

Abstract
Amyloid β (Aβ) fibrils are present as a major component in senile plaques, the hallmark of Alzheimer’s disease (AD). Diffuse plaques (non-fibrous, loosely packed Aβ aggregates) containing amorphous Aβ aggregates are also formed in brain. This work examines the influence of Cu2+ complexation by Aβ on the aggregation process in the context of charge and structural variations. Changes in the surface charges of Aβ molecules due to Cu2+ binding, measured with a zeta potential measurement device, were correlated with the aggregate morphologies examined by atomic force microscopy. As a result of the charge variation, the “colloid-like” stability of the aggregation intermediates, which is essential to the fibrillation process, is affected. Consequently Cu2+ enhances the amorphous aggregate formation. By monitoring variations in the secondary structures with circular dichroism spectroscopy, a direct transformation from the unstructured conformation to the β-sheet structure was observed for all types of aggregates observed (oligomers, fibrils, and/or amorphous aggregates). Compared to the Aβ aggregation pathway in the absence of Cu2+ and taking other factors affecting Aβ aggregation (i.e., pH and temperature) into account, our investigation indicates that formations of amorphous and fibrous aggregates diverge from the same β-sheet-containing partially folded intermediate. This study suggests that the hydrophilic domain of Aβ also plays a role in the Aβ aggregation process. A kinetic model was proposed to account for the effects of the Cu2+ binding on these two aggregation pathways in terms of charge and structural variations.
1. INTRODUCTION
Amyloid β (Aβ) peptide deposits are associated with the neuropathology of Alzheimer’s disease (AD).1–5 While Aβ fibrils are a main component in senile plaques, there exist the diffuse plaques in AD-afflicted brain. Unlike senile plaques, diffuse plaques are composed of amorphous Aβ aggregates.6 Diffuse plaques have been postulated to be benign and formed before the formation of Aβ aggregates of higher orders (i.e., oligomers, protofibrils, and fibrils containing well-ordered β-pleated sheets).7 These higher-ordered Aβ aggregates, particularly oligomers, are known to be more toxic than the amorphous aggregates.8–10 Although extensive studies have been carried out in vitro on the formation of the “main-pathway” aggregates (from monomer to oligomers, and subsequently to protofibrils and fibrils), much less efforts have been devoted to the studies of amorphous aggregate formation, which is regarded as an “off-pathway” product. Relatively scant attention has been paid to the influence of the hydrophilic domain in the Aβ peptides, which varies between 39 and 43 amino acid residues but all have the same hydrophilic domain in the first 16 residues, on the overall aggregation behavior. Therefore, it would be of great interest to know what factors dictate the formation of either type of aggregate, and the mechanisms and driving force leading to the different aggregates. Such a study may shed light on the development of effective therapeutic modality.
Recently, there has been a growing interest about the role of metal ions in modulating the Aβ aggregation11–18 and in generating reactive oxygen species.19–23 Consequently the metal coordination chemistry of Aβ peptides has been extensively studied.24–30 Such an interest stems from the fact that abnormally high concentrations of metal ions (Cu2+, Fe3+ and Zn2+) exist within the core and periphery of senile plaques31, 32 and these metals are found to be complexed by Aβ in postmortem AD brain.33 However, regarding the role of Cu2+ in the Aβ aggregation, conflicting results have been reported. While some studies have shown that copper ions can expedite Aβ aggregation,11–13 others suggest that they could inhibit Aβ aggregate formation.14, 34 Recent studies have shown that the morphology of metal-induced Aβ aggregates is largely amorphous.14–18 Most of the ionizable residues in the Aβ peptide are not in the β-strand domains that are commonly believed to be responsible for the fibrous core formation. Rather, they are located either in the hydrophilic domain or in the hairpin loop.35 In addition, the copper binding sites are also in the hydrophilic domain. The hydrophilic domain is more accessible to aqueous solution and the overall surface charge of Aβ consequently can be significantly affected by solution pH and metal binding. The fact that amorphous Aβ aggregates are formed at pH 5.5, a value close to the isoelectric point of Aβ (pI),14, 36 suggests the important role of charge variation in the Aβ aggregation process. Factors (e.g., pH, Aβ monomer concentration, temperature, and ionic strength of the solution) that are known to affect Aβ aggregation, however, have not been considered in the context of the charge states of the peptide, the kinetics of β-sheet formation and stacking, and the colloidal stability of monomers and soluble oligomers. Aβ point mutations associated with early onset AD wherein negatively charged Glu-22 and Asp-23 are replaced by neutral (glutamine or asparagine), hydrophobic (glycine), or positively charged (lysine) residues were found to aggregate faster, possibly suggesting a link of the surface charges to the pathological effect of Aβ peptides.37 Alteration of the net charge on the Aβ peptide37 may consequently accelerate the Aβ aggregation and trigger the onset of the disease. Similarly, the structural change in the hydrophilic domain of Aβ (amino acid residues 1–16) caused by Cu2+ binding could impose/alleviate steric hindrance to β-sheet stacking of the hydrophobic domains, a step believed to be essential for Aβ fibrillation.38
What are the factors determining the morphologies of Aβ aggregates under different experimental conditions? We hypothesized that charge and structural variations upon copper binding and pH change affect both the colloidal stability of the aggregation intermediates and the kinetics of the overall aggregation process. In this work, we measured the effects of Cu2+ binding on the charge variation of Aβ(1–42), which is the more aggregation-prone variant among the various Aβ peptides, and correlated the charge variation to the morphologies of the aggregates. We also studied the secondary structure transformation kinetics in the aggregation process. Based on the experimental results and considering the thermodynamic stabilities of the various intermediates and products, we proposed a two-pathway aggregation mechanism that takes into account of the charge effect, the role of the Aβ(1–42) hydrophilic domain, and the kinetics of β-sheet formation and stacking.
2. MATERIALS AND METHODS
2.1 Sample Preparation
Aβ(1–42), in the lyophilized form, was either purchased from American Peptide Company, Inc. (purity 97%, containing trifluoroacetate as the counter ion) or generously donated by Dr. C. Glabe (University of California-Irvine). All other chemicals were obtained from Sigma-Aldrich, unless otherwise stated. To ensure that no substantial aggregation occurs and to rid the solution of any aggregates, Aβ(1–42) samples were routinely prepared using a protocol similar to those developed by Teplow and coworkers.39 Briefly, the Aβ(1–42) solution was freshly prepared with 20 mM NaOH to a final concentration of 0.5 mM. The solution was then sonicated for 1 min and kept at room temperature for 5 min to completely dissolve the sample. This was followed by centrifuging at 13,000 rpm for 20 min to remove any aggregates that might have formed in the solution. The supernatant was used as the stock solution. Prior to each experiment, aliquots of the 0.5 mM stock solution were diluted with 10 mM phosphate buffer (pH 7.4 or 6.6) to 25 μM. Since it is generally believed that Cu2+ binds to Aβ(1–42) in a 1:1 stoichiometry,40–42 the Cu2+ concentration was also kept at 25 μM. Throughout the work, 1 mM CuCl2 dissolved in 1 mM H2SO4 was used as the Cu2+ stock solution. All the aqueous solutions were purified using deionized water (Millipore, 18 MΩ cm).
2.2 Atomic Force Microscopy
AFM images were obtained on a PicoScan SPM microscope (Agilent Technology Inc., Phoenix, AZ) equipped with a magnetic alternating current mode. We have used it to measure aggregates and intermediates formed with α-synuclein and SMA proteins.43, 44 For imaging, Ni2+-treated mica slides were used as the substrate to electrostatically attract negatively charged aggregates to the surface. Treatment of the mica surface with Ni2+ involves freshly cleaving mica slides and immersing them in 10 mM NiCl2 for 15 min, followed by thoroughly rinsing with deionized water and drying with N2. Aliquots of Aβ(1–42) or Aβ(1–42)/Cu2+ solutions were taken out at a predetermined incubation time, dropped onto the Ni2+-treated mica, and kept in contact with the surface in a humidified chamber for 10 min. The slides were then thoroughly rinsed with water to remove salt, and then dried with N2. Substrates immersed in Aβ(1–42)-free solution were also imaged as controls.
2.3 Circular Dichroism Spectroscopy
CD spectra were collected on a J-810 spectropolarimeter (Jasco Inc., Japan) at room temperature in a cuvette with a 0.1-cm path. The spectra were recorded at a 0.5-nm interval from 260 to 190 nm. Each spectrum is the average of eight scans. The kinetics of β-sheet formation was studied by plotting the ellipticity values at 197 nm (characteristic of the unstructured conformation) and at 217 nm (characteristic of the β-sheet structure) against the incubation time. The CD spectroscopy measures the soluble or dispersed species. During incubation, large aggregates could sink to the bottom of the cuvette out of the detection window, resulting in a loss of the CD signal. For consistency, before each measurement, the cuvette was thoroughly shaken and sonicated for 30 s for better dispersion.
2.4 Zeta Potential Measurement
Aβ(1–42) or Aβ(1–42)/Cu2+ solutions were loaded in zeta potential cells and the potential values were measured with a Zetasizer Nano ZS instrument (Malvern Instruments Ltd, UK). During aggregation the potential was found to be stable within ca. 6 h. The potentials reported are the average potentials measured in the first 6 h.
2.5 Thioflavin T (ThT) Assay
The Aβ(1–42) aggregation process was also studied using the ThT assay. At different incubation time points, 20 μL of 25 μM Aβ(1–42) incubated with and without Cu2+ (25 μM) in 10 mM phosphate buffer were withdrawn and added to 180 μL of a 10 mM phosphate buffer (pH 7.4) solution containing 20 μM ThT. Fluorescence was measured using a Fluorolog 3 spectrofluorometer (Horiba Jobin Yvon, Edison, NJ) with slit widths of 2 nm and excitation and emission wavelengths at 450 and 480 nm, respectively. Each data point is the average of three replicate measurements.
3. RESULTS
3.1 Cu2+-Binding and Solution pH both Alter the Charge State of Aβ
The tendency for a protein or peptide to aggregate is dependent on its colloidal stability in solution.45 When the adhesive force between two or more peptide molecules is less than the cumulative repulsion between them, the molecules are considered to be “stable” in solution.45 In terms of electrostatic interactions and solvation, peptides or proteins can be regarded as colloidal particles whose stability is highly dependent on their mass-to-charge ratio and ionic strength of the solution. As the sizes of the Aβ(1–42) monomer and especially the β-sheet-containing oligomers are comparable to small colloidal particles,46 experimental conditions affecting colloidal particle stability can become important to the Aβ(1–42) aggregations. Since pI of Aβ(1–42) is ca. 5.537, its charges are subject to pH variation in solution. Complexation with metal ions (e.g. Cu2+) also changes the overall charge state of the peptide. These factors will affect the colloidal stability and aggregation behavior. To our surprise, there has been no report on how the pH variation and Cu2+ binding affect the overall peptide charge, the aggregation kinetics, and the aggregate morphologies of Aβ(1–42).
Zeta potential indicates the degree of repulsion between adjacent, similarly charged particles in a dispersed solution. For molecules and particles that are small enough, a high zeta potential will confer stability, i.e., the solution or dispersion will resist agglomeration. We determined zeta potentials of Aβ(1–42) and Aβ(1–42) complexed with Cu2+ at different pH. Stable zeta potentials are shown in Table 1. The zeta potentials of Aβ(1–42) at pH 7.4 and 6.6 indicate that Aβ(1–42) is negatively charged. As pH approaches 5.5, the zeta potential becomes less negative. Stability of the Aβ(1–42) monomer and its oligomers at pH 6.6 decreases with respect to that of their counterparts at pH 7.4. As a result, the agglomeration process is accelerated and amorphous aggregates are predicted to form before unstructured Aβ(1–42) monomers (analogous to “random coils”) or β-sheets in the partially folded Aβ(1–42) oligomers are orderly assembled or stacked. Similarly, Cu2+ complexation by Aβ(1–42) also decreases the zeta potential (Table 1), and consequently the colloidal stability decreases and the amorphous aggregate formation is predicted to be more predominant.
Table 1.
Zeta potential values of Aβ(1–42) and Aβ(1–42)-Cu2+ complex at different pH
| Solution | pH | T (°C) | Zeta potential (mV) |
|---|---|---|---|
| Aβ(1–42) | 7.4 | 25 | −35.6 ±0.4 |
| 6.6 | 25 | −25.0 ±0.5 | |
| Aβ(1–42)–Cu2+ | 7.4 | 25 | −26.5±0.3 |
| 6.6 | 25 | −22.3 ±0.6 |
3.2 Decrease in Charges via Copper Complexation Inhibits Aβ(1–42) Fibrillation But Accelerates Amorphous Aggregate Formation
Although the acceleration of the Aβ aggregation by metal ions has been reported by several studies,11, 12 most studies did not differentiate the morphologies of the aggregates formed with and without metal ions and correlate morphologies with secondary structures. A few Aβ(1–42) oligomers with a height of 1–2 nm generally formed at the beginning of the incubation in the presence (Figure 1A) and absence of Cu2+ (Figure 1F). After 12-h incubation at room temperature in the presence of Cu2+, oligomers appear together with a large number of amorphous aggregates (Figure 1B). This is in sharp contrast to a Cu2+-free solution in which globular oligomers became more abundant (Figure 1G). The heights of these amorphous aggregates are 20–100 nm and their diameters range from 10 to 200 nm. As time elapsed (~24 h), most of the oligomers in the absence of Cu2+ appear to be converted into protofibrils with heights of 2–8 nm and lengths up to 400 nm (Figure 1H). A closer examination of the morphology of these protofibrils reveals that there exist the typical 4–5 nm high “aggregation units” (the smallest particles containing ordered β-sheet structures) observable for amyloidogenic proteins.47–49 Surprisingly, a similar incubation of the Cu2+-containing solution produced almost exclusively amorphous aggregates (Figure 1C). The amorphous aggregates persisted even when the incubation was prolonged to 96 h (Figure 1D). The amorphous aggregates remain essentially the same after further incubation (Figure 1E). Although there is some variability in determining the sizes of the amorphous aggregates due to interaction of AFM tips with the sticky soft aggregates, amorphous aggregates were routinely observed. In contrast, incubation of the Cu2+-free Aβ(1–42) solution over an extended period of time (e.g., 96 h) yielded fibrils that are as long as a few microns (Figure 1J). Cross-sectional contours of typical fibrils (Figure 1J) revealed that the aggregate heights are 4–5 nm with the “aggregation units” on top of the fibrils (indicated by arrows).
Figure 1.

AFM images of Aβ(1–42) solutions incubated at pH 7.4 and 25 °C in the presence of Cu2+ for (A) 0, (B) 12, (C) 48, (D) 96, and (E) 168 h and in the absence of Cu2+ for (F) 0, (G) 12, (H) 54 and (I) 96 h. Image J shows a cross-sectional contour of typical fibrils in (I). The scale bars in all images correspond to 400 nm. Aβ(1–42) concentrations in both the Cu2+-containing (25 μM) and Cu2+-free solutions were the same (25 μM).
3.3 Lowering Solution pH Close to the Aβ(1–42) Isoelectric Point Decreases Aβ(1–42) Charges and Favors Amorphous Aggregation
To probe whether the amorphous aggregate formation is solely attributable to the Cu2+ complexation, the Aβ(1–42) aggregation process in the presence of Cu2+ was compared to that in a Cu2+-free solution at pH 6.6. Such a pH value is closer to the pI of Aβ (5.537) than the physiological pH used in the aforementioned experiment. Initially, only oligomers were formed in both solutions with sizes and morphologies (Figure 2A and Figure 2D) analogous to those observed at pH 7.4. (cf. Figure 1A and Figure 1F). In the Cu2+-containing solution, 12-h incubation led to the formation of large, conglomerated amorphous aggregates (Figure 2B). Extending the incubation time up to 168 h produced more amorphous aggregates but no fibrils (Figure 2C). As for the Cu2+-free Aβ(1–42) solution, after a 12-h incubation, protofibrils were found to be intermingled with amorphous aggregates (Figure 2E). A prolonged incubation produced more amorphous aggregates (Figure 2F), but unlike the case of pH 7.4 (cf. Figure 1H and Figure 1I), no fibrils were found. Our observation of amorphous aggregates at pH 6.6 without Cu2+ is consistent with the findings by Wood et al. who produced amorphous aggregates by incubating Aβ(1–40) at pH 5.8.50 Thus it is apparent that lowering the solution pH and adding Cu2+ into the Aβ(1–42) solution both facilitate the amorphous aggregate formation. We excluded the Aβ(1–42) concentration used from being responsible for the low fibril abundance, as 25 μM is higher than the threshold concentration required for fibrillation (~10 μM 51).
Figure 2.

AFM images of Aβ(1–42) solutions incubated in the presence of Cu2+ for 0 (A), 12 (B) and 168 h (C) and in the absence of Cu2+ for 0 (D), 12 (E) and 96 h (F). The incubation temperature was 25 °C and pH was 6.6. The scale bars in all images correspond to 400 nm. Aβ(1–42) concentrations in both the Cu2+ (25 μM)-containing and Cu2+-free solutions were the same (25 μM)
3.4 Temperature Increase Favors Fibrous Aggregate Formations
We also studied Aβ(1–42) aggregation in both Cu2+-containing and -free solutions at 37°C (the physiologically relevant temperature), and compared the results to those observed at 25°C. In line with the general trend observed from Cu2+-containing Aβ(1–42) solutions at 25°C (Figures 1A–E), no fibrils emerged (Figures 3A–D). The amorphous aggregates coexisted with oligomeric species throughout this incubation period. However, the density and size of the amorphous aggregates are both smaller than those formed at 25°C. This observation suggests that a higher temperature may have slowed down the formation of larger amorphous aggregates. It is possible that the higher temperature enhances the thermal motion and/or solubility of oligomeric species, preventing larger aggregates from being produced. As expected, time-lapse AFM images collected from a Cu2+-free Aβ(1–42) solution showed globular oligomers at the beginning of the incubation (Figure 3E) and protofibrils and fibrils at later times (Figures 3F and 3G). Although the morphologies of these aggregates are similar to those shown in Figures 1F–I, the time required to form fibrils is significantly shorter. Notice that the density of fibrils in Figure 3G is greater than that in Figure 1I, even though the former was produced within a shorter time frame.
Figure 3.

AFM images of Aβ(1–42) aggregates formed via incubation at pH 7.4 and 37 °C in the presence of Cu2+ for (A) 0, (B) 12, (C) 24, and (D) 72 h, and in the absence of Cu2+ for (E) 0, (F), 12, and (G) 24 h. Panel H shows the cross sectional contour of a few fibrils intersecting the green line in (G). The scale bars in all images correspond to 400 nm. The Aβ(1–42) and Cu2+ concentrations are the same as those for Figures 1 and 2.
The fibril formation has been known to be a highly cooperative process.52 In the case of Aβ(1–42), recent studies have suggested that partially folded dimers and/or low-molecular-weight oligomers are energetically favored, and the stacking of the β-sheets of these intermediate aggregates must be in-register or self-complementary.35, 53 Based on our observation, it is apparent that the in-register stacking, generally accepted as the rate-determining step throughout the fibrillation process,38, 54 is more dependent on temperature than the process (pathway) responsible for the amorphous aggregate formation.
To assess the cumulative effects of lower pH and higher temperature on the Aβ(1–42) aggregation, we also sampled aggregates from Cu2+-containing (Figures 4A–E) and -free (Figures 4F–J) Aβ(1–42) solutions at pH 6.6 and 37°C. Again, the presence of Cu2+ resulted in the exclusive formation of amorphous aggregates, some of which were agglomerated into larger pieces (Figure 4D). Similar to the experiment conducted at a higher pH (cf. Figure 3), protofibrils and fibrils are predominant in the absence of Cu2+ (Figures 4F–J). A comparison between Figures 4F–J and Figures 3E–G clearly reveals that, at a higher temperature and in the absence of Cu2+, fibril formation is favored. A point noteworthy is that small amorphous aggregates were also present (e.g., those denoted by the arrows in Figures 4G–J). The coexistence of amorphous and fibrous aggregates suggests that there exist two competing aggregation pathways. A significant finding from our experiments is that, as long as Cu2+ is present, amorphous aggregates will be always the major, if not sole, form.
Figure 4.

AFM images of Aβ(1–42) aggregates formed in solutions incubated at pH 6.6 and 37 °C for different times. The top images were obtained from a solution containing Cu2+ at (A) 0, (B) 4, (C) 12, (D) 24, and (E) 168 h. The bottom images were acquired from a Cu2+-free solution at (F) 0, (G) 12, (H) 24, (I) 48 and (J) 96 h. The insets depict the cross sectional contour denoted by the green line. The scale bars in all images represent 400 nm. Other experimental conditions are the same as in Figures 1–3.
3.5 Amorphous Aβ(1–42) Aggregates also Contain the β-sheet Structure
Although AFM is a powerful technique for determining the aggregate morphology, it is difficult to use the data to extract structural information at the molecular level. Moreover, the AFM resolution is limited to the observation of large intermediates and/or aggregates typically formed at later stages of the aggregation. To gain insight into the initial stage of the aggregation process, we resorted to circular dichroism (CD) spectroscopy to study the transition of the natively unstructured Aβ(1–42) monomers to species possessing secondary structure(s). CD has been used in the past 55–57 to glean information about the secondary structures of Aβ(1–42) and its aggregates. For example, Kirkitadze et al. showed that β-amyloid undergoes a gradual transition from the natively unstructured state to oligomers in the α-helical conformation and ultimately to β-sheet-rich aggregates.55 Such a transition has also been demonstrated to be highly dependent on the experimental condition. In the presence of Cu2+, how the secondary structure of the Cu2+-containing Aβ(1–42) evolves and whether there exists any difference in the secondary structure between the Cu2+-containing and Cu2+-free Aβ(1–42) solutions have not been investigated in detail.
Figure 5A is an overlay of CD spectra from Aβ(1–42) solutions (pH = 7.4, 25 °C) that had been incubated for different lengths of time. The appearance of the peak at 197 nm (characteristic of the unstructured conformation) and the absence of a peak at 217 nm (characteristic of the β-sheet structure) confirm that the Aβ(1–42) monomers are essentially unstructured. After 7 h, the peak at 197 nm became difficult to discern. The peak at 217 nm grew continuously at the expense of the peak at 197 nm, suggesting that there was a transformation from the unstructured Aβ(1–42) monomer to a structure rich in β-sheets. Such a transformation is also reflected by the presence of an isodichroic point at ~ 206 nm. Recall that the AFM image of the solution incubated for 7 h shows globular Aβ(1–42) oligomers as the predominate aggregate (cf. Fig. 1G). Thus, these Aβ(1–42) oligomers must possess the β-sheet structure and the solution sampled after 7 h of incubation comprised both Aβ(1–42) monomers and oligomers or even some higher-ordered structures. The abundance of protofibrils and fibrils at longer incubation times (cf. Figures 1H and 1I, respectively) are consistent with the quantity of the β-sheet-containing structure(s) (Fig. 5A) for the similar time intervals. Therefore, most, if not all, of these structures shown by curves labeled with 26 and 148 h are in the forms of protofibrils and fibrils. A point worth mentioning is that the isodichroic point is indicative of the presence of only two conformations (secondary structures) in solution at any given time. This observation is consistent with a previously reported finding.58 Therefore, it appears that between 0 and 7 h, the monomer species (natively unstructured) always co-existed with one or more β-sheet-containing aggregate(s), viz., oligomer, protofibril and/or fibril.
Figure 5.

CD spectra of 25 μM Aβ(1–42) in a (A) Cu2+-free solution and (B) 25 μM Cu2+ solution incubated at pH 7.4 and 25 °C for different lengths of time. Panel (C) plots the ellipticity value at 197 nm (top two curves) and 217 nm (bottom two curves) against incubation time. The curves with open circles represent CD signals of Aβ(1–42) in the presence of Cu2+ and the curves with filled circles correspond to those in the absence of Cu2+.
Having characterized the Aβ(1–42) aggregates at different stages, we turned to the CD spectra of Aβ(1–42) aggregates formed in the presence of Cu2+ (Figure 5B). While the overall spectra and the position of the isodichroic point are comparable to those in Figures 5A, the higher initial ellipticity value at 197 nm in Figure 5B suggests an accelerated conversion of monomers to β-sheet-rich structures. This observation is consistent with the greater number of oligomers shown by AFM in the presence of Cu2+ (Figure 1B). A particularly noteworthy point is that, despite the similarity in the CD spectra between Figure 5A and Figure 5B, aggregates of drastically different morphologies were found by AFM (i.e., fibrils in Figure 1H vs. amorphous aggregates in Figures 1B and 1E). Thus, we conclude that, in the presence of Cu2+, the amorphous aggregates must also contain the β-sheet structure. To better represent the gradual evolvement of the β-sheet structure, we plotted the ellipticity values at 217 and 197 nm vs. incubation time for both Cu2+-free and -containing Aβ(1–42) solutions (Fig. 5C). These plots clearly illustrate that at and after ca. 80 h, a conversion of monomers to β-sheet-containing aggregates was complete. Therefore, it appears that the presence of Cu2+ is not unique in transforming the natively unstructured state of Aβ(1–42) to the β-sheet conformation. We need to stress that the conventional definition of “unstructured aggregates” is based on the morphological appearance or lack of the a-helical and β-sheet structures. Thus the comparability of the CD spectra between protofibrils/fibrils and amorphous aggregates suggests that the main and off-main pathway aggregates should be originated from the same β-sheet-containing intermediate. Unlike the continuous growth of preformed protofibrils/fibrils by incorporating monomers in solution onto the fibrillar templates,59 the attachment of the monomeric and oligomeric species onto the amorphous aggregates is random and does not require a “locking” step.59 Our observation and reasoning are consistent with the following studies. Dobson and coworkers discovered that formation of the β-sheet structure of a synthetic peptide did not necessarily lead to fibril formation, and amorphous aggregates could adopt the β-sheet structure.60 When a point mutation of Aβ(1–28) at position 16 (lysine to alanine to annihilate a positive charge) was made, the resultant amorphous aggregates were shown by X-ray diffraction to have a β-sheet structure.61 Takeuchi and colleague reported that Aβ(1–40) amorphous aggregates induced by Zn2+ and Cu2+ at pH 7.4 contain the β-sheet structure. 62 Finally, precipitation of amorphous aggregate should be partially resulted from the smaller solubility inherent in a β-sheet-containing molecule or species.63
3.6 β-Sheet Formation and Stacking Kinetics Influence the Morphologies of the Final Aβ(1–42) Aggregates
The formation of aggregates of different morphologies is at least governed in part by the kinetics of the β-sheet formation, the subsequent β-sheet stacking or even the assembly of the “aggregation units” (small particles containing pleated β-sheets). To correlate the β-sheet formation kinetics to the morphological changes of Aβ(1–42) aggregates, we also collected CD spectra of Aβ(1–42) in the absence and presence of Cu2+ at different pH values and temperatures. As summarized in Table 2, in the absence of Cu2+, maintaining the same temperature while lowering the solution pH to 6.6 accelerates the transformation from the natively unstructured monomeric species to β-sheet–containing species, with amorphous aggregates as the main product. Interestingly, increasing the temperature from 25 to 37 °C also accelerates the transformation to the β-sheet structure; but fibrils were the ultimate aggregate. Thus a higher temperature favors the fibrillation process, probably by affecting the β-sheet stacking kinetics (vide infra). The “aggregation units” in AFM images (cf. Figure 1c) are known to be rich in β-sheets 47, 64, 65 and the fast conversion of Aβ monomer into the more stable “aggregation units” shifts the equilibrium to a β-sheet-containing aggregate.
Table 2.
Aggregates observed in various solutions and time to produce the highest β-sheet content
| Solution | pH | T (°C) | Major Aggregate(s) | Time (h)# |
|---|---|---|---|---|
| Aβ(1–42) | 7.4 | 25 | Oligomers and fibrils | 70 |
| 37 | Oligomers and fibrils | 12 | ||
| 6.6 | 25 | Largely amorphous aggregates | 20 | |
| 37 | Amorphous aggregates and fibrils | 8 | ||
| Aβ(1–42)/Cu2+ | 7.4 | 25 | Amorphous aggregates | 70 h |
| 37 | Amorphous aggregates | ~12 | ||
| 6.6 | 25 | Amorphous aggregates | ~20 | |
| 37 | Amorphous aggregates | 8 |
Time for conversion from the unstructured conformation to β-sheet based on the CD spectra
The β-sheet content does not appear to be quantitatively related to fibril aggregates. Although the β-sheet formation and stacking is a prerequisite for the fibril formation, samples that displayed a β-sheet CD peak can also proceed to the amorphous aggregate, as demonstrated by the correlation of our CD data to AFM images. Overall, it is clear that the β-sheet structure is not unique to amyloid fibrils, and other forms of aggregates such as the amorphous aggregates can also contain β-sheet structure(s). 60
It is worth emphasizing that the β-sheets referred here are the secondary structure identified by CD spectroscopy, which is not necessarily detectable with the ThT assay. The ThT assay detects the orderly stacked β-sheets, which tend to be formed at later stages of the aggregation process and might not obvious in solution populated by amorphous aggregates. Indeed, incubated under the same condition as in Figure 5, an Aβ(1–42)-only solution gave rise to the ThT fluorescence during the aggregation process, while the Aβ(1–42) solution mixed with Cu2+ did not enhance the ThT fluorescence (Figure S1).
4. DISCUSSION
From our findings, it is clear that Aβ fibrillation and amorphous aggregate formation are two competitive pathways governed by experimental factors such as Cu2+ binding, pH, and temperature. Eisenberg and coworkers proposed three consecutive processes during the fibrillation of a small peptide, GNNQQNY38, viz., the alignment of β-strands to form a β-sheet, followed by self-complementation of two β-sheets 35, 53 to yield a pair of sheets, and culminated by interaction of these sheets to form fibrils.38 Aggregation of Aβ(1–42) in the presence of Cu2+ to the amorphous form is a direct result of the impact of copper complexation on these processes. The effect of Cu2+ complexation can be rationalized by considering factors such as the peptide surface charges and steric hindrance of the hydrophilic domain to the β-sheet stacking.
Taken together, the overall aggregation processes can be schematically shown in Figure 6. One Aβ(1–42) monomer combines with another via hydrogen bonding to produce a dimer or with more molecules to form a partially folded oligomer (e.g. tetramer, pentamer, etc). Glabe’s group identified stable dimers of Aβ(1–40) and proposed that dimerization is the initial event in amyloid aggregation.66, 67 Hilbich et al. reported that the β-sheet conformation is inherent in dimers of Aβ(1–42) and Aβ(1–43), and addition of salt to the incubation solution produces amorphous precipitates with β-sheets. 68 The fact that our CD spectra revealed the β-sheet structure in solutions containing different aggregates is consistent with these observations. The β-sheet formation has been noted as a rapid process,38, 52 and requires that β-strands from two or more Aβ molecules align via intermolecular hydrogen bonds. Aβ(1–42) dimers or partially folded oligomers can fold into a hairpin motif (see Figure 6) encompassing residues 23–29, and intermolecular hydrogen bonding of β-strands of two Aβ(1–42) molecules at residues 18–26 and 31–42 constitute two β-sheets.35, 53, 69 The resultant structure is stabilized by hydrophobic side-chain interactions between two β-sheets (i.e., β-sheet stacking). On the other hand, the emergence of the β-sheet-stacked hairpin-structured intermediate is a slow process.70 The fact that CD spectra of both amorphous and fibrillar aggregates exhibit the β-sheet characteristics suggests that monomer (which is unstructured) should not be the origin of these aggregates. Also, once most intermediates are converted into stacked β-sheet oligomers, no substantial energy barriers exist to prevent them from growing into fibrils (in fact the hydrophobic interaction between adjacent β-sheets will help dehydrate the otherwise unstable oligomers or intermediates). Given the relative kinetic barriers for the different steps, it is reasonable to consider that the two aggregation pathways diverge mainly from the same partially folded β-sheet structures/intermediates (e.g., dimer and small oligomers).
Figure 6.
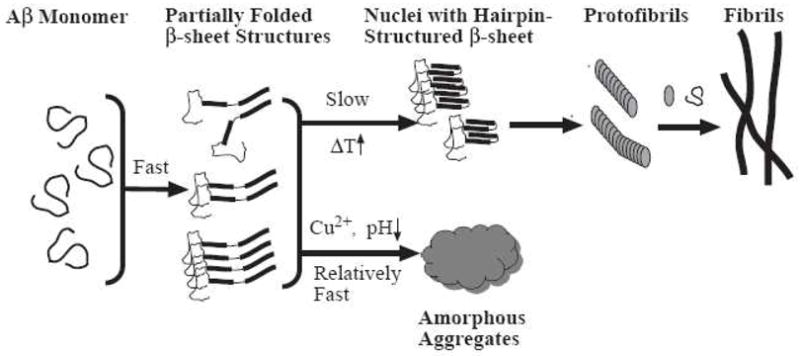
A qualitative model showing the two competing Aβ aggregation processes (pathways). The hairpin formation and β-sheet stacking process of the partially folded β-sheet structures is slow because the corresponding energy barrier is high. This process is significantly affected by temperature. In contrast, the amorphous aggregation process is a relatively fast process and largely governed by the colloidal stability of the partially folded structures or oligomeric species, which are in turn affected by copper binding and pH change.
Under certain experimental conditions, the β-sheet-containing dimers or partially folded oligomers, before developing into the hairpin-like β-sheet stacked structure, can coagulate to form amorphous aggregates (Figure 6). This is a route (“off-pathway”) that competes with the main pathway, since folding of Aβ molecules and the subsequent “in-register” stacking of β-sheets are not kinetically favored. A higher activation energy barrier has been noted by Eisenberg and coworkers,38 since this process involves a decrease in entropy and dehydration of the two β-sheets. Sciarretta et al. reported that bridging residues Asp-23 and Lys-28 of Aβ with a lactam group dramatically expedites the fibril formation.70 This further supports our suggestion that formation of a salt bridge between Asp-23 and Lys-28 and stacking two β-sheets in the hairpin configuration (see Figure 6) must be sluggish. That incubating Aβ solutions at a higher temperature favors the main pathway (fibrillation) further demonstrates that the β-sheet stacking (or the formation of nuclei) is the rate determining step. The stability of the unstacked β-sheet intermediates is the key factor determining the final aggregate form or pathway (i.e., fibrils vs. amorphous aggregates). From thermodynamic point of view, the fibrous aggregates should be more stable than amorphous counterparts. Eventually, amorphous aggregates should transform to the fibril counterpart. But within our experimental timeframe, such a transformation was not observed.
Now it becomes conceivable that deviation from the main aggregation pathway is also pH- and temperature-dependent. At pH 6.6 and 25 °C, Aβ(1–42) produced predominantly amorphous aggregates. At the same pH but an elevated temperature (37 °C), a mixture of fibrils and amorphous aggregates was observed, though the former are more abundant (cf. Figures 4F–I). The production of amorphous aggregates is in common with the coalescence of colloidal particles, a process dictated by the electrostatics and intermolecular interaction such as the Van der Waals force. Analogous to the colloidal system, electrostatic repulsion between charged protein or peptide side chains creates a kinetic barrier to the coalescence of these molecules. In general, such a barrier is not high and can be easily overcome by thermal energy. Thus, for proteins or peptides that are not highly charged, they will randomly approach to one another and interact via intermolecular interactions, leading to amorphous aggregates.
At 25 °C, lowering the pH of the incubation solution decreases the net negative charges on Aβ(1–42), as evidenced by the zeta potential decrease (Table 1). As a consequence, the colloidal stability of Aβ(1–42) monomers and partially folded oligomers is decreased. Such instability hastens the formation of amorphous aggregates containing partially folded β-sheet structures and the conversion from the unstructured conformation to the β-sheet conformation. This explains why CD spectra displayed faster transformation from the unstructured to the β-sheet conformations (cf. Table 2). Again, the dimers and oligomeric species will combine randomly and rapidly, leaving little time for the hairpin structure formation and the ordered β-sheet stacking.38, 70 As contrasted by the AFM images collected from samples incubated at two different pH values (Figures 1F–I and 2D–F), fibrillation gives way to the amorphous aggregate formation as the pH value is lowered. For Aβ(1–42) solution incubated at neutral pH, an increase in temperature can augment the rate at which β-sheet stacking proceeded and facilitate the fibrillation (Table 2 and Figures 1 and 3). In other words, the equilibrium shown in Figure 6 is favored in the direction of the main (fibrillation) pathway at an elevated temperature
A rather unique situation was encountered when the incubation pH was lowered (6.6) but the temperature was raised (37 °C). Under such a circumstance, the lower pH annihilates the Aβ(1–42) surface charges to help accelerate the amorphous aggregate formation, while the higher temperature facilitates the β-sheet stacking and favors the fibrillation process. The net effect is that the rates of the two competing processes (Figure 6) become comparable, producing a mixture of globular oligomers and amorphous aggregates (Figure 4B–E). The facts that Aβ carries a substantial amount of charges71 at very low pH (where Aβ becomes positively charged) and highly charged peptides72 assemble solely to fibril aggregates lend further support to our model.
The most interesting observation in our work is that the presence of Cu2+ in the incubation solution tips the aggregation process to the direction of amorphous aggregate formation. As reflected by Table 2, this effect is so pronounced that the effect of temperature on the fibrillation process (vide supra) is overwhelmed. At neutral and slightly acidic pH, electron paramagnetic resonance and fluorescence spectroscopy have shown that Cu2+ is bound to Aβ.42, 73 Using high-resolution mass spectrometry and voltammetry, we identified the oxidation state of copper in Aβ(1–42) to be 2+.40 Thus, incorporating Cu2+ in the more exposed hydrophilic domain of Aβ(1–42) neutralizes some negative charges on Aβ(1–42) (Table 1). Neutralization of the surface charges of Aβ(1–42) by Cu2+ binding decreases the colloidal stability of dimers and partially folded oligomers. Similar to a decrease of solution pH, the conversion of unstructured monomers by Cu2+ complexation to dimers and/or partially folded oligomers and the combination of these intermediates to produce amorphous aggregates will be expedited. In fact, the exclusive formation of amorphous Aβ(1–42) aggregates in the presence of Cu2+ again suggests that the colloid-like property of peptide is important in changing the course of aggregation.
The fact that Aβ(1–42) in the presence of Cu2+ did not form fibrils under all conditions studied suggests that, in addition to the charge variation, Cu2+ binding may have also created other adverse effect on β-sheet stacking. We hypothesize that Cu2+ complexation by Aβ(1–42) may have inhibited the hairpin structure formation and the follow-up stacking of β-sheets (Figure 6). Although the exact metal binding sites in Aβ are still in debate, it is commonly accepted that the N-terminus and two of the histidine residues (at positions 6, 13 or 14) are involved in the complexation, in addition to an oxygen-containing ligand (possibly the Glu residue at position 3).41, 73 Notice these four ligands are all located in the hydrophilic domain of Aβ(1–42), which wraps around the metal ion.73 Such a spatial rearrangement may impose greater steric hindrance to the folding of the β-sheets formed within residues 31–42, further slowing down the β-sheet stacking process. The resultant effects are a concurrent decrease of surface charges and retardation of β-sheet staking. Furthermore the intermolecular linking through copper binding74 facilitates the polymerization and the dynamically unstable low charged, non-ordered stacked oligomers adhere each other without providing enough time for β-sheets to stack. These effects would favor the “off-pathway” to produce amorphous aggregates.
5. CONCLUSION
We have demonstrated that the fibrillar (main) and amorphous (off) aggregation pathways of Aβ(1–42) are affected by Cu2+ binding. Detailed spectroscopic and morphological characterization under different incubation conditions indicate that the deviation from the main aggregation pathway is modulated by charge and structural variations resulted from the Cu2+ binding. The two competing pathways diverge from the same β-sheet-containing intermediates, evidenced by the fact that CD spectra of solutions producing amorphous aggregates and fibrils both displayed β-sheet-signifying peaks. The amorphous aggregates are the product of random agglomeration of these intermediates. The more ordered stacking of these intermediates, on the other hand, leads to ordered oligomers or nuclei, which subsequently grow into fibrils. The colloidal stability of the partially folded intermediates, which is also affected by solution pH and temperature, is important in dictating the ultimate aggregation pathway. Complexation of Cu2+ by the hydrophilic domain of Aβ(1–42), in addition to decreasing the Aβ(1–42) surface charge and the stability of the partially folded Aβ(1–42) intermediates, could also impose steric hindrance to the ordered β-sheet stacking. This further tilts the aggregation process toward accumulation of the partially folded intermediates, leading to the appearance of amorphous aggregates.
Supplementary Material
Acknowledgments
This work was supported by an NINDS grant (SC1MS0701555-01), an NSF grant (1112105), the ACS-Petroleum Research Funds (49390-UR10), and NSF-CREST (JRD-0932421). S. H. acknowledges partial support from an NIH-NCMHD grant (P20 MD001089-03).
Footnotes
Thioflavin T assay for monitoring the kinetics of aggregation process of Aβ(1–42) in the absence and presence of Cu2+ monitored. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Alzheimer’s disease-genotypes, phenotype, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 3.Turoverov KK, Kuznetsova IM, Uversky VN. The protein kingdom extended: ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog Biophys Mol Biol. 2010;102:73–84. doi: 10.1016/j.pbiomolbio.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Triulzi RC, Li C, Naistat D, Orbulescu J, Leblanc RM. A two-dimensional approach to study amyloid β-peptide fragment (25–35) J Phys Chem C. 2007;111:4661–4666. [Google Scholar]
- 5.McMasters MJ, Hammer RP, McCarley RL. Surface-induced aggregation of beta amyloid peptide by substituted alkanethiol monolayers supported on gold. Langmuir. 2005;21:4464–4470. doi: 10.1021/la047044w. [DOI] [PubMed] [Google Scholar]
- 6.Yamaguchi H, Nakazato Y, Hirai S, Shoji M, Harigaya Y. Electron micrograph of diffuse plaques. Initial stage of senile plaque formation in Alzheimer’s brain. Am J Pathol. 1989;135:593–597. [PMC free article] [PubMed] [Google Scholar]
- 7.Huang THJ, Yang DS, Fraser PE, Chakrabartty A. Alternate aggregation pathways of the Alzheimer β-amyloid peptide. J Biol Chem. 2000;275:36436–36440. doi: 10.1074/jbc.M005698200. [DOI] [PubMed] [Google Scholar]
- 8.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 9.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Pozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CC, Krafft GA, Klein WL. Diffusible, nonofibrillar ligands derived from Aβ are potent central nervous neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lambert MP, Viola KL, Chromy BA, Chang L, Morgan TE, Yu J, Venton DL, Krafft GA, Finch CE, Klein WL. Vaccination with soluble Aβ oligomers generates toxicity-neutralizing antibodies. J Neurochem. 2001;79:595–605. doi: 10.1046/j.1471-4159.2001.00592.x. [DOI] [PubMed] [Google Scholar]
- 11.Atwood CS, Moir RD, Huang X, Scarpa RC, Bacarra NME, Romano DM, Hartshorn MA, Tanzi RE, Bush AI. Dramatic aggregation of Alzheimer Aβ by Cu(II) is induced by conditions representing physiological acidosis. J Biol Chem. 1998;273:12817–12826. doi: 10.1074/jbc.273.21.12817. [DOI] [PubMed] [Google Scholar]
- 12.Bush AI, Petingell WH, Multhaup G, Paradis MD, Vonsattel JP, Gusella JF, Beyreuther K, Masters CL, Tanzi RE. Rapid induction of Alzheimer Aβ amyloid formation by zinc. Science. 1994;265:1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 13.Mantyh PW, Ghilardi JR, Rodgers S, Demaster E, Allen CJ, Stimson ER, Maggio JE. Aluminum, iron, and zinc ions promote aggregation physiological concentrations of β-amyloid peptide. J Neurochem. 1993;61:1171–1174. doi: 10.1111/j.1471-4159.1993.tb03639.x. [DOI] [PubMed] [Google Scholar]
- 14.Klug GMJA, Losic D, Subasinghe SS, Aguilar MI, Martin LL, Small DH. Amyloid protein oligomers induced by metal ions and acid pH are distinct from those generated by slow spontaneous aging at neutral pH. Eur J Biochem. 2003;270:4282–4293. doi: 10.1046/j.1432-1033.2003.03815.x. [DOI] [PubMed] [Google Scholar]
- 15.Yoshiike Y, Tanemura K, Murayama O, Akagi T, Murayama M, Sato S, Sun X, Tanaka N, Takashima A. New insights on how metals disrupt amyloid aggregation and their effects on amyloid cytotoxicity. J Biol Chem. 2001;276:32293–32299. doi: 10.1074/jbc.M010706200. [DOI] [PubMed] [Google Scholar]
- 16.Ha C, Ryu J, Park CB. Metal ions differentially influence the aggregation and deposition of Alzheimer’s β-amyloid on a solid template. Biochemistry. 2007;46:6118–6125. doi: 10.1021/bi7000032. [DOI] [PubMed] [Google Scholar]
- 17.Raman B, Ban T, Yamaguchi K-i, Sakai M, Kawai T, Naiki H, Goto Y. Metal ion-dependent effects of clioquinol on the fibril growth of an amyloid beta peptide. J Biol Chem. 2005;280:16157–16162. doi: 10.1074/jbc.M500309200. [DOI] [PubMed] [Google Scholar]
- 18.Pedersen JT, Ostergaard J, Rozlosnlk N, Gamelgaard B, Heegaard NHH. Cu(II) Mediates kinetiocally distinct, non-amyloidogenic aggregation of amyloid-β peptides. J Biol Chem. 2011;286:26952–26963. doi: 10.1074/jbc.M111.220863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.da Silva GFZ, Tay WM, Ming LJ. Catechol oxidase-like oxidation chemistry of the 1–20 and 1–16 fragments of alzheimers disease-related beta-amyloid peptide-their structure-activity correlation and the fate of hydrogen peroxide. J Biol Chem. 2005;280:16601–16609. doi: 10.1074/jbc.M411533200. [DOI] [PubMed] [Google Scholar]
- 20.de Silva GFZ, Ming LJ. Alzheimer’s disease-related copper(II)-β-amyloid peptide exhibits phenol monooxygenase and catechol oxidase activities. Angew Chem Int Ed. 2005;44:5501–5504. doi: 10.1002/anie.200501013. [DOI] [PubMed] [Google Scholar]
- 21.Jiang D, Li X, Liu L, Yagnik GB, Zhou F. Reaction rates and mechanism of the ascorbic acid oxidation by molecular oxygen facilitated by Cu(II)-containing amyloid-beta complexes and aggregates. J Phys Chem B. 2010;114:4896–4903. doi: 10.1021/jp9095375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hewitt N, Rauk A. Mechanism of hydrogen peroxide production by copper-bound amyloid beta peptide: a theoretical study. J Phys Chem B. 2009;113:1202–1209. doi: 10.1021/jp807327a. [DOI] [PubMed] [Google Scholar]
- 23.Lu Y, Prudent M, Qiao L, Mendez MA, Girault HH. Copper(I) and copper(II) binding to β-amyloid 16 (Aβ16) studied by electrospray ionization mass spectrometry. Metallomics. 2010;2:474–479. doi: 10.1039/c004693k. [DOI] [PubMed] [Google Scholar]
- 24.Karr JW, Szalai VA. Roles of aspartate-1 in Cu(II) binding to the amyloid-β peptide of Azheimer’s disease. J Am Chem Soc. 2007;129:3796–3797. doi: 10.1021/ja068952d. [DOI] [PubMed] [Google Scholar]
- 25.Karr JW, Szalai VA. Cu(II) binding to monomeric, oligomeric, and fibrillar forms of the Alzheimer’s disease amyloid-beta peptide. Biochemistry. 2008;47:5006–5016. doi: 10.1021/bi702423h. [DOI] [PubMed] [Google Scholar]
- 26.Shearer J, Callan PE, Tran T, Szalai VA. Cu K-edge X-ray absorption spectroscopy reveals differential copper coordination within amyloid-β oligomers compared to amyloid-β monomer. Chem Comm. 2010;46:9137–9139. doi: 10.1039/c0cc02446e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eury H, Bijani C, Faller P, Hureau C. Copper(II) coordination to amyloid β: murine versus human peptide. Angew Chem Int Ed. 2011;50:901–905. doi: 10.1002/anie.201005838. [DOI] [PubMed] [Google Scholar]
- 28.Parthasarathy S, Long F, Miller Y, Xiao Y, McElheny D, Thurber K, Ma B, Nussinov R, Ishii Y. Molecular-level examination of Cu2+ binding structure for amyloid fibrils of 40-residue Alzheimer’s β by solid-state NMR spectroscopy. J Am Chem Soc. 2011;133:3390–3400. doi: 10.1021/ja1072178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shin B-k, Saxena S. Substantial contribution of the two imidazole rings of the His13_ His14 dyad to Cu(II) binding in amyloid-β(1–16) at physiological pH and its significance. J Phys Chem A. 2011;115:9590–9602. doi: 10.1021/jp200379m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Youssef EK, Pierre D, Peter F, Petra H. New insights into the coordination of Cu(II) by the amyloid-β 16 peptide from Fourier Transform IR spectroscopy and isotopic labeling. J Phys Chem B. 2011;115:14812–14821. doi: 10.1021/jp207328y. [DOI] [PubMed] [Google Scholar]
- 31.Curtain CC, Ali FE, Smith DG, Bush AI, Masters CL, Barnham KJ. Metal ions, pH, and cholesterol regulate the interactions of Alzheimer’s disease amyloid-β peptide with membrane lipid. J Biol Chem. 2003;278:2977–2982. doi: 10.1074/jbc.M205455200. [DOI] [PubMed] [Google Scholar]
- 32.Liu G, Huang W, Moir RD, Vanderburg CR, Lai B, Peng Z, Tanzi RE, Rogers JT, Huang X. Metal exposure and Alzheimer’s pathogenesis. J Struct Biol. 2006;155:45–51. doi: 10.1016/j.jsb.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 33.Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR. Metal binding and oxidation of amyloid-β within isolated senile plaque cores: Raman microscopic evidence. Biochemistry. 2003;42:2768–2773. doi: 10.1021/bi0272151. [DOI] [PubMed] [Google Scholar]
- 34.Zou J, Kajita K, Sugimoto N. Cu2+ inhibits the aggregation of amyloid β-peptide(1 – 42) in vitro. Angew Chem Int Ed. 2001;40:1433–7851. doi: 10.1002/1521-3773(20010618)40:12<2274::AID-ANIE2274>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 35.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer’s amyloid-β(1–42) fibrils. Proc Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gorman PM, Yip CM, Fraser PE, Chakrabartty A. Alternate aggregation pathways of Alzheimer β-amyloid peptide: Aβ association kinetics at endosomal pH. J Mol Biol. 2003;325:743–757. doi: 10.1016/s0022-2836(02)01279-2. [DOI] [PubMed] [Google Scholar]
- 37.Antzutkin ON. Amyloidosis of Alzheimer’s Aβ peptides: solid-state nuclear magnetic resonance, electron paramagnetic resonance, transmission electron microscopy, scanning transmission electron microscopy and atomic force microscopy studies. Magn Reson Chem. 2004;42:231–246. doi: 10.1002/mrc.1341. [DOI] [PubMed] [Google Scholar]
- 38.Nelson R, Sawaya MR, Balbirnie M, Madsen AØ, Riekel C, Grothe R, Eisenberg D. Structure of the cross-β spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fezoui Y, Hartley DM, Harper JD, Khurana R, Walsh DM, Condron MM, Selkoe DJ, Lansbury PT, Jr, Fink AL, Teplow DB. An improved method of preparing the amyloid β-protein for fibrillogenesis and neurotoxicity experiments. Amyloid: Int J Exp Clin Invest. 2000;7:166–178. doi: 10.3109/13506120009146831. [DOI] [PubMed] [Google Scholar]
- 40.Jiang D, Men L, Wang J, Zhang Y, Chickenyen S, Wang Y, Zhou F. Redox reactions of copper complexes formed with different β-amyloid peptides and their neuropathalogical relevance. Biochemistry. 2007;46:9270–9282. doi: 10.1021/bi700508n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karr JW, Kaupp LJ, Szalai VA. Amyloid-β binds Cu2+ in a mononuclear metal ion binding site. J Am Chem Soc. 2004;126:13534–13538. doi: 10.1021/ja0488028. [DOI] [PubMed] [Google Scholar]
- 42.Hou L, Zagorski MG. NMR reveals anomalous copper(II) binding to the amyloid Aβ peptide of Alzheimer’s disease. J Am Chem Soc. 2006;128:9260–9261. doi: 10.1021/ja046032u. [DOI] [PubMed] [Google Scholar]
- 43.Zhu M, Han S, Zhou F, Millet IS, Fink AL. Annular oligomeric amyloid intermediates observed by in-situ AFM. J Biol Chem. 2004;279:24452–24459. doi: 10.1074/jbc.M400004200. [DOI] [PubMed] [Google Scholar]
- 44.Zhu M, Rajamani S, Kaylor J, Han S, Zhou F, Fink AL. The flavonoid baicalein inhibits fibrillation of a-synuclein and disaggregates existing fibrils. J Biol Chem. 2004;279:26846–26857. doi: 10.1074/jbc.M403129200. [DOI] [PubMed] [Google Scholar]
- 45.Lehninger AL, Nelson DL, Cox MM. Principles of Biochemistry. 4. W. H. Freeman & Co; New York: 2004. [Google Scholar]
- 46.Lomakin A, Chung DS, Benedek GB, Kirschner DA, Teplow DB. On the nucleation and growth of amyloid beta-protein fibrils: detection of nuclei and quantification of rate constants. Proc Natl Acad Sci USA. 1996;93:1125–1129. doi: 10.1073/pnas.93.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mastrangelo IA, Ahmed M, Sato T, Liu W, Wang C, Hough P, Smith SO. High-resolution atomic force microscopy of soluble Aβ(1–42) oligomers. J Mol Biol. 2006;358:106–119. doi: 10.1016/j.jmb.2006.01.042. [DOI] [PubMed] [Google Scholar]
- 48.Kowalewski T, Holtzman DM. In situ atomic force microscopy study of Alzheimer’s β-amyloid peptide on different substrates: New insights into mechanism of β-sheet formation. Proc Natl Acad Sci USA. 1999;96:3688–3693. doi: 10.1073/pnas.96.7.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harper JD, Wong SS, Lieber CM, Lansbury PT., Jr Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 50.Wood SJ, Maleeff B, Hart T, Wetzel R. Physical, morphological and functional differences between pH 5.8 and 7.4 aggregates of the Alzheimer’s amyloid peptide. J Mol Biol. 1996;256:870–877. doi: 10.1006/jmbi.1996.0133. [DOI] [PubMed] [Google Scholar]
- 51.Morgan C, Colombres M, Nunez MT, Inestrosa NC. Structure and function of amyloid in Alzheimer’s disease. Prog Neurobiol. 2004;74:323–349. doi: 10.1016/j.pneurobio.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 52.Sivaraman T, Kumar TKS, Chang DK, Lin WY, Yu C. Events in the kinetic folding pathway of a small, all beta-sheet protein. J Biol Chem. 1998;273:10181–10189. doi: 10.1074/jbc.273.17.10181. [DOI] [PubMed] [Google Scholar]
- 53.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamaguchi T, Yagi H, Goto Y, Matsuzaki K, Hoshino M. A disulfide-linked amyloid-β peptide dimer forms a protofibril-like oligomer through a distinct pathway from amyloid fibril formation. Biochemistry. 2010;49:7100–7107. doi: 10.1021/bi100583x. [DOI] [PubMed] [Google Scholar]
- 55.Kirkitadze MD, Condron MM, Teplow DB. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J Mol Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 56.Soto C, Castano EM, Frangione B, Inestrosa NC. The a-helical to β-strand transition in the amino-terminal fragment of the amyloid β-peptide modulate amyloid formation. J Biol Chem. 1995;270:3063–3067. doi: 10.1074/jbc.270.7.3063. [DOI] [PubMed] [Google Scholar]
- 57.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selko DJ, Teplow DB. Amyloid β-protein fibrillogenesis: structure and biological activity of protofibbrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 58.Barrow CJ, Zagorski MG. Solution structures of β peptide and its constituent fragments: relation to amyloid deposition. Science. 1991;253:179–182. doi: 10.1126/science.1853202. [DOI] [PubMed] [Google Scholar]
- 59.Esler WP, Stimson ER, Jennings JM, Vinters HV, Ghilardi JR, Lee JP, Mantyh PW, Maggio JE. Alzheimer’s disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry. 2000;39:6288–6295. doi: 10.1021/bi992933h. [DOI] [PubMed] [Google Scholar]
- 60.Paz MLdl, Goldie K, Zurdo J, Lacroix E, Dobson CM, Hoenger A, Serrando L. De novo designed peptide-based amyloid fibrils. Proc Natl Acad Sci USA. 2002;99:16052–16057. doi: 10.1073/pnas.252340199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kirschner DA, Inouye H, Duffy LK, Sinclair A, Lind M, Selkoe DJ. Synthetic peptide homologous to beta protein from Alzheimer disease forms amyloid-like fibrils in vitro. Proc Natl Acad Sci USA. 1987;84:6953–6957. doi: 10.1073/pnas.84.19.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miura T, Suzuki K, Kohata N, Takeuchi H. Metal binding modes of Alzheimer’s amyloid β-peptide in insoluble aggregates and soluble complexes. Biochemistry. 2000;39:7024–7031. doi: 10.1021/bi0002479. [DOI] [PubMed] [Google Scholar]
- 63.Rodger A, Norden B. Circular Dichroism and Linear Dichroism. Oxford University Press; Oxford: 1997. [Google Scholar]
- 64.Gerber R, Tahiri-Alaoui A, Hore PJ, James W. Oligomerization of the human prion protein proceeds via a molten globule intermediate. J Biol Chem. 2007;282:6300–6307. doi: 10.1074/jbc.M608926200. [DOI] [PubMed] [Google Scholar]
- 65.Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL. Self-assembly of Aβ1–42 into globular neurotoxins. Biochemistry. 2003;42:12749–12760. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- 66.Garzon-Rodriguez W, Sepulveda-Becerra M, Milton S, Glabe CG. Soluble amyloid Aβ-(1–40) exists as a stable dimer at low concentrations. J Biol Chem. 1997;272:21037–21044. doi: 10.1074/jbc.272.34.21037. [DOI] [PubMed] [Google Scholar]
- 67.Garzon-Rodriguez W, Vega A, Sepulveda-Becerrai M, Milton S, Johnson DA, Yatsimirsky AK, Glabe CG. A conformation change in the carboxyl terminus of Alzheimer’s Aβ(1–40) accompanies the transition from dimer to fibril as revealed by fluorescence quenching analysis. J Biol Chem. 2000;30:22645–22649. doi: 10.1074/jbc.M000756200. [DOI] [PubMed] [Google Scholar]
- 68.Hilbich C, Kisters-Woike B, Reed J, Masters CL, Beyreuther K. Aggregation and secondary structure of synthetic amyloid betaA4 peptides of Alzheimer’s disease. J Mol Biol. 1991;218:149–163. doi: 10.1016/0022-2836(91)90881-6. [DOI] [PubMed] [Google Scholar]
- 69.Zhao J, Wang Q, Liang G, Zheng J. Molecular dynamics simulations of low-ordered Alzheimer β-amyloid oligomers from dimer to hexamer on self-assembled monolayers. Langmuir. 2011;27:14876–14887. doi: 10.1021/la2027913. [DOI] [PubMed] [Google Scholar]
- 70.Sciarretta KL, Gordon DJ, Petkova AT, Tycko R, Meredith SC. Aβ40-lactam(D23/K28) models a conformation highly favorable for nucleation of amyloid. Biochemistry. 2005;44:6003–6014. doi: 10.1021/bi0474867. [DOI] [PubMed] [Google Scholar]
- 71.Carrotta R, Manno M, Bulone D, Martorana V, Biagio PLS. Protofibril formation of amyloid β-protein at low pH via a non-cooperative elongation mechanism. J Biol Chem. 2005;280:30001–30008. doi: 10.1074/jbc.M500052200. [DOI] [PubMed] [Google Scholar]
- 72.Zurdo J, Guijarro JI, Jiménez JL, Saibil HR, Dobson CM. Dependence on solution conditions of aggregation and amyloid formation by an SH3 domain. J Mol Biol. 2001;311:325–340. doi: 10.1006/jmbi.2001.4858. [DOI] [PubMed] [Google Scholar]
- 73.Syme CD, Nadal RC, Rigby SEJ, Viles JH. Copper binding to the amyloid-beta peptide associated with Alzheimer’s disease. J Biol Chem. 2004;279:18169–18177. doi: 10.1074/jbc.M313572200. [DOI] [PubMed] [Google Scholar]
- 74.Pedersen JT, Teilum K, Heegaard NHH, Østergaard J, Adolph HW, Hemmingsen L. Rapid formation of a preoligomeric peptide–metal–peptide complex following copper(II) binding to amyloid-β peptides. Angew Chem Int Ed. 2011;50:2532–2535. doi: 10.1002/anie.201006335. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.