

Abstract
Thyroid carcinomas, the most common endocrine tumors in humans, have an increasing incidence in the U.S. and worldwide. There are four major types of thyroid cancers: papillary, follicular, anaplastic, and medullary carcinomas. In recent years, significant progress has been made in the identification of genetic alterations in thyroid carcinomas, particularly, papillary and medullary thyroid cancers. Mouse models of thyroid cancer are valuable tools in elucidating molecular genetic changes underlying thyroid carcinogenesis and in identifying potential molecular targets for therapeutic intervention. Representative mouse models of papillary, follicular, and medullary carcinomas are reviewed here with particular emphasis on those for follicular thyroid carcinomas. Challenges for further development in the modeling of thyroid cancer will also be discussed.
Keywords: thyroid cancer, thyroid hormone, mouse models
Introduction
Thyroid carcinomas are the most common endocrine neoplasms in humans, affecting approximately 1 % of the population [1]. Thyroid cancer is one of the few cancer types with increasing incidence in the United States and around the globe, particularly among women [2]. The neoplasms originate from two distinct endocrine cell types of thyroid glands: thyroid hormone producing follicular epithelial cells and calcitonin producing parafollicular cells (C-cells). There are four major types of thyroid cancers: papillary, follicular, anaplastic, and medullary thyroid carcinoma. The majority of thyroid tumors including papillary, follicular, and anaplastic carcinomas are derived from follicular epithelial cells. Only about 3% of thyroid tumors are from parafollicular cells.
Among the thyroid carcinomas derived from follicular epithelial cells, more than 80 % of them are papillary thyroid carcinomas (PTCs). In most cases, PTCs are well-differentiated types of tumors that metastasize only to local lymph nodes and respond well to radioactive iodide treatment with a favorable 98 % 10-year survival outcome. Follicular thyroid carcinomas (FTCs) accounts for about 15 % of thyroid carcinomas. Compared to PTCs, FTCs metastasize more likely via the vascular system to distant organs and are less likely to take up therapeutic radioactive iodide. FTCs have poorer outcome than PTCs with a 93% 10-year survival rate. Although anaplastic thyroid carcinomas (ATCs) account for only 2 % of thyroid cancers, they often have an unfavorable outcome as a result of local invasion, distant metastases, and the loss of the ability to take up radioactive iodide. ATCs could develop from existing well-differentiated FTCs [1].
Of thyroid carcinomas, 2–3% are derived from parafollicular cells. Medullary thyroid carcinomas (MTCs) metastasize to local lymph nodes as well as distant organs. MTCs are treated with either surgery and/or external beam radiation. The 10-year survival rate for MTC is between 40 % and 95 %, depending mainly on whether patients have distant metastasis [3].
Morphology and Functions of the Thyroid Gland
Two types of cells responsible for the dual endocrine functions of the thyroid gland are thyroid follicular and parafollicular cells. They have distinct structures, functions, origins, and gene expression patterns. Thyroid follicular cells form the thyroid follicles, spherical structures serving as storage sites for controlled release of thyroid hormones [4]. The parafollicular cells are scattered in the interfollicular space. The two cell types originate from two different embryological structures of the thyroid anlage and the ultimobranchial bodies, respectively. The cells of the thyroid anlage and the ultimobranchial bodies migrate from their respective sites of origin and ultimately merge in the thyroid gland. The cells originating from the anlage organize the thyroid follicles, whereas the parafollicular cells scatter within the interfollicular space [5].
Thyroid follicular cells express specific proteins essential for thyroid hormone biosynthesis. Genes encoding thyroglobulin (TG), thyroperoxidase (TPO), sodium/iodide symporter (NIS), and TSH receptor (TSHR) are expressed in the thyroid. Iodine is captured and transported by NIS from the bloodstream to iodinate tyrosine residues of TG in the presence of TPO to form T3 and T4. Upon release, T4 is converted to more active T3 by deiodinases in the target tissues (Fig. 1).
Fig. 1.
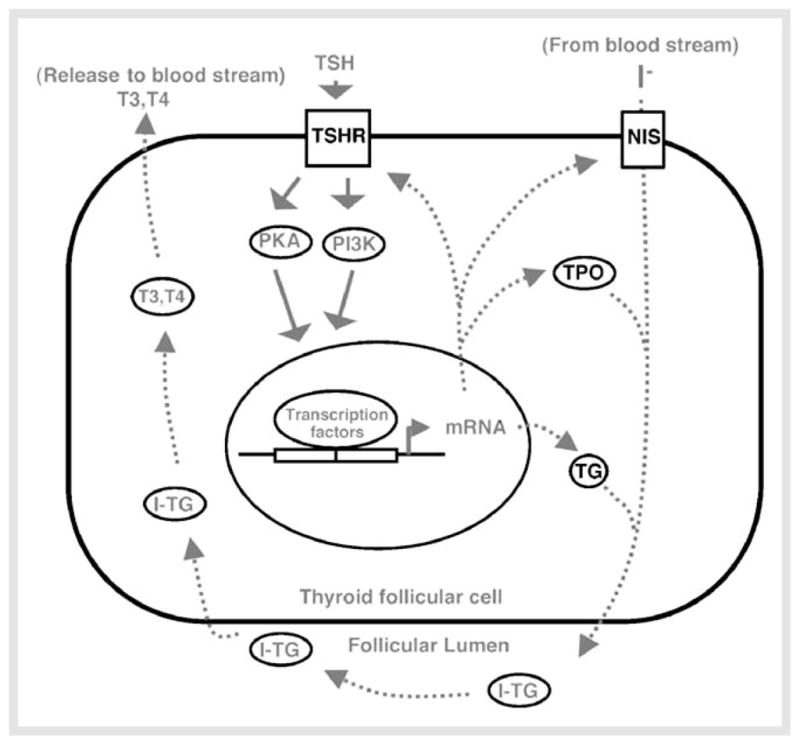
Schematic representation of PKA and PI3K signaling pathways in normal thyroid follicular cells under the control of TSH. TSH is the major growth regulator of thyroid follicular cells. Binding of TSH to TSHR activates the PKA, PI3K and other signaling pathways, leading to proliferation and differentiation of follicular cells. The differentiated follicular cells produce thyroid specific proteins such as thyroglobulin (TG), thyroid peroxidase (TPO), thyroid-stimulating hormone receptor (TSHR), and sodium/iodide symporter (NIS) to direct synthesis and release of thyroid hormones, T3 and T4. I-TG: iodinated-TG.
During embryonic development, the proteins appear at a precise stage: Tg, Tpo, and Tshr genes are expressed by E14.5 [6]; the NIS gene is detected by E16 [7]. T4 is first detected at E16.5 [8]. The signals for the proliferation and differentiation of the thyroid follicles in adults and embryos are different. Although TSH is the most important growth stimulus for thyroid follicular cells, it is not involved in embryonic development of follicular cells. Instead, the genes encoding for the transcription factors Titf1/Nkx2–1 Foxe1, Pax8, and Hhex express both in mature thyroid cells and in their precursors. These factors express at the very beginning of thyroid morphogenesis and play crucial roles in the organogenesis of the thyroid gland. These factors are also present in other embryonic tissues, but all four are co-expressed only in the thyroid anlage. The expression of these four factors is required for the early stages of thyroid morphogenesis. Among four transcription factors, PAX8 that is fused to the peroxisome proliferator-activated receptor-γ (PPARγ; PAX8-PPARγ) has been identified to be involved in tumorigenesis. The PAX8-PPARγ is frequently found in FTC, but rarely in adenomas. The Tg promoter is frequently used to specifically target exogenous genes to the follicular cells.
The development of parafollicular cells is less well understood. It seems that a membrane bound tyrosine kinase receptor RET that binds glial-derived neurotrophic factors is important for parafollicular cell development. The Ret gene is highly expressed in parafollicular cells. Ret-deficient mice have less calcitonin-immunoreactive cells than do wild-type littermates at birth [9].
Etiology and Genetic Alteration in Thyroid Carcinogenesis
Thyroid follicular cells can undergo neoplastic transformation to carcinomas of three histotypes: papillary, follicular, and anaplastic. PTCs frequently have mutations in genes involved in MAPK signaling pathways, such as RET, BRAF, and RAS. Genetic alterations in thyroid cells such as RET translocation, NTRK1 translocation, and BRAF or RAS mutations cause constitutive activation of the MAPK pathway, disrupt TSH-mediated signaling, and induce papillary thyroid tumorigenesis (Fig. 2). The RET protooncogene located on chromosome 10q11.2 encodes a receptor tyrosine kinase highly expressed in the parafollicular cells, but less in thyroid follicular cells. RET is a component of a cell-surface complex that binds glial-derived neurotrophic factor (GDNF), neurturin, artemin, and persephin. The binding of ligand causes receptor dimerization and initiation of intracellular signaling. However, in PTCs, the RET protooncogene is fused to one of the constitutively expressed genes in thyroid follicular cells [10, 11]. The paracentric inversion of the long arm of chromosome 10 leads to the fusion of RET either with the coiled-coil domain containing gene 6 or with the nuclear receptor coactivator gene-4, resulting in the formation of the RET oncogenes RET/PTC1 [12] and RET/PTC3 [13], respectively. Therefore, the RET rearrangement results in chimeric oncoproteins with constitutive tyrosine kinase activity in thyroid follicular cells.
Fig. 2.

Aberrant activation of the MAPK signaling pathway in papillary thyroid tumor cells. In normal follicular cells, binding of proliferation signal molecules to membrane receptor tyrosine kinases (RTK) such as RET and NTRK1 leads to receptor phosphorylation and activation of the downstream effector RAS. The phosphorylation cascade in the downstream effectors leads to the translocation of ERK to the nucleus to affect genes involved in cell differentiation, proliferation, apoptosis, and survival. In PTC, fusion genes such as RET/PTCs and TRK-T1 and mutated genes such as BRAFV600E and RAS constitutively activate MAPK signaling, thereby leading to the development of carcinogenesis.
It is now well recognized that ionizing radiation exposure causes RET rearrangements, particularly in children [10, 14]. After the Chernobyl nuclear reactor accident in 1986, the marked increase in the incidence of thyroid carcinomas in children in affected areas was reported to have risen to 90 per million [15, 16]. RET expression resulting from chromosome rearrangement is found in about 30 % of all PTCs, but frequently in PTC with a history of radiation exposure [16]. RET/PTC1 and RET/PTC3 account for approximate 70% and 30%, respectively, of RET rearrangements found in PTCs. For the patients without radiation exposure, RET/PTC1 is most frequently detected while RET/PTC3 is common for the patients with radiation exposure. NTRK1 is a receptor tyrosine kinase normally involved in nerve growth factor signaling [17]. Like RET rearrangements, NTRK1 can fuse to other genes and form a constitutively active oncogene, such as TRK-T1, leading to PTC [18]. However, NTRK1 rearrangements are less frequent than RET rearrangements and are not associated with radiation exposure.
Sporadic PTC unrelated to radiation exposure make up more than two thirds of all cases, and mutations of BRAF and RAS are found in about 40% and 10–20% of PTCs, respectively [1,19–21]. BRAF is an intracellular effector of MAPK signaling pathway. Upon activation triggered by RAS binding and recruitment of other proteins to the cell membrane, this kinase phosphorylates and activates MAPK/ERK kinase and downstream effectors. A point mutation of BRAF results in V600E conversion [22] that is associated with poor prognosis. In the unphosphorylated BRAF protein, the hydrophobic interactions between the activation loop and the ATP binding site maintain the protein in an inactive conformation. The V600E substitution disrupts these interactions, resulting in sustained phosphorylation of MEK and activation of the signaling pathway [23].
RAS is a guanine nucleotide binding protein that is a key intermediate in signal transduction pathways. The activation state of RAS depends on whether it binds to GTP or GDP. When it binds to GDP, it is inactive; when it binds to GTP, it initiates a downstream signaling pathway [24]. Mutations of RAS often involve codons 12, 13, and 61 of all three HRAS, KRAS, and NRAS genes [1, 20]. The impaired ability in the conversion of GTP to GDP by RAS mutations results in constitutive activation of signaling in this pathway [24]. Although BRAF mutations, RAS mutations as well as RET rearrangements are all detected in PTCs, these genetic alterations are mutually exclusive [25].
MTCs arise from the calcitonin-secreting parafollicular cells of the thyroid gland. Of thyroid carcinomas, 2 – 3% are due to MTC. Twenty to twenty-five percent of MTCs are inheritable [26]. Both inheritable and up to 50 % of sporadic MTCs have been identified with the gain-of-function point mutation of RET [27], which also undergoes rearrangements in PTC as described earlier [10]. The point mutations often occur in one of several possible codons [26,28]. RET is highly expressed in parafollicular cells. However, in unstimulated conditions, the kinase is inactive due to an inhibition of kinase domain resulting from intramolecular structures. Mutations disrupt the interaction of intramolecular domains, leading to a conformation with active kinase activity. Similar to the RET rearrangement, the mutated RET is constitutively active in the parafollicular cells or other neuroendocrine cells [9].
For FTCs, point mutations of the RAS gene and PAX8-PPARγ rearrangement are the most frequent genetic alterations. Less frequent mutations and other alterations of the genes in the PI3K/AKT signaling pathway can be found in these tumors, although their contribution to FTC remains to be elucidated. RAS mutations are found in 40–50% of conventional follicular carcinomas and in 20–40% of adenomas [29–31]. The most frequently affected hotspots are NRAS codon 61 and HRAS codon 61. The presence of a RAS mutation has been found to correlate with tumor dedifferentiation and a less favorable prognosis [32]. The more aggressive biological properties of these tumors may be due to the effect of the mutant RAS protein on promoting chromosomal instability, which has been demonstrated at least in in vitro experiments [33].
PAX8-PPARγ rearrangement occurs in approximately 35% of FTC [34–36]. Different from RAS mutations, which are frequent in both PTC and FTC, PAX8-PPARγ rearrangement occurs frequently in FTC. The rearrangement results in the translocation t(2;3)(q13;p25), leading to the fusion of the thyroid transcription factor PAX8 gene to the PPARγ gene [37]. Therefore, the rearrangement results in overexpression of the PPARγ protein [37]. Tumors harboring PAX8-PPARγ tend to occur in younger patients and more frequently reveal vascular invasion [35, 36]. However, how the fusion protein causes FTC is still not well understood [38].
As for the etiology of FTC, no link to environmental ionizing radiation exposure and inheritable cancer has been reported. Whether TSH is the initiator of FTC is still controversial. Several lines of evidence, however, have implicated the role of TSH in thyroid tumorigenesis. The follicular carcinomas are most prevalent in iodine-deficient regions [39–42]. An increase in the risk of thyroid tumorigenesis in patients with multinodular goiter has been reported [39, 43–45]. In laboratory animals, it is known that xenobiotics could result in thyroid tumors by lowering T4 levels, leading to an increase in circulating TSH and proliferation of thyroid follicular cells [46, 47]. The xenobiotics inducing a higher TSH level include hepatic Cyp2B inducers as well as inhibitors of thyroperoxidase, 5′-deiodinase and the sodium/iodide symporter [47]. In experimental mice, TSH promotes development of FTC [48, 49]. Recent evidence, however, does not support the role of TSH as an initiator of FTC [50–53]. The identification of multiple pathways associated with thyroid carcinogenesis suggests that additional genetic changes need to occur for the transformation of the TSH-stimulated hyper-proliferative thyroid cells to cancer cells [54, 55]. In support of this notion, Kato et al. reported that in a spontaneously developed mouse model of FTC (TRβPV/PV mice; see section: Follicular Thyroid Carcinoma), PPARγ insufficiency more aggressively promotes the progression of FTC as compared to TRβPV/PV mice with PPARγ. However, TRβPV/PV mice with or without PPARγ insufficiency have similarly highly elevated serum TSH levels [56]. These results indicate that it is less likely TSH alone is sufficient to induce FTC, but collaboration of multiple genetic events is necessary in conferring the cancer phenotype.
In mutant Ki-Ras expressing mice, thyroid glands developed adenomas, though at very low frequency after a long latency. It is believed that the action of an activated Ras gene alone is not sufficient for malignant transformation of follicular cells in vivo. Instead, mutant Ras expression in follicular cells probably represents a favorable genetic ground for tumor development.
Other genetic alterations are also found in thyroid carcinomas. For example, loss of p53 function is the primary genetic alteration identified in the development of anaplastic thyroid carcinomas [57–59], but β-catenin activating mutations have also been detected in these anaplastic thyroid carcinomas, where the protein likely mediates loss of cell-cell adhesions and also acts as a transcription factor in upregulating growth-promoting genes [60].
Mouse Models of Thyroid Cancer
Papillary thyroid carcinoma
The binding of proliferation signal molecules to receptors such as RET and NTRK1 leads to receptor phosphorylation and activation of downstream effectors such as RAS. As a result, BRAF is recruited to phosphorylate and to activate the MEK-ERK signaling. After the activated ERK translocates from cytoplasm into the nucleus, it regulates the transcription of genes involved in cell differentiation, proliferation, apoptosis, and survival. The genetic alteration in thyroid cells such as RET translocation, NTRK1 translocation and BRAF or RAS mutations cause constitutive activation of the MAPK pathway resulting in PTC (Fig. 2). Most mouse models are based on the genetic alterations identified in human tumor cells. The mouse models of PTCs harbor activating genes such as RET/PTC1, RET/PTC3, and TRK-T1 or mutated genes such as BRAF and RAS.
RET/PTC1
RET/PTC1 is one of the most common genetic alterations in PTC and accounts for 70% of RET rearrangement found in that carcinoma. In FVB/N mice, the fusion gene under the control of bovine thyroglobulin promoter was targeted to the thyroid. The mice expressing RET/PTC1 protein consistently developed bilateral PTCs with pathological features similar to human carcinomas [61]. The onset of hyperplasia and carcinomas varied depending on copy numbers of the RET/PTC1 fusion gene expressed in the mice. All mice with low copy number developed the bilateral thyroid carcinomas between 1 and 6 months of age. Mice with high copy numbers had dysplastic thyroid glands at birth. All mice examined developed carcinomas comparable to human PTC as young as 1 month of age [62]. The promoter used to target the gene to the thyroid also affects the manifestation of the phenotypes. For example, when a mouse line expressing RET/PTC1 was created under the control of the rat thyroglobulin promoter, less than half of the mice developed tumors even after 8–16 months [63]. Mice from the high-copy lines had profound congenital hypothyroidism, as indicated by dwarfism, low serum T4 and T3 levels, and marked elevation in serum TSH. T4 supplementation was able to correct the phenotypic dwarfism and was essential for the survival and efficient breeding of these mice. Maintaining the mice on a low iodide diet resulted in persistent elevation of TSH levels and enhanced tumorigenesis [64]. Despite the fact that local invasion was present in all mice examined, distant metastases in mice were not observed up to 5 months of age. The occurrence of metastasis was rare even in the absence of p53 alleles [65]. The lack of metastases is a limitation of this model to mimic human thyroid carcinomas.
RET/PTC3
RET/PTC3 accounts for 30% of all RET/PTC rearrangements. Thyroid-targeted RET/PTC3 gene expression led to follicular cell hyperplasia starting at 2 months of age. PTC occurs in up to 55 % of mice aged 3 months or older [66]. An examination of auxiliary lymph modes in a 10-month-old mouse identified a 1 cm3 mass with features reminiscent of human differentiated PTC with RET/PTC3 immunostaining. In all, two of six mice examined showed evidence of metastases. The dedifferentiation often seen in human carcinomas with RET/PTC3 translocation was not observed in RET/PTC3-induced thyroid tumors.
Recently, microarray gene profiling was used to assess the similarity in the altered gene expression patterns between PTC in humans and RET/PTC3 mice [67,68]. Several human PTC characteristics were observed in RET/PTC3 mice: overexpression of many immune-related genes, regulation of human PTC markers, upregulation of EGF-like growth factors and significant regulation of angiogenesis, and extracellular matrix remodeling-related genes [67, 68]. However, similarities were incomplete in the overall gene expression and the age-dependent gene expression [67]. Therefore, RET/PTC3 mice are partial and transient models of human PTC. But RET/PTC3 mice at younger age could be a useful mouse model to study the biological processes shared between human and mouse PTCs.
BRAFV600E
Mutation of the BRAF gene is the most common genetic alterations in PTCs (occurring in about 45 % of all PTCs). Though at least 20 mutations were identified in the BRAF gene, more than 90 % of all mutations involved the nucleotide 1799 and resulted in a valine-to-glutamic acid substitution at residue 600 (V600E). The expression of BRAFV600E was targeted to thyroid cells of the FVB/N mice by using a bovine thyroglobulin promoter. The mice displayed 2-fold increased TSH, but had a normal level of T3 and somatic growth. A majority of mice with a higher level of BRAFV600E expression developed multifocal bilateral PTC [69, 70]. Invasive carcinomas were frequently observed. Approximately half of the mice aged 12 to 22 weeks revealed focal areas of dedifferentiation composed of solid sheets of spindle cells lacking characteristic nuclear features of papillary carcinoma and showing no evidence of follicular architecture or colloid formation. In the transgenic line with expression of BRAFV600E at a higher level, tumors frequently invaded the vascular vessels and adjacent adipose tissue and skeletal muscle, sometimes leading to tracheal compression. The carcinomas in mice were associated with a 30 % decrease in survival at 22 weeks. However, no evidence of metastasis to distant tissues was found [69].
TRK-T1
Transgenic B6C3F1 mice expressing the fusion protein TRK-T1 were prepared by using the bovine thyroglobulin promoter. Overall, 54 % of transgenic mice with thyroid-targeted TRK-T1 expression displayed a loss of normal thyroid structure. For the mice aged 7 months or younger, 38 % of the thyroids were normal. For mice older than 7 months, all thyroids showed pathological abnormalities. Hyperplasia was evident as early as age 3 months in some mice, but could appear as late as age 10 months in others. Twenty-three percent of the mice aged 7 months or younger and 78 % of the transgenic mice older than 7 months developed thyroid carcinoma with the characteristics of human differentiated carcinomas [71]. However, no local or distant metastases were observed. Nor were lymphocytic infiltrates detected. It appears that the expression of TRK-T1 alone is not sufficient for the manifestation of these advanced tumor phenotypes in the mice. However, the mice provide a model to further study the role of NTRK1 rearrangement in local thyroid tumorigenesis.
RAS
Although HRAS mutations were frequently detected in PTCs, mice that have stable integration of mutated RAS transgenes in the genome are not yet available [72,73]. Transgenic mice that harbor the human mutant RASG12V specifically expressed in the thyroid follicular cells directed by the bovine thyroglobulin promoter were reported [73]. Four independent founder lines of transgenic mice designed as M1, M2, M3, and M4 were generated. Except the M3 mice, the founder mice developed unifocal or unilateral papillary thyroid tumors similar to those observed in the patients with PTC in a range of size between 0.5 and 1.0 cm. The lung metastatses were also observed in two founder mice. But intriguingly, none of them could transmit the phenotypes to offspring. Therefore, the utility of these mice for the study of thyroid cancer is limited.
Recently, a transgenic mouse model expressing a mutated human NRAS(Gln61Lys) specifically in thyroid follicular cells was also established directed by using thyroglobulin promoter [74]. These mice developed carcinomas with mixed papillary-follicular malignant characteristics. The detailed description of this mouse model is provided in the section of follicular thyroid carcinomas (see Section Follicular Thyroid Carcinoma).
Adenosine A2 receptor/papillomavirus type 16 E7 oncogene
Cyclic AMP (cAMP) is an intracellular second messenger to mediate TSH stimulated growth and differentiation of thyroid follicular cells. The adenosine A2 receptor, which activates adenylyl cyclase via coupling to the stimulating G protein (Gs), has been shown to promote constitutive activation of the cAMP cascade when transfected into various cell types. Transgenic mice expressing the canine adenosine A2 receptor were generated under control of the bovine thyroglobulin gene promoter. Thyroid-specific expression of the adenosine A2 receptor promotes thyroid hyperplasia, goiter, and severe hyperthyroidism causing premature death of these mice [50].
The oncogenic functions of human papillomavirus E7 protein are attributed to its interaction with the retinoblastoma susceptibility gene product, RB1, and other related proteins. Mouse model expressing the E7 oncogene was generated under the control of the bovine thyroglobulin gene promoter. When these mice aged, they developed well-differentiated follicular and papillary carcinomas [49].
Interbreeding two transgenic lines generated mice expressing both the adenosine A2 receptor and the E7 oncogene. These mice developed larger goiters and more severe hyperthyroidism as compared with the respective parental mice. The mice rapidly developed malignant thyroid tumors similar to those of human PTC and disseminated through the blood stream, generating multiple differentiated tumors in the lung. These metastatic tumors appeared as early as 2 months after birth and their frequency increased to 75 % in mice older than 3 months [75]. Due to the high incidence of metastatic tumors at the earlier stage, these mice may be potentially useful to dissect the multiple signaling pathways required for distant metastasis.
Follicular Thyroid Carcinoma
Most models for FTC were established based on the genetic alterations discovered in human tumors. Ras mutations and PAX8-PPARγ rearrangement are common genetic alterations observed in FTC. However, mouse models based on these genetic alterations have not been documented to fully recapitulate human FTC. Instead, the models established from several mutant genes such as the gene encoding α1B-adrenergic receptor, Rap1bG12V, or thyroid hormone receptor β mutant (TRβPV) have been used to study the development of FTC.
Mutant α1B-adrenergic receptor
The main regulator of thyroid function and growth is TSH. The signals from three pathways control the TSH-stimulated proliferation of thyroid follicular cells: cAMP cascade, phospholipase C pathway, and thyrosine kinase/RAS/mitogen-activated protein kinase pathway. TSH receptor activation leads to the stimulation of the cAMP cascade. It is well established that activation of the cAMP cascade maintains differentiation of thyrocytes and leads to the activation of both their function and proliferation. A second cascade that has been shown to promote proliferation while inducing dedifferentiation in thyroid cells in culture is the diacylglycerol (DAG)/protein kinase C branch of the phospholipase C pathway. Phospholipase C can be stimulated in rodent cells by α1-adrenergic agonists. The third cascade stimulating proliferation is the thyrosine kinase/RAS/mitogen-activated protein kinase pathway.
In a transgenic model, the bovine thyroglobulin gene promoter was used to direct the expression of a constitutively active mutant of the α1B-adrenergic receptor gene that stimulated both the adenylyl cyclase and phospholipase C pathways in thyroid follicular cells [48]. The thyroids were increased in size in the first few weeks, and some reached a weight of 130 mg by 12 months. The enlarged gland preserved the general organization of follicles at the early stage. The mean size and number of the nodules increased as mice aged. The organization of the gland became increasingly heterogeneous, with the appearance of actively proliferating nodules. In larger nodules, foci of cells were found with large nuclei with morphological criteria that characterize human papillary carcinomas. In a few cases, all morphological criteria of thyroid follicular cell differentiation were lost; spindle cell foci were found without formation of identifiable follicles. They frequently acquired structural characteristics considered malignant criteria for human thyroid tumors. Some growing nodules invaded muscular tissues. Some nodules appeared highly vascularized. Differentiated lung metastases were observed at an overall frequency estimated to be 20% in mice older than12 months. The manifestation of the advanced tumor features in this mouse model may be useful for the studies of the invasion and metastasis mediated by cAMP and phospholipase C signaling pathways in FTC.
RAS
RAS is one of the frequent mutations detected in FTC. A mouse model was generated bearing a fusion construct of rat thyroglobulin promoter and the mutant KRAS gene. Up to age 20 months, all mice examined were apparently healthy with a normal growth rate and serum levels of thyroid hormones, although histological examination identified some abnormalities such as disorganized follicles and adenomas. The treatment of the mice with goitrogen increased the number of adenomas. One mass with the characteristic of carcinomas was also identified in the mouse treated with goitrogen for 6 months [76], suggesting that while RAS mutation alone is not sufficient to induce follicular thyroid carcinomas, it acts as a predisposing factor [76]. TSH could promote the manifestation of RAS genotypes in these mice. The observations in this mouse model suggest that multiple genetic alterations could contribute to the development of FTC. The mice could be used for the study of the genetic actors that promote the development of FTC.
Recently, a transgenic mouse model expressing a mutated human NRAS(Gln61Lys) specifically in thyroid follicular cells was also established directed by the thyroglobulin promoter [74]. Thyroid histology of transgenic mice was compared to that of wild type littermate controls at 6, 12, and 18 months of age. Progressive lesions from follicular cell hyperplasia to adenoma and carcinoma were observed in these transgenic mice. Among the 88 mice examined, 26 mice developed follicular carcinomas and 9 had invasive carcinomas with large poorly differentiated areas closely resembling those observed in human patients. Mice with carcinomas displayed well-differentiated follicular or mixed papillary-follicular malignant features. Furthermore, distant metastases were observed in the livers of three mice, lung of two mice, and the right femur of one mouse. Although the aggressive features require long latency, the mice are potentially useful for the studies of thyroid carcinogenesis.
Rap1bG12V
Rap1 GTPases are members of the Ras superfamily of G-proteins, which regulate cell proliferation and differentiation. cAMP signaling leads to activation and phosphorylation of Rap1b. In the model, the expression of Cre-floxed active Rap1bG12V or the dominant negative mutant Rap1bS17N was driven by the thyroglobulin promoter. These mice express Rap1bG12V specifically in thyrocytes. They were crossed with CRE-ER-Tx mice to generate a tamoxifen-inducible switch model. Upon stimulation of Cre activity by tamoxifen, these mice ceased to express active Rap1bG12V and instead produced the inactive Rap1bS17N [77].
Mice expressing Rap1bG12V showed no signs of hypo- or hyper-thyroidism and exhibited normal growth profiles, indicating normal physiological levels of thyroidal, growth, and gonadal hormones. Further analysis of these mice did not detect signifficant differences in TSH levels and thyroid histology. Thus, under basal low thyroidal cAMP signaling, expression of Rap1bG12V does not interfere with differentiation of the gland and does not produce any mitogenic advantage in either heterozygous or homozygous mice. However, chronic elevation of thyroidal cAMP by increasing circulating levels of TSH produced a switch to mitogenic action in Rap1bG12V heterozygous mice. Exposure of Rap1bG12V mice to a 6-month goitrogen program led to marked thyroid enlargements with nodular lesions. The hyperplastic and nodular phenotype observed in the Rap1bG12V mouse thyroids after 6 months of sustained elevation of thyroidal cAMP was completely reversed after removal of the goitrogenic stimulus for 2 months. The serum TSH concentrations and thyroid hormones returned to normal levels, followed by re-establishment of typical follicular structures. However, after long-term goitrogenic treatment, all Rap1bG12V transgenic but not the WT mice developed very large multi-lobular and hyperemic glands. These glands exhibited signs of FTC characterized by the presence of invasion of the thyroid capsule, surrounding tissues, and blood vessels. Both extracapsular invasion and vascular invasion were verified by tagged HA staining, but no evidence of metastasis could be found despite a complete necropsy. The absence of correlation between invasion and metastasis is not well explained. Despite initial vascular invasion, it seems that most of the malignant cells are destroyed in the circulation, though any evidence of apoptotic cells or HA stain in vessels of several potential metastatic targets were not established. When the Rap1b “switch” was made from constitutively active Rap1bG12V to dominant negative Rap1bS17N, the thyroid gland size was reduced by 50 % in the face of continued goitrogen treatment. Ki67, BrdU, and PCNA labeling indices and terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling indicated that decreased cellular proliferation, rather than increased apoptosis, accounted for the decrease in thyroid hyperplasia. It seems that Rap1b is oncogenic in the presence of elevated TSH-mediated signaling. The model might be useful in studies of multiple factors for their contribution to the development of thyroid cancer.
TRβPV/PV
The TRβPV/PV knockin mouse was initially created to model an inheritable disease called resistance to thyroid hormone (RTH). RTH is a syndrome of reduced tissue sensitivity to thyroid hormone. The hallmark of RTH is elevated serum thyroid hormone levels associated with non-suppressible TSH. Other clinical signs are goiter, short stature, decreased weight, tachycardia, hearing loss, attention-deficit hyperactivity disorder, decreased IQ, and dyslexia. Most patients are heterozygous and the clinical symptoms are mild [78]. Only one patient homozygous for mutant THRB has been reported [79]. This patient, who died at a young age, displayed an extraordinary and complex phenotype of extreme RTH with high levels of thyroid hormone and TSH [79]. Thyroid hormone receptors (TRs) are members of the ligand-dependent transcription factor family encoded by the THRA and THRB genes. Alternative splicing of the TRβ gene transcript gives rise to three T3-binding proteins (β1, β2 and β3). TRβPV/PV mice were created by targeting the PV mutation to the TRβ gene locus via homologous recombination and the Cre-lox system [80]. The PV mutation was identified in a patient with RTH [81] with a frameshift mutation in the C-terminal 14 amino acids of TRβ, resulting in a complete loss of T3 binding and transcriptional capacity. The loss of negative feedback by T3 on the hypothalamus-pituitary-thyroid axis due to PV mutation led to persistent upregulation of TSH and elevated thyroid hormones [80].
Remarkably, as TRβPV/PV mice aged, a significant increase in their morbidity was observed when compared with wild-type and TRβPV/+ mice. The cumulative survival analysis showed that less than 10% of the TRβPV/PV mice survived at the age of 14 months whereas 95% of TRβPV/+ heterozygous mice survived. Detailed histopathological examination of thyroid glands led to the discovery that TRβPV/PV mice spontaneously develop FTC resembling the pathological progression of human thyroid cancer. In homozygous (TRβPV/PV) mice, follicular cell hyperplasia appears around 3 weeks of age. As early as 2 months of age, the mutant mice exhibited papillary hyperplasia. By 4–5 months of age, the extent of hyperplasia was increased and signs of FTC such as capsular and vascular invasion were already apparent. Distant metastases via vascular vessels to the lungs and heart were common in the mice over 5 months of age. The pathological progression was sequentially from hyperplasia (100 % of all mice examined), capsular invasion (91%), vascular invasion (74%), anaplasia (35%) to eventual distal metastasis (30%) (Fig. 3). Metastasis involves mainly the lung and occasionally the endocardium, but not the local lymph nodes. The small, round, and dark nuclei of the developed thyroid carcinomas are characteristic of follicular carcinoma. These characteristics are significantly different from that of papillary carcinoma [82]. The findings that TRβPV/PV mice spontaneously develop FTC similar to human cancer indicate that these mice could be used as a model to elucidate the molecular genetic changes underlying FTC and to identify potential molecular targets for treatment and diagnosis. Indeed, consistent with human FTC, PPARγ was shown to be a tumor suppressor in TRβPV/PV mice [56,83]. Treatment of TRβPV/PV mice with a PPARγ agonist, rosiglitazone, delays the progression of thyroid carcinogenesis, indicating the PPARγ agonists could be tested as a therapeutic agent for FTC [56]. Moreover, similar to human FTC, activation of the PI3K-AKT signaling was demonstrated in thyroid tumors of TRβPV/PV mice [84–86], suggesting that PI3K could be a potential molecular target. Indeed, treatment of TRβPV/PV mice with a specific PI3K inhibitor, LY294002, delays tumor progression and blocks metastatic spread [87]. Thus, PI3K and its downstream effectors could be explored as therapeutic targets for the treatment of human FTC. In addition, consistent with observations in studies of human thyroid cancer, PTTG1 was found to be highly elevated in the thyroid tumors of TRβPV/PV mice. Further studies indicate that overexpression of PTTG1 not only inhibits mitotic progression and causes chromosomal aberrations [88, 89], but also promotes angiogenesis to contribute to thyroid carcinogenesis [90].
Fig. 3.
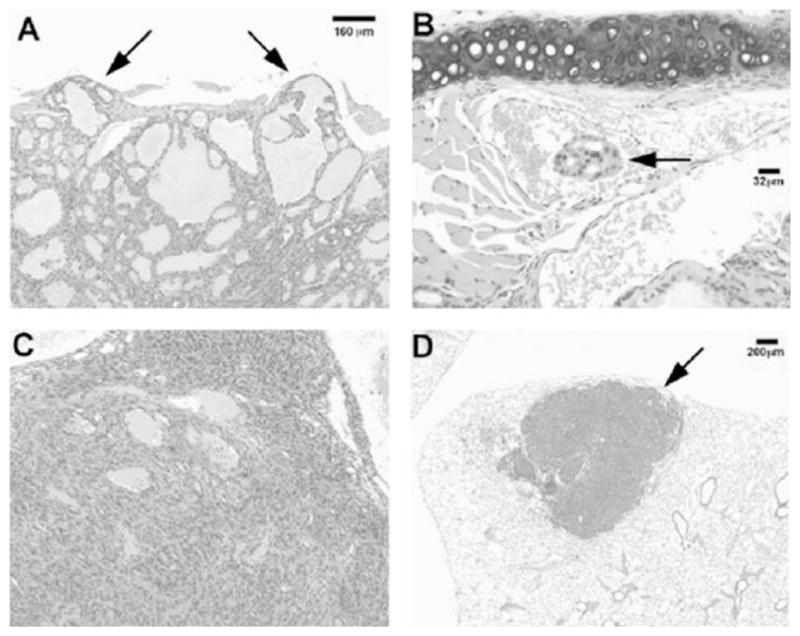
TR3PV/PV mice spontaneously develop follicular thyroid carcinogenesis with pathological progression similar to human thyroid cancer. Panel A shows capsular invasion (arrows) in the thyroid, panel B shows a focus of vascular invasion (arrow), panel C shows an area of anaplasia (formation of spindle cells) in the thyroid, and panel D shows a low magnification of a metastatic lesion (arrow) in the lung.
TRβPV/PV mice proved to be a useful mouse model to uncover novel genetic changes during FTC. Using TRβPV/PV mice, Kim et al. found that gelsolin, a novel TRβ interacting protein, exhibits the function of modulating tumor progression [91]. Importantly, using TRβPV/PV mice, Ying et al. showed that the steroid hormone receptor coactivtor-3, an oncogene in breast cancer [92], is a tumor promoter in thyroid carcinogenesis [93]. Moreover, with the use of TRβPV/PV mice, novel nongenomic actions of TRβ mutants were discovered, including the regulation of the stability of key cellular regulators, β-catenin and PTTG1, and the identification of AKT as a novel interacting protein of TRβ [88,94,95].
Beside the TRβPV/PV mouse, spontaneous development of FTC was also consistently observed in the mice with one mutated TRβ (TRβPV), but in the absence of the wild-type TRβ allele [96]. This TRβPV/− mouse was generated by cross-breeding TRβPV/+ mice with TRβ knockout mice [96]. The pathological progression of thyroid carcinomas in TRβPV/− mice was indistinguishable from that in TRβPV/PV mice. These results indicate that in the absence of a wild-type allele, the mutation of one TRβ allele is sufficient for the mutant mice to spontaneously develop follicular thyroid carcinoma. These results provide, for the first time, in vivo evidence to suggest that the TRβ gene could function as a tumor suppressor gene. Importantly, these findings present the possibility that the TRβ receptor could serve as a novel therapeutic target in thyroid cancer.
Anaplastic Thyroid Carcinoma
SV40 Large T antigen
Anaplastic thyroid carcinoma was modeled in mice expressing an oncogene derived from the polyomavirus SV40. SV40 large T antigen (TAg) is an oncoprotein capable of transforming a variety of cell types. The transforming activity of TAg is due to its perturbation of activities of the retinoblastoma protein (pRB), p53 tumor suppressor, and other cellular factors, including transcriptional co-activators p300 and CBP. The coding region for the simian virus-40 large T-antigens fused to bovine thyroglobulin gene promoter was directed to its expression in thyroid follicular cells. The mice developed a dramatic enlargement of the thyroid gland accompanied by compression of trachea and esophagus, dyspnea, inspiratory stridor, dysphagia and hypothyroidism, frequently leading to premature death. The thyroid of these transgenic mice exhibited hyperplasia at birth, and the mice developed poorly differentiated thyroid adenocarcinomas at an early age. The cultured follicular cells derived from these large T-expressing thyroids lost expression of both thyroglobulin and thyroperoxidase, though low levels of TSH receptors were detected. Local invasion was frequently observed in these mice with rare distant lung metastasis [97]. Due to the lack of the fully developed thyroid follicles, mice were severely hypothyroid and required supplementation of T4 for growth and survival to produce offspring. Although the severe hypothyroidism observed in this model is not common in human thyroid carcinomas, the mouse model is useful for studying the mechanism of the dedifferentiation of aggressive thyroid cancer.
Medullary Thyroid Carcinoma
RET is highly expressed in parafollicular cells. Binding of its ligands, the glial-derived neurotrophic factors, triggers RET homodimerization and tyrosine phosphorylation and subsequent activation to increase cell proliferation [3]. In parafollicular cells, mutations of RET have been found to constitutively activate cellular signaling, resulting in persistent cell proliferation and parafollicular carcinomas. The models for MTCs were generated by over-expressing oncogene v-Ha-ras or mutated oncogenic RET in transgenic mice. Mice deficient in the expression of tumor suppressor Rb and/or p53 were also reported as models of medullary thyroid carcinoma.
RETC634R
Both inheritable and sporadic MTCs were found to have the mutations in the RET gene. In patients with MEN-2A, a point mutation in the RET gene (C634R) was identified [98]. A transgenic mouse model was generated that targeted the expression of RETC643R in the C cells under the control of the C cell-specific calcitnonin/calcitonin gene-related peptide promoter (CGRP) [28]. Mice in two established transgenic lines developed MTC at age 14 months with elevated circulating calcitonin levels three times higher than that of nontransgenic mice. Of interest is that one of the two lines (designated as #5319) developed bilateral C cell hyperplasia as early as 3 weeks of age and subsequently progressed to multifocal and bilateral medullary thyroid carcinomas [28]. Microscopic foci of MTC were detected in the mice between 2 and 7 months of age. In the mice older than 8 months, bilateral and multifocal MTC were readily detected with the tumor size ranging from 5 to 30 mm. In this line, distant metastasis to liver with multiple nodes as observed in the advanced stage of human MTC was detected in one of eight mice examined [28]. These tumors were morphologically indistinguishable from patients with MEN-2A. This model could be useful to further study the mechanism of the tumorigenesis induced by the mutant RET gene.
Rb and p53
The retinoblastoma susceptibility (RB) and p53 genes encode tumor suppressors involved in the control of cell growth. Loss or mutations of these tumor suppressor genes have been reported in many types of human cancers. It was found that almost all examined single Rb mutant allele developed pituitary adenoma. However, the heterozygous Rb mice with the combination of p53 homozygous or heterozygous deficiency developed multiple endocrine tumors in addition to lymphomas and sarcomas, with a 40% incidence of medullary carcinomas. Approximately 37% of mice with simultaneous heterozygous deletional mutation of both tumor suppressors developed C-cell hyperplasia and medullary thyroid carcinomas [99]. Tumors were well differentiated and distant metastases were rare [100, 101]. The tumor types seen in this model are similar to those observed in MEN-2. Although the mice with a deficiency of tumor suppressors are not the correct genetic model for this syndrome, they may be useful to further study the inheritable endocrine tumorigenesis.
v-Ha-ras
Though mutated RAS have not been detected in MTC, it is known that v-Has-ras mutated at residues 12 and 69 induce neuroendocrine differentiation of MTC cell lines in vitro [102]. The majority of hemizygous mice expressing v-Ha-ras in C cells under control of the CGRP promoter developed uni- or bilateral medullary thyroid carcinoma within one year of age [103]. The tumors grew from small nodules of C-cell hyperplasia into locally invasive and hemorrhagic metastatic foci. Two of 22 thyroid tumors were associated with lung metastases; another thyroid tumor was associated with local nodal metastases; no other metastases were evident. Tumors in these mice secreted calcitonin. However, the roles of RAS mutations in MTC remain to be elucidated.
Perspectives and Future Challenges in Modeling Thyroid Cancer
In the last decade, significant progress has been made in the creation of rodent models of thyroid cancers. Still, modeling of thyroid cancer could be further improved to faithfully recapitulate human thyroid cancer not only to elucidate molecular genetic changes underlying cancer development and progression, but also to develop novel agents for diagnosis and treatment.
The transgenic mouse models of PTC expressing activated kinases such as RET, BRAF or RAS, capture many pathological characteristics of human thyroid cancer. Unfortunately, these models often lack the desirable advanced features of dedifferentiation and metastasis that are the main causes for failed radioactive iodine treatment [61, 69].
MEN-2A is a subtype of multiple endocrine neoplasia type 2 characterized by familial MTC associated with pheochromocytomas and hyperparathyroidism. The features of bilateral and multicenteric tumors with hyperparathyroidism of the inheritable MEN-2A syndrome were reproduced in the mice using RETC634R though pheochromocytoma was not observed [28]. In the RETC634R model, variations in tumor penetrance between mouse background strains were observed [104]. While tumors were observed in 98% of CBA/ca mice, the FVB/N mice expressing RETC634R backcrossed from the same founder did not develop any tumor [104]. This study highlights the concerns about the impact of mouse background strains on the manifestation of phenotypes.
Many features of human FTC have been recapitulated in the TRβPV/PV mouse. The TRβPV/PV mouse is an unique mouse model as it closely resembles human FTC in terms of pathological progression and its underlying genetic alterations [55,56,82–91], yet it is not known whether the only reported patient with homozygous mutation of the TRβ gene [79] developed thyroid cancer. The other concern for this model is the presence of hyperthyroidism and highly elevated TSH due to thyroid hormone resistance displayed in TRβPV/PV mice. Currently, the roles of thyroid hormones and TSH in thyroid tumorigenesis remain to be fully elucidated. Since FTC are often associated with dietary iodide deficiency or perturbations of the pituitary-thyroid axis [46, 47], it is important to understand whether chronic TSH stimulation and disturbance of the pituitary-thyroid axis could contribute to thyroid carcinogenesis. Thus, the TRβPV/PV mouse presents an opportunity to dissect the crosstalk in the pathways of a TRβ mutant-and TSH-signaling and to clarify whether TSH is involved in the initiation of thyroid carcinogenesis.
Current models for thyroid papillary or medullary carcinomas are based on the genetic alterations discovered in human thyroid tumors. By using target tissue specific-promoters, mutant genes are directed to the target tissues of transgenic mice. One of the perplexing observations pertaining to these transgenic mouse models is that they frequently lack advanced tumor features such as dedifferentiation. One possible explanation is that transgenes are under the control of a cell-specific promoter such as the thyroglobulin promoter. Since the cell-specific promoter is not active in undifferentiated cells, the expression of a transgene would be suppressed once a cell becomes undifferentiated. A possible approach to overcome this difficulty would be to use a promoter that could be activated in undifferentiated cells or a promoter that could be independent of the status of cell differentiation. The promoter that could be induced by an exogenous agent such as an inducible promoter in the undifferentiated cells might also be useful.
For mouse models that were generated using genetic alterations found in human cancer, the altered gene is expected to express and initiate phenotypic changes at the early stages of development. Therefore, this approach is suitable for mouse models for an inheritable thyroid cancer such as MEN-2A. Indeed, in the mouse model expressing RETC634R transgene, the development of MTC and hyperparathyroidism was similar to that observed in the patient with MEN-2A [28]. However, inheritable medullary cancer only accounts for less than 3% of all thyroid carcinomas [1,3,9]. More than 97% of naturally occurring human thyroid tumors result from somatic mutations that occur in a person born with a normal functional thyroid gland [1]. Unfortunately, for most of the transgenic mice, normal development of thyroids was disturbed, often with abnormal thyroid functions such as hyperthyroidism or hypothyroidism since the expression of the activated oncogenes is turned on at an early embryonic stage [61, 97]. To model thyroid cancer derived from somatic mutations, it is logical that the mutation be introduced at a single cell to initiate carcinogenesis at a specific stage after the full development of the thyroid gland.
Recent publications have demonstrated several approaches that could be used to introduce a somatic mutation into cells at a specific developmental stage. One strategy is to use a recombinase-mediated activation [105, 106]. In this approach, an oncogene placed downstream of a stop codon signal is introduced into the mouse embryonic cell genome by using a microinjection gene transfer procedure. Although an oncogene is integrated into the mouse genome at an early embryonic stage, the mRNA of this introduced gene is transcribed, but it cannot be translated into proteins unless the signal to suppress the gene translation is removed. This approach was exemplified in a model of lung cancer [105]. The sequence containing K-Ras was first introduced into the site downstream of a stop signal within recombinase recognizing sequences. The silent K-Ras oncogene was then activated by a Cre-recombinase in an adenovirus vector locally delivered to the lung [105]. The advantages of this approach over transgenic embryonic activation are that the number of the activated cells, activation sites and timing could be well controlled according the purposes of the studies. As compared with the mouse model in which mutant RAS is expressed throughout the lung, this model provides a closer recapitulation of human lung cancer, in which tumor forms and progresses from a single individual somatic cell harboring an activated oncogene gene [105]. However, the activation of the oncogene in the recombinase-mediated model does not naturally occur, in that the recombinase expressing adenovirus is required to remove the stop codon sequence element. Another mouse model for lung cancer that closely mimics sporadic somatic mutations occurring in human tumors has also been reported [107]. In this approach, endogenous intra-chromosomal recombination mechanisms were utilized to simulate the random somatic activation of K-Ras oncogene. To make the chromosome susceptible for a mutagenic recombination event, one additional nonfunctional partial K-Ras gene copy was inserted into the genomic site of the endogenous K-Ras allele in tandem. Intra-chromosomal recombination between homologous sequences was estimated to occur at a frequency between 10−3 and 10−7 per cell generation and generated a single functional oncogenic K-Ras allele. In the model, 100 % of these mice developed early onset lung adenocarcinomas [105].
In thyroid cancer, gene translocation/fusion is commonly observed. Several mouse models were established based on the activated oncogene resulting from gene translocation such as RET/PTC1. In these models, copies of the activated oncogene were integrated into the mouse genome. A problem associated with transgenic mouse models is the marked variability in the manifestation of the phenotypes of the different founder mouse lines [61–63,69]. The different copies of the inserted oncogene or different integration sites of the oncogene could explain the markedly variable phenotypes in lines derived from different founders. One approach to reduce the variability would be to mimic the translocation to direct the expression of the oncogene as observed in human thyroid carcinomas. The induced translocation could be experimentally achieved in the transgenic model [108–110]. To induce translocation, sequences such as loxP sites were inserted into two separate nonhomologous chromosomal sites to make them susceptible to the homologous recombination. Recombinase expression activates the translocation event [108]. This approach has been used to generate better models of human leukemia-associated translocations, such as the t(8;21)(q22;q22) [109] and t(9;11)(p22;q23) [110] translocations, which cause acute leukemia [109, 110].
Thyroid cancer patients have better 5-year and 10-year survival rates than patients with other cancers. However, thyroid cancer is one of the few cancer types with increasing incidence in the United States and around the globe, particularly among women [111]. Though surgery or radiation therapy or both are effective against local differentiated carcinomas, the choices of successful treatments are limited for dedifferentiated, invasive, or meta-static cancers. The need for further therapeutic development for such cancer is clear. Small molecules of the inhibitors against RET, RAS, BRAF, and ERK in the proliferation signaling pathways in tumors of follicular and C cell origin have been extensively examined. These inhibitors are usually tested in mouse xenograft models of thyroid cancer before investigators move on to clinical trials. The most commonly used cell lines in SCID mouse models of thyroid cancer are papillary thyroid carcinoma cell lines: TPC-1, FB2, K1, B-CPCP and NPA; follicular thyroid carcinoma cell lines: FRO, RTC-R2, FTC-133 and FTC-238; anaplastic thyroid carcinoma cell lines: DRO, WRO, KAT-4 and ARO; and medullary thyroid carcinoma cell lines: TT and rMTC. Although these xenograft models are easy to produce and the progression of tumors can be easily monitored, increasing evidence indicates that the xenograft models might not be adequate for the prediction of a clinical response. While it seems that transgenic models are valid alternative systems for preclinical tests to evaluate the feasibility, efficacy, and safety of new therapeutic candidates against thyroid cancer, some important issues should thoroughly be taken into consideration: (1) One obstacle is the difficulty in evaluating the tumor growth in a transgenic mouse except for a surface tumor such as skin cancer. One way to circumvent this difficulty is to use an in vivo imaging system such as fluorescence imaging or bioluminescence imaging. (2) Another obstacle is the high cost involved in developing new drugs. Therefore, more convenient and less expensive approaches would be desirable to assess tumor formation. One possible approach would be to integrate enzymes such as β-galactosidase into the genome of tumor cells so that the growth of the tumor and the efficacy of the treatment against the tumorigenesis could be monitored simply by β-galactosidase activity in the serum. (3) One of the most important issues for preclinical application of mouse models is that the potential targets for therapeutic intervention should be humanized so that the therapeutic agents such as monoclonal antibodies or small molecular kinase inhibitors could be tested in the mouse model and applied directly to treat human subjects. Only then would the transgenic models be more likely to become part of effective screening procedures for drugs against cancer.
Acknowledgments
We regret any reference omissions due to length limitation. We wish to thank all colleagues and collaborators who have contributed to the work described in this review. The present research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. We are deeply indebted to Drs. Pilar Santisteban, Sissy Jhaing, and Mark Willingham for the critical reading of the manuscript and valuable suggestions.
References
- 1.Nikiforova MN, Nikiforov YE. Molecular genetics of thyroid cancer: implications for diagnosis, treatment and prognosis. Expert Rev Mol Diagn. 2008;8:83–95. doi: 10.1586/14737159.8.1.83. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 3.Lodish MB, Stratakis CA. RET oncogene in MEN2, MEN2B, MTC and other forms of thyroid cancer. Expert Rev Anticancer Ther. 2008;8:625–632. doi: 10.1586/14737140.8.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mauchamp J, Mirrione A, Alquier C, Andre F. Follicle-like structure and polarized monolayer: role of the extracellular matrix on thyroid cell organization in primary culture. Biol Cell. 1998;90:369–380. [PubMed] [Google Scholar]
- 5.De Felice M, Di Lauro R. Thyroid development and its disorders: genetics and molecular mechanisms. Endocr Rev. 2004;25:722–746. doi: 10.1210/er.2003-0028. [DOI] [PubMed] [Google Scholar]
- 6.Lazzaro D, Price M, de Felice M, Di Lauro R. The transcription factor TTF-1 is expressed at the onset of thyroid and lung morphogenesis and in restricted regions of the foetal brain. Development. 1991;113:1093–1104. doi: 10.1242/dev.113.4.1093. [DOI] [PubMed] [Google Scholar]
- 7.Postiglione MP, Parlato R, Rodriguez-Mallon A, Rosica A, Mithbaokar P, Maresca M, Marians RC, Davies TF, Zannini MS, De Felice M, Di Lauro R. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proc Natl Acad Sci USA. 2002;99:15462–15467. doi: 10.1073/pnas.242328999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meunier D, Aubin J, Jeannotte L. Perturbed thyroid morphology and transient hypothyroidism symptoms in Hoxa5 mutant mice. Dev Dyn. 2003;227:367–378. doi: 10.1002/dvdy.10325. [DOI] [PubMed] [Google Scholar]
- 9.Castellone MD, Santoro M. Dysregulated RET signaling in thyroid cancer. Endocrinol Metab Clin North Am. 2008;37:363–374. viii. doi: 10.1016/j.ecl.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Nikiforov YE. RET/PTC rearrangement in thyroid tumors. Endocr Pathol. 2002;13:3–16. doi: 10.1385/ep:13:1:03. [DOI] [PubMed] [Google Scholar]
- 11.Nikiforova MN, Stringer JR, Blough R, Medvedovic M, Fagin JA, Nikiforov YE. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science. 2000;290:138–141. doi: 10.1126/science.290.5489.138. [DOI] [PubMed] [Google Scholar]
- 12.Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Della Porta G, Fusco A, Vecchio G. PTC is a novel rearranged form of the ret protooncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell. 1990;60:557–563. doi: 10.1016/0092-8674(90)90659-3. [DOI] [PubMed] [Google Scholar]
- 13.Bongarzone I, Butti MG, Coronelli S, Borrello MG, Santoro M, Mondellini P, Pilotti S, Fusco A, Della Porta G, Pierotti MA. Frequent activation of ret protooncogene by fusion with a new activating gene in papillary thyroid carcinomas. Cancer Res. 1994;54:2979–2985. [PubMed] [Google Scholar]
- 14.Boice JD., Jr Radiation-induced thyroid cancer – what’s new? J Natl Cancer Inst. 2005;97:703–705. doi: 10.1093/jnci/dji151. [DOI] [PubMed] [Google Scholar]
- 15.Cardis E, Kesminiene A, Ivanov V, Malakhova I, Shibata Y, Khrouch V, Drozdovitch V, Maceika E, Zvonova I, Vlassov O, Bouville A, Goulko G, Hoshi M, Abrosimov A, Anoshko J, Astakhova L, Chekin S, Demidchik E, Galanti R, Ito M, Korobova E, Lushnikov E, Maksioutov M, Masyakin V, Nerovnia A, Parshin V, Parshkov E, Piliptsevich N, Pinchera A, Polyakov S, Shabeka N, Suonio E, Tenet V, Tsyb A, Yamashita S, Williams D. Risk of thyroid cancer after exposure to 131I in childhood. J Natl Cancer Inst. 2005;97:724–732. doi: 10.1093/jnci/dji129. [DOI] [PubMed] [Google Scholar]
- 16.Klugbauer S, Lengfelder E, Demidchik EP, Rabes HM. High prevalence of RET rearrangement in thyroid tumors of children from Belarus after the Chernobyl reactor accident. Oncogene. 1995;11:2459–2467. [PubMed] [Google Scholar]
- 17.Tallini G. Molecular pathobiology of thyroid neoplasms. Endocr Pathol. 2002;13:271–288. doi: 10.1385/ep:13:4:271. [DOI] [PubMed] [Google Scholar]
- 18.Bongarzone I, Fugazzola L, Vigneri P, Mariani L, Mondellini P, Pacini F, Basolo F, Pinchera A, Pilotti S, Pierotti MA. Age-related activation of the tyrosine kinase receptor protooncogenes RET and NTRK1 in papillary thyroid carcinoma. J Clin Endocrinol Metab. 1996;81:2006–2009. doi: 10.1210/jcem.81.5.8626874. [DOI] [PubMed] [Google Scholar]
- 19.Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63:1454–1457. [PubMed] [Google Scholar]
- 20.Namba H, Rubin SA, Fagin JA. Point mutations of ras oncogenes are an early event in thyroid tumorigenesis. Mol Endocrinol. 1990;4:1474–1479. doi: 10.1210/mend-4-10-1474. [DOI] [PubMed] [Google Scholar]
- 21.Benvenga S. Update on thyroid cancer. Horm Metab Res. 2008;40:323–328. doi: 10.1055/s-2008-1073155. [DOI] [PubMed] [Google Scholar]
- 22.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 23.Ciampi R, Nikiforov YE. RET/PTC rearrangements and BRAF mutations in thyroid tumorigenesis. Endocrinology. 2007;148:936–941. doi: 10.1210/en.2006-0921. [DOI] [PubMed] [Google Scholar]
- 24.Saxena N, Lahiri SS, Hambarde S, Tripathi RP. RAS: target for cancer therapy. Cancer Invest. 2008;26:948–955. doi: 10.1080/07357900802087275. [DOI] [PubMed] [Google Scholar]
- 25.Riesco-Eizaguirre G, Santisteban P. New insights in thyroid follicular cell biology and its impact in thyroid cancer therapy. Endocr Relat Cancer. 2007;14:957–977. doi: 10.1677/ERC-07-0085. [DOI] [PubMed] [Google Scholar]
- 26.Quayle FJ, Moley JF. Medullary thyroid carcinoma: including MEN 2A and MEN 2B syndromes. J Surg Oncol. 2005;89:122–129. doi: 10.1002/jso.20184. [DOI] [PubMed] [Google Scholar]
- 27.Gimm O, Dralle H. C-cell cancer – prevention and treatment. Langenbecks Arch Surg. 1999;384:16–23. doi: 10.1007/s004230050168. [DOI] [PubMed] [Google Scholar]
- 28.Michiels FM, Chappuis S, Caillou B, Pasini A, Talbot M, Monier R, Lenoir GM, Feunteun J, Billaud M. Development of medullary thyroid carcinoma in transgenic mice expressing the RET protooncogene altered by a multiple endocrine neoplasia type 2A mutation. Proc Natl Acad Sci USA. 1997;94:3330–3335. doi: 10.1073/pnas.94.7.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Esapa CT, Johnson SJ, Kendall-Taylor P, Lennard TW, Harris PE. Prevalence of Ras mutations in thyroid neoplasia. Clin Endocrinol (Oxf) 1999;50:529–535. doi: 10.1046/j.1365-2265.1999.00704.x. [DOI] [PubMed] [Google Scholar]
- 30.Motoi N, Sakamoto A, Yamochi T, Horiuchi H, Motoi T, Machinami R. Role of ras mutation in the progression of thyroid carcinoma of follicular epithelial origin. Pathol Res Pract. 2000;196:1–7. doi: 10.1016/S0344-0338(00)80015-1. [DOI] [PubMed] [Google Scholar]
- 31.Suarez HG, du Villard JA, Severino M, Caillou B, Schlumberger M, Tubiana M, Parmentier C, Monier R. Presence of mutations in all three ras genes in human thyroid tumors. Oncogene. 1990;5:565–570. [PubMed] [Google Scholar]
- 32.Basolo F, Pisaturo F, Pollina LE, Fontanini G, Elisei R, Molinaro E, Iacconi P, Miccoli P, Pacini F. N-ras mutation in poorly differentiated thyroid carcinomas: correlation with bone metastases and inverse correlation to thyroglobulin expression. Thyroid. 2000;10:19–23. doi: 10.1089/thy.2000.10.19. [DOI] [PubMed] [Google Scholar]
- 33.Saavedra HI, Knauf JA, Shirokawa JM, Wang J, Ouyang B, Elisei R, Stambrook PJ, Fagin JA. The RAS oncogene induces genomic instability in thyroid PCCL3 cells via the MAPK pathway. Oncogene. 2000;19:3948–3954. doi: 10.1038/sj.onc.1203723. [DOI] [PubMed] [Google Scholar]
- 34.Dwight T, Thoppe SR, Foukakis T, Lui WO, Wallin G, Hoog A, Frisk T, Larsson C, Zedenius J. Involvement of the PAX8/peroxisome proliferator-activated receptor gamma rearrangement in follicular thyroid tumors. J Clin Endocrinol Metab. 2003;88:4440–4445. doi: 10.1210/jc.2002-021690. [DOI] [PubMed] [Google Scholar]
- 35.French CA, Alexander EK, Cibas ES, Nose V, Laguette J, Faquin W, Garber J, Moore F, Jr, Fletcher JA, Larsen PR, Kroll TG. Genetic and biological subgroups of low-stage follicular thyroid cancer. Am J Pathol. 2003;162:1053–1060. doi: 10.1016/S0002-9440(10)63902-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikiforova MN, Lynch RA, Biddinger PW, Alexander EK, Dorn GW, 2nd, Tallini G, Kroll TG, Nikiforov YE. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. J Clin Endocrinol Metab. 2003;88:2318–2326. doi: 10.1210/jc.2002-021907. [DOI] [PubMed] [Google Scholar]
- 37.Kroll TG, Sarraf P, Pecciarini L, Chen CJ, Mueller E, Spiegelman BM, Fletcher JA. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma [corrected] Science. 2000;289:1357–1360. doi: 10.1126/science.289.5483.1357. [DOI] [PubMed] [Google Scholar]
- 38.Placzkowski KA, Reddi HV, Grebe SK, Eberhardt NL, McIver B. The role of the PAX8/PPARgamma fusion oncogene in thyroid cancer. PPAR Res. 2008 Oct 29; doi: 10.1155/2008/672829. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gandolfi PP, Frisina A, Raffa M, Renda F, Rocchetti O, Ruggeri C, Tombolini A. The incidence of thyroid carcinoma in multinodular goiter: retrospective analysis. Acta Biomed. 2004;75:114–117. [PubMed] [Google Scholar]
- 40.Lawal O, Agbakwuru A, Olayinka OS, Adelusola K. Thyroid malignancy in endemic nodular goitres: prevalence, pattern and treatment. Eur J Surg Oncol. 2001;27:157–161. doi: 10.1053/ejso.2000.1085. [DOI] [PubMed] [Google Scholar]
- 41.Rivas M, Santisteban P. TSH-activated signaling pathways in thyroid tumorigenesis. Mol Cell Endocrinol. 2003;213:31–45. doi: 10.1016/j.mce.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 42.Ward JM, Ohshima M. The role of iodine in carcinogenesis. Adv Exp Med Biol. 1986;206:529–542. doi: 10.1007/978-1-4613-1835-4_37. [DOI] [PubMed] [Google Scholar]
- 43.Mack WJ, Preston-Martin S, Bernstein L, Qian D, Xiang M. Reproductive and hormonal risk factors for thyroid cancer in Los Angeles County females. Cancer Epidemiol Biomarkers Prev. 1999;8:991–997. [PubMed] [Google Scholar]
- 44.Rios A, Rodriguez JM, Canteras M, Galindo PJ, Balsalobre MD, Parrilla P. Risk factors for malignancy in multinodular goitres. Eur J Surg Oncol. 2004;30:58–62. doi: 10.1016/j.ejso.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 45.Truong T, Orsi L, Dubourdieu D, Rougier Y, Hemon D, Guenel P. Role of goiter and of menstrual and reproductive factors in thyroid cancer: a population-based case-control study in New Caledonia (South Pacific), a very high incidence area. Am J Epidemiol. 2005;161:1056–1065. doi: 10.1093/aje/kwi136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Capen CC. Overview of structural and functional lesions in endocrine organs of animals. Toxicol Pathol. 2001;29:8–33. doi: 10.1080/019262301301418829. [DOI] [PubMed] [Google Scholar]
- 47.Capen CC. Mechanistic data and risk assessment of selected toxic end points of the thyroid gland. Toxicol Pathol. 1997;25:39–48. doi: 10.1177/019262339702500109. [DOI] [PubMed] [Google Scholar]
- 48.Ledent C, Denef JF, Cottecchia S, Lefkowitz R, Dumont J, Vassart G, Parmentier M. Costimulation of adenylyl cyclase and phospholipase C by a mutant alpha 1B-adrenergic receptor transgene promotes malignant transformation of thyroid follicular cells. Endocrinology. 1997;138:369–378. doi: 10.1210/endo.138.1.4861. [DOI] [PubMed] [Google Scholar]
- 49.Ledent C, Marcotte A, Dumont JE, Vassart G, Parmentier M. Differentiated carcinomas develop as a consequence of the thyroid specific expression of a thyroglobulin-human papillomavirus type 16 E7 transgene. Oncogene. 1995;10:1789–1797. [PubMed] [Google Scholar]
- 50.Ledent C, Dumont JE, Vassart G, Parmentier M. Thyroid expression of an A2 adenosine receptor transgene induces thyroid hyperplasia and hyperthyroidism. EMBO J. 1992;11:537–542. doi: 10.1002/j.1460-2075.1992.tb05084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mazzaferri EL. Management of a solitary thyroid nodule. N Engl J Med. 1993;328:553–559. doi: 10.1056/NEJM199302253280807. [DOI] [PubMed] [Google Scholar]
- 52.Michiels FM, Caillou B, Talbot M, Dessarps-Freichey F, Maunoury MT, Schlumberger M, Mercken L, Monier R, Feunteun J. Oncogenic potential of guanine nucleotide stimulatory factor alpha subunit in thyroid glands of transgenic mice. Proc Natl Acad Sci USA. 1994;91:10488–10492. doi: 10.1073/pnas.91.22.10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zeiger MA, Saji M, Gusev Y, Westra WH, Takiyama Y, Dooley WC, Kohn LD, Levine MA. Thyroid-specific expression of cholera toxin A1 subunit causes thyroid hyperplasia and hyperthyroidism in transgenic mice. Endocrinology. 1997;138:3133–3140. doi: 10.1210/endo.138.8.5347. [DOI] [PubMed] [Google Scholar]
- 54.Chevillard S, Ugolin N, Vielh P, Ory K, Levalois C, Elliott D, Clayman GL, El-Naggar AK. Gene expression profiling of differentiated thyroid neoplasms: diagnostic and clinical implications. Clin Cancer Res. 2004;10:6586–6597. doi: 10.1158/1078-0432.CCR-04-0053. [DOI] [PubMed] [Google Scholar]
- 55.Ying H, Suzuki H, Furumoto H, Walker R, Meltzer P, Willingham MC, Cheng SY. Alterations in genomic profiles during tumor progression in a mouse model of follicular thyroid carcinoma. Carcinogenesis. 2003;24:1467–1479. doi: 10.1093/carcin/bgg111. [DOI] [PubMed] [Google Scholar]
- 56.Kato Y, Ying H, Zhao L, Furuya F, Araki O, Willingham MC, Cheng SY. PPARgamma insufficiency promotes follicular thyroid carcinogenesis via activation of the nuclear factor-kappaB signaling pathway. Oncogene. 2006;25:2736–2747. doi: 10.1038/sj.onc.1209299. [DOI] [PubMed] [Google Scholar]
- 57.Hosal SA, Apel RL, Freeman JL, Azadian A, Rosen IB, LiVolsi VA, Asa SL. Immunohistochemical Localization of p53 in Human Thyroid Neoplasms: Correlation with Biological Behavior. Endocr Pathol. 1997;8:21–28. doi: 10.1007/BF02739704. [DOI] [PubMed] [Google Scholar]
- 58.Nikiforov YE. Genetic alterations involved in the transition from well-differentiated to poorly differentiated and anaplastic thyroid carcinomas. Endocr Pathol. 2004;15:319–327. doi: 10.1385/ep:15:4:319. [DOI] [PubMed] [Google Scholar]
- 59.Quiros RM, Ding HG, Gattuso P, Prinz RA, Xu X. Evidence that one subset of anaplastic thyroid carcinomas are derived from papillary carcinomas due to BRAF and p53 mutations. Cancer. 2005;103:2261–2268. doi: 10.1002/cncr.21073. [DOI] [PubMed] [Google Scholar]
- 60.Garcia-Rostan G, Tallini G, Herrero A, D’Aquila TG, Carcangiu ML, Rimm DL. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999;59:1811–1815. [PubMed] [Google Scholar]
- 61.Jhiang SM, Sagartz JE, Tong Q, Parker-Thornburg J, Capen CC, Cho JY, Xing S, Ledent C. Targeted expression of the ret/PTC1 oncogene induces papillary thyroid carcinomas. Endocrinology. 1996;137:375–378. doi: 10.1210/endo.137.1.8536638. [DOI] [PubMed] [Google Scholar]
- 62.Cho JY, Sagartz JE, Capen CC, Mazzaferri EL, Jhiang SM. Early cellular abnormalities induced by RET/PTC1 oncogene in thyroid-targeted transgenic mice. Oncogene. 1999;18:3659–3665. doi: 10.1038/sj.onc.1202709. [DOI] [PubMed] [Google Scholar]
- 63.Santoro M, Chiappetta G, Cerrato A, Salvatore D, Zhang L, Manzo G, Picone A, Portella G, Santelli G, Vecchio G, Fusco A. Development of thyroid papillary carcinomas secondary to tissue-specific expression of the RET/PTC1 oncogene in transgenic mice. Oncogene. 1996;12:1821–1826. [PubMed] [Google Scholar]
- 64.Sagartz JE, Jhiang SM, Tong Q, Capen CC. Thyroid-stimulating hormone promotes growth of thyroid carcinomas in transgenic mice with targeted expression of the ret/PTC1 oncogene. Lab Invest. 1997;76:307–318. [PubMed] [Google Scholar]
- 65.La Perle KM, Jhiang SM, Capen CC. Loss of p53 promotes anaplasia and local invasion in ret/PTC1-induced thyroid carcinomas. Am J Pathol. 2000;157:671–677. doi: 10.1016/S0002-9440(10)64577-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Powell DJ, Jr, Russell J, Nibu K, Li G, Rhee E, Liao M, Goldstein M, Keane WM, Santoro M, Fusco A, Rothstein JL. The RET/PTC3 oncogene: meta-static solid-type papillary carcinomas in murine thyroids. Cancer Res. 1998;58:5523–5528. [PubMed] [Google Scholar]
- 67.Burniat A, Jin L, Detours V, Driessens N, Goffard JC, Santoro M, Rothstein J, Dumont JE, Miot F, Corvilain B. Gene expression in RET/PTC3 and E7 transgenic mouse thyroids: RET/PTC3 but not E7 tumors are partial and transient models of human papillary thyroid cancers. Endocrinology. 2008;149:5107–5117. doi: 10.1210/en.2008-0531. [DOI] [PubMed] [Google Scholar]
- 68.Jin L, Burniat A, Dumont JE, Miot F, Corvilain B, Franc B. Human thyroid tumours, the puzzling lessons from E7 and RET/PTC3 transgenic mice. Br J Cancer. 2008;99:1874–1883. doi: 10.1038/sj.bjc.6604740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Knauf JA, Ma X, Smith EP, Zhang L, Mitsutake N, Liao XH, Refetoff S, Nikiforov YE, Fagin JA. Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Res. 2005;65:4238–4245. doi: 10.1158/0008-5472.CAN-05-0047. [DOI] [PubMed] [Google Scholar]
- 70.Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer. 2005;12:245–262. doi: 10.1677/erc.1.0978. [DOI] [PubMed] [Google Scholar]
- 71.Russell JP, Powell DJ, Cunnane M, Greco A, Portella G, Santoro M, Fusco A, Rothstein JL. The TRK-T1 fusion protein induces neoplastic transformation of thyroid epithelium. Oncogene. 2000;19:5729–5735. doi: 10.1038/sj.onc.1203922. [DOI] [PubMed] [Google Scholar]
- 72.Feunteun J, Michiels F, Rochefort P, Caillou B, Talbot M, Fournes B, Mercken L, Schlumberger M, Monier R. Targeted oncogenesis in the thyroid of transgenic mice. Horm Res. 1997;47:137–139. doi: 10.1159/000185456. [DOI] [PubMed] [Google Scholar]
- 73.Rochefort P, Caillou B, Michiels FM, Ledent C, Talbot M, Schlumberger M, Lavelle F, Monier R, Feunteun J. Thyroid pathologies in transgenic mice expressing a human activated Ras gene driven by a thyroglobulin promoter. Oncogene. 1996;12:111–118. [PubMed] [Google Scholar]
- 74.Vitagliano D, Portella G, Troncone G, Francione A, Rossi C, Bruno A, Giorgini A, Coluzzi S, Nappi TC, Rothstein JL, Pasquinelli R, Chiappetta G, Terracciano D, Macchia V, Melillo RM, Fusco A, Santoro M. Thyroid targeting of the N-ras(Gln61Lys) oncogene in transgenic mice results in follicular tumors that progress to poorly differentiated carcinomas. Oncogene. 2006;25:5467–5474. doi: 10.1038/sj.onc.1209527. [DOI] [PubMed] [Google Scholar]
- 75.Coppee F, Gerard AC, Denef JF, Ledent C, Vassart G, Dumont JE, Parmentier M. Early occurrence of metastatic differentiated thyroid carcinomas in transgenic mice expressing the A2a adenosine receptor gene and the human papillomavirus type 16 E7 oncogene. Oncogene. 1996;13:1471–1482. [PubMed] [Google Scholar]
- 76.Santelli G, de Franciscis V, Portella G, Chiappetta G, D’Alessio A, Califano D, Rosati R, Mineo A, Monaco C, Manzo G, Pozzi L, Vecchio G. Production of transgenic mice expressing the Ki-ras oncogene under the control of a thyroglobulin promoter. Cancer Res. 1993;53:5523–5527. [PubMed] [Google Scholar]
- 77.Ribeiro-Neto F, Leon A, Urbani-Brocard J, Lou L, Nyska A, Altschuler DL. cAMP-dependent oncogenic action of Rap1b in the thyroid gland. J Biol Chem. 2004;279:46868–46875. doi: 10.1074/jbc.M406858200. [DOI] [PubMed] [Google Scholar]
- 78.Yen PM. Molecular basis of resistance to thyroid hormone. Trends Endocrinol Metab. 2003;14:327–333. doi: 10.1016/s1043-2760(03)00114-0. [DOI] [PubMed] [Google Scholar]
- 79.Ono S, Schwartz ID, Mueller OT, Root AW, Usala SJ, Bercu BB. Homozygosity for a dominant negative thyroid hormone receptor gene responsible for generalized resistance to thyroid hormone. J Clin Endocrinol Metab. 1991;73:990–994. doi: 10.1210/jcem-73-5-990. [DOI] [PubMed] [Google Scholar]
- 80.Kaneshige M, Kaneshige K, Zhu X, Dace A, Garrett L, Carter TA, Kazlauskaite R, Pankratz DG, Wynshaw-Boris A, Refetoff S, Weintraub B, Willingham MC, Barlow C, Cheng S. Mice with a targeted mutation in the thyroid hormone beta receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci USA. 2000;97:13209–13214. doi: 10.1073/pnas.230285997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parrilla R, Mixson AJ, McPherson JA, MacClaskey JH, Weintraub BD. Characterization of seven novel mutations of the c-erbA beta gene in unrelated kindreds with generalized thyroid hormone resistance. Evidence for two “hot spot” regions of the ligand binding domain. J Clin Invest. 1991;88:2123–2130. doi: 10.1172/JCI115542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Suzuki H, Willingham MC, Cheng SY. Mice with a mutation in the thyroid hormone receptor beta gene spontaneously develop thyroid carcinoma: a mouse model of thyroid carcinogenesis. Thyroid. 2002;12:963–969. doi: 10.1089/105072502320908295. [DOI] [PubMed] [Google Scholar]
- 83.Ying H, Suzuki H, Zhao L, Willingham MC, Meltzer P, Cheng SY. Mutant thyroid hormone receptor beta represses the expression and transcriptional activity of peroxisome proliferator-activated receptor gamma during thyroid carcinogenesis. Cancer Res. 2003;63:5274–5280. [PubMed] [Google Scholar]
- 84.Kim CS, Vasko VV, Kato Y, Kruhlak M, Saji M, Cheng SY, Ringel MD. AKT activation promotes metastasis in a mouse model of follicular thyroid carcinoma. Endocrinology. 2005;146:4456–4463. doi: 10.1210/en.2005-0172. [DOI] [PubMed] [Google Scholar]
- 85.Furuya F, Guigon CJ, Zhao L, Lu C, Hanover JA, Cheng SY. Nuclear receptor corepressor is a novel regulator of phosphatidylinositol 3-kinase signaling. Mol Cell Biol. 2007;27:6116–6126. doi: 10.1128/MCB.00900-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Furuya F, Hanover JA, Cheng SY. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone beta receptor. Proc Natl Acad Sci USA. 2006;103:1780–1785. doi: 10.1073/pnas.0510849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Furuya F, Lu C, Willingham MC, Cheng SY. Inhibition of phosphatidylinositol 3-kinase delays tumor progression and blocks metastatic spread in a mouse model of thyroid cancer. Carcinogenesis. 2007;28:2451–2458. doi: 10.1093/carcin/bgm174. [DOI] [PubMed] [Google Scholar]
- 88.Ying H, Furuya F, Zhao L, Araki O, West BL, Hanover JA, Willingham MC, Cheng SY. Aberrant accumulation of PTTG1 induced by a mutated thyroid hormone beta receptor inhibits mitotic progression. J Clin Invest. 2006;116:2972–2984. doi: 10.1172/JCI28598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zimonjic DB, Kato Y, Ying H, Popescu NC, Cheng SY. Chromosomal aberrations in cell lines derived from thyroid tumors spontaneously developed in TRbetaPV/PV mice. Cancer Genet Cytogenet. 2005;161:104–109. doi: 10.1016/j.cancergencyto.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 90.Kim CS, Ying H, Willingham MC, Cheng SY. The pituitary tumor-transforming gene promotes angiogenesis in a mouse model of follicular thyroid cancer. Carcinogenesis. 2007;28:932–939. doi: 10.1093/carcin/bgl231. [DOI] [PubMed] [Google Scholar]
- 91.Kim CS, Furuya F, Ying H, Kato Y, Hanover JA, Cheng SY. Gelsolin: a novel thyroid hormone receptor-beta interacting protein that modulates tumor progression in a mouse model of follicular thyroid cancer. Endocrinology. 2007;148:1306–1312. doi: 10.1210/en.2006-0923. [DOI] [PubMed] [Google Scholar]
- 92.Torres-Arzayus MI, Font de Mora J, Yuan J, Vazquez F, Bronson R, Rue M, Sellers WR, Brown M. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell. 2004;6:263–274. doi: 10.1016/j.ccr.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 93.Ying H, Willingham MC, Cheng SY. The steroid receptor coactivator-3 is a tumor promoter in a mouse model of thyroid cancer. Oncogene. 2008;27:823–830. doi: 10.1038/sj.onc.1210680. [DOI] [PubMed] [Google Scholar]
- 94.Furuya F, Ying H, Zhao L, Cheng SY. Novel functions of thyroid hormone receptor mutants: beyond nucleus-initiated transcription. Steroids. 2007;72:171–179. doi: 10.1016/j.steroids.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Guigon CJ, Zhao L, Lu C, Willingham MC, Cheng SY. Regulation of beta-catenin by a novel nongenomic action of thyroid hormone beta receptor. Mol Cell Biol. 2008;28:4598–4608. doi: 10.1128/MCB.02192-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kato Y, Ying H, Willingham MC, Cheng SY. A tumor suppressor role for thyroid hormone beta receptor in a mouse model of thyroid carcinogenesis. Endocrinology. 2004;145:4430–4438. doi: 10.1210/en.2004-0612. [DOI] [PubMed] [Google Scholar]
- 97.Ledent C, Dumont J, Vassart G, Parmentier M. Thyroid adenocarcinomas secondary to tissue-specific expression of simian virus-40 large T-antigen in transgenic mice. Endocrinology. 1991;129:1391–1401. doi: 10.1210/endo-129-3-1391. [DOI] [PubMed] [Google Scholar]
- 98.Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, Love DR, Mole SE, Moore JK, Papi L, Ponder MA, Telenius H, Tunnacliffe A, Ponder BAJ. Germ-line mutations of the RET protooncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363:458–460. doi: 10.1038/363458a0. [DOI] [PubMed] [Google Scholar]
- 99.Harvey M, Vogel H, Lee EY, Bradley A, Donehower LA. Mice deficient in both p53 and Rb develop tumors primarily of endocrine origin. Cancer Res. 1995;55:1146–1151. [PubMed] [Google Scholar]
- 100.Ziebold U, Lee EY, Bronson RT, Lees JA. E2F3 loss has opposing effects on different pRB-deficient tumors, resulting in suppression of pituitary tumors but metastasis of medullary thyroid carcinomas. Mol Cell Biol. 2003;23:6542–6552. doi: 10.1128/MCB.23.18.6542-6552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Coxon AB, Ward JM, Geradts J, Otterson GA, Zajac-Kaye M, Kaye FJ. RET cooperates with RB/p53 inactivation in a somatic multi-step model for murine thyroid cancer. Oncogene. 1998;17:1625–1628. doi: 10.1038/sj.onc.1202381. [DOI] [PubMed] [Google Scholar]
- 102.Nakagawa T, Mabry M, de Bustros A, Ihle JN, Nelkin BD, Baylin SB. Introduction of v-Ha-ras oncogene induces differentiation of cultured human medullary thyroid carcinoma cells. Proc Natl Acad Sci USA. 1987;84:5923–5927. doi: 10.1073/pnas.84.16.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Johnston D, Hatzis D, Sunday ME. Expression of v-Ha-ras driven by the calcitonin/calcitonin gene-related peptide promoter: a novel transgenic murine model for medullary thyroid carcinoma. Oncogene. 1998;16:167–177. doi: 10.1038/sj.onc.1201478. [DOI] [PubMed] [Google Scholar]
- 104.Cranston AN, Ponder BA. Modulation of medullary thyroid carcinoma penetrance suggests the presence of modifier genes in a RET transgenic mouse model. Cancer Res. 2003;63:4777–4780. [PubMed] [Google Scholar]
- 105.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lakso M, Sauer B, Mosinger B, Jr, Lee EJ, Manning RW, Yu SH, Mulder KL, Westphal H. Targeted oncogene activation by site-specific recombination in transgenic mice. Proc Natl Acad Sci USA. 1992;89:6232–6236. doi: 10.1073/pnas.89.14.6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 108.Smith AJ, De Sousa MA, Kwabi-Addo B, Heppell-Parton A, Impey H, Rabbitts P. A site-directed chromosomal translocation induced in embryonic stem cells by Cre-loxP recombination. Nat Genet. 1995;9:376–385. doi: 10.1038/ng0495-376. [DOI] [PubMed] [Google Scholar]
- 109.Buchholz F, Refaeli Y, Trumpp A, Bishop JM. Inducible chromosomal translocation of AML1 and ETO genes through Cre/loxP-mediated recombination in the mouse. EMBO Rep. 2000;1:133–139. doi: 10.1093/embo-reports/kvd027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Collins EC, Pannell R, Simpson EM, Forster A, Rabbitts TH. Inter-chromosomal recombination of Mll and Af9 genes mediated by cre-loxP in mouse development. EMBO Rep. 2000;1:127–132. doi: 10.1093/embo-reports/kvd021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sheils O. Molecular classification and biomarker discovery in papillary thyroid carcinoma. Expert Rev Mol Diagn. 2005;5:927–946. doi: 10.1586/14737159.5.6.927. [DOI] [PubMed] [Google Scholar]