

Summary
In all cells, protein degradation is a constant, ongoing process that is critical for cell survival and repair. The ubiquitin/proteasome pathway (UPP) is the major proteolytic pathway that degrades intracellular proteins in a regulated manner. It plays critical roles in many cellular processes and diseases. Disruption of the UPP is particularly relevant to pathophysiological conditions that provoke the accumulation of aberrant proteins, such as in aging as well as in a variety of neurodegenerative disorders including Alzheimer’s and Parkinson’s diseases. For unknown reasons, most of these neurodegenerative disorders that include familial and sporadic cases exhibit a late onset. It is possible that these neurodegenerative conditions exhibit a late onset because proteasome activity decreases with aging. Aging-dependent impairment in proteolysis mediated by the proteasome may have profound ramifications for cell viability. It can lead to the accumulation of modified, potentially toxic proteins in cells and can cause cell injury or premature cell death by apoptosis or necrosis. While it is accepted that aging affects UPP function, the question is why does aging cause a decline in regulated protein degradation by the UPP? Herein, we review some of the properties of the UPP and mechanisms mediating its age-dependent impairment. We also discuss the relevance of these findings leading to a model that proposes that UPP dysfunction may be one of the milestones of aging.
Introduction
The ubiquitin (Ub)/proteasome pathway (UPP) is the major pathway for regulated nonlysosomal degradation of intracellular proteins in eukaryotes. Short-lived proteins such as cell cycle regulators and transcription factors, as well as abnormal proteins from the cytosol, nucleus and endoplasmic reticulum are specifically recognized and degraded through this pathway (Chen & Hochstrasser, 1996; Coux et al., 1996; Bader et al., 2007). The UPP is of vital importance for maintaining homeostasis and normal function in eukaryotic cells (Heinemeyer et al., 1993). In addition, the UPP degrades mutant and structurally abnormal proteins, thus preventing their accumulation and aggregation.
The UPP plays a critical role in aging and in the pathogenesis of most neurodegenerative diseases. High levels of oxidized proteins detected in the aging brain are an indication of proteasome impairment, as this proteolytic complex degrades the majority of oxidatively modified proteins (Grune et al., 2004). Moreover, the accumulation and aggregation of ubiquitinated proteins detected in most neurodegenerative disorders, such as Alzheimer’s and Parkinson’s diseases, is also a sign of UPP dysfunction, as this pathway degrades ubiquitinated proteins (Hyun et al., 2004).
The UPP
The UPP requires most proteins to be tagged by Ub to target them for degradation (Fig. 1). Proteolysis by the UPP involves two major steps: ubiquitination and degradation. A deubiquitination step also plays important roles in this pathway as it edits the protein state of ubiquitination and removes the Ub tag for recycling.
Fig. 1.

Ubiquitination and degradation of proteins by the ubiquitin (Ub)/proteasome pathway (UPP). Protein ubiquitination is a complex ATP-dependent process in which Ub is sequentially activated by Ub-activating enzymes (E1), transferred to Ub-conjugating enzymes (E2) and ligated to protein substrates by Ub ligases (E3). Poly-Ub chains are formed by isopeptide bonds between Gly76 and Lys48 on adjacent Ub molecules. Once a protein is polyubiquitinated, it is degraded rapidly by the 26S proteasome. Deubiquitinating enzymes (DUB) remove and disassemble poly-Ub chains.
Ubiquitination
Ub is a small protein of 76 amino acids crucial to the degradation of many cytosolic, nuclear and endoplasmic reticulum proteins (Hochstrasser, 1995). It is ubiquitous to every eukaryotic cell. There are at least three human Ub genes, two of which, the poly-Ub B and C genes, contain heat-shock promoters (Mayer et al., 1991). Ubiquitination is a complex process involving the following sequence of events (Fig. 1): (i) formation of a high energy thioester bond between Ub and a Ub-activating enzyme (E1) in a reaction that requires adenosine triphosphate (ATP) hydrolysis; (ii) formation of a thioester bond between the activated Ub and Ub-conjugating enzymes (E2); (iii) covalent attachment of the carboxyl terminal of Ub, most often to the ε-amino group of a lysine residue on protein substrates via an isopeptide bond; this reaction is mediated by Ub ligases (E3), which confer substrate specificity to the UPP; and (iv) assembly of multi-Ub chains carried out by ubiquitination factors (E4), which promote the production of longer Ub chains. In some cases, Ub can be transferred directly to the protein substrate by Ub-conjugating enzymes (E2) complexed with E3s (Hershko & Ciechanover, 1998).
At least four molecules of Ub forming a tetra-Ub chain need to be attached to the substrate to ensure efficient recognition and degradation by the 26S proteasome machinery (Cook et al., 1994). These chains are formed by successive attachment of monomers by an isopeptide bond, most frequently formed between the side chain of Lys 48 in one Ub and the carboxyl group of the C-terminal Gly76 of a neighboring Ub. Attachment of poly-Ub chains to lysine residues on a protein results in at least a tenfold increase in its degradation rate (Beal et al., 1996). Longer poly-Ub chains exhibit increased binding affinity to the 26S proteasome, thus enhancing the degradation of the poly-ubiquitinated substrate (Pickart, 1997). Together with K48, there are seven lysine residues in Ub that can be utilized to form poly-Ub chains: K6, K11, K27, K29, K33, K48 and K63. The least common of the ‘linkage’ sites are K27 and K29. K63 linkage is used by several pathways including cell signaling, ribosomal function, DNA repair, activation of the nuclear factor κB (NFκB) signaling complex and mitochondrial morphogenesis. K29 has similar characteristics as K48. K6 linkage seems to counteract proteasomal degradation (Shang et al., 2005).
Deubiquitination
Ub is removed from ubiquitinated proteins by deubiquitinating enzymes (DUB), which also disassemble poly-Ub chains. More than 90 genes encoding DUBs have been identified, making them one of the largest families of enzymes involved in the Ub pathway (Chung & Baek, 1999). DUBs are cysteine proteases that hydrolyze the amide bond immediately after the COOH-terminal Gly76 (Wilkinson, 1997). There are two major classes of DUBs: (i) Ub carboxyl-terminal hydrolases (UCH) are smaller and remove small amides, esters, peptides and small proteins at the carboxyl terminus of Ub, and (ii) the larger Ub-specific processing proteases (UBP), which disassemble the poly-Ub chains and edit the ubiquitination state of proteins (Wilkinson, 1997).
The proteasome
Covalent binding of Ub to proteins marks them for degradation by the 26S proteasome, a Ub/ATP-dependent proteinase that is a multicomponent enzymatic complex with a native molecular mass of ~2000 kDa (Coux et al., 1996). The 26S proteasome is composed of two major particles. The 20S proteasome, a cylinder-like structure located in the center, comprises the proteolytic core of the enzyme. The 19S particle (PA700) is the regulatory component and may be attached to each end of the cylinder-like 20S proteasome (DeMartino & Slaughter, 1999).
The 19S particle is a multicomponent complex itself containing at least 17 subunits and can be further divided into two sub-complexes: the base and the lid. The base confers adenosine triphosphatase (ATPase) activity and consists of six AAA ATPase subunits (Rpt1–Rpt6) and two non-ATPase subunits (Rpn1 and Rpn2), whereas the lid is made up of eight non-ATPase subunits (Rpn3, Rpn5–9, Rpn11 and Rpn 12), which can specifically bind poly-Ub-tagged substrates and also deubiquitinate them (DeMartino & Slaughter, 1999). Rpn10 helps to tether the lid and the base and is a receptor for polyubiquitinated proteins (van Nocker et al., 1996; Glickman et al., 1998). The 19S components confer Ub/ATP dependency to proteolysis by the 26S proteasome (Glickman et al., 1999).
The eukaryotic 20S proteasome consists of 28 subunits arranged in four heptameric-stacked rings forming a barrel-like structure, each consisting of seven protein subunits. The α-type subunits, comprising the two outer rings, provide binding sites for regulatory particles and form a gated channel leading to the inner proteolytic chamber. The β-type subunits contain the active sites. The whole particle is a dimer with an α7β7β7α7 subunit arrangement (DeMartino & Slaughter, 1999).
The 20S particle may be alternatively capped by other complexes, including the 11S activator (PA28). PA28 is a cytoplasmic complex formed by equal, stoichiometric amounts of two different 28-kDa subunits, PA28α and PA28β, forming a 200-kDa heterohexamer. PA28-capped 20S proteasomes prefer substrates that are partially degraded proteins and peptides, rather than intact polyubiquitinated molecules (Dubiel et al., 1992; Gray et al., 1994).
Proteasome assembly
The 19S particle guides proteins into the 20S proteasome chamber where unfolded proteins are degraded into short peptides (of eight to nine amino acids). Association between the two particles is a dynamic process and requires ATP-hydrolysis (Liu et al., 2006). During degradation of one substrate molecule, it was calculated that 300–400 ATP molecules are hydrolyzed. Substrate binding activates ATP hydrolysis, which promotes three processes: substrate unfolding, gate opening in the 20S particle and protein translocation (Benaroudj et al., 2003). ATP hydrolysis is also necessary for the rapid dissociation of the 26S proteasome into the 19S and 20S particles when PIPs (proteasome-interacting protein) are released from the proteasome (Babbitt et al., 2005).
Upon ATP binding and hydrolysis each ATPase plays its own functional role. Inactivation of the Rpt2 ATPase alone inhibits the opening of the gating channel on the 20S proteasome (Kohler et al., 2001). The Rpt5 ATPase interacts with substrate-attached poly-Ub chains, suggesting a role in substrate recruitment (Lam et al., 2002). The Rpt1 and Rpt6 ATPases bind Ubr1 and Ufd4, which are E3 ligases, implying that these ATPases play a role in the recruitment of the ubiquitination machinery (Xie & Varshavsky, 2000).
Processive proteolysis and endoproteolysis by the proteasome
Substrate proteolysis by the proteasome involves sequential catalytic steps suggesting a ‘bite-chew’ processive model (Kisselev et al., 1999). An alternative nonprocessive model of degradation was proposed comprising multiple, independent cleavages with dissociation of the degradation intermediates (Wang et al., 1999).
Recognition of Ub conjugates by the 26S proteasome does not always lead to complete degradation. In some cases the proteasome only degrades specific protein segments and leaves other parts of the substrate intact. This process, termed ‘regulated Ub/proteasome-dependent processing’ or RUP, is essential for the regulation of certain transcription factors (Rape & Jentsch, 2004). Proteolysis by the proteasome is a highly processive event indicating that RUP must invoke specific mechanisms that restrict activity of the proteasome. Studies from two different groups suggest that proteolysis by the proteasome does not always start at the N- or C-terminal ends of the substrate, but can also occur at internal polypeptide loops (Liu et al., 2003; Rape & Jentsch, 2004). To integrate the two aspects of protein processing into a coherent model, it was suggested that protein processing by the proteasome is initiated by the translocation of flexible domains into the proteasome. These flexible domains could be either the N- or C-terminal ends of the polypeptide chain or internal protein loops. Degradation then proceeds towards both ends of the polypeptide chain but will come to a halt when the proteasome reaches tightly folded protein domains. Segments not restricted by tightly folded barriers are completely degraded, whereas the folded domains (and sequences beyond these domains) are spared from degradation (Rape & Jentsch, 2004; Piwko & Jentsch, 2006). The proteasome thus possesses endoproteolytic activity, which refers to its ability to cleave substrates in an internal region even when the ends are unable to enter the proteolytic chamber (Liu et al., 2003).
Access to the proteolytic chamber of the proteasome
The conventional view of how substrates enter the proteasome is that it is regulated by activators. The catalytic active sites of the proteasome can hydrolyze most sequences, and it is essential that its substrates be protected from indiscriminate proteolysis. This is ensured by the architecture of the 20S proteasome, which has the active sites in the central chamber of the 20S particle protected by the compartments of the α-subunits in the antechambers. To enter the 20S proteasome, the substrate must pass the α-subunits, which exclude folded proteins. The N-terminal residues of the α-subunits form a gate. Substrates are delivered to the interior of the 20S proteasome by the 19S particle, which recognizes the substrates through their post-translational ligation to Ub. The 19S particle binds the poly-Ub chain, cleaves the bond connecting poly-Ub and substrate, unfolds the substrate, opens the gate to the 20S particle and translocates the substrate into the catalytic chamber.
In vitro studies indicated that some substrates enter the 20S particle without the assistance of an activator (Liu et al., 2003). Chimeras of natively unfolded proteins were generated consisting of p21 or α-synuclein with a stably folded green fluorescent protein (GFP). Only the flexible parts of the chimeric substrates were degraded when the GFP was either at the N- or C- terminus, as the unfolded chains could enter the 20S proteasome. These studies clearly demonstrate that some untagged substrates can pass the α-annulus in either direction. Interestingly, the unfolded domains of p21 and α-synuclein were proteolysed even when they were generated as covalently closed circular constructs with no free termini. These observations strongly argue that flexible regions of proteasome substrates can pass through the α-annulus in a hairpin conformation (Liu et al., 2003).
The three peptidase activities of the 20S proteasome
Among the 14 different subunits (7α and 7β) of the 20S proteasome, only three of them exhibit active sites for peptide bond hydrolysis, namely, β1 (caspase-like), β2 (trypsin-like) and β5 (chymotrypsin-like). The chymotrypsin-like activity cleaves after amino acids with large or hydrophobic side chains, the trypsin-like activity cleaves after basic residues and the caspase-like activity is a postglutamyl activity that recognizes acidic amino acid residues (Orlowski & Wilk, 2000).
The β5-associated chymotrypsin-like activity seems to be the initial and rate-limiting step in protein degradation by the 20S proteasome (Kisselev et al., 1999). During this initial step, protein substrates are cleaved into large peptide fragments. In the following steps, the caspase-like and the trypsin-like activities further breakdown the fragments generated in the initial step. Studies with yeast (Heinemeyer et al., 1993) and Drosophila (Belote & Fortier, 2002) demonstrated that a functional β5 subunit is essential for survival.
The 20S proteasome is a threonine protease in which the nucleophilic attack is mediated by the N-terminal Thr (Groll et al., 1997). The three β subunits (β1, β2 and β5) bearing the active sites are first synthesized as precursor proteins each containing a propeptide at the N-terminus, which must be cleaved off for the subunits to become catalytically active (Schmidtke et al., 1996). Processing of the three catalytically active subunits into mature forms occurs only after their incorporation into the 20S proteasomes.
Aging and the UPP
One of the most accepted theories of aging is the loss of quality control in protein turnover with the concomitant build-up of oxidatively modified proteins. As proteasomes selectively degrade oxidatively damaged as well as ubiquitinated proteins it is postulated that proteasome activity declines with aging. No apparent changes in the levels of Ub or ubiquitinating enzymes (E1, E2 and E3) with aging are reported in the literature. However, one must keep in mind that due to the vast numbers and widely divergent substrates of E2 and E3 classes of enzymes, age-dependent changes in many of these enzymes remain to be assessed.
Age-dependent decrease in proteasome activity
A loss of proteasome activity with aging is supported by decreased subunit expression, alterations and/or replacement of proteasome subunits and formation of inhibitory cross-linked proteins (Carrard et al., 2002; Keller et al., 2002; Ferrington et al., 2005; Friguet, 2006). For example, the chymotrypsin-like activity was reported to be significantly lower in the hearts, lungs, kidneys, livers, spinal cords, hippocampi and cerebral cortexes of 24- and 28-month-old Fisher 344 rats compared to 3-week and/or 3-month-old animals (Keller et al., 2000). Furthermore, microarray analysis of age-related variations in gene expression patterns were reported for both mitotic (human fibroblasts) and postmitotic (rat skeletal myocytes) cells (Ly et al., 2000). Less than 2% of the monitored genes were affected by age under either condition. The expression of several genes encoding for 26S proteasome subunits declined with age in both situations. In human epidermal cells and rat myocardiac cells, the accumulation of oxidatively modified proteins was associated with decreased proteasome activity and content, implying that proteasome expression is down-regulated with age (Carrard et al., 2002). Other investigators demonstrated that proteasome activity and expression changes with age but there is no consensus because the ages and cell types tested vary from study to study (Keller et al., 2002). Clear mechanisms explaining the observed changes are still lacking. Food restriction, which is currently the only experimental paradigm that halts the aging process, prevents the age-dependent changes in proteasome function and structure in mice and rats, further supporting the notion that the proteasome plays a role in the aging process (Lee et al., 1999; Gaczynska et al., 2001).
Age-dependent decreases in ATP levels and proteasome assembly – a potential link
Our recent studies with Drosophila established that aging perturbs 26S proteasome assembly (Vernace et al., 2007). The activity of the 26S proteasome declined significantly when the flies reached an age at which overall ATP levels were highly reduced. This suggests a tight correlation between ATP concentrations and 26S proteasome activity, as the latter requires ATP for its functional assembly. Throughout most of the fly lifespan, 26S proteasome activity seems to be maintained at a more or less constant level. However, a steep decline in 26S proteasome activity was observed when the flies reached an ‘old’ age (43–47 days) coinciding with a drop in their climbing ability. In the ‘old’ flies, the sharp deficit in 26S proteasome activity increased their sensitivity to proteotoxic stress. Their lifespan was significantly shortened when they were fed sublethal concentrations of a proteasome inhibitor. This increased susceptibility was also manifested by the accumulation of ubiquitinated proteins, apparent only in ‘old’ but not ‘young’ flies. These results provide strong support for considering ATP reduction and 26S proteasome impairment as aging hallmarks (Fig. 2). In addition, they suggest that an ‘old’ age deficit in 26S proteasome activity may contribute to the late onset observed in most human neurodegenerative disorders.
Fig. 2.
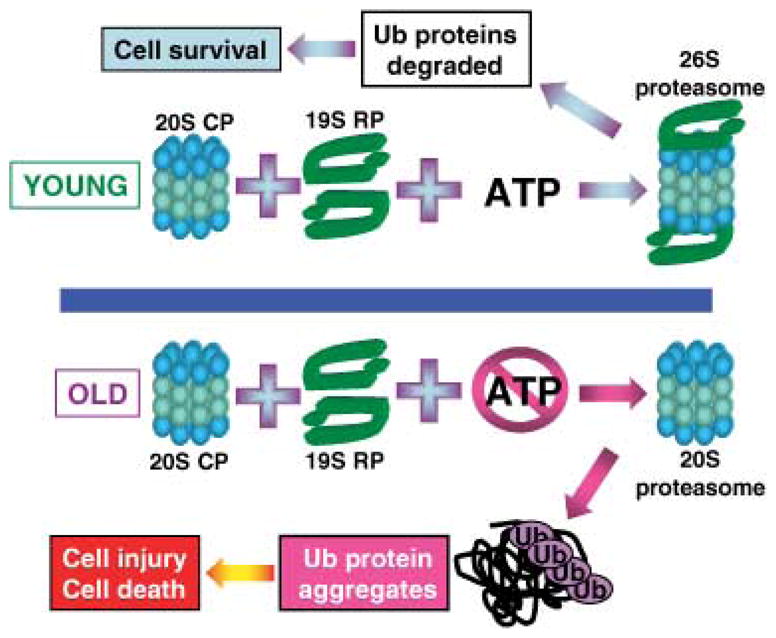
Model of the effects of aging on 26S proteasome assembly (see text for explanation). 20S CP, 20S proteasome and core particle of the 26S proteasome; 19S RP, regulatory particle of the 26S proteasome; Ub, ubiquitin.
Senescence-dependent decrease in proteasome activity
Human fibroblasts in culture have a limited proliferative capacity and after several passages enter a state of senescence. Senescent cells are viable and functional, but have altered genetic and biochemical characteristics when compared with young cells. Fibroblast senescence is a good model for studying aging at the cellular level and is used in many studies to address the role of the proteasome in aging.
Human senescent fibroblasts exhibit a reduction in the levels of all three proteasome activities, proteasome content and proteasome subunit expression levels when compared to young passage cells (Chondrogianni et al., 2003). In addition, senescent fibroblasts contained increased levels of oxidized as well as ubiquitinated proteins.
Early and late passage fibroblasts responded differently to treatment with γ-interferon (IFN-γ). Following treatment with IFN-γ, young cells exhibited increases in chymotrypsin-like and caspase-like activities as well as in the levels of the immuno (not constitutive) subunits. No such changes were detected in senescent cells. The inability to respond to IFN-γ was observed only when cells became irreversibly growth arrested (Stratford et al., 2006).
To establish the effect of proteasome inhibition on cell proliferation and lifespan, fibroblasts were treated with proteasome inhibitors, such as epoxomicin and MG132. Epoxomicin is an irreversible inhibitor that mainly blocks the chymotrypsin-like and trypsin-like activities of the proteasome (Meng et al., 1999), whereas MG132 is a reversible inhibitor that acts mostly on the chymotrypsin-like activity of the proteasome (Lee & Goldberg, 1996). These proteasome inhibitors reduced the chymotrypin-like activity right after treatment and low levels were still recorded one week later. The caspase-like activity was not immediately affected but neither inhibitor specifically blocks this activity (Chondrogianni & Gonos, 2004). Fibroblast treatment with proteasome inhibitors led to shortened lifespan, induction of a senescent-like phenotype and a dose-dependent impairment of proliferative potential (Torres et al., 2006).
To overcome the decrease in proteasome activity observed in senescent fibroblasts, the β5 subunit was stably transfected into fibroblasts. Overexpression of the β5 subunit resulted in higher levels of the β1 and β2 subunits as well as increases in all three proteasome activities. Compared to vector-transfected cell lines, all clones overexpressing the β5 subunit exhibited higher survival rates when treated with the proteasome inhibitors epoxomicin or MG132 or with various oxidants and stressors (Chondrogianni et al., 2005).
Another strategy used to prevent the decrease in proteasome activity associated with senescence involved overexpressing the proteasome chaperone POMP (proteasome maturation protein). UMP1 and POMP proteins (yeast and human, respectively) are key molecules in the process of proteasome assembly. UMP1 is required for autocatalytic active site maturation and assembly (Ramos et al., 1998). POMP protein interacts with the 20S proteasome and is the human homolog of UMP1 (Witt et al., 2000). POMP overexpression in fibroblasts led to increased levels of assembled as well as functional proteasomes, and a greater ability to effectively cope with oxidants and other stressors (Chondrogianni & Gonos, 2007).
Interestingly, fibroblast cultures from healthy centenarians exhibited proteasome characteristics (expression levels and activity) similar to younger rather than elderly individuals (Chondrogianni et al., 2000). These studies strengthen the prospect of genetically manipulating the proteasomal system as a promising antiaging therapeutical strategy.
The UPP in age-dependent disorders
The UPP plays a role in age-related disorders such as sarcopenia and progeroid syndromes. Sarcopenia is an age-related form of muscle wasting. It is coupled to a decrease in insulin-like growth factor 1 (IGF-1) signaling and to higher levels of free Ub in human and rat skeletal muscle of aged individuals compared to young ones (Cai et al., 2000). Injecting free Ub into young healthy rats induces muscle degeneration and mimics the muscle wasting of old rats, showing that increases in free Ub contribute to muscle wasting (Cai et al., 2004).
Components of the UPP interact with disease-causing mutated proteins in at least two segmental progeroid syndromes. Werner’s syndrome (WS) is an autosomal recessive disease manifested by the premature onset of age-related phenotypes. Individuals with WS are characterized by a shorter than normal stature together with characteristics of normal aging, such as cataracts, osteoporosis, arteriosclerosis, hair graying and skin aging, but they manifest these characteristics at an earlier age (Davis & Kipling, 2006). The gene that is defective in WS is the WRN (Werner’s syndrome helicase) gene, which is a member of the RecQ protein-like 4 (RECQL) family of helicases (Yu et al., 1996; Balajee et al., 1999). The helicase WRN associates with Cdc48/p97, an AAA ATPase implicated in the UPP (Wojcik et al., 2004).
RECQL4 is the dysfunctional protein in Rothmund–Thomson syndrome, another progeroid syndrome characterized by growth retardation, skin and bone defects, as well as predisposition to cancer. RECQL4 interacts with UBR1 and UBR2, which are E3 ligases involved in the N-end rule pathway, although RECQL4 is not a substrate for these enzymes (Yin et al., 2004).
Together these findings clearly demonstrate that there is a relationship between the UPP and the age-related diseases described above.
Overall relevance
A deficient UPP, in particular a decline in proteasome activity, will have a major impact on overall intracellular protein turnover, as it is considered to be the major system involved in regulated protein degradation within cells. A decline in proteasome activity will affect not only normal protein turnover within cells but also their ability to effectively cope with proteotoxic damages caused by life long environmental and/or genetic factors. Overall, the studies reviewed herein imply that intracellular protein degradation is impaired in ‘older’ individuals and may play a critical role in aging as well as in the late onset of many diseases.
The following model is proposed (Fig. 3): in ‘young’ individuals a fully active UPP provides them with a constant, ongoing process to degrade intracellular proteins that are no longer needed or that are defective; in ‘old’ individuals this steady state is disrupted by many factors. This hurdle in protein degradation will cause the abnormal build-up of intracellular protein deposits, leading to cell injury and in many cases to cell death. In conclusion, UPP dysfunction could be considered as one of the major milestones of the aging process.
Fig. 3.
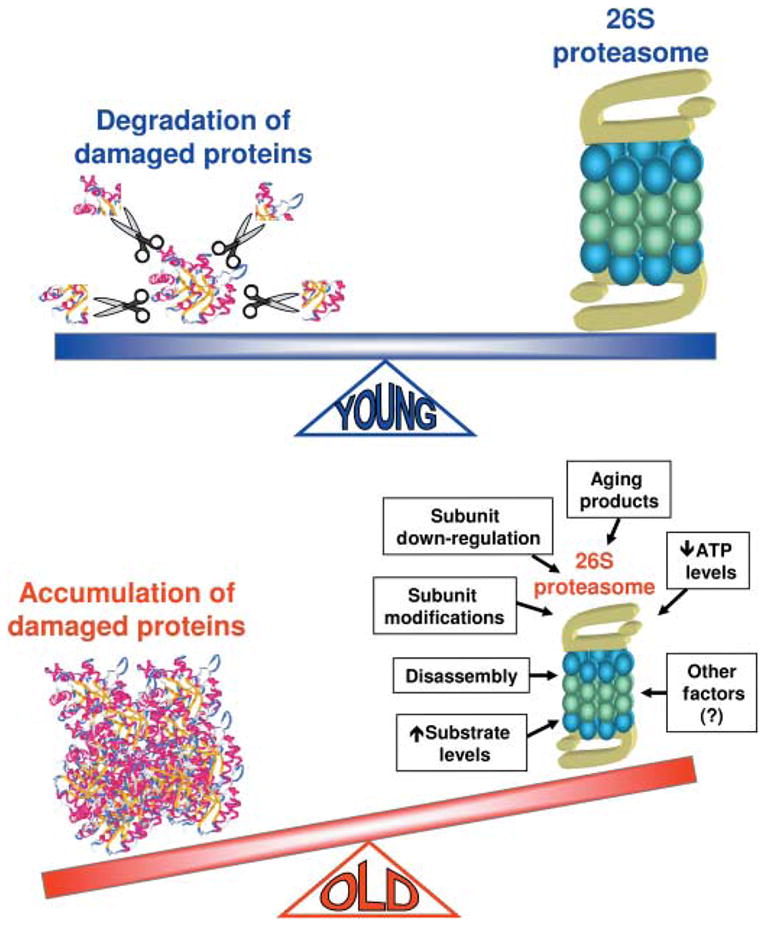
In all cells, proteasomal protein degradation is a constant, ongoing process that is critical for cell survival and repair. It ensures that proteins that are no longer needed as well as damaged proteins are degraded (scissors). Aging-dependent impairment in proteolysis mediated by the proteasome may have profound ramifications for cell viability. It can lead, for example, to the accumulation of modified, potentially toxic proteins in cells and can cause premature cell death by apoptosis or necrosis.
Acknowledgments
Please note that this review is not intended to be comprehensive and we apologize to the authors whose work is not mentioned. This work was supported by National Institutes of Health (NIH) [GM60654 (SCORE) to Thomas Schmidt-Glenewinkel and Maria E. Figueiredo-Pereira (heads of subproject) from National Institute of General Medical Sciences (NIGMS); NS41073 (Specialized Neuroscience Research Programs) to Maria E. Figueiredo-Pereira (head of subproject) from National Institute of Neurological Disorders and Stroke; and RR03037 to Hunter College (Maria E. Figueiredo-Pereira; head of subproject) from NIGMS/RCMI (Research Centers in Minority Institutions)].
References
- Babbitt SE, Kiss A, Deffenbaugh AE, Chang YH, Bailly E, Erdjument-Bromage H, Tempst P, Buranda T, Sklar LA, Baumler J, Gogol E, Skowyra D. ATP hydrolysis-dependent disassembly of the 26S proteasome is part of the catalytic cycle. Cell. 2005;121:553–565. doi: 10.1016/j.cell.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Bader N, Jung T, Grune T. The proteasome and its role in nuclear protein maintenance. Exp Gerontol. 2007 doi: 10.1016/j.exger.2007.03.010. in press. [DOI] [PubMed] [Google Scholar]
- Balajee AS, Machwe A, May A, Gray MD, Oshima J, Martin GM, Nehlin JO, Brosh R, Orren DK, Bohr VA. The Werner syndrome protein is involved in RNA polymerase II transcription. Mol Biol Cell. 1999;10:2655–2668. doi: 10.1091/mbc.10.8.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal R, Deveraux Q, Xia G, Rechsteiner M, Pickart C. Surface hydrophobic residues of multiubiquitin chains essential for proteolytic targeting. Proc Natl Acad Sci USA. 1996;93:861–866. doi: 10.1073/pnas.93.2.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belote JM, Fortier E. Targeted expression of dominant negative proteasome mutants in Drosophila melanogaster. Genesis. 2002;34:80–82. doi: 10.1002/gene.10131. [DOI] [PubMed] [Google Scholar]
- Benaroudj N, Zwickl P, Seemuller E, Baumeister W, Goldberg AL. ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol Cell. 2003;11:69–78. doi: 10.1016/s1097-2765(02)00775-x. [DOI] [PubMed] [Google Scholar]
- Cai D, Lee KK, Li M, Tang MK, Chan KM. Ubiquitin expression is up-regulated in human and rat skeletal muscles during aging. Arch Biochem Biophys. 2004;425:42–50. doi: 10.1016/j.abb.2004.02.027. [DOI] [PubMed] [Google Scholar]
- Cai D, Li M, Lee K, Lee K, Wong W, Chan K. Age-related changes of aqueous protein profiles in rat fast and slow twitch skeletal muscles. Electrophoresis. 2000;21:465–472. doi: 10.1002/(SICI)1522-2683(20000101)21:2<465::AID-ELPS465>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Carrard G, Bulteau A, Petropoulos I, Friguet B. Impairment of proteasome structure and function in aging. Int J Biochem Cell Biol. 2002;34:1461. doi: 10.1016/s1357-2725(02)00085-7. [DOI] [PubMed] [Google Scholar]
- Chen P, Hochstrasser M. Autocatalytic subunit processing couples active site formation in the 20S proteasome to completion of assembly. Cell. 1996;86:961–972. doi: 10.1016/s0092-8674(00)80171-3. [DOI] [PubMed] [Google Scholar]
- Chondrogianni N, Gonos ES. Proteasome inhibition induces a senescence-like phenotype in primary human fibroblasts cultures. Biogerontology. 2004;5:55–61. doi: 10.1023/b:bgen.0000017687.55667.42. [DOI] [PubMed] [Google Scholar]
- Chondrogianni N, Gonos ES. Overexpression of hUMP1/POMP proteasome accessory protein enhances proteasome-mediated anti-oxidant defense. Exp Gerontol. 2007 doi: 10.1016/j.exger.2007.01.012. in press. [DOI] [PubMed] [Google Scholar]
- Chondrogianni N, Petropoulos I, Franceschi C, Friguet B, Gonos ES. Fibroblast cultures from healthy centenarians have an active proteasome. Exp Gerontol. 2000;35:721–728. doi: 10.1016/s0531-5565(00)00137-6. [DOI] [PubMed] [Google Scholar]
- Chondrogianni N, Stratford FL, Trougakos IP, Friguet B, Rivett AJ, Gonos ES. Central role of the proteasome in senescence and survival of human fibroblasts: induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J Biol Chem. 2003;278:28026–28037. doi: 10.1074/jbc.M301048200. [DOI] [PubMed] [Google Scholar]
- Chondrogianni N, Tzavelas C, Pemberton AJ, Nezis IP, Rivett AJ, Gonos ES. Overexpression of proteasome β5 assembled subunit increases the amount of proteasome and confers ameliorated response to oxidative stress and higher survival rates. J Biol Chem. 2005;280:11840–11850. doi: 10.1074/jbc.M413007200. [DOI] [PubMed] [Google Scholar]
- Chung CH, Baek SH. Deubiquitinating enzymes: their diversity and emerging roles. Biochem Biophys Res Commun. 1999;266:633–640. doi: 10.1006/bbrc.1999.1880. [DOI] [PubMed] [Google Scholar]
- Cook WJ, Jeffrey LC, Kasperek E, Pickart CM. Structure of tetraubiquitin shows how multiubiquitin chains can be formed. J Mol Biol. 1994;236:601–609. doi: 10.1006/jmbi.1994.1169. [DOI] [PubMed] [Google Scholar]
- Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- Davis T, Kipling D. Werner Syndrome as an example of inflamm-aging: possible therapeutic opportunities for a progeroid syndrome? Rejuvenation Res. 2006;9:402–407. doi: 10.1089/rej.2006.9.402. [DOI] [PubMed] [Google Scholar]
- DeMartino GN, Slaughter CA. The proteasome, a novel protease regulated by multiple mechanisms. J Biol Chem. 1999;274:22123–22126. doi: 10.1074/jbc.274.32.22123. [DOI] [PubMed] [Google Scholar]
- Dubiel W, Pratt G, Ferrell K, Rechsteiner M. Purification of an 11 S regulator of the multicatalytic protease. J Biol Chem. 1992;267:22369–22377. [PubMed] [Google Scholar]
- Ferrington DA, Husom AD, Thompson LV. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005;19:644–646. doi: 10.1096/fj.04-2578fje. [DOI] [PubMed] [Google Scholar]
- Friguet B. Oxidized protein degradation and repair in ageing and oxidative stress. FEBS Lett. 2006;580:2910–2916. doi: 10.1016/j.febslet.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Gaczynska M, Osmulski PA, Ward WF. Caretaker or undertaker? The role of the proteasome in aging. Mech Ageing Dev. 2001;122:235–254. doi: 10.1016/s0047-6374(00)00246-3. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, Cjeka Z, Baumeister W, Fried VA, Finley D. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell. 1998;94:615–623. doi: 10.1016/s0092-8674(00)81603-7. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Rubin DM, Fu H, Larsen CN, Coux O, Wefes I, Pfeifer G, Cjeka Z, Vierstra R, Baumeister W, Fried V, Finley D. Functional analysis of the proteasome regulatory particle. Mol Biol Rep. 1999;26:21–28. doi: 10.1023/a:1006928316738. [DOI] [PubMed] [Google Scholar]
- Gray CW, Slaughter CA, DeMartino GN. PA28 activator protein forms regulatory caps on proteasome stacked rings. J Mol Biol. 1994;236:7–15. doi: 10.1006/jmbi.1994.1113. [DOI] [PubMed] [Google Scholar]
- Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 A resolution (see comments) Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- Grune T, Jung T, Merker K, Davies KJ. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int J Biochem Cell Biol. 2004;36:2519–2530. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Heinemeyer W, Gruhler A, Mohrle V, Mahe Y, Wolf DH. PRE2, highly homologous to the human major histocompatibility complex-linked RING10 gene, codes for a yeast proteasome subunit necessary for chymotryptic activity and degradation of ubiquitinated proteins. J Biol Chem. 1993;268:5115–5120. [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr Opin Cell Biol. 1995;7:215–223. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- Hyun DH, Gray DA, Halliwell B, Jenner P. Interference with ubiquitination causes oxidative damage and increased protein nitration: implications for neurodegenerative diseases. J Neurochem. 2004;90:422–430. doi: 10.1111/j.1471-4159.2004.02493.x. [DOI] [PubMed] [Google Scholar]
- Keller JN, Gee J, Ding Q. The proteasome in brain aging. Ageing Res Rev. 2002;1:279–293. doi: 10.1016/s1568-1637(01)00006-x. [DOI] [PubMed] [Google Scholar]
- Keller JN, Huang FF, Markesbery WR. Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience. 2000;98:149–156. doi: 10.1016/s0306-4522(00)00067-1. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, Akopian TN, Castillo V, Goldberg AL. Proteasome active sites allosterically regulate each other, suggesting a cyclical bite-chew mechanism for protein breakdown. Mol Cell. 1999;4:395–402. doi: 10.1016/s1097-2765(00)80341-x. [DOI] [PubMed] [Google Scholar]
- Kohler A, Cascio P, Leggett DS, Woo KM, Goldberg AL, Finley D. The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release. Mol Cell. 2001;7:1143–1152. doi: 10.1016/s1097-2765(01)00274-x. [DOI] [PubMed] [Google Scholar]
- Lam YA, Lawson TG, Velayutham M, Zweier JL, Pickart CM. A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal. Nature. 2002;416:763–767. doi: 10.1038/416763a. [DOI] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL. Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J Biol Chem. 1996;271:27280–27284. doi: 10.1074/jbc.271.44.27280. [DOI] [PubMed] [Google Scholar]
- Lee CK, Klopp RG, Weindruch R, Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- Liu CW, Corboy MJ, DeMartino GN, Thomas PJ. Endoproteolytic activity of the proteasome. Science. 2003;299:408–411. doi: 10.1126/science.1079293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CW, Li X, Thompson D, Wooding K, Chang TL, Tang Z, Yu H, Thomas PJ, DeMartino GN. ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol Cell. 2006;24:39–50. doi: 10.1016/j.molcel.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly DH, Lockhart DJ, Lerner RA, Schultz PG. Mitotic misregulation and human aging (see comments) Science. 2000;287:2486–2492. doi: 10.1126/science.287.5462.2486. [DOI] [PubMed] [Google Scholar]
- Mayer RJ, Arnold J, Laszlo L, Landon M, Lowe J. Ubiquitin in health and disease. Biochim Biophys Acta. 1991;1089:141–157. doi: 10.1016/0167-4781(91)90002-4. [DOI] [PubMed] [Google Scholar]
- Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci USA. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nocker S, Sadis S, Rubin DM, Glickman M, Fu H, Coux O, Wefes I, Finley D, Vierstra RD. The multiubiquitin-chain-binding protein Mcb1 is a component of the 26S proteasome in Saccharomyces cerevisiae and plays a nonessential, substrate-specific role in protein turnover. Mol Cell Biol. 1996;16:6020–6028. doi: 10.1128/mcb.16.11.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski M, Wilk S. Catalytic activities of the 20S proteasome, a multicatalytic proteinase complex. Arch Biochem Biophys. 2000;383:1–16. doi: 10.1006/abbi.2000.2036. [DOI] [PubMed] [Google Scholar]
- Pickart CM. Targeting of substrates to the 26S proteasome. FASEB J. 1997;11:1055–1066. doi: 10.1096/fasebj.11.13.9367341. [DOI] [PubMed] [Google Scholar]
- Piwko W, Jentsch S. Proteasome-mediated protein processing by bidirectional degradation initiated from an internal site. Nat Struct Mol Biol. 2006;13:691–697. doi: 10.1038/nsmb1122. [DOI] [PubMed] [Google Scholar]
- Ramos PC, Hockendorff J, Johnson ES, Varshavsky A, Dohmen RJ. Ump1p is required for proper maturation of the 20S proteasome and becomes its substrate upon completion of the assembly. Cell. 1998;92:489–499. doi: 10.1016/s0092-8674(00)80942-3. [DOI] [PubMed] [Google Scholar]
- Rape M, Jentsch S. Productive RUPture: activation of transcription factors by proteasomal processing. Biochim Biophys Acta. 2004;1695:209–213. doi: 10.1016/j.bbamcr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Schmidtke G, Kraft R, Kostka S, Henklein P, Frommel C, Lowe J, Huber R, Kloetzel PM, Schmidt M. Analysis of mammalian 20S proteasome biogenesis: the maturation of beta- subunits is an ordered two-step mechanism involving autocatalysis. EMBO J. 1996;15:6887–6898. [PMC free article] [PubMed] [Google Scholar]
- Shang F, Deng G, Liu Q, Guo W, Haas AL, Crosas B, Finley D, Taylor A. Lys6-modified ubiquitin inhibits ubiquitin-dependent protein degradation. J Biol Chem. 2005;280:20365–20374. doi: 10.1074/jbc.M414356200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratford FL, Chondrogianni N, Trougakos IP, Gonos ES, Rivett AJ. Proteasome response to interferon-gamma is altered in senescent human fibroblasts. FEBS Lett. 2006;580:3989–3994. doi: 10.1016/j.febslet.2006.06.029. [DOI] [PubMed] [Google Scholar]
- Torres C, Lewis L, Cristofalo VJ. Proteasome inhibitors shorten replicative life span and induce a senescent-like phenotype of human fibroblasts. J Cell Physiol. 2006;207:845–853. doi: 10.1002/jcp.20630. [DOI] [PubMed] [Google Scholar]
- Vernace VA, Arnaud L, Schmidt-Glenewinkel T, Figueiredo-Pereira ME. Aging perturbs 26S proteasome assembly in Drosophila melanogaster. FASEB J. 2007 doi: 10.1096/fj.06-6751com. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Chait BT, Wolf I, Kohanski RA, Cardozo C. Lysozyme degradation by the bovine multicatalytic proteinase complex (Proteasome): evidence for a nonprocessive mode of degradation. Biochemistry. 1999;38:14573–14581. doi: 10.1021/bi990826h. [DOI] [PubMed] [Google Scholar]
- Wilkinson KD. Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J. 1997;11:1245–1256. doi: 10.1096/fasebj.11.14.9409543. [DOI] [PubMed] [Google Scholar]
- Witt E, Zantopf D, Schmidt M, Kraft R, Kloetzel PM, Kruger E. Characterization of the newly identified human Ump1 homologue POMP and analysis of LMP7 (β 5i) incorporation into 20S proteasomes. J Mol Biol. 2000;301:1–9. doi: 10.1006/jmbi.2000.3959. [DOI] [PubMed] [Google Scholar]
- Wojcik C, Yano M, DeMartino GN. RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis. J Cell Sci. 2004;117:281–292. doi: 10.1242/jcs.00841. [DOI] [PubMed] [Google Scholar]
- Xie Y, Varshavsky A. Physical association of ubiquitin ligases and the 26S proteasome. Proc Natl Acad Sci USA. 2000;97:2497–2502. doi: 10.1073/pnas.060025497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Kwon YT, Varshavsky A, Wang W. RECQL4, mutated in the Rothmund–Thomson and RAPADILINO syndromes, interacts with ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Hum Mol Genet. 2004;13:2421–2430. doi: 10.1093/hmg/ddh269. [DOI] [PubMed] [Google Scholar]
- Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD. Positional cloning of the Werner’s syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]