

Abstract
Apert syndrome (AS) is characterized by craniosynostosis (premature fusion of cranial sutures) and severe syndactyly of the hands and feet. Two activating mutations, Ser-252 → Trp and Pro-253 → Arg, in fibroblast growth factor receptor 2 (FGFR2) account for nearly all known cases of AS. To elucidate the mechanism by which these substitutions cause AS, we determined the crystal structures of these two FGFR2 mutants in complex with fibroblast growth factor 2 (FGF2) . These structures demonstrate that both mutations introduce additional interactions between FGFR2 and FGF2, thereby augmenting FGFR2–FGF2 affinity. Moreover, based on these structures and sequence alignment of the FGF family, we propose that the Pro-253 → Arg mutation will indiscriminately increase the affinity of FGFR2 toward any FGF. In contrast, the Ser-252 → Trp mutation will selectively enhance the affinity of FGFR2 toward a limited subset of FGFs. These predictions are consistent with previous biochemical data describing the effects of AS mutations on FGF binding. Alterations in FGFR2 ligand affinity and specificity may allow inappropriate autocrine or paracrine activation of FGFR2. Furthermore, the distinct gain-of-function interactions observed in each crystal structure provide a model to explain the phenotypic variability among AS patients.
The fibroblast growth factor receptor (FGFR) signaling pathway is involved in a variety of critical physiological and pathological processes (1). Activating mutations in the extracellular domains of FGFR1–3 are responsible for many human skeletal disorders (2), including the craniosynostosis syndromes (3). The craniosynostosis syndromes are mostly caused by gain-of-function mutations in FGFR2 and include Apert syndrome (AS), Crouzon syndrome, Crouzon syndrome with acanthosis nigricans, coronal craniosynostosis, Pfeiffer syndrome, Jackson–Weiss syndrome, Antley–Bixler syndrome, and Beare–Stevenson cutis gyrata. AS [ref. 4; Online Mendelian Inheritance in Man database at http://www3.ncbi.nlm.nih.gov/omim/], the most severe of the craniosynostosis syndromes, is unique among the craniosynostosis syndromes in that it is also characterized by severe syndactyly (bony and cutaneous) of the hands and feet. Numerous other anomalies, including central nervous system abnormalities, cardiovascular defects, urogenital anomalies, and dermatologic manifestations, also have been observed in AS patients. Interestingly, statistically significant differences were described for severity of syndactyly and presence of cleft palate between two subgroups of AS patients with Ser-252 → Trp (Ser252Trp) or Pro-253 → Arg (Pro253Arg) mutations (5, 6), which account for ≈67% and 32% of AS patients (6–9), respectively.
Most mutations in the extracellular domain of FGFR2 cause receptor activation by facilitating dimerization. For example, Crouzon syndrome is caused by mutations that create an unpaired cysteine residue either by gain or loss of an ectodomain cysteine residue or by destabilizing disulfide bond formation in Ig-like domain 3 (D3). Free cysteines then form intermolecular disulfide bridges between two FGFRs, leading to ligand-independent receptor dimerization and activation (10–12). In contrast, AS is caused by substitution of one of two adjacent residues, Ser252Trp or Pro253Arg, in the highly conserved linker region between Ig-like domain 2 (D2) and D3 (7). Because of the nature and location of these mutations, it is unlikely that constitutive activation via intermolecular disulfide bond formation is the etiology of AS. Nevertheless, it is still probable that AS results from FGFR2 gain-of-function because these mutations decrease the dissociation rate of FGFR2 from FGFs in vitro (13, 14) and cause ligand-dependent FGFR2 activation in vivo (14, 15). A common theme in models put forth for FGFR2 hyperactivation by AS mutations suggests that AS mutations cause a conformational change in FGFR2 that increases ligand-binding affinity (13, 14, 16, 17). However, based on the proximity of Ser-252 and Pro-253 to FGF2 in the crystal structure of FGF2–FGFR2 complex, we recently proposed that AS mutations could create additional interactions between FGFR and FGF that may lead to increased ligand-binding affinity (18). In this report, we describe the crystal structures of these mutant FGFR2s in complex with FGF2. Based on these structures and sequence alignment analysis of the FGF family, we propose a model that accounts for the phenotypic differences observed between the two subgroups of AS patients.
Materials and Methods
Protein Expression and Purification.
The point mutations Ser252Trp and Pro253Arg were introduced into the ligand-binding domain of human FGFR2(IIIc) (residues 147–366) by using the Quik Change site-directed mutagenesis kit (Stratagene) and using a previously reported bacterial expression construct of wild-type FGFR2(IIIc) (residues 147–366) as template (18). The wild-type and mutant FGFR2(IIIc) proteins were expressed in Escherichia coli, refolded in vitro, and complexed with full-length FGF2 immobilized on a heparin-Sepharose column by using a previously reported protocol (18). The resulting complexes were further purified by size exclusion chromatography on Superdex 200 column (Amersham Pharmacia), using previously reported conditions (18).
DNA fragments generated by PCR of full-length human FGF2 cDNA (residues 1–155) were subcloned into the pET-28a bacterial expression vector by using NcoI and XhoI cloning sites. To avoid intermolecular disulfide-bridge formation that could hamper crystallization, two solvent-exposed cysteines in FGF2 (Cys-78 and Cys-96) were replaced by serines. After transformation of the BL21 (DE3) E. coli strain, cells containing the FGF2-expression plasmid were induced with 1 mM isopropyl 1-thio-β-d-galactopyranoside for 5 h. The bacteria were then centrifuged and subsequently lysed in a 25 mM Hepes–NaOH buffer (pH 7.5) containing 250 mM NaCl, by using a French cell press (Sim Aminco, Rochester, NY). After centrifugation, soluble FGF2 was loaded onto a Source S column (Amersham Pharmacia). Bound FGF2 was eluted by a linear gradient of NaCl to 1 M in a 25 mM Hepes–NaOH buffer (pH 7.5). Further purification of FGF2 was achieved by size exclusion chromatography on Superdex 75 (Amersham Pharmacia) equilibrated with 25 mM Hepes–NaOH buffer (pH 7.5) containing 1 M NaCl.
Crystallization and Data Collection.
Crystals of wild-type and AS mutant FGFR2(IIIc) in complex with full-length FGF2 (residues 1–155) were generated by using previously reported crystallization conditions (18) for wild-type FGFR2(IIIc) complexed with N-terminal truncated FGF2 (residues 24–155). Two microliters of protein solution (10 mg/ml in 25 mM Hepes–NaOH, pH 7.5/150 mM NaCl) were mixed with 2 μl of the crystallization buffer, consisting of 10–15% polyethylene glycol 4000 and 10% isopropyl alcohol in 0.1 M Hepes–NaOH (pH 7.5). All three complexes formed triclinic crystals, space group P1, with four molecules of FGF2 and four molecules of FGFR2 in the unit cell and a solvent content of ≈58%. The unit cell dimensions are as follows: a = 71.681 Å, b = 73.567 Å, c = 90.828 Å, α = 90.009°, β = 90.164°, and γ = 90.005° for the wild-type complex; a = 70.970 Å, b = 72.657 Å, c = 89.739 Å, α = 89.985°, β = 89.696°, and γ = 89.999° for the Ser252Trp mutant complex, and a = 70.796 Å, b = 72.490 Å, c = 89.919 Å, α = 90.011°, β = 89.816°, and γ = 89.996° for the Pro253Arg mutant complex.
Crystals were flash-frozen in dry nitrogen stream by using mother liquor containing 10% glycerol as cryoprotectant. Diffraction data for the Ser252Trp mutant FGFR2–FGF2 complex were collected on an R-Axis IIC image plate detector at home institution x-ray generator [Rigaku RU-200 rotating anode source (Cu Kα) equipped with double-focusing mirrors] operated at 50 kV and 100 mA. Diffraction data for the wild-type and Pro253Arg mutant FGFR2(IIIc)–FGF2 complexes were collected on a charge-coupled device detector at beamline X4A at the National Synchrotron Light Source, Brookhaven National Laboratory. All data were processed by using denzo and scalepack (19).
Structure Determination and Refinement.
Molecular replacement solutions for the four copies of FGFR2(IIIc)–FGF2 complex in the unit cells of wild-type and mutant FGFR2(IIIc)–FGF2 crystals were found with amore (20) by using the structure of wild-type FGFR2(IIIc) complexed with the N-terminal truncated FGF2 (Protein Data Bank identification code 1EV2) (18) as the search model. Tight noncrystallographic symmetry restraints were imposed throughout the refinement for the backbone atoms of FGF2, D2 and D3. Simulated annealing and positional/B factor refinement were performed by using cns (21). Model building into 2Fo − Fc and Fo − Fc electron density maps was performed with program o (22). The average B factors for all of the protein atoms are 33.4 Å2 for the wild-type, 39.2 Å2 for the Ser252Trp mutant, and 43.3 Å2 for the Pro253Arg mutant complexes. The refined model for the wild-type FGF2–FGFR2(IIIc) structure is composed of four FGF2 molecules (residues 25–154) and four FGFR2(IIIc) molecules (residues 150–361). The βC–βC′ loop in D3 (residues 295–306) in all four copies of FGFR2 has a poor electron-density map and is not included in the final model. The refined model for the Ser252Trp mutant FGFR2(IIIc)–FGF2 structure contains four FGF2 molecules (residues 21–154) and four FGFR2(IIIc) molecules (residues 150–361). As in the wild-type structure, the βC-βC′ loop in D3 (residues 295–306) in all of the four copies of the Ser252Trp FGFR2(IIIc) has poor electron density map and are not included in the final model. The refined model for the Pro253Arg mutant FGFR2(IIIc)–FGF2 structure is composed of four FGF2 molecules (residues 25–154), four FGFR2(IIIc) molecules (residues 150–361), and 77 water molecules. In this structure, residues 295–306 (βC-βC′ loop in D3) in FGFR2(IIIc) and residues 22–24 at the N terminus of FGF2 have sufficient 2Fo − Fc electron density in two copies of FGFR2(IIIc) and FGF2, respectively, and are modeled. However, the B values for these residues are high (above 80.0 Å2).
Results
Wild-type and AS mutant FGFR2 ectodomains were produced in E. coli, refolded in vitro, and complexed with full-length FGF2 (residues 1–155). The resulting complexes were then purified and crystallized by using previously reported crystallization conditions for wild-type FGFR2 complexed with an N-terminal truncated FGF2 (residues 24–155) (18). All three FGFR2–FGF2 complexes formed triclinic crystals with unit cell dimensions similar to those of previously reported FGFR2–FGF2(24–155) crystals (18). Crystal structures of the wild-type and mutant complexes were solved by molecular replacement by using the structure of the FGFR2–FGF2(24–155) complex as the search model and were refined to 2.6 (wild-type), 2.7 (Ser252Trp), and 2.3 (Pro253Arg) Å. The atomic model for each complex consists of four FGF2 and four FGFR2 molecules. In addition, the Pro253Arg mutant FGFR2–FGF2 structure contains 77 ordered water molecules. Data collection and refinement statistics are given in Table 1.
Table 1.
Summary of crystallographic analysis
| Structure | Data collection statistics
|
Refinement statistics*
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Resolution, Å | Reflections (total/unique) | Completeness, % | Rsym,† % | Signal, 〈I/σI〉 | Resolution, Å | Reflections | Rcryst/Rfree,‡ % | Root-mean square deviations
|
|||
| Bonds, Å | Angles, ° | B factors,§ Å2 | |||||||||
| Wild type | 30.0–2.5 | 150,523/65,847 | 96.4 (78.9)¶ | 4.9 (18.4)¶ | 18.6 | 25.0–2.6 | 54,637 | 24.8/27.0 | 0.008 | 1.4 | 1.1 |
| Ser252Trp | 30.0–2.7 | 74,426/46,770 | 91.6 (79.7)¶ | 5.0 (24.5)¶ | 12.1 | 25.0–2.7 | 46,161 | 23.8/26.7 | 0.008 | 1.4 | 1.2 |
| Pro253Arg | 30.0–2.3 | 321,836/85,442 | 97.8 (95.9)¶ | 4.1 (13.7)¶ | 26.1 | 25.0–2.3 | 75,473 | 23.7/26.0 | 0.008 | 1.5 | 1.4 |
Atomic model: 10,059 protein atoms (wild type); 10,280 protein atoms (Ser252Trp); 10,669 protein atoms and 77 water molecules (Pro253Arg).
Rsym = 100 × ΣhklΣi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl).
Rcryst/Rfree = 100 × Σhkl∥Fo(hkl)| − |Fc(hkl)∥/Σhkl|Fo(hkl)|, where Fo (>0σ) and Fc are the observed and calculated structure factors, respectively. Five percent of the reflections were used for calculation of Rfree.
For bonded protein atoms.
Value in parentheses is for the highest resolution shell: 2.59–2.50 Å (wild type), 2.80–2.70 Å (Ser252Trp), and 2.38–2.30 Å (Pro253Arg).
Overall Structures.
The overall structure of each AS mutant FGFR2–FGF2 complex is similar to the wild-type FGF2–FGFR2 structure. Superimposition of the Cα traces of mutant FGFR2s with that of wild-type FGFR2 gives a root-mean-square deviation of only 0.39 Å (for Ser252Trp) and 0.31 Å (for Pro253Arg), demonstrating that the relative disposition of the D2 and D3 domains is not affected by the mutations (Fig. 1). This observation contradicts a previous model proposing that AS mutations augment FGF-binding affinity by increasing the rigidity of the D2-D3 linker region and accentuating a conformational change in FGFR2 that occurs upon FGF binding (13–14).
Figure 1.
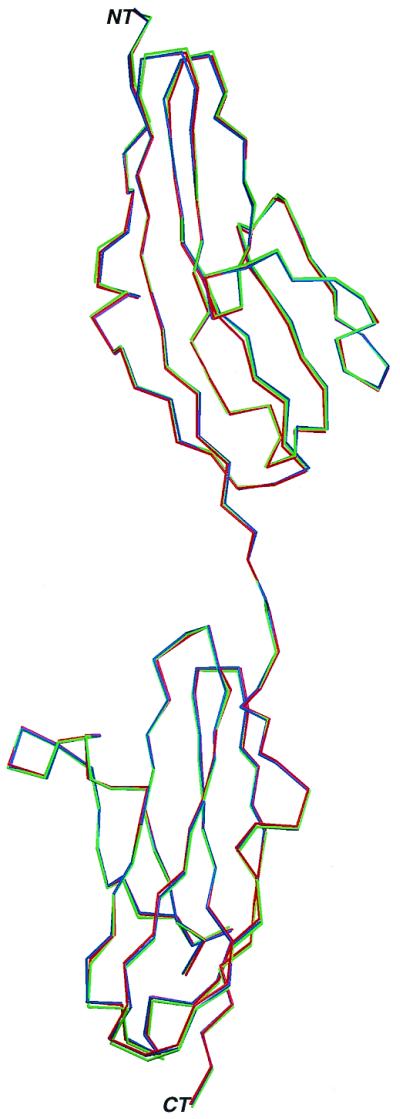
AS mutations do not affect the relative disposition of D2 and D3 in FGFR2. Cα traces of wild-type (green), mutant Ser252Trp (blue), and Pro253Arg (red) FGFR2s are superimposed. The N and C termini are denoted by the letters NT and CT. This figure was made by using programs MOLSCRIPT (23) and RASTER3D (24). The Cα traces of Ser252Trp and Pro253Arg mutant FGFR2s deviate by only 0.39 Å and 0.31 Å, respectively, from the Cα trace of wild-type FGFR2.
Of interest, a recent report of a ternary FGF1–FGFR2-heparin complex suggested that the Ser252Trp mutation activates FGFR2 by facilitating a trans- to cis-isomerization of Pro-253 that occurs upon heparin binding (17). However, in the Ser252Trp mutant structure, as in the wild-type structure, Pro-253 clearly remains in a trans configuration. Moreover, in all previously reported binary FGF–FGFR structures (18, 25–26) or ternary FGF2–FGFR1-heparin structure (27), Pro-253 is found only in a trans configuration. The trans- to cis-isomerization of Pro-253 observed in the FGF1–FGFR2-heparin structure (17) may be the result of partial refolding of FGFR2 rather than a consequence of a heparin-catalyzed conformational change. Instead, the crystal structures presented here unequivocally demonstrate that AS mutations lead to aberrant FGFR2 activation solely by altering ligand affinity and specificity.
FGF2 in Complex with Ser252Trp Mutant FGFR2.
In the crystal structure of wild-type FGF2–FGFR2, the N-terminal residues before His-25 in FGF2 are disordered and not included in the atomic model. However, in the Ser252Trp mutant FGF2–FGFR2 structure, the difference Fourier electron density map allows for the modeling of four additional N-terminal residues (Phe21-Pro-Pro-Gly24) of FGF2. The favorable ordering of these residues is due to the introduction of a unique set of interactions between the Ser252Trp mutant FGFR2 and FGF2 (Fig. 2A). These interactions can be attributed to the formation of a hydrophobic patch composed of Trp-252, Tyr-281, and Ile-257 of FGFR2 that is engaged by Phe-21 of FGF2. Although Tyr-281 and Ile-257 are inherently present in wild-type FGFR2, this hydrophobic patch cannot form efficiently in the absence of a hydrophobic residue at position 252. This is further evident by the wild-type FGF2–FGFR2 structure, where the side chain of Tyr-281 is observed to exist in two alternating rotamer positions (data not shown). The stabilization of this hydrophobic patch by Trp-252 allows the side chain hydroxyl group of Tyr-281 to form a hydrogen bond with the FGF backbone residue Pro-22 (Fig. 2A). In addition, the FGF backbone carbonyl oxygen of Gly-24 engages in an oxygen-aromatic interaction with phenyl ring of Tyr-281. The presence of these additional interactions will likely strengthen FGF2–FGFR2 affinity and is consistent with data demonstrating that, compared to wild-type FGFR2, the Ser252Trp mutant has an approximate 6.5-fold decrease in the rate of dissociation (koff) from FGF2 (13). Of interest, there are two instances of AS patients with a Ser252Phe mutation (6, 8) that is likely to activate FGFR2 through a similar mechanism.
Figure 2.

Gain-of-function interactions between the AS mutant FGFR2s and FGF2. (A) Additional contacts between the N terminus of FGF2 and the Ser252Trp mutant FGFR2. (B) Hydrogen bonds between FGF2 and Arg-253 of the Pro253Arg mutant FGFR2. D2 and D3 of FGFR2 are shown in green and cyan, respectively. The short linker that connects D2 and D3 is colored gray. FGF2 is shown in orange. In addition, the FGF2 N-terminal region (Phe21-Pro-Pro-Gly24), which is ordered only in the Ser252Trp mutant FGFR2–FGF2 structure, is colored purple. Oxygen atoms are red, nitrogen atoms blue, and carbon atoms have the same coloring as the molecules to which they belong. Dotted lines represent hydrogen bonds. The hydrogen-bonding distances are indicated. (Right) Views of whole structure in the exact orientation as in the detailed views are shown, and the region of interest is boxed. This figure was created by using the programs MOLSCRIPT and RASTER3D.
FGF2 in Complex with Pro253Arg Mutant FGFR2.
Activation of FGFR2 by the Pro253Arg mutation is due to the presence of a set of FGFR2–FGF2 interactions that is not observed in either the Ser252Trp mutant or in the wild-type FGFR2–FGF2 structures (Fig. 2B). The guanidinium group of Arg-253 in FGFR2 makes three hydrogen bonds with FGF2, two with backbone carbonyl oxygens of Leu-107 and Glu-108 and a third with the side-chain amide group of Asn-111. These additional hydrogen bonds are also predicted to increase ligand-receptor affinity. These additional interactions will also likely strengthen FGF2–FGFR2 affinity and are consistent with data demonstrating that the Pro253Arg mutant has an almost 1.5-fold decrease in koff from FGF2, relative to wild-type FGFR2. It is also noteworthy that homologous mutations in FGFR1 (Pro252Arg) and FGFR3 (Pro250Arg) are responsible for Pfeiffer syndrome (28) and Muenke craniosynostosis (29), respectively. Hence, these mutant receptors are also likely to engage in similar additional contacts with FGF.
Discussion
Implications for Ligand Binding.
Receptor-binding specificity is an essential mechanism in the regulation of FGF responses and is primarily achieved through alternative splicing of two exons, IIIb and IIIc, in the second half of D3 in FGFRs (30). The critical role of second half of D3 in conferring specificity is best exemplified by the FGFR2(IIIc)/FGFR2(IIIb) system. FGFR2(IIIc) binds FGF1 and FGF2 but does not bind FGF7. In contrast, FGFR2(IIIb) binds FGF1 and FGF7 but binds FGF2 with negligible affinity. Because AS mutations are located in the D2-D3 linker region, they affect both the FGFR2(IIIb) and FGFR2(IIIc) splice variants.
The basis for increased affinity of FGF2 for the Ser252Trp mutant FGFR2(IIIc) is the hydrophobic contact between Phe-21 of FGF2 and Trp-252, Tyr-281, and Ile-257 of FGFR2(IIIc). Therefore, the Ser252Trp mutant FGFR2(IIIc) is predicted to exhibit increased affinity toward FGFs with a hydrophobic residue at a position corresponding to Phe-21 of FGF2. A survey of the 22 human FGF sequences reported to date (18, 31) reveals that the N-terminal region outside the β-trefoil core of FGFs is highly divergent and that only a limited subset of FGFs, most notably FGF7 (KGF), fulfill this criterion (Fig. 3A). Interestingly, we have also detected an increased interaction between FGF10 and the Ser252Trp mutant FGFR2 by size-exclusion chromatography (M.M., unpublished data). FGF10 has an asparagine (Asn-71) at a position corresponding to Phe-21 of FGF2 (Fig. 3A) and is therefore not predicted to engage in additional hydrophobic interactions with the Ser252Trp mutant FGFR2. It is possible that the inherent flexibility of the N terminus of FGFs preceding the rigid β-trefoil core may permit additional interactions to take place between Tyr-70 (adjacent to Asn-71) of FGF10 and the Ser252Trp mutant FGFR2.
Figure 3.

Sequence alignment of all 22 known FGFs at regions that are involved in additional contacts with the Ser252Trp (A) or Pro253Arg (B) mutant FGFR2s. The secondary structure assignments were obtained by using the program PROCHECK (32). The location and length of the β-strands are shown on the top of sequence alignments. The numbering of β-strands is according to the published nomenclature (33). A period represents sequence identity to FGF2. FGF2 residues whose side-chain or main-chain atoms are engaged in additional contacts with AS FGFR2s are highlighted with red and green, respectively. The corresponding identical residues in other FGFs also are indicated by using the same coloring scheme. In addition, hydrophobic residues in other FGFs at the position homologous to Phe-21 of FGF2 are highlighted with purple and yellow.
In contrast, the three hydrogen bonds between Arg-253 and FGF2 in the Pro253Arg mutant FGFR2(IIIc)–FGF2 involve residues within the conserved β-trefoil core of FGFs. Because two of these hydrogen bonds are made with the backbone atoms of FGF2, we predict the Pro253Arg mutant FGFR2(IIIc) should display a general increase in affinity toward all 22 known human FGFs. Interestingly, the third hydrogen bond with FGF2 is mediated by the side chain of Asn-111, which is also conserved in FGF4, FGF6, FGF8, FGF17, and FGF18 (Fig. 3B). These FGFs are predicted to show a more marked increase in affinity for the Pro253Arg mutant FGFR2(IIIc) in comparison to other FGFs.
Of interest, each AS mutation is not only predicted to increase the affinity of FGFR2(IIIc) for its specific ligands but also may enable FGFR2(IIIc) to bind ligands that it does not normally bind. For example, the presence of a tyrosine residue (Tyr-58) in FGF7 in a homologous position to Phe-21 of FGF2 (Fig. 3A) may allow FGF7 to bind to the Ser252Trp mutant FGFR2(IIIc). Similarly, the Pro253Arg mutant FGFR2(IIIc) may bind to FGF7 via two additional nonspecific hydrogen bonds as observed in the crystal structure of Pro253Arg FGFR2(IIIc) in complex with FGF2 (Fig. 2B). Indeed, our structural predictions are supported by recent biochemical experiments that showed that both AS mutant FGFR2s can bind to and be activated by FGF7 (14). These data strongly support our structure- and sequence-based prediction that AS mutations can affect FGFR2 specificity.
Analysis of the crystal structures provides a potential explanation for the differential effects of AS mutations on FGF-binding kinetics. Anderson et al. (13) demonstrated that both AS mutant FGFR2s exhibit a decrease in koff from FGFs. The relative decreases in koff values of the wild-type and Ser252Trp mutant FGFR2 for FGF1, FGF2, and FGF4 are entirely consistent with structure- and sequence-based predictions. The profound decrease in koff (6.5-fold) of FGF2 for the Ser252Trp mutant can be attributed to the hydrophobic interactions between Phe-21 of FGF2 and Trp-252 and Tyr-281 of the mutant receptor (Fig. 2A). The aromatic ring of Phe-21 packs optimally against Trp-252 and Tyr-281 of the Ser252Trp mutant FGFR2. Sequence alignment of FGFs (Fig. 3A) shows that the aliphatic side chain of Leu-18 in FGF1 can also engage in hydrophobic interactions with Trp-252 and Tyr-281, although this interaction would be expected to be much weaker than the interaction between FGF2 and the Ser252Trp FGFR2 mutant. This is concurrently reflected by the significantly smaller decrease in koff (1.65-fold) of FGF1 for the Ser252Trp FGFR2 mutant relative to wild-type FGFR2.
Unlike the profound decrease in koff of FGF2 for the Ser252Trp FGFR2 mutant, the reduction in koff of FGF2 for the Pro253Arg FGFR2 mutant is modest. Nevertheless, this decrease is relevant, because a similar decrease was found in the off-rate of FGF1 binding to the same mutant receptor (13). The universal decrease in koff values is consistent with the structural observations and reflects the nature of conserved interactions predicted to occur between the Pro253Arg FGFR2 mutant and FGFs.
A Model for Clinical Variability.
Changes in ligand affinity and specificity of AS mutants present an attractive model for the phenotypic variability observed between the two AS mutants. Two reports have documented that AS patients with the Ser252Trp mutation present more frequently with cleft palate, whereas patients with the Pro253Arg mutations exhibit a more severe syndactyly (5, 6). Normal human limb development is controlled by an intricate epithelial–mesenchyme paracrine signaling loop, in which the mesenchymally expressed FGFR2 splice variant, FGFR2(IIIc), is activated by epithelially expressed FGFs (FGF2, FGF4, FGF6, FGF8, and FGF9). Similarly, the epithelially expressed splice form, FGFR2(IIIb)/keratinocyte growth factor receptor, is normally activated by mesenchymally expressed FGFs (FGF7 and FGF10). Recently, two rare cases of AS have been reported in which de novo Alu-element insertions result in the ectopic expression of wild-type FGFR2(IIIb) in mesenchymal cells (8). Consequently, it may be possible that syndactyly results from FGF7- or FGF10-mediated autocrine activation of ectopic mesenchymal FGFR2(IIIb)/keratinocyte growth factor receptor.
Based on the structural data presented in this report, we propose that both AS mutations can allow mesenchymal FGFR2(IIIc) to bind mesenchymally expressed FGF7 and FGF10. Because of loss of FGFR2(IIIc)-binding specificity, the pathology of AS may result from inappropriate autocrine activation of FGFR2(IIIc). Moreover, relative to Ser252Trp AS patients, the phenotypically more severe syndactyly in Pro253Arg AS patients can be accounted for by the higher affinity of the latter mutant FGFR2(IIIc) for FGF7 and FGF10 (ref. 14 and M.M., unpublished data).
Conversely, an increase in specific ligand affinity may also account for phenotypic differences among AS patients. FGF2, which is expressed in both facial ectoderm and mesenchyme, has been shown to increase outgrowth of facial mesenchyme (34). Cleft palate formation, which is more common in AS patients with the Ser252Trp mutation (5, 6), may result from an increase in FGFR2(IIIc) affinity for a specific ligand such as FGF2. This is consistent with the observation that the Ser252Trp mutant FGFR2(IIIc) has a more profound decrease in koff (≈4.5-fold greater) for FGF2 than the Pro253Arg mutant FGFR2(IIIc) (13). In addition to furnishing a structural basis for AS and a model for phenotypic variability, these crystal structures also establish a framework for the engineering of high-affinity FGFs that may have therapeutic value for a variety of physiological and pathological conditions.
Acknowledgments
M.M. acknowledges X.-P. Kong and S. R. Hubbard for comments and helpful discussions and C. Ogata for synchrotron beamline assistance. We also thank B. K. Yeh for critically reading the manuscript. Beamline X4A at the National Synchrotron Light Source, a Department of Energy facility, is supported by the Howard Hughes Medical Institute. This work was supported by the National Institutes of Health Grants R01 DE13686 (to M.M.) and R01 HD35692 (to D.M.O.).
Abbreviations
- AS
Apert syndrome
- FGF
fibroblast growth factor
- FGFR
FGF receptor
- D2 and D3
Ig-like domains 2 and 3
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1II4 and 1IIL).
References
- 1.McIntosh I, Bellus G A, Jabs E W. Cell Struct Funct. 2000;25:85–96. doi: 10.1247/csf.25.85. [DOI] [PubMed] [Google Scholar]
- 2.Gorlin R J. J Craniomaxillofac Surg. 1997;25:69–79. doi: 10.1016/s1010-5182(97)80048-0. [DOI] [PubMed] [Google Scholar]
- 3.Hehr U, Muenke M. Mol Genet. 1999;68:139–151. doi: 10.1006/mgme.1999.2915. [DOI] [PubMed] [Google Scholar]
- 4.Apert M E. Bull Soc Med Hôp Paris. 1906;23:1310–1313. [Google Scholar]
- 5.Slaney S F, Oldridge M, Hurst J A, Moriss-Kay G M, Hall C M, Poole M D, Wilkie A O. Am J Hum Genet. 1996;58:923–932. [PMC free article] [PubMed] [Google Scholar]
- 6.Lajeunie E, Cameron R, El-Ghouzzi V, de Parseval N, Journeau P, Gonzales M, Delezoide A L, Bonaventure J, Le Merrer M, Renier D. J Neurosurg. 1999;90:443–447. doi: 10.3171/jns.1999.90.3.0443. [DOI] [PubMed] [Google Scholar]
- 7.Wilkie A O M, Slaney S F, Oldridge M, Poole M D, Ashworth G J, Hockley A D, Hayward R D, David D J, Pulleyn L J, Rutland P, et al. Nat Genet. 1995;9:165–172. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- 8.Oldridge M, Zackai E H, McDonald-McGinn D M, Iseki S, Morriss-Kay G M, Twigg S R, Johnson D, Wall S A, Jiang W, Theda C, et al. Am J Hum Genet. 1999;64:446–461. doi: 10.1086/302245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsai F J, Tsai C H, Peng C T, Lin S P, Hwu W L, Wang T R, Lee C C, Wu J Y. Acta Paediatr Taiwan. 1999;40:31–33. [PubMed] [Google Scholar]
- 10.Neilson K M, Friesel R E. J Biol Chem. 1995;270:26037–26040. doi: 10.1074/jbc.270.44.26037. [DOI] [PubMed] [Google Scholar]
- 11.Neilson K M, Friesel R. J Biol Chem. 1996;271:25049–25057. doi: 10.1074/jbc.271.40.25049. [DOI] [PubMed] [Google Scholar]
- 12.Robertson S C, Meyer A N, Hart K C, Galvin B D, Webster M K, Donoghue D J. Proc Natl Acad Sci USA. 1998;95:4567–4572. doi: 10.1073/pnas.95.8.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson J, Burns H D, Enriquez-Harris P, Wilkie A O M, Heath J K. Hum Mol Genet. 1998;7:1475–1479. doi: 10.1093/hmg/7.9.1475. [DOI] [PubMed] [Google Scholar]
- 14.Yu K, Herr A B, Waksman G, Ornitz D M. Proc Natl Acad Sci USA. 2000;97:14536–14541. doi: 10.1073/pnas.97.26.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mansukhani A, Bellosta P, Sahni M, Basilico C. J Cell Biol. 2000;149:1297–1308. doi: 10.1083/jcb.149.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang F, Lu W, McKeehan K, Mohamedali K, Gabriel J L, Kan M, McKeehan W L. Biochemistry. 1999;38:160–171. doi: 10.1021/bi981758m. [DOI] [PubMed] [Google Scholar]
- 17.Pellegrini L, Burke D F, von Delft F, Mulloy B, Blundell T L. Nature (London) 2000;407:1029–1034. doi: 10.1038/35039551. [DOI] [PubMed] [Google Scholar]
- 18.Plotnikov A N, Hubbard S R, Schlessinger J, Mohammadi M. Cell. 2000;101:413–424. doi: 10.1016/s0092-8674(00)80851-x. [DOI] [PubMed] [Google Scholar]
- 19.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 20.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 21.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 22.Jones T A, Zou J Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 23.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 24.Merritt E A, Bacon D J. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 25.Plotnikov A N, Schlessinger J, Hubbard S R, Mohammadi M. Cell. 1999;98:641–650. doi: 10.1016/s0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]
- 26.Stauber D J, DiGabriele A D, Hendrickson W A. Proc Natl Acad Sci USA. 2000;97:49–54. doi: 10.1073/pnas.97.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schlessinger J, Plotnikov A N, Ibrahimi O A, Eliseenkova A V, Yeh B K, Yayon A, Linhardt R J, Mohammadi M. Mol Cell. 2000;6:743–750. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 28.Meunke M, Schell U, Hehr A, Robin N H, Losken H W, Schinzel A, Pulleyn L J, Rutland P, Reardon W, Malcolm S, et al. Nat Genet. 1994;8:269–274. doi: 10.1038/ng1194-269. [DOI] [PubMed] [Google Scholar]
- 29.Bellus G A, Gaudenz K, Zackai E H, Clarke L A, Szabo J, Francomano C A, Muenke M. Nat Genet. 1996;14:174–176. doi: 10.1038/ng1096-174. [DOI] [PubMed] [Google Scholar]
- 30.Johnson D E, Williams L T. Adv Cancer Res. 1993;60:1–41. doi: 10.1016/s0065-230x(08)60821-0. [DOI] [PubMed] [Google Scholar]
- 31.Venkataraman G, Raman R, Sasisekharan V, Sasisekharan R. Proc Natl Acad Sci USA. 1999;96:3658–3663. doi: 10.1073/pnas.96.7.3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 33.Faham S, Linhardt R J, Rees D C. Curr Opin Struct Biol. 1998;8:578–586. doi: 10.1016/s0959-440x(98)80147-4. [DOI] [PubMed] [Google Scholar]
- 34.Richman J M, Herbert M, Matovinopvic E, Walin J. Dev Biol. 1997;189:135–147. doi: 10.1006/dbio.1997.8656. [DOI] [PubMed] [Google Scholar]