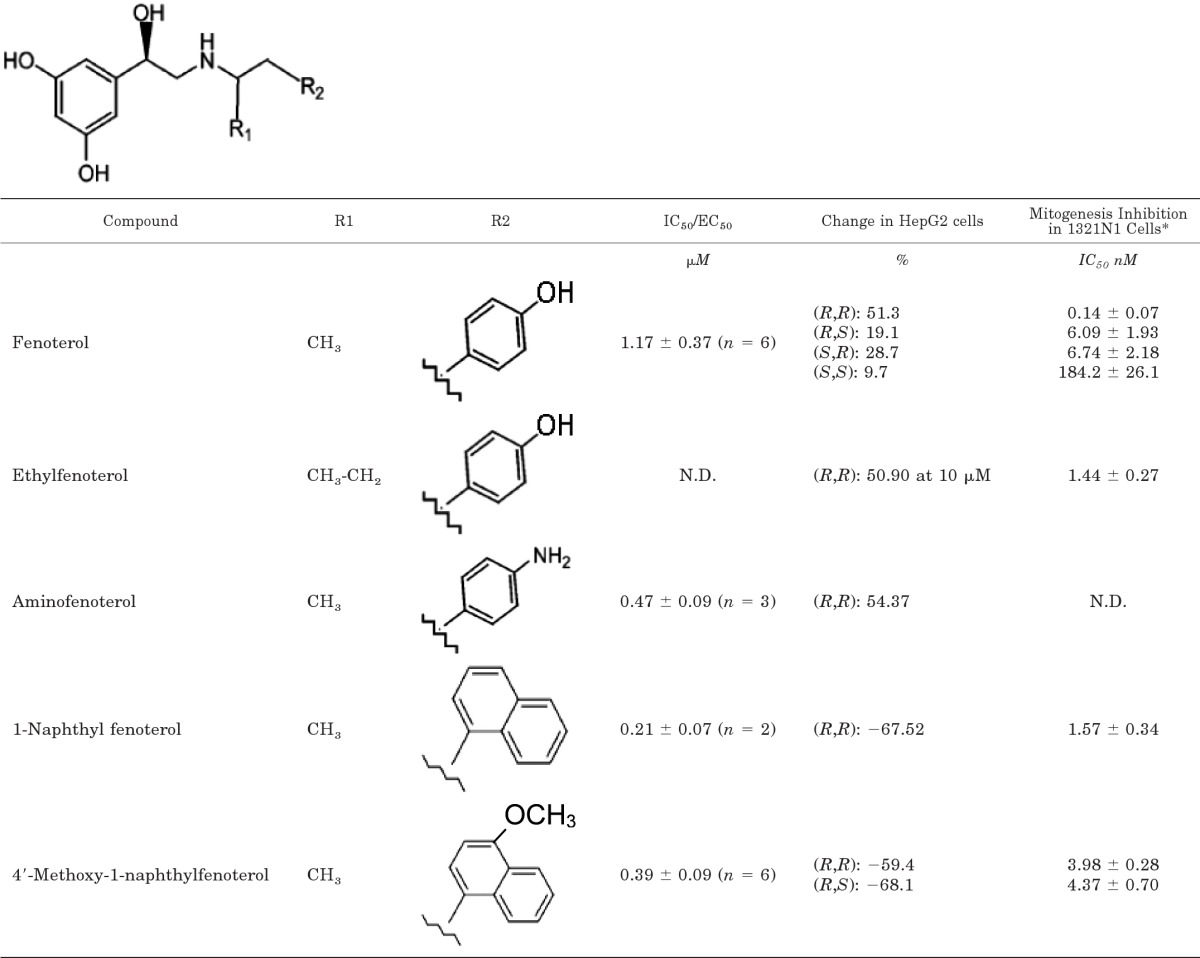
N.D., not determined.
* From Table 2 in Toll et al. (2011).
Fig. 2.

The effects of isoproterenol, Fen, and fenoterol derivatives on cell growth are cell type specific. A and B, serum-starved HepG2 cells were incubated with vehicle or the indicated concentrations of Iso, Fen, or (R,R′)-aminofenoterol (NH2-fen) (A) or MNF (B) for 24 h, and the levels of [3H]thymidine incorporation were measured; see Materials and Methods for experimental details. Representative concentration-response curves are shown. C, HepG2 cells in serum-depleted medium and 1321N1 cells in complete medium were treated with compounds at 1 μM for 24 h. D, HepG2 and 1321N1 cells were incubated without (SFM) or with serum (CM) in the presence of the indicated concentrations of Iso or Fen. Quantification of percentage change in [3H]thymidine incorporation versus control is expressed as means ± S.E. and represents results from two to six independent experiments, each performed in triplicate dishes. In most instances, error bars are smaller than the symbols.
Fen has two chiral centers and four possible steroisomeric forms: (R,R′), (R,S′), (S,R′), and (S,S′). The effect of the stereochemistry on the proliferative effect of Fen was determined by using a concentration of 1 μM of each isomer. The data indicate that all of the isomers induced an increase in [3H]thymidine incorporation, and stereochemistry had only a quantitative effect on this process, with Fen producing the greatest increase (51.3%) and (S,S′)-fenoterol producing the lowest (9.7%) (Table 1). This result was consistent with the previously reported inhibitory effect of Fen stereoisomers on mitogenesis in 1321N1 cells in which the inhibitory potency was (R,R′) > (R,S′) ≈ (S,R′) ≫ (S,S′) (Toll et al., 2011) (see Table 1). The effect of the change of the N-alkyl methyl group to an ethyl moiety {(R,R′)-ethylfenoterol} and the substitution of an 4′-amino group for the 4′-hydroxyl group {(R,R′)-aminofenoterol} were also investigated. Neither alteration changed the direction of the effect on [3H]thymidine incorporation, and (R,R′)-aminofenoterol seemed to be 3-fold more active than Fen with an EC50 value of 0.47 ± 0.09 μM (Table 1; Fig. 2A).
In a previous study, we reported that the incorporation of a naphthyl moiety into the Fen molecule reduced the potency of the resulting compound, but not the inhibitory effect on mitogenesis in 1321N1 cells (Toll et al., 2011). In this study, the opposite effect was observed as MNF and 1-naphthylfenoterol inhibited [3H]thymidine incorporation with IC50 values of 0.39 ± 0.09 and 0.21 ± 0.07 μM, respectively (Table 1; Fig. 2B). The change in the stereochemistry of the chiral center on the N-alkyl portion of the MNF molecule had no effect on the antiproliferative response because 1 μM concentrations of MNF and (R,S′)-4-methoxy-1-naphythylfenoterol produced equivalent decreases in [3H]thymidine incorporation of −59.4 and −68.1%, respectively (Table 1).
MNF is a full and potent β2-AR agonist in respect to the stimulation of cAMP expression in human embryonic kidney cells stably transfected with β2-AR and 1321N1 cells, with EC50 values of 3.9 and 68.9 nM, respectively (Jozwiak et al., 2010; Toll et al., 2011). Because HepG2 cells displayed substantial sensitivity to (R,R′)-aminofenoterol (EC50 = 0.47 ± 0.09 μM) and MNF (IC50 = 0.39 ± 0.09 μM) with regard to [3H]thymidine incorporation, the responsiveness of 1321N1 cells to the two compounds was determined and found to be markedly lower (Fig. 2C). The specificity of the observed β2-AR response to Fen and MNF in the HepG2 and 1321N1 cells was tested by using the U87MG cells, which have been shown to be devoid of β2-AR binding activity (Toll et al., 2011). In this cell line, MNF produced a potent inhibition of cellular proliferation, whereas Fen had no effect (Supplemental Fig. 2).
In the previous study of the effect of isoproterenol and Fen on mitogenesis in 1321N1 cells, the experiments were conducted by using complete medium. To determine whether the presence of serum or its absence significantly influenced the extent of mitogenesis in response to isoproterenol and Fen, the experiments were repeated using both experimental protocols. The results indicated that HepG2 cells exhibited a better sensitivity in serum-depleted medium, whereas the sensitivity was greater in 1321N1 cells maintained in complete medium (Fig. 2D). These data suggest that there are contrasting mitogenic responses to β2-AR agonists in HepG2 and 1321N1 cells.
β2-AR Antagonism Does Not Inhibit the Antiproliferative Action of MNF While Preventing the Growth-Promoting Effects of Fen in HepG2 Cells.
The divergent actions mediated by Fen and MNF are consistent with activation of distinct signaling pathways with opposite effects on cell proliferation. To test this hypothesis, HepG2 cells were pretreated with ICI 118,551, followed by incubation in the presence of Fen or MNF for 24 h. Although ICI 118,551 alone showed a modest, but significant, concentration-dependent increase in cell proliferation, up to ∼16% at 1 μM (Fig. 3A), its addition markedly blocked Fen-stimulated mitogenesis (Fig. 3, B and C). However, the antiproliferative effect of MNF was refractory to ICI 118,551 pretreatment (Fig. 3, B and D).
Fig. 3.

β2-AR antagonist does not inhibit the antiproliferative action of MNF in HepG2 cells. A and B, serum-depleted HepG2 cells were incubated with the indicated concentrations of the β-AR antagonist ICI-118,551 (ICI) for 1 h followed by the addition of vehicle (A), Fen (B, left), or MNF (B, right) for 24 h, and levels of [3H]thymidine incorporation were measured. *, p < 0.05. Representative concentration-response curves for Fen and MNF are shown in B. C and D, quantification of percentage change in [3H]thymidine incorporation versus control is expressed as means ± S.E. and represents results from three independent experiments, each performed in triplicate dishes. C, Fen. D, MNF.
We then tested the possibility that the action of Fen could be hampered by the coaddition of MNF. The results showed clearly a mitogenic response in HepG2 cells that was intermediate between Fen and MNF alone, and the pretreatment with ICI 118,551 partially restored the antiproliferative effects of MNF (Supplemental Fig. 2, top). However, characteristics of the cell proliferation profile elicited by Fen in 1321N1 cells and MNF in U87MG cells were maintained by the cotreatment with Fen and MNF (Supplemental Fig. 2, middle and bottom). As expected, pretreatment with ICI 118,551 blocked Fen signaling in 1321N1 cells while being inactive against the antiproliferative action of MNF in U87MG cells (Supplemental Fig. 2). These results indicate that the effects of Fen and MNF on cell proliferation are cell type-specific and may require the activation of distinct receptors.
MNF Induces Apoptosis in HepG2 Cells.
The proliferation of HepG2 cells was assessed by flow cytometry analysis using propidium iodide staining to examine the cell cycle. Fen produced no significant alterations of the cell cycle, but MNF caused a temporal decrease in the G2/M- and S-phase cell populations (G2/M, 13.8 ± 1.1% in control versus 10.2 ± 0.9% after 6 h, 14.6 ± 1.8% after 12 h, and 8.9 ± 1.6% after 24 h; S, 34.7 ± 0.3% in control versus 34.1 ± 0.9% after 6 h, 13.7 ± 1.2% after 12 h, and 24.6 ± 4.2% after 24 h) in HepG2 cells treated with 1 μM MNF (Fig. 4). The treatment with MNF also yielded a time-dependent increase in the number of sub-G1 events, reaching a maximum of 21.5 ± 0.7% by 12 h (Fig. 4, bottom right). No significant increase in sub-G1 events was observed when cells were treated with Fen (1 μM) for up to 24 h.
Fig. 4.

MNF increases the number of sub-G1 events in HepG2 cells. Serum-depleted HepG2 cells were harvested after 6-, 12-, and 24-h treatment with vehicle (top left), Fen (1 μM) (top right), or MNF (1 μM) (bottom left). Cells were fixed, stained and then analyzed for DNA content by using flow cytometry. Representative DNA content analysis in various phases of the cell cycle after 24-h treatment with vehicle, Fen, or MNF is shown. The number of sub-G1 events, which displays cells in late-stage apoptosis or already dead, in function of treatment duration was quantified and represents results from two independent experiments, each performed in duplicate dishes (bottom right). Data are expressed as means ± S.E. (n = 4).
Sub-G1 events occur when cells have proceeded to the late stage of apoptosis or are already dead. To directly measure apoptosis, flow cytometry analysis with Annexin V/PI staining was carried out in HepG2 cells. The percentage of apoptotic cells induced by a 24-h treatment with MNF (1 μM) was increased 5.7-fold compared with control (p < 0.01). However, Fen treatment reduced apoptosis compared with control untreated cells (Fig. 5).
Fig. 5.

MNF induces apoptosis in HepG2 cells. Serum-depleted HepG2 cells were treated with vehicle (top left), Fen (1 μM) (top right), or MNF (1 μM) (bottom left) for 24 h, stained with Annexin V and PI, and then analyzed by flow cytometry. Representative profiles are shown. The fraction of annexin V-positive HepG2 cells that were apoptotic was quantitated and represents results from two independent experiments, each performed in duplicate dishes (lower right). Data are expressed as means ± S.E. (n = 4).
Role of Cannabinoid Receptors in the Control of Cell Proliferation of MNF and Fen.
Because of the coexpression of β2-AR and CBRs and their impact on HepG2 cell proliferation (Wu et al., 2010; Yuan et al., 2010), we assessed whether the regulation of mitogenesis in response to Fen and MNF could occur through CBR signaling mechanisms.
The mRNA levels of CB1R and CB2R were determined by reverse transcription-PCR in HepG2, 1321N1, and U87MG cells (Fig. 6A). The results indicated that HepG2 and U87MG cells expressed CB1R and CB2R, whereas 1321N1 cells had no detectable levels of CBR mRNAs. Therefore, one should expect cell type-specific differences in responsiveness to CBR ligands. Indeed, potent regulatory effects of synthetic cannabinoid compounds were observed in cells treated with Fen and MNF compared with controls. Similar to MNF, treatment of HepG2 cells with the cannabinoid receptor agonist WIN 55,212-2 (1 μM) reduced cell proliferation and canceled out the growth-promoting action of Fen (Fig. 6B). AM251 and AM630 are synthetic inverse agonists for CB1R and CB2R, respectively (Pertwee, 2005). We observed that cell pretreatment with AM251 or AM630 had no impact on the mitogenic responses of Fen (Fig. 6C), indicating that basal-level activity of these two CBRs does not play a major role in Fen's proliferative action. However, preincubation with AM251 or AM630 completely inhibited the antiproliferative effects of MNF in HepG2 cells (Fig. 6C), which is consistent with the involvement of CBRs in MNF signaling. In support of this hypothesis, we found that 1321N1 cells, which were unresponsive to MNF, were refractory to CBR ligands, when added alone or combined with Fen (Supplemental Fig. 3, A and B). However, the antiproliferative effects of MNF were partially blocked by AM251 and AM630 in the β2-AR-deficient U87MG cells (Supplemental Fig. 3, C and D).
Fig. 6.

Role of CBR activation in the antiproliferative action of MNF in HepG2 cells. A, total RNA was extracted from HepG2, 1321N1, and U87MG cells, and then analyzed semiquantitatively by PCR. A nontemplate control (NTC) has been included (lane 1). GAPDH, glyceraldehyde-3-phosphate dehydrogenase. B and C, serum-depleted HepG2 cells were incubated with the CBR agonist WIN 55,212-2 (Win; 1 μM) (B) or antagonists AM251 (1 μM) or AM630 (0.5 μM) (C) for 1 h followed by the addition of vehicle, Fen (0.5 μM), or MNF (0.25 μM) for 24 h. D, serum-depleted HepG2 cells were pretreated without or with pertussis toxin (PTX; 50 ng/ml) for 16 h followed by the addition of vehicle, Fen (0.5 μM), MNF (0.5 μM), or WIN 55,212-2 (0.5 μM) for 24 h. In B to D, the levels of [3H]thymidine incorporation were measured. Quantification of percentage change in [3H]thymidine incorporation versus control is expressed as means ± S.D. and represents results from three (B and C) or two (D) independent experiments, each performed in triplicate dishes. *, p < 0.05.
Coupling of MNF to cell proliferation seemed to depend on Gαi/o proteins, because the MNF and WIN 55,212-2 antiproliferative responses were inhibited by an 18-h pretreatment with pertussis toxin (50 ng/ml) in HepG2 cells (Fig. 6D). Under these conditions, pertussis toxin had no effect on Fen-induced cell proliferation.
Discussion
β-AR agonists and antagonists influence cell growth and function (Evans et al., 2010), which can lead to inhibition or induction of malignant diseases. The effect is cell specific because both β2-AR antagonists and agonists have been shown to attenuate cell growth (Carie and Sebti, 2007; Zhang et al., 2010; Toll et al., 2011). In the case of β2-AR agonists, the effect seems to be through cAMP-dependent pathways and is subject to antagonism by ICI 118,551 (Carie and Sebti, 2007; Toll et al., 2011). However, β2-AR agonism has also been shown to elicit growth and survival of several different cancer cell types (Sastry et al., 2007; Yuan et al., 2010), such as the isoproterenol-induced increase in HepG2 cell proliferation (Yuan et al., 2010).
To explore the intercellular differences in the prosurvival and antisurvival pathways activated by β2-AR agonists, we examined the effect of Fen and its analogs on the proliferation of HepG2 cells, with the objective of comparing the data with the compounds' previously observed attenuation of 1321N1 cellular growth (Toll et al., 2011). In this study, Fen increased HepG2 cell proliferation, consistent with the previously reported effect of isoproterenol (Yuan et al., 2010), and the fact that ICI 118,551 blocked this response indicates the involvement of β2-ARs. However, neither Fen nor isoproterenol induced cAMP accumulation in HepG2 cells, although treatment with forskolin demonstrated that the cells express functional adenylyl cyclase. The lack of effect of β2-AR agonists on cAMP accumulation may be caused by the low level of β2-AR protein expression in these cells. It is also possible that the intracellular accumulation of cAMP may be reduced by MRP4/5-mediated export (Cheng et al., 2010). In this study, the presence of the MRP inhibitor probenecid had no effect on isoproterenol-mediated increase in cAMP accumulation in HepG2 cells, and the overall expression levels for MRP4/5 proteins in these cells were found to be below the detection limit. Another possibility is that although the β2-ARs are poorly coupled to the stimulatory Gα protein, they may exhibit functional interactions with other signaling intermediates that promote cell growth. Our results demonstrate that Fen activated the phosphatidylinositol 3-kinase/Akt and ERK pathways in HepG2 cells. In support of our findings, the nontyrosine kinase c-Src has been implicated in the switching of signaling of β2-AR from adenylyl cyclase coupling to the mitogen-activated protein kinase/ERK pathway (Ahn et al., 1999; Luttrell et al., 1999). It should also be noted that scaffold proteins dock c-Src to the membrane-bound β2-AR and lead to β2-AR-mediated cell proliferation through the activation of ERK1/2 (Tao and Malbon, 2008; Zhang et al., 2011).
Our previous studies have examined the effect of Fen stereochemistry and structure on β2-AR stimulation of cAMP accumulation and inhibition of mitogenesis in 1321N1 cells. The data demonstrated that changes in the molecule's two chiral centers produce only quantitative changes in β2-AR agonism (Jozwiak et al., 2010; Toll et al., 2011). A similar effect was observed in the HepG2 cells because all of the Fen stereoisomers produced an increase in [3H]thymidine incorporation (reported as percentage change) with (R,R′) ≫ (S,R′) ≈ (R,S′) ≫ (S,S′) (Table 1). The same result was produced by the Fen analogs (R,R′)-ethylfenoterol and (R,R′)-aminofenoterol (Table 1; Fig. 2A). The results indicate that when the Fen molecule contains a 4′-substituted phenyl ring the compound stimulates [3H]thymidine incorporation in HepG2 cells and the stereochemistry of the molecule influences this effect, but does not qualitatively change it. A full structure-activity relationship study has been initiated, and the results will be reported elsewhere.
Previous studies have also demonstrated that the substitution of a naphthyl moiety for the phenyl ring on the N-alkyl portion of Fen, to generate naphthylfenoterol (NF) analogs, does not affect the β2-AR agonist activity with respect to the stimulation of cAMP accumulation, inhibition of mitogenesis in 1321N1 cells, or cardiomyocyte contractility (Jozwiak et al., 2010; Toll et al., 2011). Thus, the incubation of HepG2 cells with (R,R′)-1-naphthylfenoterol and MNF was expected to produce a stimulation of [3H]thymidine incorporation. However, a qualitative difference was observed as MNF and (R,R′)-1-naphthylfenoterol inhibited [3H]thymidine incorporation (Table 1; Fig. 2B). In addition, unlike Fen, a change in the stereochemistry of the chiral center on the N-alkyl portion of the MNF molecule had no effect on the antiproliferative response (Table 1).
Because the Fen and NF analogs used in this study are potent and selective β2-AR agonists, a potential explanation for the effect produced by replacing a phenyl ring with a naphthyl ring is “ligand-directed signaling” or “biased agonism” (Seifert and Dove, 2009). It has been demonstrated that β2-AR binds ligands in multiple conformations and binding to different receptor conformations can lead to differences in signal transduction (Wisler et al., 2007; Seifert and Dove, 2009). In respect to the Fen and NF molecules, initial comparative molecular field analysis studies of the interaction of the Fen analogs with the β2-AR have indicated that the naphthyl substituent of the NF molecules can interact with the β2-AR through a series of π-π and π-hydrogen bond interactions unavailable to the phenyl moiety on the Fen molecule (Jozwiak et al., 2007, 2010). It is doubtful that the binding of NF analogs to the β2-AR is the primary mechanism responsible for the decrease in [3H]thymidine incorporation, because the MNF response was insensitive to ICI 118,551 (Fig. 3). Moreover, MNF treatment of U87MG cells, which lack β2-AR binding activity, reduced cell growth, whereas Fen had no effect (Supplemental Fig. 2). These data do not eliminate the possibility that NFs bind to and stabilize a conformation of the β2-ARs expressed endogenously in HepG2 cells that is distinct from the conformation stabilized by Fen. However, such conformation may not be critical for the initiation of cell proliferation.
The data suggest that other GPCRs may be involved in the antiproliferative effects of MNF. One potential target is the CBRs because previous studies have suggested that CBRs modulate β2-AR activity (Gardiner et al., 2005). Thus, although the effect of MNF on CBR function was never anticipated, the molecular structure of MNF does share some similarities with recently reported CBR ligands. In particular, binding and molecular modeling studies indicate that the presence of a naphthyl moiety increases binding to the CB2R through π-π and hydrophobic interactions (Osman et al., 2010). It is interesting to note that the CB2R model used in the study by Osman et al. (2010) was constructed based on the structure of the β2-AR. Therefore, it is likely that the extent of β2-AR and CBR expression within a given cell line may influence the overall growth-promoting properties and survival function of Fen versus MNF.
We now report pharmacological evidence to suggest that MNF mediates its antiproliferative effects through activation of the CBRs. First, WIN 55,212-2 mimicked this MNF response, and the combination of MNF plus WIN 55,212-2 showed a lack of additive effect. Moreover, selective inhibition of the CB1R and CB2R showed suppression of MNF signaling. The inability of WIN 55,212-2 to modulate cell proliferation in 1321N1 cells may be best explained by their substantially lower CBR expression levels compared with HepG2 and U87MG cell lines (Biswas et al., 2003; Aguado et al., 2007 Curran et al., 2005; Wu et al., 2010) (Fig. 6A). Moreover, our data demonstrate that the Fen-mediated increase in HepG2 cell proliferation is neutralized by WIN 55,212-2; the most plausible mechanism accounting for this observation is that stimulation of Gαi-linked CBRs in response to WIN 55,212-2 inhibits cell growth primarily by negatively targeting the phosphatidylinositol 3-kinase/Akt and/or ERK pathways (Wang et al., 1999). This observation is supported by the inhibition of the MNF and WIN 55,212-2 responses in HepG2 cells pretreated with pertussis toxin. Taken together, our findings indicate a complex cell type-specific involvement of CBRs in the antimitogenic and proapoptotic activities of the β2-AR agonist MNF through a mechanism that does not require β2-AR activation.
In the last several years, development of new CBR ligands has become an intense area in cancer research because of the role of the endocannabinoid system in the regulation of cell proliferation and apoptosis (Oesch and Gertsch, 2009; Guindon and Hohmann, 2011; Hermanson and Marnett, 2011). In considering the overall influence of CBRs on MNF signaling, it is clear that CBR-directed signaling has the potential to be much more complex than initially thought, because it depends not only on which CBRs are present (CB1R versus CB2R), but also on the multiplicity of active and intermediate states of each receptor. Further complicating the issue is our demonstration, using selective inverse agonists, that the inhibition of either CB1R or CB2R antagonizes MNF response, which probably results from the ability of these receptors to form functional heteromers (Callén et al., 2012). The coexpression of β2-AR with CBRs in many tissues and cancer cell lines and their propensity to heterodimerize (Milligan, 2009; Hudson et al., 2010) suggest an even more complex picture, whereby MNF and other potential dually acting β2-AR-CBR ligands may yield a new class of compounds capable of having an unique affinity and/or selectivity profile. The therapeutic potential for synthetic small bivalent ligands holds great promise as new lead compounds in a wide range of disparate diseases (for review, see Valant et al., 2012).
Supplementary Material
Acknowledgments
We thank Sutapa Kole for expert technical assistance.
This research was supported in part by the Intramural Research Program of the National Institutes of Health and the National Institutes of Health National Institute on Aging under Contract N01AG-3-1009.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
- Fen
- (R,R′)-fenoterol
- β2-AR
- β2-adrenergic receptor
- MNF
- (R,R′)-4-methoxy-1-naphthylfenoterol
- WIN 55,212-2
- (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-napthalenylmethanone
- AM251
- 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-(1-piperidyl)pyrazole-3-carboxamide
- AM630
- 1-[2-(morpholin-4-yl)ethyl]-2-methyl-3-(4-methoxybenzoyl)-6-iodoindole
- ICI 118,551
- 3-(isopropylamino)-1-[(7-methyl-4-indanyl)oxy]butan-2-ol
- CBR
- cannabinoid receptor
- ERK
- extracellular-regulated kinase
- GPCR
- G protein-coupled receptor
- NF
- naphthylfenoterol
- IBMX
- 3-isobutyl-1-methylxanthine
- MRP
- multidrug resistance protein
- PBS
- phosphate-buffered saline
- FBS
- fetal bovine serum
- PCR
- polymerase chain reaction
- siRNA
- short interfering RNA
- PI
- propidium iodide
- Iso
- isoproterenol.
Authorship Contributions
Participated in research design: Paul, Wersto, Toll, Bernier, and Wainer.
Conducted experiments: Paul, Ramamoorthy, Scheers, and Jimenez.
Contributed new reagents or analytic tools: Wersto.
Performed data analysis: Paul, Ramamoorthy, Wersto, Bernier, and Wainer.
Wrote or contributed to the writing of the manuscript: Paul, Wersto, Bernier and Wainer.
References
- Agarwal D, Glasel JA. (1999) Differential effects of opioid and adrenergic agonists on proliferation in a cultured cell line. Cell Prolif 32:215–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguado T, Carracedo A, Julien B, Velasco G, Milman G, Mechoulam R, Alvarez L, Guzmán M, Galve-Roperh I. (2007) Cannabinoids induce glioma stem-like cell differentiation and inhibit gliomagenesis. J Biol Chem 282:6854–6862 [DOI] [PubMed] [Google Scholar]
- Ahn S, Maudsley S, Luttrell LM, Lefkowitz RJ, Daaka Y. (1999) Src-mediated tyrosine phosphorylation of dynamin is required for β2-adrenergic receptor internalization and mitogen- activated protein kinase signaling. J Biol Chem 274:1185–1188 [DOI] [PubMed] [Google Scholar]
- Audet M, Bouvier M. (2008) Insights into signaling from the β2-adrenergic receptor structure. Nat Chem Biol 4:397–403 [DOI] [PubMed] [Google Scholar]
- Biswas KK, Sarker KP, Abeyama K, Kawahara K, Iino S, Otsubo Y, Saigo K, Izumi H, Hashiguchi T, Yamakuchi M, et al. (2003) Membrane cholesterol but not putative receptors mediates anandamide-induced hepatocyte apoptosis. Hepatology 38:1167–1177 [DOI] [PubMed] [Google Scholar]
- Callén L, Moreno E, Barroso-Chinea P, Moreno-Delgado D, Cortés A, Mallol J, Casadó V, Lanciego JL, Franco R, Lluis C, et al. (2012) Cannabinoid receptors CB1 and CB2 form functional heteromers in brain. J Biol Chem 287:20851–20865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carie AE, Sebti SM. (2007) A chemical biology approach identifies a β-2 adrenergic receptor agonist that causes human tumor regression by blocking the Raf-1/Mek-1/Erk1/2 pathway. Oncogene 26:3777–3788 [DOI] [PubMed] [Google Scholar]
- Cheng D, Ren J, Jackson EK. (2010) Multidrug resistance protein 4 mediates cAMP efflux from rat preglomerular vascular smooth muscle cells. Clin Exp Pharmacol Physiol 37:205–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copsel S, Garcia C, Diez F, Vermeulem M, Baldi A, Bianciotti LG, Russel FG, Shayo C, Davio C. (2011) Multidrug resistance protein 4 (MRP4/ABCC4) regulates cAMP cellular levels and controls human leukemia cell proliferation and differentiation. J Biol Chem 286:6979–6988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran NM, Griffin BD, O'Toole D, Brady KJ, Fitzgerald SN, Moynagh PN. (2005) The synthetic cannabinoid R(+)WIN 55,212-2 inhibits the interleukin-1 signaling pathway in human astrocytes in a cannabinoid receptor-independent manner. J Biol Chem 280:35797–35806 [DOI] [PubMed] [Google Scholar]
- Evans BA, Sato M, Sarwar M, Hutchinson DS, Summers RJ. (2010) Ligand-directed signalling at β-adrenoceptors. Br J Pharmacol 159:1022–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Shumay E, Malbon CC, Wang H. (2001) c-Src tyrosine kinase binds the β2-adrenergic receptor via phospho-Tyr-350, phosphorylates G-protein-linked receptor kinase 2, and mediates agonist-induced receptor desensitization. J Biol Chem 276:13240–13247 [DOI] [PubMed] [Google Scholar]
- Gardiner SM, March JE, Kemp PA, Bennett T. (2005) Involvement of CB1-receptors and β-adrenoceptors in the regional hemodynamic responses to lipopolysaccharide infusion in conscious rats. Am J Physiol Heart Circ Physiol 288:H2280–H2288 [DOI] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG. (2011) The endocannabinoid system and cancer: therapeutic implication. Br J Pharmacol 163:1447–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack KK, Tougas MR, Jones KT, El-Dahr SS, Radhakrishna H, McCarty NA. (2010) A novel bioassay for detecting GPCR heterodimerization: transactivation of β2 adrenergic receptor by bradykinin receptor. J Biomol Screen 15:251–260 [DOI] [PubMed] [Google Scholar]
- Hermanson DJ, Marnett LJ. (2011) Cannabinoids, endocannabinoids, and cancer. Cancer Metastasis Rev 30:599–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson BD, Hébert TE, Kelly ME. (2010) Physical and functional interaction between CB1 cannabinoid receptors and β2-adrenoceptors. Br J Pharmacol 160:627–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozwiak K, Khalid C, Tanga MJ, Berzetei-Gurske I, Jimenez L, Kozocas JA, Woo A, Zhu W, Xiao RP, Abernethy DR, et al. (2007) Comparative molecular field analysis of the binding of the stereoisomers of fenoterol and fenoterol derivatives to the β2 adrenergic receptor. J Med Chem 50:2903–2915 [DOI] [PubMed] [Google Scholar]
- Jozwiak K, Woo AY, Tanga MJ, Toll L, Jimenez L, Kozocas JA, Plazinska A, Xiao RP, Wainer IW. (2010) Comparative molecular field analysis of fenoterol derivatives: a platform towards highly selective and effective β2-adrenergic receptor agonists. Bioorg Med Chem 18:728–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahsai AW, Xiao K, Rajagopal S, Ahn S, Shukla AK, Sun J, Oas TG, Lefkowitz RJ. (2011) Multiple ligand-specific conformations of the β2-adrenergic receptor. Nat Chem Biol 7:692–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp WC, Wersto RP. (1992) Flow cytometry in the monitoring of therapeutic trials, in Manual of Clinical Laboratory Immunology, 4th ed (Rose NR, DeMacario EC, Fahey JL, Friedman H, Penn GM, eds) pp 933–941, ASM Press, Washington, DC [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, et al. (1999) β-Arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science 283:655–661 [DOI] [PubMed] [Google Scholar]
- Milligan G. (2009) G protein-coupled receptor hetero-dimerization: contribution to pharmacology and function. Br J Pharmacol 158:5–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas C, Lacasa D, Giudicelli Y, Demarne Y, Agli B, Lecourtier MJ, Lhuillery C. (1991) Dietary (n-6) polyunsaturated fatty acids affect β-adrenergic receptor binding and adenylate cyclase activity in pig adipocyte plasma membrane. J Nutr 121:1179–1186 [DOI] [PubMed] [Google Scholar]
- Oesch S, Gertsch J. (2009) Cannabinoid receptor ligands as potential anticancer agents–high hopes for new therapies? J Pharm Pharmacol 61:839–853 [DOI] [PubMed] [Google Scholar]
- Osman NA, Mahmoud AH, Allarà M, Niess R, Abouzid KA, Di Marzo V, Abadi AH. (2010) Synthesis, binding studies and molecular modeling of novel cannabinoid receptor ligands. Bioorg Med Chem 18:8463–8477 [DOI] [PubMed] [Google Scholar]
- Palazuelos J, Aguado T, Egia A, Mechoulam R, Guzmán M, Galve-Roperh I. (2006) Non-psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J 20:2405–2407 [DOI] [PubMed] [Google Scholar]
- Pertwee RG. (2005) Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci 76:1307–1324 [DOI] [PubMed] [Google Scholar]
- Pertwee RG. (2006) The pharmacology of cannabinoid receptors and their ligands: an overview. Int J Obes (Lond) 30 (Suppl 1):S13–S18 [DOI] [PubMed] [Google Scholar]
- Sastry KS, Karpova Y, Prokopovich S, Smith AJ, Essau B, Gersappe A, Carson JP, Weber MJ, Register TC, Chen YQ, et al. (2007) Epinephrine protects cancer cells from apoptosis via activation of cAMP-dependent protein kinase and BAD phosphorylation. J Biol Chem 282:14094–14100 [DOI] [PubMed] [Google Scholar]
- Seifert R, Dove S. (2009) Functional selectivity of GPCR ligand stereoisomers: new pharmacological opportunities. Mol Pharmacol 75:13–18 [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. (2006) β-Arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J Biol Chem 281:1261–1273 [DOI] [PubMed] [Google Scholar]
- Tao J, Malbon CC. (2008) G-protein-coupled receptor-associated A-kinase anchoring proteins AKAP5 and AKAP12: differential signaling to MAPK and GPCR recycling. J Mol Signal 3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L, Jimenez L, Waleh N, Jozwiak K, Woo AY, Xiao RP, Bernier M, Wainer IW. (2011) β2-Adrenergic receptor agonists inhibit the proliferation of 1321N1 astrocytoma cells. J Pharmacol Exp Ther 336:524–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uberti MA, Hague C, Oller H, Minneman KP, Hall RA. (2005) Heterodimerization with β2-adrenergic receptors promotes surface expression and functional activity of α1D-adrenergic receptors. J Pharmacol Exp Ther 313:16–23 [DOI] [PubMed] [Google Scholar]
- Valant C, Robert Lane J, Sexton PM, Christopoulos A. (2012) The best of both worlds? Bitopic orthosteric/allosteric ligands of G protein-coupled receptors. Annu Rev Pharmacol Toxicol 52:153–178 [DOI] [PubMed] [Google Scholar]
- Wang XQ, Lindberg FP, Frazier WA. (1999) Integrin-associated protein stimulates 21-dependent chemotaxis via Gi-mediated inhibition of adenylate cyclase and extracellular-regulated kinases. J Cell Biol 147:389–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielinga PR, van der Heijden I, Reid G, Beijnen JH, Wijnholds J, Borst P. (2003) Characterization of the MRP4- and MRP5-mediated transport of cyclic nucleotides from intact cells. J Biol Chem 278:17664–17671 [DOI] [PubMed] [Google Scholar]
- Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, Shenoy SK, Lefkowitz RJ. (2007) A unique mechanism of β-blocker action: carvedilol stimulates β-arrestin signaling. Proc Natl Acad Sci U S A 104:16657–16662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo AY, Wang TB, Zeng X, Zhu W, Abernethy DR, Wainer IW, Xiao RP. (2009) Stereochemistry of an agonist determines coupling preference of β2-adrenoceptor to different G proteins in cardiomyocytes. Mol Pharmacol 75:158–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrzal PK, Devost D, Pétrin D, Goupil E, Iorio-Morin C, Laporte SA, Zingg HH, Hébert TE. (2012) Allosteric interactions between the oxytocin receptor and the β2-adrenergic receptor in the modulation of ERK1/2 activation are mediated by heterodimerization. Cell Signal 24:342–350 [DOI] [PubMed] [Google Scholar]
- Wu WJ, Yang Q, Cao QF, Zhang YW, Xia YJ, Hu XW, Tang WX. (2010) [Membrane cholesterol mediates the endocannabinoids-anandamide affection on HepG2 cells.] Zhonghua Gan Zang Bing Za Zhi 18:204–208 [DOI] [PubMed] [Google Scholar]
- Yang Q, Liu HY, Zhang YW, Wu WJ, Tang WX. (2010) Anandamide induces cell death through lipid rafts in hepatic stellate cells. J Gastroenterol Hepatol 25:991–1001 [DOI] [PubMed] [Google Scholar]
- Yuan A, Li Z, Li X, Yi S, Wang S, Cai Y, Cao H. (2010) The mitogenic effectors of isoproterenol in human hepatocellular carcinoma cells. Oncol Rep 23:151–157 [PubMed] [Google Scholar]
- Zhang D, Ma QY, Hu HT, Zhang M. (2010) β2-Adrenergic antagonists suppress pancreatic cancer cell invasion by inhibiting CREB, NFκB and AP-1. Cancer Biol Ther 10:19–29 [DOI] [PubMed] [Google Scholar]
- Zhang P, He X, Tan J, Zhou X, Zou L. (2011) β-Arrestin2 mediates β-2 adrenergic receptor signaling inducing prostate cancer cell progression. Oncol Rep 26:1471–1477 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.