

Abstract
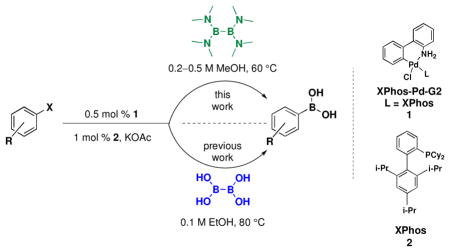
The palladium-catalyzed borylation of aryl and heteroaryl halides with a novel borylating agent, tetrakis(dimethylamino)diboron [(Me2N)2B-B(NMe2)2], is reported. The method is complementary to the previously reported method utilizing bis-boronic acid (BBA) in that certain substrates perform better under one set of optimized reaction conditions than the other. Because tetrakis(dimethylamino)diboron is the synthetic precursor to both BBA and bis(pinacolato)diboron (B2Pin2), the new method represents a more atom economical and efficient approach to current borylation methods.
The discovery of the palladium-catalyzed borylation of aryl halides with bis(pinacolato)diboron (B2Pin2) by Miyaura and coworkers in 1995 revolutionized the synthesis of arylborons used for cross-coupling reactions.1 For the first time, pinacol boronates could be prepared without the use of harsh organometallic reagents, providing access to numerous aryl- and heteroaryl boron derivatives possessing sensitive functional groups. With greater access to diverse borylated species, exploration of the Suzuki-Miyaura reaction exploded. Since then, many groups have focused on improving the scope of this important C-C bond-forming reaction. Surprisingly, few have worked to develop new ways to access the requisite borylated species. The methods that have emerged still rely largely on B2Pin2 or modified versions of this reagent.2–13
Recently, we reported a user-friendly, more environmentally sound method that provides direct access to boronic acids utilizing bis-boronic acid (BBA).14,15 With the crude boronic acid provided after Celite filtration and aqueous work-up, one can isolate the boronic acid itself or synthesize myriad boronate esters and trifluoroborates. It is also possible to skip the work-up and isolation altogether and instead perform a one-pot, two-step borylation/Suzuki reaction, providing cross-coupled products from two aryl halides (Scheme 1). This method represents a breakthrough on the existing Miyaura borylation using B2Pin2. Through the use of BBA, pinacol is eliminated from the entire process, thus making access to the boronic acid much easier and more efficient.
Scheme 1.

Synthesis of Trifluoroborates, Boronate Esters, and One-pot Cross-coupled Products from the Same Boronic Acid Intermediate
The use of BBA had appeared only infrequently in the chemical literature at the start of our research, in part because it was not commercially available, and thus its synthesis was required.16–18 The synthetic sequence for the production of BBA is shared with that of B2Pin2 up to the precursor tetrakis(dimethylamino)diborane [(Me2N)2BB(NMe2)2].19 However, instead of adding pinacol in the last step, (Me2N)2BB(NMe2)2 is hydrolyzed with aqueous acid at low temperatures. The product BBA, a white solid, precipitates out of solution and can be filtered off and dried (Scheme 2).20,21
Scheme 2.

Synthesis of B2Pin2 and BBA From Shared Precursor (Me2N)2BB(NMe2)2.
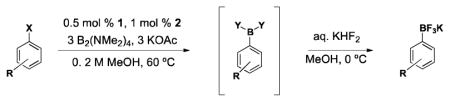
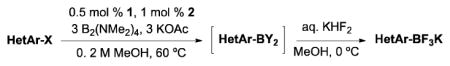
(Me2N)2BB(NMe2)2 has appeared in the chemical literature as a precursor to other boronate esters,22–29 but not as a high-yielding, general borylating agent.30,31 In this article, we report the first broadly successful borylation of aryl- and heteroaryl halides and pseudo halides using (Me2N)2BB(NMe2)2. Through the use of this method, the necessity to convert (Me2N)2BB(NMe2)2 into BBA or B2Pin2 is avoided, reducing both time and chemical waste. These savings are especially noteworthy when one considers the inefficiency of B2Pin2 in general. Pinacol makes up >90% of the mass of B2Pin2. Once synthesized, many pinacol boronates are converted to the corresponding boronic acids or trifluoroborates with often harsh, inefficient, or tedious methods, while needlessly disposing of pinacol.23,32–41 As with our first borylation method with BBA, (Me2N)2BB(NMe2)2 virtually eliminates this demasking step, providing more efficient and greener access to desired compounds.
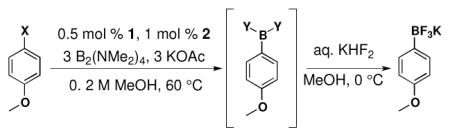
The new method utilizing (Me2N)2BB(NMe2)2 is performed with only slight modifications to our recently optimized method using BBA and Buchwald’s second generation palladium precatalyst (XPhos-Pd-G2, L = XPhos, Scheme 3).15,42 The same low catalyst load of 0.5 mol % with 1.0 mol % of XPhos (3:1 ligand/catalyst) is used as well as 3 equivalents of KOAc. Although the BBA method is performed in EtOH, the current method produces higher yields when run in MeOH. Aside from the fact that BBA is a solid and (Me2N)2BB(NMe2)2 is a liquid, operationally speaking, the set-up of the reactions is identical. As all reagents used are air stable, the solids are first weighed on a bench top balance. The vessel is then sealed and placed under an atmosphere of argon. Degassed MeOH is added to the reaction via syringe, followed by (Me2N)2BB(NMe2)2 in a similar manner. The reaction is then heated for the time required. When BBA was used in our previous method, a very distinct color change occurred, indicating the completion of the reaction. Although a color change is observed with (Me2N)2BB(NMe2)2, it is slightly less distinct with some substrates, and therefore a GC analysis is performed to confirm consumption of starting material.
Scheme 3.

Comparison of (Me2N)2BB(NMe2)2 and BBA Methods
In our first disclosure of the borylation of aryl chlorides with BBA, we demonstrated the isolation of the boronic acid product but found it difficult to obtain the pure crystalline form without some loss in yield. The same was found with the current method. Although the trifluoroborate of 4-fluorobenzene was obtained in an excellent yield of 97% (Table 1, entry 8), isolation of the corresponding boronic acid afforded the crystalline solid in 85% yield (~95% pure, Table 1, entry 8f). As we seek to demonstrate the efficiency of the present method, crude boronic acids were conveniently converted to the corresponding trifluoroborates. This transformation also preserves the C-B bond more effectively if the compounds are to be stored on the bench top for prolonged periods of time.
Table 1.
Trifluoroborate Synthesis from Aryl Chlorides and Bromides
| ||||
|---|---|---|---|---|
| entry | X | product | time (h) | % isolated yield |
| 1 | Cl |
|
5 | 94, 94a, 93b |
| 2 | Cl |
|
3 | 96, 86b, 93c |
| 3 | Cl |
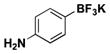
|
7 | 39, 68b |
| 4 | Br |
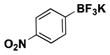
|
3.5 | 56, 64b |
| 5 | Cl |
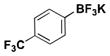
|
2.5 | 97, 91b, 98d |
| 6 | Cl |
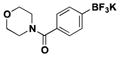
|
5.5 | 85, 81b |
| 7 | Cl |
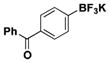
|
2.5 | 92, 91b |
| 8 | Cl |
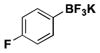
|
3 | 97, 98a 98b, 87e, 85f |
| 9 | Br |
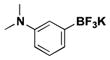
|
6 | 90, 94b |
| 10 | Cl |
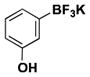
|
5 | 84, 97b |
| 11 | Br |
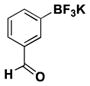
|
5 | 96g, 87b,g |
| 12 | Br |
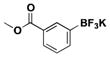
|
3 | 84, 97b |
| 13 | Br |
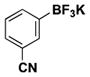
|
6 | 91, 94b |
| 14 | Br |
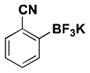
|
7 | 4, 27b |
| 15 | Br |
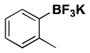
|
26 | 75, 80b |
| 16 | Cl |
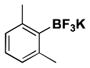
|
22 | 81, 53b |
| 17 | Cl |
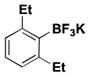
|
22 | 43 |
| 18 | Cl |
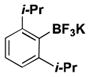
|
22 | 0 |
General conditions: 0.5 mol % of 1, 1.0 mol % of 2, 3.0 equiv of KOAc, 3.0 equiv of B2(NMe2)4, MeOH (0.2 M), 60 °C for time indicated.
0.5 M MeOH.
Yield from previous method with B2(OH)4 as boron source (ref 15).
Reaction run in a round bottom flask with reflux condensor under argon.
10 mmol reaction run under general reaction conditions.
(1)5.0 mol % of Pd(OAc)2, 10 mol % of 2, 3.0 equiv of KOAc, 60 °C in 2 mL MeOH for 20 min. (2) 3.0 equiv of B2(NMe2)4 dissolved in 5.5 mL of MeOH, 1-chloro-4-fluorobenzene, 60 °C for time indicated.
Yield of isolated boronic acid.
From the acetonide.
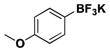
The scope of the borylation of aryl chlorides and bromides is outlined in Table 1. The method tolerates a wide range of functional groups, providing most trifluoroborates in good to excellent yield. It should also be noted that increasing the solvent concentration from 0.2 M MeOH to 0.5 M did not appear to affect the yield in the substrates attempted (Table 1, entries 1a and 8a). A reaction was performed in a round bottom flask fitted with a reflux condenser to determine whether a sealed reaction vessel affected the reaction. There was no apparent difference in carrying out the reaction under these conditions (Table 1, entry 2c). The reaction can also be efficiently scaled to 10 mmol with no loss in yield (Table 1, entry 5d).
Although not as operationally simple, the less expensive Pd(OAc)2 can be used in place of the preformed precatalyst 1, providing the 4-fluorophenyltrifluoroborate in 87% yield (Table 1, entry 8e). (Me2N)2BB(NMe2)2 appears in our hands to be more stable to the borylation reaction conditions than BBA, but preconversion of Pd(OAc)2 to Pd(0) or the use of the preformed Pd-XPhos-G2 still provide far superior conversion to product than reactions using Pd(OAc)2 directly. Preformation to Pd(0) is adequately achieved by heating Pd(OAc)2 with XPhos and KOAc in MeOH for 30 min at 60 °C before addition of (Me2N)2BB(NMe2)2 and the aryl halide.
For comparison, yields of trifluoroborates obtained through the use of BBA are also included in the tables throughout.15 In general, yields are comparable across both synthetic platforms (Table 1, entries 1, 6–9, 13, and 15). In a few instances, BBA provides yields 10% or above that of (Me2N)2BB(NMe2)2 (Table 1, entries 3, 4, 10, and 12). Noteworthy are the cases where (Me2N)2BB(NMe2)2 significantly outperforms BBA (Table 1, entries 2, 5, and 11), with the 2,6-dimethylphenyltrifluoroborate (Table 1, entry 16) providing the most impressive example of reactivity differences. Even the 2,6-diethylphenyltrifluoroborate was obtained in a reasonable yield of 43% (Table 1, entry 17). The complementary nature of the two methods now provides more synthetic options for the borylation of aryl halides.
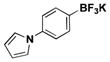
The method was also extended to heteroaryl halides, further demonstrating that certain substrates provide superior results employing one method over the other. For example, both the isoxazole- and benzoxazole-substituted phenyltrifluoroborates are obtained in superior yield when BBA is used as the borylating source (Table 2, entries 4 and 7).15 However, (Me2N)2BB(NMe2)2 can be utilized to afford 3-thienyltrifluoroborate, a material that could not be accessed in the previously developed BBA method. With respect to the indoles synthesized, the use of a Boc protecting group provides significantly higher yields than when it is not used (Table 2, entries 5 and 6), with (Me2N)2BB(NMe2)2 providing good to excellent results for both. Most noteworthy when comparing methods is the significant difference in catalyst load when borylating heteroaryl compounds (Table 2, entries 3 and 5). When using BBA, 5 mol % palladium was required.15 With (Me2N)2BB(NMe2)2, a 10-fold decrease in catalyst loading could be achieved (0.5 mol %).
Table 2.
Trifluoroborate Synthesis from Heteroaryl Chlorides and Bromides
| ||||
|---|---|---|---|---|
| entry | X | product | time (h) | % isolated Yield |
| 1 | Cl |
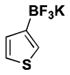
|
6 | 45 |
| 2 | Cl |
|
5 | 5 |
| 3 | Cl |
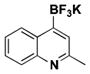
|
11 | 58, 47a |
| 4 | Br |
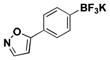
|
5.5 | 37, 85a |
| 5 | Br |
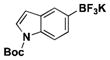
|
4.5 | 96, 93a |
| 6 | Br |
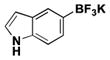
|
6 | 70 |
| 7 | Cl |
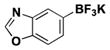
|
5 | 36, 81a |
| 8 | Br |
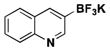
|
5 | 64, 68a |
General conditions: 0.5 mol % of 1, 1.0 mol % of 2, 3.0 equiv of KOAc, 3.0 equiv of B2(NMe2)4, MeOH (0.2 M), 60 °C for time indicated.
Yield from previous method with B2(OH)4 as boron source (ref 15).
Finally, we explored the scope of electrophilic partners that could be utilized in the reaction, using 4-substituted anisoles for direct comparison. The bromo, chloro, and triflate-substituted anisole all performed well with either borylation method. Iodides, however, provide the best results with BBA as the borylating agent.
In summary, the first palladium-catalyzed direct borylation of aryl and heteroaryl halides utilizing (Me2N)2BB(NMe2)2 as a general borylating agent has been demonstrated. The method tolerates a wide range of functional groups, providing the corresponding trifluoroborates in good to excellent yields.
As (Me2N)2BB(NMe2)2 is the common precursor to both BBA and B2Pin2, the novel method represents an even more atom economical and efficient approach to previously reported methods utilizing BBA or other derivatives. However, the methods are complementary, and in some cases superior results can be obtained when one borylating agent [BBA or (Me2N)2BB(NMe2)2] is used instead of the other. Also, because BBA is a solid and (Me2N)2BB(NMe2)2 is a liquid, there is now a choice of dosing if desired. All reagents in the method are easily handled outside of a glovebox, and degassed MeOH is used. Low catalyst loads, low temperatures, and high solvent concentrations provide an efficient and easy to use method for borylation.
Supplementary Material
Table 3.
Scope of Electrophiles in the Borylation Reaction with (Me2N)2BB(NMe2)2
General conditions: 0.5 mol % of 1, 1.0 mol % of 2, 3.0 equiv of KOAc, 3.0 equiv of B2(NMe2)4, MeOH (0.2 M), 60 °C for time indicated.
Yield from previous method with B2(OH)4 as boron source (ref 15).
Acknowledgments
We thank the National Institutes of Health (NIGMS R01 GM081376) for its generous support of this research. SLJT is funded through a Merck Doctoral Fellowship. SLJT would like to thank Merck West Point Medicinal Chemistry for their generous financial support and encouragement during her doctoral program. Simon Berritt (University of Pennsylvania) is acknowledged for his dedication to the UPenn/Merck HTE Laboratory. We thank BASF for a generous donation of (Me2N)2BB(NMe2)2.
Footnotes
Supporting Information Available. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ishiyama T, Murata M, Miyaura N. J Org Chem. 1995;60:7508–7510. [Google Scholar]
- 2.Billingsley K, Barder T, Buchwald S. Angew Chem Int Ed. 2007;46:5359–5363. doi: 10.1002/anie.200701551. [DOI] [PubMed] [Google Scholar]
- 3.Billingsley KL, Buchwald SL. J Org Chem. 2008;73:5589–5591. doi: 10.1021/jo800727s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow WK, So CM, Lau CP, Kwong FY. Chem Eur J. 2011;17:6913–6917. doi: 10.1002/chem.201100361. [DOI] [PubMed] [Google Scholar]
- 5.Fürstner A, Seidel G. Org Lett. 2002;4:541–543. doi: 10.1021/ol0171463. [DOI] [PubMed] [Google Scholar]
- 6.Ishiyama T, Ishida K, Miyaura N. Tetrahedron. 2001;57:9813–9816. [Google Scholar]
- 7.Kawamorita S, Ohmiya H, Iwai T, Sawamura M. Angew Chem Int Ed. 2011;50:8363–8366. doi: 10.1002/anie.201103224. [DOI] [PubMed] [Google Scholar]
- 8.Lu J, Guan Z, Gao J, Zhang Z. Appl Organometal Chem. 2011;25:537–541. [Google Scholar]
- 9.Moldoveanu C, Wilson D, Wilson C, Corcoran P, Rosen B, Percec V. Org Lett. 2009;11:4974–4977. doi: 10.1021/ol902155e. [DOI] [PubMed] [Google Scholar]
- 10.Moldoveanu C, Wilson D, Wilson C, Leowananwat P, Resmerita A, Lui C, Rosen B, Percec V. J Org Chem. 2010;75:5438–5452. doi: 10.1021/jo101023t. [DOI] [PubMed] [Google Scholar]
- 11.Rosen B, Huang C, Percec V. Org Lett. 2008;10:2597–2600. doi: 10.1021/ol800832n. [DOI] [PubMed] [Google Scholar]
- 12.Tang W, Keshipeddy S, Zhang Y, Wei X, Savoie J, Patel ND, Yee NK, Senanayake CH. Org Lett. 2011;13:1366–1369. doi: 10.1021/ol2000556. [DOI] [PubMed] [Google Scholar]
- 13.Wilson D, Wilson C, Moldoveanu C, Resmerita A, Corcoran P, Hoang L, Rosen B, Percec V. J Am Chem Soc. 2010;132:1800–1801. doi: 10.1021/ja910808x. [DOI] [PubMed] [Google Scholar]
- 14.Molander GA, Trice SLJ, Dreher SD. J Am Chem Soc. 2010;132:17701–17703. doi: 10.1021/ja1089759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molander GA, Trice SLJ, Kennedy SM, Dreher SD, Tudge MT. J Am Chem Soc. 2012;134:11667–11673. doi: 10.1021/ja303181m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Pilarski LT, Szabo K. Angew Chem Int Ed. 2011;50:8230–8232. doi: 10.1002/anie.201102384. [DOI] [PubMed] [Google Scholar]; b) Sebelius S, Olsson V, Szabo K. J Am Chem Soc. 2005;127:10478–10479. doi: 10.1021/ja052885q. [DOI] [PubMed] [Google Scholar]
- 17.Selander N, Kipke A, Sebelius S, Szabo K. J Am Chem Soc. 2007;129:13723–12731. doi: 10.1021/ja074917a. [DOI] [PubMed] [Google Scholar]
- 18.Selander N, Szabo K. Chem Commun. 2008:3420–3422. doi: 10.1039/b804920c. [DOI] [PubMed] [Google Scholar]
- 19.Ishiyama T, Murata M, Ahiko T, Miyaura N. Org Synth Coll Vol X. 2004;10:115–119. [Google Scholar]
- 20.McCloskey AL, Brotherton RJ, Boone JL. J Am Chem Soc. 1961;83:4750–4754. [Google Scholar]
- 21.BBA is now commercially available. CAS 13675-18-8.
- 22.Clegg W, Lawlor F, Lesley G, Marder T, Norman N, Orpen G, Quayle M, Rice C, Scott A, Souza F. J Organomet Chem. 1997;550:183–192. [Google Scholar]
- 23.Zaidlewicz M, Wolan A. J Organomet Chem. 2002;657:129–135. [Google Scholar]
- 24.Nguyen P, Lesley G, Taylor NTM. Inorg Chem. 1994;33:4623–4624. [Google Scholar]
- 25.Boller TM, Murphy JM, Hapke M, Ishiyama T, Miyuaura N, Hartwig JF. J Am Chem Soc. 2005;127:14263–14278. doi: 10.1021/ja053433g. [DOI] [PubMed] [Google Scholar]
- 26.Clegg W, Johann T, Marder T, Norman N, Orpen G, Peakman T, Quayle M, Rice C, Scott A. J Chem Soc, Dalton Trans. 1998:1431–1438. [Google Scholar]
- 27.Moezzi A, Olmstead M, Power P. Organometallics. 1992;11:2383–2388. [Google Scholar]
- 28.Moezzi A, Olmstead M, Power P. J Chem Soc, Dalton Trans. 1992:2429–2434. [Google Scholar]
- 29.Lawlor F, Norman N, Pickett N, Robins EG, Nguyen P, Lesley G, Marder T, Ashmore J, Green J. Inorg Chem. 1998;37:5282–5288. [Google Scholar]
- 30.Ishiyama T, Matsuda N, Murata M, Ozawa F, Suzuki A, Miyaura N. Organometallics. 1996;15:713–720. [Google Scholar]
- 31.Ishiyama T, Itoh Y, Kitano T, Miyaura N. Tetrahedron Lett. 1997;38:3447–3450. [Google Scholar]
- 32.Nakamura H, Fujiwara M, Yamamoto Y. J Org Chem. 1998;63:7529–7530. doi: 10.1021/jo980818r. [DOI] [PubMed] [Google Scholar]
- 33.Falck JR, Bondlela M, Venkataraman SK. J Org Chem. 2001;66:7148–7150. doi: 10.1021/jo015838z. [DOI] [PubMed] [Google Scholar]
- 34.Deng H, Jung JK, Liu T, Kuntz KW, Snapper ML, Hoveyda AH. J Am Chem Soc. 2003;125:9032–9034. doi: 10.1021/ja030249r. [DOI] [PubMed] [Google Scholar]
- 35.Song YL, Morin C. Synlett. 2001:266–268. [Google Scholar]
- 36.Jung ME, Lazarova TI. J Org Chem. 1999;64:2976–2977. doi: 10.1021/jo9902751. [DOI] [PubMed] [Google Scholar]
- 37.Ma D, Wu Q. Tetrahedron Lett. 2001;42:5279–5281. [Google Scholar]
- 38.Yang W, He H, Drueckhammer DG. Angew Chem Int Ed. 2001;40:1714–1718. [PubMed] [Google Scholar]
- 39.Pennington T, Kardiman C, Hutton C. Tetrahedron Lett. 2004;45:6657–6660. [Google Scholar]
- 40.Yuen A, Hutton C. Tetrahedron Lett. 2005;46:7899–7903. [Google Scholar]
- 41.Bagutski V, Ros A, Aggarwal VK. Tetrahedron. 2009;65:9956–9960. [Google Scholar]
- 42.Kinzel T, Zhang Y, Buchwald SL. J Am Chem Soc. 2010;132:14073–14075. doi: 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.