

Background: RhoA GTPase regulates airway smooth muscle (SM) contraction and actin polymerization by an unknown mechanism.
Results: Agonist stimulation induces the RhoA-dependent recruitment of paxillin, vinculin, FAK, and cdc42 GEFs to adhesomes, which catalyze cdc42 and N-WASP activation.
Conclusion: RhoA regulates actin polymerization and SM contraction by regulating the adhesome assembly.
Significance: Rho-mediated adhesome assembly is a novel mechanism for the regulation of agonist-induced SM contraction.
Keywords: Adhesion, Cdc42, Excitation-Contraction Coupling, Focal Adhesion Kinase, Small GTPases, Smooth Muscle, N-Wasp, Adhesome, Paxillin, Vinculin
Abstract
The activation of the small GTPase RhoA is necessary for ACh-induced actin polymerization and airway smooth muscle (ASM) contraction, but the mechanism by which it regulates these events is unknown. Actin polymerization in ASM is catalyzed by the actin filament nucleation activator, N-WASp and the polymerization catalyst, Arp2/3 complex. Activation of the small GTPase cdc42, a specific N-WASp activator, is also required for actin polymerization and tension generation. We assessed the mechanism by which RhoA regulates actin dynamics and smooth muscle contraction by expressing the dominant negative mutants RhoA T19N and cdc42 T17N, and non-phosphorylatable paxillin Y118/31F and paxillin ΔLD4 deletion mutants in SM tissues. Their effects were evaluated in muscle tissue extracts and freshly dissociated SM cells. Protein interactions and cellular localization were analyzed using proximity ligation assays (PLA), immunofluorescence, and GTPase and kinase assays. RhoA inhibition prevented ACh-induced cdc42 activation, N-WASp activation and the interaction of N-WASp with the Arp2/3 complex at the cell membrane. ACh induced paxillin phosphorylation and its association with the cdc42 GEFS, DOCK180 and α/βPIX. Paxillin tyrosine phosphorylation and its association with βPIX were RhoA-dependent, and were required for cdc42 activation. The ACh-induced recruitment of paxillin and FAK to the cell membrane was dependent on RhoA. We conclude that RhoA regulates the contraction of ASM by catalyzing the assembly and activation of cytoskeletal signaling modules at membrane adhesomes that initiate signaling cascades that regulate actin polymerization and tension development in response to contractile agonist stimulation. Our results suggest that the RhoA-mediated assembly of adhesome complexes is a fundamental step in the signal transduction process in response to agonist -induced smooth muscle contraction.
Introduction
The small GTPase RhoA is widely recognized as an important regulator of cytoskeletal dynamics in many cell types (1–5). RhoA activation can stimulate phosphorylation of the regulatory light chain of myosin II through its inhibitory effects on the catalytic activity of smooth muscle myosin light chain (MLC)2 phosphatase or by activating kinases such as ROCK that directly phosphorylate MLC (6–9). In smooth muscle, the RhoA-mediated effects on smooth muscle (SM) myosin II activation have been widely recognized to play an important role in the regulation of contractility and shortening (8, 9). However, in airway smooth muscle tissues, the inhibition of RhoA activation profoundly depresses agonist-induced tension development with little effect on SM MLC phosphorylation; thus RhoA appears to regulate the contractility of airway smooth muscle by a mechanism that is largely independent of its effects on SM myosin II regulatory light chain phosphorylation (10).
Actin polymerization and cytoskeletal reorganization play a key role in the regulation of active tension in many smooth muscle tissues and cells, including airway smooth muscle (11–15). The inhibition of actin polymerization depresses tension development in response to contractile stimulation with little or no effect on SM myosin II regulatory light chain phosphorylation and crossbridge cycling (11, 12, 16, 17). RhoA inactivation dramatically depresses stimulus-induced actin polymerization in airway smooth muscle tissues, suggesting that the role of RhoA in regulating active tension development in this tissue is primarily due to its effects on actin cytoskeletal dynamics. The objective of this study was to evaluate the molecular mechanisms by which RhoA regulates cytoskeletal dynamics and tension development in airway smooth muscle.
In airway smooth muscle, agonist-stimulated actin polymerization requires activation of the actin nucleation promoting protein, N-WASp (neuronal Wiskott-Aldrich syndrome protein) (17). When N-WASp is activated, it undergoes a change in conformation that enables it to bind to the actin-related protein complex (Arp2/3 complex), which forms a template for the addition of monomeric actin (G-actin) to existing F-actin filaments resulting in formation of new F-actin (18–20). N-WASp activation is directly and specifically regulated by the small GTPase, cdc42, which binds to its CRIB domain (20–23). Neither Rac nor Rho GTPases can bind directly to WASP family proteins, thus RhoA cannot directly regulate N-WASp activity (24, 25). N-WASp is activated by cdc42 during the contractile stimulation of airway smooth muscle tissues, and cdc42 activation is necessary for actin polymerization and active tension development in this tissue (26).
Proteins that localize to extracellular matrix-cytoskeletal membrane junctions (adhesomes) have been implicated in the activation of N-WASp and actin polymerization during the contractile simulation of airway smooth muscle: The scaffolding/adaptor protein paxillin (27, 28) is recruited to the membrane and undergoes tyrosine phosphorylation in response to agonist stimulation (29, 30). Paxillin phosphorylation is required for agonist-stimulated actin polymerization and contraction in airway smooth muscle (31). The paxillin-binding protein vinculin (27) also localizes to adhesome complexes during the contractile stimulation airway smooth muscle and contributes to the regulation of actin polymerization (29, 32), as does focal adhesion kinase (FAK) (29), which induces the tyrosine phosphorylation of paxillin (33, 34).
Our present results provide evidence that RhoA regulates tracheal smooth muscle contraction by catalyzing the recruitment of paxillin, vinculin, and FAK to an adhesion junction associated signaling complex, and that this promotes the association of paxillin with the cdc42 guanine nucleotide exchange factors (GEFs), PIX (PAK-interacting exchange factor) and DOCK180 (Dedicator of Cytokinesis) (35). This adhesome signaling complex mediates the activation cdc42, which regulates activation of the actin polymerization catalysts, N-WASp and the Arp 2/3 complex and the polymerization of cortical actin. These results document a novel mechanism for the regulation of smooth muscle contraction by RhoA that is distinct from its previously documented role in regulating the phosphorylation of the regulatory light chain of SM myosin II. Our observations suggest that the RhoA-mediated assembly of adhesome signaling complexes is an essential step in excitation-contraction coupling and tension development in smooth muscle tissues during agonist-induced contractile activation.
EXPERIMENTAL PROCEDURES
Preparation of Smooth Muscle Tissues Measurement of Force
Mongrel dogs (20–25 kg) were anesthetized with pentobarbital sodium (30 mg/kg, iv.) and quickly exsanguinated in accordance with procedures approved by the Institutional Animal Care and Use Committee (IACUC), Indiana University School of Medicine. A tracheal segment was immediately removed and immersed in physiological saline solution (PSS) (composition in mm: 110 NaCl, 3.4 KCl, 2.4 CaCl2, 0.8 MgSO4, 25.8 NaHCO3, 1.2 KH2PO4, and 5.6 glucose, bubbled with 95% O2, 5% CO2). Strips of tracheal smooth muscle (1.0 × 0.2–0.5 × 15 mm) were dissected free of connective and epithelial tissues and maintained within a tissue bath in PSS at 37 °C. Contractile force was measured isometrically by attaching the tissues to Grass force-displacement transducers. Prior to the beginning of each experimental protocol, muscle length was increased to maintain a preload of ∼1 gm, and tissues were stimulated repeatedly with acetylcholine (ACh) until stable responses were obtained. The force of contraction to ACh was determined before and after treatment with plasmids or other reagents.
Transfection of Smooth Muscle Tissues
The use of constructs for the hemagglutinin (HA) human RhoA Asn-19, human cdc42 Asn-17, and chicken paxillin Y118/31F in tracheal smooth muscle tissues have been previously described (10, 26, 31). Constructs for chicken paxillin ΔLD4 (36, 37), chicken paxillin Y118/31F were generously provided by Dr. Christopher Turner, SUNY Upstate Medical University. The cDNAs encoding these constructs were subcloned into the mammalian expression vector pcDNA3.1. Escherichia coli (Bluescript) transformed with these plasmids were grown in LB medium, and plasmids were purified by alkaline lysis with SDS using a purification kit from Qiagen Inc.
Plasmids were introduced into tracheal smooth muscle strips by the method of reversible permeabilization (10, 17, 26, 31). After equilibration of the tissues and establishing a muscle length for the generation of maximal isometric force, muscle strips were attached to metal mounts to maintain them isometrically at constant length. The strips were incubated successively in each of the following solutions: Solution 1 (at 4 °C for 120 min) containing (in mm): 10 EGTA, 5 Na2ATP, 120 KCl, 2 MgCl2, and 20 N-Tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (TES); Solution 2 (at 4 °C overnight) containing (in mm): 0.1 EGTA, 5 Na2ATP, 120 KCl, 2 MgCl2, 20 TES, and 10 μg/ml plasmids. Solution 3 (at 4 °C for 30 min) containing (in mm): 0.1 EGTA, 5 Na2ATP, 120 KCl, 10 MgCl2, 20 TES; and Solution 4 (at 22 °C for 60 min) containing (in mm): 110 NaCl, 3.4 KCl, 0.8 MgSO4, 25.8 NaHCO3, 1.2 KH2PO4, and 5.6 dextrose. Solutions 1–3 were maintained at pH 7.1 and aerated with 100% O2. Solution 4 was maintained at pH 7.4 and was aerated with 95% O2-5% CO2. After 30 min in Solution 4, CaCl2 was added gradually to reach a final concentration of 2.4 mm. The strips were then incubated in a CO2 incubator at 37 °C for 2 days in serum-free DMEM containing 5 mm Na2ATP, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10 μg/ml plasmids.
Assessment of RhoA and cdc42 Activation
The activation of RhoA or cdc42 was determined using pull-down assays for activated RhoA or activated cdc42 (10, 26). Pulverized muscle tissues were mixed with lysis buffer (1% Np-40, 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 10% glycerol, 2 mm EDTA, phosphatase inhibitors (in mm: 2 sodium orthovanadate, 2 molybdate, and 2 sodium pyrophosphate), and protease inhibitors (in mm: 2 benzamidine, 0.5 aprotinin, and 1 phenylmethylsulfonyl fluoride) for 2 h at 4 °C. For the analysis of RhoA activation, the extracted proteins were reacted with a GST-peptide for the RBD region of rhotekin, which has a high affinity for GTP-Rho. For the analysis of cdc42 activation, the extracted proteins were reacted with GST-p21-activated kinase (PAK) binding domain (PBD). Activated GTP-bound RhoA or cdc42 were affinity-precipitated by glutathione beads and quantified by immunoblot.
Cell Dissociation and Immunofluorescence Analysis
Freshly dissociated primary cells were used for these studies to avoid the morphological changes in cytoskeletal organization and changes in phenotype that occur during the culture of smooth muscle cells. Smooth muscle cells were enzymatically dissociated from tracheal muscle strips (17). For immunofluorescence analysis, freshly dissociated cells were plated onto glass coverslips and allowed to adhere for 30–60 min, were stimulated with 10−5 m ACh or left unstimulated, and then fixed, permeabilized, and incubated with primary antibodies against proteins of interest.
In Situ Proximity Ligation Assay
DuolinkTM in situ proximity ligation assays (PLA) were performed to detect cellular interactions between N-WASp and Arp2, vinculin and paxillin, FAK and paxillin, or PIX and paxillin (32, 38). PLA yields a fluorescent signal when the target proteins are localized within 40 nm of each other. Smooth muscle cells were freshly dissociated from sham-treated or transfected canine tracheal smooth muscle tissues as previously described (17, 29). Dissociated smooth muscle cells were stimulated with 10−5 m ACh or left unstimulated and then fixed, permeabilized and incubated with primary antibodies followed by secondary antibodies conjugated to PLA probes. Duolink hybridization, ligation, amplification, and detection media were administered according to the manufacturer's instructions. Randomly selected cells from both unstimulated and ACh-stimulated groups were analyzed for N-WASP-Arp2, vinculin, and paxillin, FAK and paxillin or paxillin, and β-PIX interactions by visualizing Duolink fluorescent spots using a Zeiss LSM 510 confocal microscope. The total number of Duolink fluorescent spots for the N-WASp-Arp2 and paxillin-FAK interactions were counted using Olink Bioscience Image Tools software. The localization of the vinculin-paxillin complex was evaluated by determining the ratio of pixel intensity between the cell periphery and the cell interior using Metamorph software (Molecular Devices, Inc. Sunnyvale, CA) (see supplemental Fig. S1 for details).
Immunoblots
Pulverized muscle strips were mixed with extraction buffer containing: 20 mm Tris-HCl at pH 7.4, 2% Triton X-100, 0.2% SDS, 2 mm EDTA, phosphatase inhibitors (2 mm sodium orthovanadate, 2 mm molybdate, and 2 mm sodium pyrophosphate), and protease inhibitors (2 mm benzamidine, 0.5 mm aprotinin, and 1 mm phenylmethylsulfonyl fluoride). Each sample was centrifuged, and the supernatant was then boiled in sample buffer (1.5% dithiothreitol, 2% SDS, 80 mm Tris-HCl, pH 6.8, 10% glycerol, and 0.01% bromphenol blue) for 5 min. Proteins were separated by SDS-PAGE and transferred to nitrocellulose. The nitrocellulose membrane was blocked with 5% milk for 1 h and probed with primary antibodies against proteins of interest followed by horseradish peroxidase-conjugated IgG. Proteins were visualized by enhanced chemiluminescence (ECL).
Immunoprecipitation of Proteins
Pulverized muscle tissues were mixed with extraction lysis buffer (see “Assessment of RhoA and cdc42 Activation”). Each sample was centrifuged (14,000 × g) for 30 min for the collection of supernatant. Muscle extracts containing equal amounts of protein were precleared for 30 min with 30 μl of 10% protein A/G-Sepharose and then incubated overnight with primary antibodies. Samples were then incubated for 2 h with 50 μl of a 10% suspension of protein A/G-Sepharose beads. Immunocomplexes were washed three times in a buffer containing 50 mm Tris HCl, pH 7.6, 150 mm NaCl, and 0.1% Triton X-100. All procedures of immunoprecipitation were performed at 4 °C.
Analysis of F-actin and G-actin
The relative proportions of F-actin and G-actin in smooth muscle tissues were analyzed using an assay kit from Cytoskeleton as previously described (10, 17, 26, 39). Briefly, each of the tracheal smooth muscle strips was homogenized in 200 μl of F-actin stabilization buffer (50 mm PIPES, pH 6.9, 5 0 mm NaCl, 5 mm MgCl2, 5 mm EGTA, 5% glycerol, 0.1% Triton X-100, 0.1% Nonidet P-40, 0.1% Tween-20, 0.1% β-mercaptoethanol, 0.001% antifoam, 1 mm ATP, 1 μg/ml pepstatin, 1 μg/ml leupeptin, 10 μg/ml benzamidine, and 500 μg/ml tosyl arginine methyl ester). Supernatants of the protein extracts were collected after centrifugation at 150,000 × g for 60 min at 37 °C. The pellets were resuspended in 200 μl of ice-cold water containing 10 μm cytochalasin D and then incubated on ice for 1 h to depolymerize F-actin. The resuspended pellets were gently mixed every 15 min. Four microliters of supernatant (G-actin) and pellet (F-actin) fractions were subjected to immunoblot analysis using anti-actin antibody (clone AC-40; Sigma). The relative amounts of F-actin and G-actin were determined using densitometry.
Reagents and Antibodies
RhoA and cdc42 activation assay kits were obtained from Cytoskeleton (Denver, CO). ROCK inhibitors, H-1152P and Y27632 were obtained from Calbiochem. The ROCK activity assay kit was obtained from Cell Biolabs (San Diego, CA). The FAK inhibitor FP 573228 was obtained from Tocris Bioscience (Bristol, UK). The DuolinkTM in situ proximity ligation assay kit was purchased from Olink Bioscience (Uppsala, Sweden). All other reagents were obtained from Sigma.
Sources of antibodies are as follows: monoclonal RhoA, Cytoskeleton; monoclonal paxillin and monoclonal cdc42, monoclonal anti-CrkII, BD Transduction; polyclonal paxillin tyrosine 118, BIOSOURCE; monoclonal N-WASp, polyclonal Arp2, N-WASp tyrosine 256, Abcam; polyclonal anti-FAK, polyclonal anti-β-PIX and anti-α-PIX, polyclonal anti-GIT1 and GIT2, polyclonal anti-DOCK180, polyclonal anti-PAK1, Cell Signaling; monoclonal anti-FAK and monoclonal anti-FAK Y397, BD Biosciences; monocloncal anti-ELMO1, horseradish peroxidase-conjugated IgG, Amersham Biosciences; polyclonal vinculin antibody (against canine cardiac vinculin) was custom made by BABCO (Richmond, CA).
Statistical Analysis
Comparisons between two groups were performed using paired or unpaired Student's t tests. Comparisons among multiple groups were performed using one-way ANOVA. Values refer to the number of cells or tissue strips used to obtain mean values. p < 0.05 was considered statistically significant.
RESULTS
RhoA Inactivation Inhibits ACh-induced N-WASp Activation in Smooth Muscle Tissues
We expressed RhoA T19N protein in tracheal muscle tissues to determine whether RhoA activation is necessary for the ACh-induced activation of N-WASp, which is required for ACh-induced actin polymerization and tension development (17). The expression of the dominant-negative RhoA T19N protein in tracheal muscle tissues inhibits endogenous RhoA activity and suppresses actin polymerization and active tension generation in response to stimulation with ACh with little effect on SM myosin light chain phosphorylation (10).
When N-WASp undergoes activation, it localizes at the cell membrane and undergoes a conformational change that enables it to bind to the Arp2/3 complex and undergo tyrosine phosphorylation at residue 256 (19, 20, 40). To assess N-WASp activation in tissue extracts, we measured N-WASp tyrosine 256 phosphorylation and evaluated the interaction of N-WASp with the Arp2/3 complex. Our results show that expression of RhoA T19N inhibits the ACh-induced increase in N-WASp tyrosine 256 phosphorylation (Fig. 1A), and that this inhibition of N-WASp activation is associated with a marked depression of ACh-induced active tension development (Fig. 1B). The inhibition of actin polymerization by RhoA T19N was confirmed in a separate set of tissues (Fig. 1C). Co-immunoprecipitation analysis demonstrated that the RhoA T19 mutant also inhibited the increased interaction of Arp2 and N-WASp that occurs in response to ACh stimulation (Fig. 1D). The ACh-induced increase in N-WASp Y256 phosphorylation, the association of N-WASp with the Arp2/3 complex, actin polymerization and active tension development were unaffected by sham treatment of the tissues and/or by the expression of WT RhoA.
FIGURE 1.

RhoA inactivation inhibits ACh induced N-WASp activation in smooth muscle tissues. A, N-WASp tyrosine 256 phosphorylation measured by immunoblot in extracts of 6 muscle tissues transfected with wild type RhoA (WT), RhoA T19N, or sham-treated. RhoA T19N significantly inhibited ACh-induced N-WASp phosphorylation relative to WT RhoA or sham-treated tissues (n = 5). B, RhoA T19N significantly inhibited ACh-induced tension development relative to WT RhoA or sham-treated tissues (n = 10). (All force measurements normalized to sham response.) C, immunoblot of soluble (G, globular) and insoluble (F, filamentous) actin in fractions from extracts of unstimulated or ACh-stimulated muscle tissues treated with RhoA T19N or with no treatment (sham). RhoA T19N significantly inhibited the increase in the F actin to G-actin ratio in response to 10−5 m ACh (n = 4). D, ACh induced a significant increase in Arp2 coimmunoprecipitation with N-WASp in sham-treated tissues but not in RhoA T19N-treated tissues (n = 4). IP: immunoprecipitate; IB, immunoblot. E, duolink in situ PLA shows the interaction of N-WASp and Arp2 in freshly dissociated differentiated canine tracheal smooth muscle cells stimulated with ACh, but not in unstimulated cells. Fluorescence and phase contrast images are shown for each cell. F, in cells from sham-treated tissues, mean DuoLink PLA spots were significantly higher in ACh-stimulated cells than in unstimulated cells (n = 27, 25). In cells from RhoA T19N-treated tissues, the mean number of DuoLink PLA spots was very small and did not change significantly with ACh-stimulation (n = 24, 32 respectively). (Cells dissociated from tissues obtained from three separate experiments.) All tissues and cells were incubated with ACh for 5 min. All values are means ± S.E. *, significantly different (p < 0.05).
A Duolink in situ PLA using probes targeted to N-WASp, and Arp2 was also used to assess the interaction of N-WASp and Arp2 in freshly dissociated tracheal smooth muscle cells from untreated tissues and tissues treated with RhoAT19N (Fig. 1, E and F and supplemental Fig. S2). In sham-treated cells, stimulation with ACh resulted in a dramatic increase in the number of fluorescent spots along the membrane, indicating many interactions between N-WASp and the Arp2/3 complex. In contrast, ACh-stimulated cells expressing RhoA T19N showed few fluorescent spots. These data provide further evidence that RhoA activation is required for the ACh-induced activation of N-WASp in airway smooth muscle.
RhoA Inactivation Inhibits the ACh-induced Activation of cdc42 in Canine Tracheal Smooth Muscle Tissues
Activation of the actin regulatory protein N-WASp is directly and specifically regulated by the small GTPase, cdc42 (20–23). Neither rac nor Rho GTPases can bind directly to WASP family proteins, thus RhoA cannot directly regulate N-WASp activity (24, 25). We therefore questioned whether RhoA might mediate N-WASp activation in tracheal muscle tissues by regulating the activation of cdc42.
We found that the inhibition of RhoA activity by RhoA T19N or cell permeable C3 exoenzyme inhibited the increase in cdc42 activation that occurs in response to ACh stimulation, and that it also inhibited tension development (Fig. 2, A, B, and supplemental Fig. S3A). In contrast, when cdc42 was inactivated by the expression of the inactive cdc42 mutant, cdc42 T17N, RhoA activation was not inhibited (Fig. 2C). The inactivation of either cdc42 or RhoA inhibited the increase in tension stimulated by ACh. Our results demonstrate that RhoA activation is prerequisite to the activation of cdc42 by ACh in tracheal smooth muscle, but that cdc42 does not regulate RhoA activity.
FIGURE 2.

RhoA inactivation inhibits the ACh-induced activation of cdc42 in canine tracheal smooth muscle tissues. A, activated cdc42 (cdc42-GTP) was affinity-precipitated from extracts of 3 unstimulated and 3 ACh-stimulated muscle strips, and the amount of activated cdc42 precipitated from each extract was quantified by immunoblot. Activated cdc42 was significantly higher in extracts from 10−5 m ACh stimulated sham-treated tissues than from ACh stimulated tissues expressing RhoA T19N or cdc42 T17N (n = 8). Blot shows non-contiguous lanes from a single gel. B, RhoA T19N and cdc42 T17N significantly inhibited tension development in response to ACh (n = 16). C, activated GTP-bound RhoA (RhoA-GTP) was affinity-precipitated from extracts of 6 unstimulated or ACh stimulated muscle strips and quantifitated by immunoblot. RhoA activation was significantly inhibited in tissues expressing RhoA T19N but was unaffected by the expression of cdc42 T17N (n = 6). Blot shows non-contiguous lanes from a single gel. All values are means ± S.E. *, significantly different, p < 0.05.
RhoA Regulates cdc42 and N-WASp Activation by Regulating Paxillin Phosphorylation
We next evaluated the mechanism by which RhoA regulates the activity of cdc42 and N-WASp. In tracheal smooth muscle, the activation of N-WASp and cdc42 are mediated by the interaction of N-WASp with the SH2/SH3 adaptor protein, CrkII, which binds to tyrosine-phosphorylated paxillin via its SH2 domain and to N-WASp via its SH3 domain (34, 39, 41). Paxillin undergoes tyrosine phosphorylation at tyrosine 31 and tyrosine 118 in response to the contractile stimulation of tracheal smooth muscle (31, 42). The phosphorylation of paxillin at these sites is necessary for actin polymerization and tension development (31).
We first assessed the roles of RhoA and cdc42 in the regulation of paxillin tyrosine phosphorylation. The inactivation of RhoA by RhoA T19N or cell permeable C3 exoenzyme inhibited the tyrosine phosphorylation of paxillin induced by ACh, whereas the inactivation of cdc42 had no effect on paxillin phosphorylation (Fig. 3A and supplemental Fig. S3B); thus the activation of RhoA but not cdc42 activation is prerequisite for paxillin tyrosine phosphorylation.
FIGURE 3.

Rho regulates cdc42 and N-WASp activation by regulating paxillin phosphorylation. A, paxillin Tyr-118 phosphorylation in response to 10−5 m ACh stimulation was significantly inhibited in tissues expressing RhoA T19N, but not in tissues expressing cdc42 T17N (n = 10). Blot shows non-contiguous lanes from a single gel. B, Thr-696 phosphorylation of MYPT1, a substrate of activated ROCK, was measured by immunobot in tissue extracts from ACh stimulated tissues. MYPT1 phosphorylation was significantly lower in tissues treated with the ROCK inhibitor, H-1152P (1 μm) (n = 4). C, there are no significant effects of the ROCK inhibitor on paxillin phosphorylation in ACh-stimulated or unstimulated tissues (n = 4). All values are means ± S.E. *, significantly different, p < 0.05.
We then investigated the possibility that Rho kinase (ROCK) might be an effector of RhoA-mediated paxillin phosphorylation. The ROCK inhibitor, H-1152P, effectively inhibited phosphorylation of the ROCK substrate MYPT1 at threonine 696 in tracheal tissues (Fig. 3B), and also inhibited the in vitro phosphorylation of a synthetic MYPT peptide (data not shown). ACh stimulation of canine tracheal smooth muscle tissues increased ROCK activation (Fig. 3B). However, ROCK inhibition did not inhibit ACh-induced paxillin phosphorylation (Fig. 3C). Similar results were obtained using the ROCK inhibitor, Y27632 (data not shown). Thus, RhoA activation is necessary for paxillin tyrosine phosphorylation in response to ACh, but RhoA mediated paxillin phosphorylation is not regulated by ROCK.
Paxillin Tyrosine Phosphorylation Is Required for cdc42 and N-WASP Activation
We evaluated the role of paxillin phosphorylation in regulating the ACh-induced activation of cdc42 and N-WASp by expressing the non-phosphorylatable paxillin mutant, paxillin Y118/31F, in tracheal muscle tissues (31). The expression of paxillin Y118/31F prevented the tyrosine phosphorylation of endogenous paxillin and tension development in response to ACh (Fig. 4A). The suppression of paxillin phosphorylation by paxillin Y118/31F inhibited N-WASp phosphorylation and its association with the Arp2/3 complex in response to ACh (Fig. 4, B and C). As expected, cdc42 inactivation also inhibited the activation of N-WASp (Fig. 4B). The inhibition of paxillin phosphorylation also suppressed the activation of cdc42, but it did not affect the activation of RhoA (Fig. 4D). We conclude that paxillin phosphorylation is prerequisite for cdc42 and N-WASp activation, but that paxillin does not regulate RhoA activity.
FIGURE 4.

Paxillin tyrosine phosphorylation is required for cdc42 and N-WASP activation, but not for RhoA activation. A, increase of paxillin Tyr-118 phosphorylation in response to 10−5 m ACh was significantly lower in tissues expressing paxillin Y118/Y31F (n = 5). Blot shows non-contiguous lanes from a single gel. Paxillin Y118/31F significantly inhibited tension development relative to sham-treated tissues (n = 8). B, expression of paxillin Y118/31F (n = 4) or cdc42 T17N (n = 5) significantly inhibited N-WASp tyrosine 256 phosphorylation in response to ACh stimulation. C, increase in co-immunoprecipitation of Arp2 with N-WASp in response to ACh stimulation was significantly inhibited in tissues expressing paxillin Y118/31F (n = 3). Blot show non-contiguous lanes from a single gel. D, immunoblots against activated GTP-bound RhoA or GTP-bound cdc42 affinity-precipitated from extracts of 4 sham or 4 paxillin Y118/31F-treated muscle strips. Expression of paxillin Y118/31F did not significantly affect RhoA activation in response to 10−5 m ACh (n = 6), but it significantly inhibited cdc42 activation (n = 4). All values are means ± S.E. *, significantly different (p < 0.05).
RhoA Regulates the Recruitment of Paxillin to the Cell Membrane in Response to Contractile Stimulation
Paxillin can bind to vinculin (27) and it co-localizes with vinculin in differentiated airway smooth muscle cells (29). The stimulation of airway smooth muscle cells increases the localization of both paxillin and vinculin at the membrane (29). We therefore investigated the possibility that RhoA regulates the interaction of paxillin with vinculin or the recruitment of paxillin to the membrane.
The effect of contractile stimulation and RhoA inactivation on the interaction between paxillin and vinculin was assessed by co-immunoprecipitation analysis (Fig. 5). Paxillin co-precipitated with vinculin from unstimulated and ACh-stimulated tissues at the same ratio regardless of the phosphorylation state of paxillin, which suggests that paxillin and vinculin form a stable molecular complex that is unaffected by ACh stimulation. RhoA inactivation by RhoA T19N had no effect on the co-immunoprecipitation of paxillin with vinculin, indicating that the interaction of paxillin with vinculin is not RhoA dependent.
FIGURE 5.

The association between paxillin and vinculin is unaffected by ACh stimulation or RhoA inhibition. A and B, vinculin was immunoprecipitated from extracts from sham-treated and RhoA-treated muscles and immunoprecipitates were blotted for vinculin, paxillin, and paxillin Tyr-118. There were no significant differences between unstimulated and ACh-stimulated tissues, or between sham and RhoA T19N-treated tissues in the amount of paxillin that coprecipitated with vinculin (n = 4). Phospho-paxillin at Tyr-118 increased significantly in vinculin immunoprecipitates from 10−5 m ACh stimulated in sham-treated tissues, but not in RhoA T19N-treated tissues. All values are means ± S.E. *, significantly different (p < 0.05).
Duolink PLA was used to determine the effect of RhoA activation on the cellular localization of paxillin/vinculin complexes (Fig. 6, A and B and supplemental Fig. S4). In sham-treated tissues, paxillin/vinculin complexes were observed throughout the cytoplasm of unstimulated cells but localized predominantly at the membrane of ACh-stimulated cells. In cells expressing RhoAT19N, paxillin/vinculin complexes did not localize at the membrane of ACh-stimulated cells; a diffuse distribution throughout the cytoplasm was observed in both stimulated and unstimulated cells. Neither ACh stimulation nor RhoA inactivation significantly affected the total fluorescence intensity of paxillin-vinculin complexes in the smooth muscle cells, indicating that the association between paxillin and vinculin is unaffected by ACh stimulation or RhoA activation (Fig. 6C). We conclude that RhoA regulates the recruitment of a protein complex containing both paxillin and vinculin to membrane adhesion junctions in response to stimulation with ACh.
FIGURE 6.

Rho regulates the paxillin-vinculin complex recruitment to the cell membrane in response to contractile stimulation. A, Duolink PLA was used to evaluate the interaction of paxillin and vinculin in freshly dissociated cells stimulated with 10−4 m ACh for 5 min or left unstimulated. DuoLink spots were observed throughout the cytoplasm of unstimulated cells but primarily at the cell periphery of ACh stimulated cells from sham treated tissues. In cells from RhoA T19N-treated tissues, DuoLink spots were distributed throughout the cytoplasm of both unstimulated and ACh-stimulated cells. B, localization of paxillin-vinculin complex was quantified by calculating ratio of fluorescence intensity between the cell periphery and the cell interior. (See supplemental Fig. S1 for details.) In cells from sham-treated tissues, ACh stimulation significantly increased the membrane to cytoplasm ratio. A significant increase in the membrane to cytoplasm ratio in response to ACh stimulation was not observed in cells treated with RhoA T19N (n = 14–23). C, there were no significant differences in the total cellular fluorescence intensity of paxillin-vinculin complexes in cells from sham-treated tissues and RhoA T19N-treated tissues with or without ACh (n = 14–23). All values are means ± S.E. *, significantly different, p < 0.05.
RhoA Regulates the Association of Paxillin with FAK at the Cell Membrane and Paxillin Tyrosine Phosphorylation in Response to Contractile Stimulation
FAK binds to paxillin and can induce the phosphorylation of paxillin at tyrosines 31 and 118 (33, 34). We used immunofluorescence analysis and Duolink PLA to evaluate the effect of RhoA on FAK activation and on the interaction between FAK and paxillin. RhoA inactivation inhibited the recruitment of both FAK and paxillin to cell membrane and prevented the phosphorylation and activation of FAK in response to ACh stimulation (Fig. 7, A and B). Tyrosine-phosphorylated paxillin was observed only at the membrane in both unstimulated and ACh-stimulated smooth muscle cells (Fig. 7C).
FIGURE 7.

The phosphorylation of paxillin occurs at the cell membrane and requires the RhoA-dependent recruitment of FAK. A, cells freshly dissociated from sham-treated or RhoA T19N treated muscle tissues were stimulated with 10−4 m ACh or left unstimulated, fixed, and double-stained for FAK and paxillin. Both proteins were distributed throughout the cytoplasm of unstimulated cells and localized to the cell membrane of ACh-stimulated cells. Neither FAK nor paxillin significantly increased at the cell membrane in response to ACh stimulation in cells dissociated from RhoA T19N-treated tissues. B, FAK phosphorylation at tyrosine 397 was significantly increased by ACh in sham-treated tissues but not RhoA T19N-inhibited tissues (n = 11). C, Tyr-118 paxillin was observed at the cell membrane of both unstimulated and ACh-stimulated cells, but the intensity of paxillin Tyr-118 phosphorylation was higher in ACh-stimulated cells. D and E, interaction of paxillin and FAK in freshly dissociated tracheal smooth muscle cells was detected using the Duolink PLA. In cells from sham-treated tissues, few or no spots are detected in unstimulated cells; whereas many spots are seen at the membrane of the ACh-stimulated cell. The total number of DuoLink spots was significantly higher in ACh-stimulated smooth muscle cells than in unstimulated cells (cells from 5 separate experiments, n = 34, 30). In cells from RhoA T19N-treated smooth muscle tissues, few or no spots were detected in unstimulated cells or ACh-stimulated cells. Cells from three separate experiments, (n = 27, 15). All values are means ± S.E. *, significantly different (p < 0.05).
We assessed the cellular location of the interaction between FAK and paxillin using Duolink PLA and evaluated whether RhoA regulated the interaction of FAK and paxillin. In sham-treated muscles, FAK interacted with paxillin in ACh-stimulated cells but not in unstimulated cells, and the interaction between FAK and paxillin occurred at the cell membrane (Fig. 7, D and E).
The Inhibition of FAK Kinase Activity Suppresses ACh-induced Cdc42 Activation and Actin Polymerization
We assessed the effect of inhibiting FAK activation on the regulation of paxillin phosphorylation, cdc42 activation and actin polymerization by treating tracheal muscle tissues with the synthetic FAK inhibitor, FP 573228 (43). We first confirmed the effectiveness of FP 573228 at inhibiting FAK activation in tracheal muscle tissues by measuring its effect on the ACh-induced phosphorylation of FAK at its activation site, Tyr-397. Treatment of the tissues with 30 μm FP 573228 prevented the increase in FAK Tyr-397 phosphorylation induced by ACh, and also suppressed the ACh-induced increase in paxillin phosphorylation at Tyr-118 (Fig. 8, A and B; n = 4, p < 0.05). The inhibition of FAK activation inhibited cdc42 activation and actin polymerization induced by stimulation with ACh (Fig. 8, C and D). The inhibition of FAK also suppressed force by more than 50% (data not shown).
FIGURE 8.

The inhibition of FAK prevents ACh-induced cdc42 activation and actin polymerization. A and B, treatment of tracheal smooth muscle tissues with 30 μm FAK inhibitor, FP 573228, inhibits phosphorylation of the FAK activation site Tyr-397 and paxillin phosphorylation at Tyr-118 in response to 10−5 m ACh (n = 4). Blot of paxillin phosphorylation shows non-contiguous lanes from a single gel C, FAK inhibitor, FP 573228, significantly inhibited the increase in the F actin to G-actin ratio in response to 10−5 m ACh (n = 4). Immunoblot of soluble (G, globular) and insoluble (F, filamentous) actin in fractions from extracts of unstimulated or ACh-stimulated muscle tissues treated with FAK inhibitor or with no inhibitor. D, treatment with FAK inhibitor, FP 573228 inhibits ACh- induced cdc42 activation (n = 3). For each assay, activated cdc42 (cdc42-GTP) was affinity-precipitated from extracts of 2 unstimulated and 2 ACh-stimulated muscle strips, and the amount of activated cdc42 precipitated from each extract was quantified by immunoblot.
These results suggest that FAK and paxillin are independently recruited to the membrane by a RhoA-dependent mechanism, and that after FAK is recruited to the membrane, it undergoes activation and induces the phosphorylation of paxillin. Thus, RhoA appears to regulate paxillin phosphorylation indirectly by mediating the recruitment of both FAK and paxillin to membrane complexes where they can interact.
The Association of Paxillin with cdc42 GEFs Is Increased by ACh Stimulation and Inhibited by RhoA Inhibition
We evaluated several GEFs as potential mediators of cdc42 activation by paxillin during contractile stimulation. The PIX proteins bind to PAK and have GEF activity toward both cdc42 and rac (35, 44). PIX proteins form a complex with GIT1 (G-protein receptor kinase-interacting tyrosine phosphorylated) and GIT2 (also called paxillin kinase linker or PKL), which are ArfGAPS that also regulate cdc42 and rac (45). During the adhesion of cultured cells to a substrate, the localization of PAX-GIT complexes to focal adhesion sites is mediated by the regulated binding of GIT molecules to the LD4 domain of paxillin (36, 37).
DOCK (Dedicator of Cytokinesis) family proteins also have GEF activity toward rac and cdc42 (35). DOCK180 interacts with Crk, a paxillin-binding adaptor protein (39, 41) and partners with ELMO (engulfment and cell motility protein) to catalyze nuclear exchange (46, 47).
We analyzed muscle tissue extracts by co-immunoprecipitation and found that the association of paxillin, CrkII, and ELMO1 with DOCK180 increases in muscles after contractile stimulation with ACh (Fig. 9A). The association of PIX proteins, GIT1, CrkII, and Pak with paxillin also increased in cells activated with ACh (Fig. 9, B and C). Similar analysis using Duolink PLA technology revealed extensive complexes between β-PIX and paxillin at the membrane of muscle cells after contractile stimulation, that were observed only minimally in unstimulated muscle cells (Fig. 9, D and E). The formation of membrane complexes between β-PIX, GIT1, and paxillin was inhibited by the inactivation of RhoA (Fig. 9, C–E).
FIGURE 9.

RhoA activation regulates the association of paxillin with cdc42 GEF. A, co-immunoprecipitation of ELMO1, paxillin, and CrkII with DOCK180 increased in ACh-stimulated tissues. Result typical of six independent experiments. B, co-immunoprecipitation of α-PIX, PAK1 and CrkII with paxillin increased after contractile stimulation with ACh. Result typical of four independent experiments. C, co-immunoprecipitation of GIT1 and β-PIX with paxillin increased in ACh-stimulated tissues and was significantly inhibited in tissues expressing RhoA T19N (n = 4). D, Duolink PLA reveals interaction between β-PIX and paxillin at the membrane of muscle cells after contractile stimulation, whereas they are not observed in unstimulated muscle cells. RhoA T19N inhibited the formation of paxillin/β-PIX complexes. E, in sham-treated tissues, the mean number of DuoLink PLA spots was significantly higher in ACh-stimulated cells than in unstimulated cells. (Cells dissociated from tissues from 3 separate experiments (n = 19, 18 cells.) In cells from RhoA T19N-treated tissues, the mean number of DuoLink PLA spots was small in both unstimulated and ACh-stimulated cells and was not significantly different (n = 18). All values are means ± S.E. *, significantly different (p < 0.05).
We confirmed the role of the paxillin/GIT/PIX complex in the activation of cdc42 by expressing a paxillin mutant with a deletion of the LD4 domain in the muscle tissues (paxillin ΔLD4) (36, 37). The LD4 domain of paxillin regulates its binding to GIT proteins and is required for the localization of GIT and PIX proteins to membrane adhesomes (36, 37). The expression of paxillin ΔLD4 in the muscle tissues prevented the interaction of paxillin with GIT1 and β-PIX in response to stimulation with ACh, as indicated by co-immunoprecipitation analysis of muscle tissue extracts and Duolink PLA assays in freshly dissociated cells (Fig. 10, A–C). Expression of paxillin ΔLD4 also inhibited the ACh-induced activation of cdc42 and force development (Fig. 10, D and E). These results indicate that contractile activation stimulates the recruitment of GEF signaling complexes to adhesomes via a RhoA-dependent mechanism where they couple paxillin to the activation of cdc42.
FIGURE 10.

The association of paxillin with β-PIX regulates cdc42 activation. A, co-immunoprecipitation of GIT1 and β-PIX with paxillin was significantly inhibited in tissues expressing paxillin ΔLD4 (n = 4). B, Duolink PLA reveals interaction between β-PIX and paxillin. Paxillin ΔLD4 inhibited the formation of paxillin/β-PIX complexes. C, in sham-treated tissues, the mean number of DuoLink PLA spots was significantly higher in ACh-stimulated cells than in unstimulated cells. (Cells dissociated from tissues from 3 separate experiments (n = 19, 18 cells.)) In cells from paxillin ΔLD4-treated tissues, the mean number of DuoLink PLA spots was small in both unstimulated and ACh-stimulated cells and was not significantly different (n = 18). All values are means ± S.E. *, significantly different (p < 0.05). D, expression of paxillin ΔLD4 inhibits ACh-induced cdc42 activation. (n = 4). For each assay, activated cdc42 (cdc42-GTP) was affinity-precipitated from extracts of 2 unstimulated and 2 ACh-stimulated muscle strips. E, paxillin ΔLD4 significantly inhibited tension development relative to sham-treated tissues (n = 8). All values are means ± S.E. *, significantly different (p < 0.05).
DISCUSSION
Our studies demonstrate that the activation of RhoA GTPase during the contractile stimulation of airway smooth muscle tissues catalyzes the assembly of signaling complexes at integrin adhesion sites that are necessary for the regulation of actin polymerization and tension development (summarized in Fig. 11). The adaptor/scaffolding protein complex that is assembled in response to RhoA activation includes paxillin and vinculin, FAK, which induces paxillin tyrosine phosphorylation, and the α/β-PIX and DOCK180 GEF signaling modules, which can regulate the activity of cdc42 and rac. We show that paxillin couples to these GEF signaling modules and regulates the activity of cdc42, which activates N-WASp to catalyze actin polymerization by the Arp2/3 complex. The association of paxillin with these GEFs is also RhoA-dependent. Thus, RhoA activation is prerequisite to all of the steps in the signaling pathway that regulates N-WASp activation in response to contractile stimulation in airway smooth muscle. These studies describe a novel mechanism for the regulation of smooth muscle contraction by RhoA that is independent of its well-known role in regulating the phosphorylation of the regulatory light chain of smooth muscle myosin II. We demonstrate that RhoA-mediated adhesome assembly plays a critical role in the process of excitation-contraction coupling during agonist-induced smooth muscle contraction.
FIGURE 11.
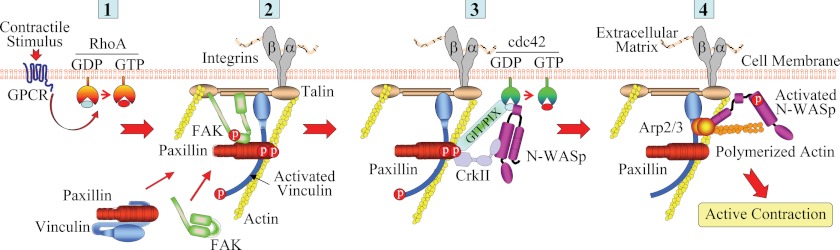
Model for proposed mechanism for the regulation of ACh induced RhoA activation on the assembly of an adhesome signaling complex in ASM. 1, ACh stimulation activates RhoA, which induces the independent recruitment of paxillin-vinculin complexes and FAK to cell adhesomes; 2, FAK and paxillin interact at the adhesome and activated FAK induces the phosphorylation of paxillin, which remains bound to activated vinculin; 3, phosphorylation of paxillin facilitates the formation of a complex containing paxillin and Crk II with DOCK180 and PIX GEFs. This complex induces the activation of cdc42. 4, cdc42 activation catalyzes the activation of N-WASp, which interacts with the Arp2/3 complex to induce actin polymerization in the cortical region of the smooth muscle cell. This enables tension generation by the smooth muscle contractile apparatus.
The regulation of airway smooth muscle contraction by RhoA has been ascribed to its inhibitory effects on the catalytic activity of smooth muscle MLC phosphatase (48, 49). The inhibition of MLC phosphatase can augment agonist-induced MLC phosphorylation, which increases smooth muscle contractility (8, 9). However, our results demonstrate that this is not the primary mechanism by which RhoA regulates the contractility of airway smooth muscle.
Studies of adhesome assembly during substrate adhesion in cultured cell lines have documented an important role for RhoA in the assembly and maturation of focal adhesions (2, 50, 51). These studies suggest that RhoA activation induces phosphorylation of the light chain of non-muscle myosin II, and that this is prerequisite to the formation of focal adhesions and stress fibers (52). In these cells, inhibiting non-muscle myosin II activation disrupts the formation of Rho-induced stress fibers and results in focal adhesion disassembly. It is possible that RhoA regulates adhesome assembly and excitation contraction coupling during the contraction of airway smooth muscle tissues by an analogous mechanism.
In airway smooth muscle, adhesion protein activation, and actin polymerization are independent of the phosphorylation of SM myosin light chain by myosin light chain kinase (MLCK) (12, 17, 32). The MLC kinase inhibitor, ML-7, suppresses myosin light chain phosphorylation and tension development in response to contractile stimulation, but it has no effect on ACh-induced actin polymerization (10). The inhibition of MLCK also does not prevent paxillin phosphorylation in response to ACh (32). Thus, if myosin II is involved in the recruitment of proteins to adhesomes in airway smooth muscle, its activation must occur independently of MLCK. One possibility is that non-muscle rather than smooth muscle isoforms of myosin II are involved in mediating adhesome assembly in airway smooth muscle, and that non-muscle myosin II activation but not smooth muscle myosin II activation occurs via a RhoA-dependent mechanism. If so, our results would suggest that the RhoA-mediated phosphorylation of non-muscle myosin II light chain would be critical for excitation-contraction coupling and smooth muscle contraction. Non-muscle myosin II is expressed in cultured human airway smooth muscle cells (53) and has also been documented in vascular smooth muscle tissues (54).
Studies of other cell types have implicated RhoA in other mechanisms that contribute to the regulation of cytoskeletal and actin dynamics. RhoA can activate LIM kinase, which phosphorylates and inactivates the actin-dynamizing protein cofilin (2, 55). However, it seems unlikely that RhoA activation could regulate actin dynamics in airway smooth muscle by regulating LIM kinase, because in this tissue contractile stimulation causes the dephosphorylation and activation of cofilin, and cofilin activation is necessary for actin polymerization and contraction (56).
RhoA is also known to regulate activity of the formins, actin nucleating proteins that associate with the fast-growing (barbed) ends of actin filaments and are involved in the formation of stress fibers in adhesive cells (57, 58). The role of formins in agonist-induced actin polymerization and contraction in smooth muscle is unknown. The possibility that RhoA-mediated formin activation plays a role in actin polymerization and contraction in smooth muscle cannot be ruled out.
Focal adhesion kinase can induce the phosphorylation of paxillin in a src-dependent manner (59, 60). We therefore considered the possibility that Rho might regulate paxillin phosphorylation and actin dynamics in airway smooth muscle cells by regulating the activity of FAK. Tyrosine phosphorylated paxillin increased in response to ACh stimulation and was found only at the membrane. FAK was also recruited to the membrane in response to ACh stimulation, and this recruitment was inhibited by RhoA inactivation (Fig. 7, B and C). Duolink PLA analysis demonstrated that paxillin and FAK only interact at the membrane of stimulated airway smooth muscle cells, and that the interaction between paxillin and FAK is inhibited by RhoA inactivation. The inhibition of FAK activation prevented ACh-induced paxillin phosphorylation, cdc42 activation, and actin polymerization. Thus, we conclude that FAK is recruited to the membrane by a RhoA-dependent mechanism independently of paxillin, and that FAK forms a complex with paxillin at the adhesome and induces its phosphorylation.
The Rho effector, ROCK, has also been implicated in the regulation of paxillin phosphorylation in several studies of non-muscle cell lines (61–63). However, in airway tissues the inhibition of ROCK did not suppress ACh-induced paxillin tyrosine phosphorylation, indicating that ROCK is unlikely to play a role in the regulation of paxillin phosphorylation in this tissue.
Studies of adhesion and spreading in several substrate-adherent non-muscle cell lines have demonstrated that the activation of cdc42 and rac can cause a transient suppression of RhoA activity, and that this suppression of RhoA activity is followed by RhoA activation and cell contraction. In cultured NMuMG cells, phosphorylated paxillin inhibits RhoA activation following integrin engagement by regulating the activity of p190RhoGAP, which suppresses RhoA activity during cell adhesion (64). Arthur and DeMali have proposed that cdc42 and RhoA are activated sequentially in response to integrin engagement, with cdc42 activation occurring initially independently of RhoA activity (65). Our observations clearly demonstrate that this paradigm is not applicable to airway smooth muscle during contractile stimulation, as we found that RhoA inhibition suppresses the ACh-induced activation of cdc42 (Fig. 2).
Our observations that contractile stimulation does not affect the ratio of co-immunoprecipitation of paxillin and vinculin or the number of paxillin/vinculin complexes reported by PLA analysis suggests that these proteins are stably associated in airway smooth muscle. PLA analysis revealed paxillin/vinculin complexes to be localized throughout the cytoplasm of unstimulated cells but concentrated at the membrane of the stimulated cells, suggesting that these proteins are recruited to the membrane as a stable complex in response to ACh stimulation. In previous studies using immunofluorescence, we found that paxillin and vinculin colocalized in airway smooth muscle cells under all conditions (29). In the present study, RhoA inhibition did not affect the interaction of paxillin and vinculin, but it prevented the recruitment of the paxillin/vinculin complexes to the membrane. Thus RhoA activation appears to be required for the process that regulates the movement of the paxillin-vinculin complex.
The large multiprotein “adhesome” complexes that connect cells within tissues to the extracellular matrix at adhesion junctions are widely known to play critical functions in cells that extend far beyond their structural role. While they provide mechanical coupling between cells and their matrix environment, they also enable cells to sense and respond to changes in the properties of their surrounding milieu (66, 67). Adhesome constituents and their associated cytoskeletal networks can mediate cellular responses to a variety of stimuli, including growth factors, inflammatory mediators, and mechanical forces (15, 67, 68).
Our current observations suggest that RhoA-mediated adhesome complex assembly is a fundamental step in the process of signal transduction initiated by the contractile agonist stimulation of airway smooth muscle. Both actin polymerization and contraction depend on the RhoA-mediated recruitment of paxillin-vinculin complexes to adhesome junctions. Paxillin has been implicated in mediating signal transduction pathways activated by growth hormones and inflammatory mediators in multiple cell types (66), and may also be important for responses to these mediators in airway smooth muscle. Thus, in airway smooth muscle, RhoA mediated assembly of adhesome complexes may be a fundamental step in the process of signal transduction in response to diverse extracellular stimuli.
This work was supported, in whole or in part, by NHLBI, National Institutes of Health (NIH) Grants HL-29289 and HL074099; and an American Lung Association and an NIH T32 postdoctoral fellowship (to W. Z.).

This article contains supplemental Figs. S1–S4.
- MLC
- smooth muscle myosin light chain
- SM
- smooth muscle
- ASM
- airway smooth muscle
- PLA
- proximity ligation assay
- N-WASp
- neuronal Wiskott-Aldrich syndrome protein
- GEF
- guanine nucleotide exchange factor
- PIX
- PAK-interacting exchange factor
- DOCK
- Dedicator of Cytokinesis
- ACh
- acetylcholine
- ROCK
- Rho kinase
- PAK
- p21-activated kinase
- GIT
- G-protein receptor kinase-interacting tyrosine phosphorylated.
REFERENCES
- 1. Allen W. E., Jones G. E., Pollard J. W., Ridley A. J. (1997) Rho, Rac, and Cdc42 regulate actin organization and cell adhesion in macrophages. J. Cell Sci. 110, 707–720 [DOI] [PubMed] [Google Scholar]
- 2. Burridge K., Wennerberg K. (2004) Rho and Rac take center stage. Cell 116, 167–179 [DOI] [PubMed] [Google Scholar]
- 3. DeMali K. A., Wennerberg K., Burridge K. (2003) Integrin signaling to the actin cytoskeleton. Curr. Opin. Cell Biol. 15, 572–582 [DOI] [PubMed] [Google Scholar]
- 4. Ridley A. J. (1999) Rho family proteins and regulation of the actin cytoskeleton. Prog. Mol. Subcellul. Biol. 22, 1–22 [DOI] [PubMed] [Google Scholar]
- 5. Ridley A. J. (2006) Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 16, 522–529 [DOI] [PubMed] [Google Scholar]
- 6. Amano M., Ito M., Kimura K., Fukata Y., Chihara K., Nakano T., Matsuura Y., Kaibuchi K. (1996) Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271, 20246–20249 [DOI] [PubMed] [Google Scholar]
- 7. Kimura K., Ito M., Amano M., Chihara K., Fukata Y., Nakafuku M., Yamamori B., Feng J., Nakano T., Okawa K., Iwamatsu A., Kaibuchi K. (1996) Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science 273, 245–248 [DOI] [PubMed] [Google Scholar]
- 8. Puetz S., Lubomirov L. T., Pfitzer G. (2009) Regulation of smooth muscle contraction by small GTPases. Physiology 24, 342–356 [DOI] [PubMed] [Google Scholar]
- 9. Somlyo A. P., Somlyo A. V. (2003) Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83, 1325–1358 [DOI] [PubMed] [Google Scholar]
- 10. Zhang W., Du L., Gunst S. J. (2010) The effects of the small GTPase RhoA on the muscarinic contraction of airway smooth muscle result from its role in regulating actin polymerization. Am. J. Physiol. 299, C298–C306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Corteling R. L., Brett S. E., Yin H., Zheng X. L., Walsh M. P., Welsh D. G. (2007) The functional consequence of RhoA knockdown by RNA interference in rat cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 293, H440–H447 [DOI] [PubMed] [Google Scholar]
- 12. Gunst S. J., Zhang W. (2008) Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am. J. Physiol. Cell Physiol. 295, C576–C587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim H. R., Gallant C., Leavis P. C., Gunst S. J., Morgan K. G. (2008) Cytoskeletal remodeling in differentiated vascular smooth muscle is actin isoform dependent and stimulus dependent. Am. J. Physiol. Cell Physiol. 295, C768–C778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rembold C. M., Tejani A. D., Ripley M. L., Han S. (2007) Paxillin phosphorylation, actin polymerization, noise temperature, and the sustained phase of swine carotid artery contraction. Am. J. Physiol. Cell Physiol. 293, C993–C1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang W., Gunst S. J. (2008) Interactions of airway smooth muscle cells with their tissue matrix: implications for contraction. Proc. Am. Thorac. Soc. 5, 32–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mehta D., Gunst S. J. (1999) Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J. Physiol. 519, 829–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang W., Wu Y., Du L., Tang D. D., Gunst S. J. (2005) Activation of the Arp2/3 complex by N-WASp is required for actin polymerization and contraction in smooth muscle. Am. J. Physiol. Cell Physiol. 288, C1145–C1160 [DOI] [PubMed] [Google Scholar]
- 18. Higgs H. N., Pollard T. D. (2001) Regulation of actin filament network formation through ARP2/3 complex: activation by a diverse array of proteins. Annu. Rev. Biochem. 70, 649–676 [DOI] [PubMed] [Google Scholar]
- 19. Mullins R. D. (2000) How WASP-family proteins and the Arp2/3 complex convert intracellular signals into cytoskeletal structures. Curr. Opin. Cell Biol. 12, 91–96 [DOI] [PubMed] [Google Scholar]
- 20. Rohatgi R., Ma L., Miki H., Lopez M., Kirchhausen T., Takenawa T., Kirschner M. W. (1999) The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97, 221–231 [DOI] [PubMed] [Google Scholar]
- 21. Carlier M. F., Ducruix A., Pantaloni D. (1999) Signaling to actin: the Cdc42-N-WASP-Arp2/3 connection. Chem. Biol. 6, R235–R240 [DOI] [PubMed] [Google Scholar]
- 22. Higgs H. N., Pollard T. D. (2000) Activation by Cdc42 and PIP(2) of Wiskott-Aldrich syndrome protein (WASp) stimulates actin nucleation by Arp2/3 complex. J. Cell Biol. 150, 1311–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rohatgi R., Ho H. Y., Kirschner M. W. (2000) Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4, 5-bisphosphate. J. Cell Biol. 150, 1299–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li R., Debreceni B., Jia B., Gao Y., Tigyi G., Zheng Y. (1999) Localization of the PAK1-, WASP-, and IQGAP1-specifying regions of Cdc42. J. Biol. Chem. 274, 29648–29654 [DOI] [PubMed] [Google Scholar]
- 25. Suzuki T., Mimuro H., Miki H., Takenawa T., Sasaki T., Nakanishi H., Takai Y., Sasakawa C. (2000) Rho family GTPase Cdc42 is essential for the actin-based motility of Shigella in mammalian cells. J. Exp. Med. 191, 1905–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang D. D., Gunst S. J. (2004) The small GTPase Cdc42 regulates actin polymerization and tension development during contractile stimulation of smooth muscle. J. Biol. Chem. 279, 51722–51728 [DOI] [PubMed] [Google Scholar]
- 27. Turner C. E., Glenney J. R., Jr., Burridge K. (1990) Paxillin: a new vinculin-binding protein present in focal adhesions. J. Cell Biol. 111, 1059–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Turner C. E., Kramarcy N., Sealock R., Burridge K. (1991) Localization of paxillin, a focal adhesion protein, to smooth muscle dense plaques, and the myotendinous and neuromuscular junctions of skeletal muscle. Exp. Cell Res. 192, 651–655 [DOI] [PubMed] [Google Scholar]
- 29. Opazo Saez A., Zhang W., Wu Y., Turner C. E., Tang D. D., Gunst S. J. (2004) Tension development during contractile stimulation of smooth muscle requires recruitment of paxillin and vinculin to the membrane. Am. J. Physiol. Cell Physiol. 286, C433–C447 [DOI] [PubMed] [Google Scholar]
- 30. Wang Z., Pavalko F. M., Gunst S. J. (1996) Tyrosine phosphorylation of the dense plaque protein paxillin is regulated during smooth muscle contraction. Am. J. Physiol. 271, C1594–C602 [DOI] [PubMed] [Google Scholar]
- 31. Tang D. D., Turner C. E., Gunst S. J. (2003) Expression of non-phosphorylatable paxillin mutants in canine tracheal smooth muscle inhibits tension development. J. Physiol. 553, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang Y., Zhang W., Gunst S. J. (2011) Activation of vinculin induced by cholinergic stimulation regulates contraction of tracheal smooth muscle tissue. J. Biol. Chem. 286, 3630–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bellis S. L., Miller J. T., Turner C. E. (1995) Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J. Biol. Chem. 270, 17437–17441 [DOI] [PubMed] [Google Scholar]
- 34. Schaller M. D., Parsons J. T. (1995) pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol. Cell Biol. 15, 2635–2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sinha S., Yang W. (2008) Cellular signaling for activation of Rho GTPase Cdc42. Cell Signal. 20, 1927–1934 [DOI] [PubMed] [Google Scholar]
- 36. West K. A., Zhang H., Brown M. C., Nikolopoulos S. N., Riedy M. C., Horwitz A. F., Turner C. E. (2001) The LD4 motif of paxillin regulates cell spreading and motility through an interaction with paxillin kinase linker (PKL). J. Cell Biol. 154, 161–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Turner C. E., Brown M. C., Perrotta J. A., Riedy M. C., Nikolopoulos S. N., McDonald A. R., Bagrodia S., Thomas S., Leventhal P. S. (1999) Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: A role in cytoskeletal remodeling. J. Cell Biol. 145, 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Söderberg O., Leuchowius K. J., Gullberg M., Jarvius M., Weibrecht I., Larsson L. G., Landegren U. (2008) Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45, 227–232 [DOI] [PubMed] [Google Scholar]
- 39. Tang D. D., Zhang W., Gunst S. J. (2005) The adapter protein CrkII regulates neuronal Wiskott-Aldrich syndrome protein, actin polymerization, and tension development during contractile stimulation of smooth muscle. J. Biol. Chem. 280, 23380–23389 [DOI] [PubMed] [Google Scholar]
- 40. Torres E., Rosen M. K. (2006) Protein-tyrosine kinase and GTPase signals cooperate to phosphorylate and activate Wiskott-Aldrich syndrome protein (WASP)/neuronal WASP. J. Biol. Chem. 281, 3513–3520 [DOI] [PubMed] [Google Scholar]
- 41. Petit V., Boyer B., Lentz D., Turner C. E., Thiery J. P., Vallés A. M. (2000) Phosphorylation of tyrosine residues 31 and 118 on paxillin regulates cell migration through an association with CRK in NBT-II cells. J. Cell Biol. 148, 957–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tang D., Mehta D., Gunst S. J. (1999) Mechanosensitive tyrosine phosphorylation of paxillin and focal adhesion kinase in tracheal smooth muscle. Am. J. Physiol. 276, C250–8 [DOI] [PubMed] [Google Scholar]
- 43. Slack-Davis J. K., Martin K. H., Tilghman R. W., Iwanicki M., Ung E. J., Autry C., Luzzio M. J., Cooper B., Kath J. C., Roberts W. G., Parsons J. T. (2007) Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 282, 14845–14852 [DOI] [PubMed] [Google Scholar]
- 44. Rosenberger G., Jantke I., Gal A., Kutsche K. (2003) Interaction of alphaPIX (ARHGEF6) with β-parvin (PARVB) suggests an involvement of alphaPIX in integrin-mediated signaling. Hum. Mol. Genet. 12, 155–167 [DOI] [PubMed] [Google Scholar]
- 45. Lynch E. A., Stall J., Schmidt G., Chavrier P., D'Souza-Schorey C. (2006) Proteasome-mediated degradation of Rac1-GTP during epithelial cell scattering. Mol. Biol. Cell 17, 2236–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Côté J. F., Vuori K. (2002) Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J. Cell Sci. 115, 4901–4913 [DOI] [PubMed] [Google Scholar]
- 47. Gumienny T. L., Brugnera E., Tosello-Trampont A. C., Kinchen J. M., Haney L. B., Nishiwaki K., Walk S. F., Nemergut M. E., Macara I. G., Francis R., Schedl T., Qin Y., Van Aelst L., Hengartner M. O., Ravichandran K. S. (2001) CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell 107, 27–41 [DOI] [PubMed] [Google Scholar]
- 48. Bai Y., Sanderson M. J. (2006) Modulation of the Ca2+ sensitivity of airway smooth muscle cells in murine lung slices. Am. J. Physiol. Lung Cell Mol. Physiol. 291, L208–L221 [DOI] [PubMed] [Google Scholar]
- 49. Sanderson M. J., Delmotte P., Bai Y., Perez-Zogbhi J. F. (2008) Regulation of airway smooth muscle cell contractility by Ca2+ signaling and sensitivity. Proc. Am. Thorac. Soc. 5, 23–31 [DOI] [PubMed] [Google Scholar]
- 50. Chrzanowska-Wodnicka M., Burridge K. (1996) Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 133, 1403–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pasapera A. M., Schneider I. C., Rericha E., Schlaepfer D. D., Waterman C. M. (2010) Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 188, 877–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vicente-Manzanares M., Ma X., Adelstein R. S., Horwitz A. R. (2009) Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 10, 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Milton D. L., Schneck A. N., Ziech D. A., Ba M., Facemyer K. C., Halayko A. J., Baker J. E., Gerthoffer W. T., Cremo C. R. (2011) Direct evidence for functional smooth muscle myosin II in the 10S self-inhibited monomeric conformation in airway smooth muscle cells. Proc. Natl. Acad. Sci. U.S.A. 108, 1421–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yuen S. L., Ogut O., Brozovich F. V. (2009) Nonmuscle myosin is regulated during smooth muscle contraction. Am. J. Physiol. Heart Circ. Physiol. 297, H191–H199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Maekawa M., Ishizaki T., Boku S., Watanabe N., Fujita A., Iwamatsu A., Obinata T., Ohashi K., Mizuno K., Narumiya S. (1999) Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285, 895–898 [DOI] [PubMed] [Google Scholar]
- 56. Zhao R., Du L., Huang Y., Wu Y., Gunst S. J. (2008) Actin depolymerization factor/cofilin activation regulates actin polymerization and tension development in canine tracheal smooth muscle. J. Biol. Chem. 283, 36522–36531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zigmond S. H. (2004) Formin-induced nucleation of actin filaments. Curr. Opin. Cell Biol. 16, 99–105 [DOI] [PubMed] [Google Scholar]
- 58. Kovar D. R. (2006) Molecular details of formin-mediated actin assembly. Curr. Opin. Cell Biol. 18, 11–17 [DOI] [PubMed] [Google Scholar]
- 59. Klinghoffer R. A., Sachsenmaier C., Cooper J. A., Soriano P. (1999) Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J. 18, 2459–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Richardson A., Parsons T. (1996) A mechanism for regulation of the adhesion-associated protein-tyrosine kinase pp125FAK. Nature 380, 538–540 [DOI] [PubMed] [Google Scholar]
- 61. Sinnett-Smith J., Lunn J. A., Leopoldt D., Rozengurt E. (2001) Y-27632, an inhibitor of Rho-associated kinases, prevents tyrosine phosphorylation of focal adhesion kinase and paxillin induced by bombesin: dissociation from tyrosine phosphorylation of p130(CAS). Exp. Cell Res. 266, 292–302 [DOI] [PubMed] [Google Scholar]
- 62. Tsuji T., Ishizaki T., Okamoto M., Higashida C., Kimura K., Furuyashiki T., Arakawa Y., Birge R. B., Nakamoto T., Hirai H., Narumiya S. (2002) ROCK and mDia1 antagonize in Rho-dependent Rac activation in Swiss 3T3 fibroblasts. J. Cell Biol. 157, 819–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Worthylake R. A., Burridge K. (2003) RhoA and ROCK promote migration by limiting membrane protrusions. J. Biol. Chem. 278, 13578–13584 [DOI] [PubMed] [Google Scholar]
- 64. Tsubouchi A., Sakakura J., Yagi R., Mazaki Y., Schaefer E., Yano H., Sabe H. (2002) Localized suppression of RhoA activity by Tyr31/118-phosphorylated paxillin in cell adhesion and migration. J. Cell Biol. 159, 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Arthur W. T., Noren N. K., Burridge K. (2002) Regulation of Rho family GTPases by cell-cell and cell-matrix adhesion. Biol. Res. 35, 239–246 [DOI] [PubMed] [Google Scholar]
- 66. Zaidel-Bar R., Itzkovitz S., Ma'ayan A., Iyengar R., Geiger B. (2007) Functional atlas of the integrin adhesome. Nat. Cell Biol. 9, 858–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zaidel-Bar R., Geiger B. (2010) The switchable integrin adhesome. J. Cell Sci. 123, 1385–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Weber G. F., Bjerke M. A., DeSimone D. W. (2011) Integrins and cadherins join forces to form adhesive networks. J. Cell Sci. 124, 1183–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]