

Background: An increase in remyelination in multiple sclerosis (MS) lesions is a possible therapeutic approach.
Results: Gemfibrozil, an FDA-approved drug for hyperlipidemia, stimulates the expression of myelin genes in primary human oligodendroglia via PPAR-β.
Conclusion: These results delineate a novel myelinogenic property of gemfibrozil.
Significance: Gemfibrozil may be of therapeutic benefit in MS and other demyelinating diseases.
Keywords: Gene Regulation, Myelin, Oligodendrocytes, Peroxisome Proliferator-activated Receptor (PPAR), Signal Transduction, Gemfibrozil, Human Oligodendroglia
Abstract
An increase in CNS remyelination and a decrease in CNS inflammation are important steps to halt the progression of multiple sclerosis. Earlier studies have shown that gemfibrozil, a lipid-lowering drug, has anti-inflammatory properties. The current study identified another novel property of gemfibrozil in stimulating the expression of myelin-specific genes (myelin basic protein, myelin oligodendrocyte glycoprotein, 2′,3′-cyclic-nucleotide 3′-phosphodiesterase, and proteolipid protein (PLP)) in primary human oligodendrocytes, mixed glial cells, and spinal cord organotypic cultures. Although gemfibrozil is a known activator of peroxisome proliferator-activated receptor-α (PPAR-α), we were unable to detect PPAR-α in either gemfibrozil-treated or untreated human oligodendrocytes, and gemfibrozil increased the expression of myelin genes in oligodendrocytes isolated from both wild type and PPAR-α(−/−) mice. On the other hand, gemfibrozil markedly increased the expression of PPAR-β but not PPAR-γ. Consistently, antisense knockdown of PPAR-β, but not PPAR-γ, abrogated the stimulatory effect of gemfibrozil on myelin genes in human oligodendrocytes. Gemfibrozil also did not up-regulate myelin genes in oligodendroglia isolated from PPAR-β(−/−) mice. Chromatin immunoprecipitation analysis showed that gemfibrozil induced the recruitment of PPAR-β to the promoter of PLP and myelin oligodendrocyte glycoprotein genes in human oligodendrocytes. Furthermore, gemfibrozil treatment also led to the recruitment of PPAR-β to the PLP promoter in vivo in the spinal cord of experimental autoimmune encephalomyelitis mice and suppression of experimental autoimmune encephalomyelitis symptoms in PLP-T cell receptor transgenic mice. These results suggest that gemfibrozil stimulates the expression of myelin genes via PPAR-β and that gemfibrozil, a prescribed drug for humans, may find further therapeutic use in demyelinating diseases.
Introduction
Myelination is a complex process that requires proliferation of oligodendrocyte progenitors, migration to various brain regions, and synthesis and deposition of the myelin membrane. There are a number of human disorders (multiple sclerosis, spinal cord injury, X-linked adrenoleukodystrophy, adrenomyeloneuropathy, neurotrauma, and HIV encephalomyelopathy) in which the loss of myelin is observed. The major approach to the management of demyelinating disorders involves an increase in remyelination of axons, resulting in clinical improvement. Oligodendrocytes are the only myelin-synthesizing cells in the CNS that express several myelin-specific genes like myelin basic protein (MBP),2 2′,3′-cyclic-nucleotide 3′-phosphodiesterase (CNPase), myelin oligodendrocyte glycoprotein (MOG), and proteolipid protein (PLP) (1–5). Therefore, an increase in the expression of myelin-specific genes is a prerequisite to an increase in CNS remyelination. However, despite intense investigation, very few drugs are available that may increase the expression of myelin genes and help in remyelination.
Peroxisome proliferator-activated receptors (PPARs), transcription factors belonging to the nuclear hormone receptor superfamily, have been reported to be expressed in brain (5–9). To date, three different PPAR subtypes, PPAR-α, PPAR-γ, and PPAR-β/δ, have been identified in vertebrate species. PPAR-α is implicated in the regulation of lipid metabolism, whereas PPAR-γ plays important roles in proliferation and differentiation of adipose cells and regulation of inflammation in many cell types including brain cells (10–14). On the other hand, PPAR-β participates in diabetes, dyslipidemia, cell proliferation, and tumorigenesis (6, 15–19).
Gemfibrozil, an activator of PPAR-α, often has been prescribed in patients to lower the level of triglycerides (6, 20), to increase the level of high density lipoprotein (HDL) cholesterol, and to decrease the level of low density lipoprotein (LDL) cholesterol (20, 21). Earlier we have demonstrated that gemfibrozil attenuates the expression of inducible nitric-oxide synthase in human astrocytes (22). Others have also shown an anti-inflammatory effect of gemfibrozil in other cells (23, 24). In the current work, we present evidence that gemfibrozil markedly stimulated the expression of myelin-specific genes (MOG, PLP, CNPase, and MBP) in human primary oligodendrocytes, mixed glial cells, and spinal cord organotypic cultures. On the other hand, gemfibrozil was unable to stimulate the expression of either astroglia-specific glial fibrillary acidic protein (GFAP) or microglia-specific CD11b. Furthermore, we demonstrate that gemfibrozil executed this novel myelinogenic property via PPAR-β but not via either PPAR-α or PPAR-γ. These results raise the possibility that this lipid-lowering drug may be of therapeutic value in human demyelinating diseases.
EXPERIMENTAL PROCEDURES
Reagents
Fetal bovine serum, Hanks' balanced salt solution, trypsin, and DMEM/F-12 were from Mediatech (Washington, D. C.). Gemfibrozil, WY14643, Solvent Blue 38, cresyl violet acetate, and lithium carbonate were obtained from Sigma. Heat-killed Mycobacterium tuberculosis (H37RA) was purchased from Difco. Incomplete Freund's adjuvant was obtained from Calbiochem. Proteolipid protein (PLP139–151) was purchased from Tocris Bioscience (Ellisville, MO). Antibodies against human PLP and MOG were obtained from Millipore (Billerica, MA). Antibodies against PPAR-α, PPAR-β, and PPAR-γ were purchased from Abcam (Cambridge, MA). Phosphorothioate-labeled antisense and scrambled oligodeoxynucleotides were synthesized in the DNA-synthesizing facility of Invitrogen. The following antisense (ASOs) and scrambled (ScOs) oligonucleotides for different PPAR genes were used: PPAR-β: ASO, 5′-GCC TTG TCC CCG CAC ACC CG-3′; ScO, 5′-GCC TCC GCA CAC CGT TCC CG-3′; PPAR-γ: ASO, 5′-TAC GGA GAG ATC CAC GGA G-3′; ScO, 5′-GGA GAT CCA CGG AGT ACA G-3′; PPAR-α, ASO: 5′-GGC CTC GAG TGG GGA GAG GGG-3′; ScO, 5′-TGC GCA CAC CCG CCC TGG CCT-3′.
Isolation of Human Mixed Glial Cultures and Primary Oligodendrocytes
Human fetal brain tissues were obtained from the Human Embryology Laboratory, University of Washington (Seattle, WA). All of the experimental protocols were reviewed and approved by the Institutional Review Board of the Rush University Medical Center. Briefly, 14–16-week-old fetal brains obtained from the Human Embryology Laboratory, University of Washington were dissociated by trituration and trypsinization (0.25% trypsin in PBS at 37 °C for 15 min). The trypsin was inactivated with 10% heat-inactivated FBS (Mediatech). The dissociated cells were filtered successively through 380- and 140-μm mesh (Sigma) and pelleted by centrifugation. The resulting suspension was centrifuged for 10 min at 1500 rpm and then resuspended in DMEM/F-12 supplemented with 20% heat inactivated FBS. Cells were plated on poly-d-lysine-coated 75-cm2 flasks and incubated at 37 °C with 5% CO2 in air. Culture medium was changed after every 3 days. The initial mixed glial cultures (grown for 9 days) were placed on a rotary shaker at 240 rpm at 37 °C for 2 h to remove loosely attached microglia. The oligodendrocytes were detached after shaking for 18 h at 200 rpm at 11 days. To purify oligodendrocytes from astrocytes and microglia, the detached cell suspension was plated in tissue culture dishes (2 × 106 cells/100 mm) for 60 min at 37 °C. This step was repeated twice for non-adherent cells to minimize the contamination. The non-adhering cells (mostly oligodendrocytes) were seeded onto poly-d-lysine-coated culture plates in complete medium (DMEM/F-12 supplemented with 10% heat inactivated FBS) at 37 °C with 5% CO2 in air. Earlier we (1, 2) have shown that oligodendrocytes isolated through this procedure are more than 98% pure.
Isolation of Primary Mouse Oligodendrocytes
Oligodendrocytes were isolated from brains of 2–3-day-old pups of wild type, PPAR-α(−/−), and PPAR-β(−/−) mice as described above. Briefly, on the 9th day, mixed glial cultures were placed on a rotary shaker at 240 rpm at 37 °C for 2 h to remove loosely attached microglia. Then on the 11th day, flasks were shaken again at 180 rpm at 37 °C for 18 h to obtain oligodendroglia. These cells were allowed to adhere on uncoated plates for 1 h to remove any residual microglia and astroglia. Non-adherent cells were mostly oligodendroglia (∼98% pure), which were centrifuged and plated on poly-d-lysine-coated plates.
Treatment of Primary Oligodendrocytes with Gemfibrozil (Gem)
Cells were initially cultured on poly-d-lysine-coated plates in DMEM/F-12 containing 10% FBS for 24 h. Then cells were treated with different concentrations of gem under serum-free conditions.
Immunostaining of GalC, CNPase, MOG, PLP, and PPAR-β
Immunostaining was performed as described earlier (1, 2). Briefly, coverslips containing 200–300 cells/mm2 were fixed with 4% paraformaldehyde for 15 min followed by treatment with cold ethanol (−20 °C) for 5 min and two rinses in PBS. Samples were blocked with 3% BSA in PBS containing Tween 20 (PBST) for 30 min and incubated in PBST containing 1% BSA and rabbit anti-PPAR-β (1:50), goat anti-GalC (1:50), goat anti-MOG (1:50), rabbit anti-PLP (1:70), and rabbit anti-CNPase (1:50). After three washes in PBST (15 min each), slides were further incubated with Cy5, Cy3, and Cy2 (Jackson ImmunoResearch Laboratories, West Grove, PA). For negative controls, a set of culture slides was incubated under similar conditions without the primary antibodies. The samples were mounted and observed under a Bio-Rad MRC1024ES confocal laser-scanning microscope.
Measurement of Oligodendrocyte Process Length
Analyses of oligodendrocyte process length were performed using the ImageJ program (National Institutes of Health, Bethesda, MD). The length of the each process was measured from the base of the process to its tip.
Semiquantitative RT-PCR Analysis
Total RNA was isolated using the Qiagen RNEasy kit following the manufacturer's protocol. To remove any contaminating genomic DNA, total RNA was digested with DNase. Semiquantitative RT-PCR was carried out as described earlier using oligo(dT)12–18 as primer and Moloney murine leukemia virus reverse transcriptase (Clontech) in a 20-μl reaction mixture. The resulting cDNA was appropriately diluted, and diluted cDNA was amplified using Titanium Taq polymerase and the following primers: human primers: hMBP: sense, 5′-GGA AAC CAC GCA GGC AAA CGA GA-3′; antisense, 5′-GAA AAG AGG CGG ATC AAG TGG GG-3′; hPLP: sense, 5′-CTT CCC TGG TGG CCA CTG GAT TGT-3; antisense, 5′-TGA TGT TGG CCT CTG GAA CCC CTC-3′; hMOG: sense, 5′-TCC TCC TCC TCC TCC AAG TGT CT-3′; antisense, 5′-AGT GGG GAT CAA AAG TCC GGT GG-3′; hCNPase: sense, 5′-GGC CAC GCT GCT AGA GTG CAA GAC-3′; antisense, 5′-GGT ACT GGT ACT GGT CGG CCA TTT-3′; hGFAP: sense, 5′-TGA GTC GGT GGA GGA GGA GAT-3′; antisense, 5′-TAG TCG TTG GCT TCG TGC TTG-3′; hCD11b: sense, 5′-CAA CCA AAG GGG CAG CCT CTA CCA G-3′; antisense, 5′-CTG GGA TGA TGC TAC CAG ACC ATC-3′; hPPAR-α: sense, 5′-CCA GTA TTT AGG ACG CTG TCC-3′; antisense, 5′-AAG TTC TTC AAG TAG GCC TCG-3′; PPAR-γ: sense, 5′-TCT CTC CGT AAT GGA AGA CC-3′; antisense, 5′-GCA TTA TGA GAC ATC CCC AC-3; hPPAR-β: sense, 5′-CTG AGG TCC GGG AAG AGG AGG AGA A-3′; antisense, 5′-GCG CTC ACA CTT CTC GTA CTC CAG C-3′; hGAPDH: sense: 5′-GTT GTC ATT GTT GTG AGC CG-3; antisense, 5′-CAT CAC AGC CAT GAT GTT GC-3′; mouse primers: mouse MOG: sense, 5′-CCT CTC CCT TCT CCT CCT TC-3′; antisense, 5′-AGA GTC AGC ACA CCG GGG TT-3′; mPLP: sense, 5′-CTT CCC TGG TGG CCA CTG GAT TGT-3′; antisense, 5′-CCG CAG ATG GTG GTC TTG TAG TCG-3′; mGAPDH: sense, 5′-CAG GGG ATG ATC ATG GCT TCT CC-3′; antisense, 5′-GAT GCT CAC AAG AGC CCC GTT AGC-3′; mGAPDH: sense, 5′-GGT GAA GGT CGG AGT CAA CG-3′; antisense, 5′-GTG AAG ACG CCA GTG GAC TC-3′. Amplified products were electrophoresed on a 1.8% agarose gel and visualized by ethidium bromide staining. The message for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used to ascertain that an equivalent amount of cDNA was synthesized from different samples.
Real Time PCR Analysis
Real time PCR analysis was performed using the ABI PRISM 7700 sequence detection system (Applied Biosystems, Foster City, CA) as described earlier (1, 25). All primers and fluorescein amidite-labeled probes were obtained from Applied Biosystems. The mRNA expression of myelin genes was normalized to the label of GAPDH mRNA. Data were processed by ABI Sequence Detection System 1.6 software and analyzed by analysis of variance.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assays were performed using a kit (Upstate Biotechnology, Lake Placid, NY) as described earlier (26). Briefly, after 3 h of stimulation, cells were fixed by adding formaldehyde, and cross-linked adducts were resuspended and sonicated. ChIP was performed on the cell lysate by overnight incubation at 4 °C with 2 μg of either anti-PPAR-β antibodies or control IgG followed by incubation with protein G-agarose (Santa Cruz Biotechnology) for 2 h. The beads were washed and incubated with elution buffer. To reverse the cross-linking and purify the DNA, precipitates were incubated in a 65 °C incubator overnight and digested with proteinase K. DNA samples were then purified and precipitated, and precipitates were washed with 75% ethanol, air-dried, and resuspended in Tris-EDTA buffer. The following primers were used for amplification of chromatin fragments of human MOG and PLP genes: MOG promoter (spanning proximal PPRE; 144 bp): sense, 5′-TCC AAC CTT GCC TCT CTG AGC A-3′; antisense, 5′-TCT TCT ACC GCG AAT CCA AAC T-3′; MOG promoter (spanning distal PPRE; 163 bp): sense: 5′-TGG AAC CTG CTA CCA ATG CTG GTC T-3′; antisense: 5′-CAG AGC CAG CAT ACA GGA CAC GCT C-3′; human PLP promoter (spanning both PPREs; 315 bp): sense, 5′-CCA CCC TCA ATC CAC ATT TCC AGA-3′; antisense, 5′-TCC AGC CTC CTT CTT CGG TCT TTT-3′.
Spinal Cord Organotypic Culture
Spinal cord organotypic cultures were prepared from spinal cord tissues from 11- to 13-week-old human embryos that were obtained from the Human Embryology Laboratory, University of Washington. Fetal spinal cords (thoracic part) were cut using a scalpel blade into ∼1.0-mm-thick slices. The slices were subsequently placed in 6-well sterile plastic culture plates. Each well contained 2 ml of complete medium. The cultures were maintained at 37 °C in an atmosphere of 5% CO2 in air for 2 days. After 2 days, slices were washed three times with serum-free medium and then treated with gem for 96 h under serum-free conditions. After 96 h, tissues were thoroughly washed with PBS three times.
Fixation and Sectioning
The cultured thoracic spinal cord slices were fixed with 4% paraformaldehyde in PBS overnight followed by three PBS washes (15 min each). Paraformaldehyde-fixed tissues were then embedded in OCT (TissueTek, Elkhart, IN) at −80 °C and processed for conventional cryosectioning. Frozen longitudinal sections (6 μm) were immunostained as described above.
Cell Viability Measurement
Mitochondrial activity was measured with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma).
Lactate Dehydrogenase Measurement
The activity of lactate dehydrogenase was measured using a direct spectrophotometric assay using an assay kit from Sigma.
Induction of EAE in PLP-TCR Transgenic Mice
PLP-TCR (5B6) transgenic mice (a kind gift from Prof. Vijay Kuchroo of the Harvard Medical School) were genotyped by FACS analysis of Thy1.1, CD4, and Vβ6. These Tg mice develop EAE spontaneously in all backcross generations at an overall average occurrence of 40%. However, upon immunization with a low dose of PLP139–151, these Tg mice exhibit severe EAE (27). Therefore, these Tg mice were immunized subcutaneously with 10 μg of PLP139–151 and 60 μg of M. tuberculosis in incomplete Freund's adjuvant. Animal maintenance and experimental protocols were approved by the Rush University Medical Center. Animals were observed daily for clinical symptoms of EAE as described by us (25, 28–30). Experimental animals were scored by a masked investigator as follows: 0, no clinical disease; 0.5, piloerection; 1, tail weakness; 1.5, tail paralysis; 2, hind limb weakness; 3, hind limb paralysis; 3.5, forelimb weakness; 4, forelimb paralysis; 5, moribund or death.
Staining for Myelin
Spinal cord sections were stained with Luxol fast blue for myelin as described earlier (28, 30). Slides were assessed in a blinded fashion for demyelination by three examiners using the following scale: 0, normal white matter; 1, rare foci; 2, a few areas of demyelination; and 3, large areas of demyelination. At least six sections of each spinal cord from each of five mice per group were scored and statistically analyzed by analysis of variance.
RESULTS
Gem Promotes the Differentiation of Human Fetal Oligodendrocytes
As reported earlier (1, 2), oligodendrocytes isolated from human fetal brains were highly pure (Fig. 1). These cells were positive for GalC, MBP, MOG, PLP, and CNPase (1, 2). However, we were unable to detect any GFAP-positive astroglia and CD11b-positive microglia in this oligodendroglial preparation (1, 2). Rarely did we observe one GFAP-positive astrocyte of 50 or more GalC-positive oligodendrocytes (1, 2). To investigate whether gem prevents the death of oligodendrocytes, we treated human oligodendrocytes with different doses of gem under serum-free conditions. As expected, after serum withdrawal, oligodendrocytes lost processes and died (Fig. 1, A and B). However, gem markedly prevented serum deprivation-induced oligodendroglial death (Fig. 1, A and B). Although gem was not very effective at a dose of 5 μm, the protective effect was visible at 10 μm (data not shown),and the effect was maximal at a dose of either 25 or 50 μm (Fig. 1A). This protection was confirmed further by MTT/lactate dehydrogenase assay. Serum withdrawal reduced cell viability as evidenced by a decrease in MTT metabolism (Fig. 1C) and an increase in lactate dehydrogenase release (Fig. 1D). Interestingly, gem effectively prevented serum withdrawal-induced loss of MTT metabolism (Fig. 1C) and stimulation of lactate dehydrogenase release (Fig. 1D). However, gem at a concentration of 200 μm or higher was toxic to oligodendrocytes (Fig. 1A). In addition to protecting oligodendrocytes from serum withdrawal-mediated death, gem also induced oligodendroglial differentiation under serum-free conditions as evidenced by elaboration and extension of processes in gem-treated cells compared with the serum-free control (Fig. 1, A and B). These results suggest that gem is capable of promoting oligodendroglial differentiation and preventing oligodendrocyte death.
FIGURE 1.

Gem stimulates the differentiation of human fetal oligodendrocytes and protects these cells from serum deprivation-induced death. After separation from other glial cells, fetal oligodendrocytes were cultured on poly-d-lysine-coated plates with DMEM/F-12 containing 10% FBS for 24 h. Then cells were treated with different concentrations of gem for 24 h under serum-free conditions followed by phase-contrast imaging (A). Process length (B) was monitored as described under “Experimental Procedures.” Three individual experiments were performed, and the data were summarized and analyzed using the Mann-Whitney test. A total of 50 oligodendrocytes per treatment was analyzed for each assay. MTT reduction activity (C) and lactate dehydrogenase (LDH) release (D) assays were performed under similar treatment conditions. Values obtained from the control group (+serum −gem) served as 100%, and data obtained in the other groups were calculated as a percentage of control accordingly. a, p < 0.001 versus control (+serum −gem) group; b, p < 0.001 versus (−serum −gem) group. Error bars represent S.D.
Dose-dependent Effect of Gem on the Expression of Myelin Genes in Human Fetal Oligodendrocytes
How does gem stimulate differentiation? One of the prerequisites of enhanced oligodendroglial differentiation is increased expression of myelin-specific genes (e.g. MBP, MOG, CNPase, and PLP) in oligodendrocytes. Therefore, to investigate the effect of gem on the expression of myelin-specific genes in primary oligodendrocytes, cells were treated with different concentrations of gem for 18 h. The expression of MBP, MOG, PLP, and CNPase was analyzed by semiquantitative RT-PCR and real time PCR. Semiquantitative RT-PCR results in Fig. 2A clearly show that gem increased the mRNA expression of MBP, CNPase, MOG, and PLP in a dose-dependent manner. Quantitative real time PCR data in Fig. 2B also support this conclusion and demonstrate that the stimulatory effect of gem was more prominent for PLP and MOG than for MBP and CNPase.
FIGURE 2.

Effect of gem on the expression of myelin genes in human fetal oligodendrocytes. Cells were treated with different concentrations of gem for 18 h under serum-free condition followed by analysis of MBP, PLP, MOG, and CNPase mRNAs by semiquantitative RT-PCR (A) and quantitative real time PCR (B). Results are means ± S.D. of three different experiments. a, p < 0.001 versus control (Cont). After 24 h of gem treatment, the protein level of PLP, MOG, and MBP was examined by Western blot (C). Cells were also immunostained with antibodies against PLP and MOG (D). The setting of the confocal microscope was kept unaltered during the entire study. Figures are representative of three independent experiments. Scale bars represent 20 μm. Cells were treated with 25 μm gemfibrozil for different time intervals followed by monitoring the mRNA expression of PLP, MOG, and MBP by semiquantitative RT-PCR (E) and quantitative real time PCR (F). Results are means ± S.D. of three different experiments. a, p < 0.001 versus control. At different time intervals, the protein level of PLP, MOG, and MBP was examined by Western blot (G). Results represent three independent experiments. Error bars represent S.D.
Next we determined the effect of gem on the expression of myelin proteins in primary oligodendrocytes. As evident from the Western blot in Fig. 2C, gem dose-dependently increased the expression of PLP, MOG, and MBP proteins in human oligodendroglia. Similarly, immunofluorescence analyses also show that gem markedly increased the protein expression of MOG and PLP in human oligodendroglia (Fig. 2D). Next we performed a time-dependent analysis of gem-treated oligodendroglia. Gem was able to increase the mRNA expression of PLP, MOG, and MBP as early as 3 h (Fig. 2, E and F). Similarly, gem also increased the protein expression of PLP, MOG, and MBP in a time-dependent manner (Fig. 2G). Taken together, these results suggest that gem alone is capable of enhancing the expression of MOG, PLP, MBP, and CNPase in primary human oligodendrocytes.
Effect of Gem on the Expression of Myelin Genes in Human Fetal Brain Mixed Glial Cultures
Next we examined whether gem was capable of enhancing the expression of myelin genes in human fetal brain mixed glial cultures. It is clearly evident from semiquantitative RT-PCR in Fig. 3A and real time PCR in Fig. 3B that gem was capable of enhancing the mRNA expression of MOG, PLP, and CNPase in mixed glial cells. Consistent with marked stimulation of MOG and PLP by gem in purified oligodendrocytes, the increase of these two myelin genes by gem was more prominent than that of CNPase in mixed glial cultures (Fig. 3, A and B). Apart from oligodendrocytes, mixed glial cultures contained other glial cells like astroglia and microglia. Although GFAP is a marker of astrocytes, microglia are identified by CD11b. Similarly, the expression of GFAP increases during activation of astrocytes and astrogliosis, whereas the expression of CD11b goes up during activation of microglia and microgliosis (31, 32). Therefore, to understand whether the effect of gem was specific for oligodendrocytes, we analyzed the mRNA expression of GFAP and CD11b in gem-treated mixed glial cells. In contrast to the stimulation of oligodendrocyte-specific genes, gem had no effect on mRNA expression of either astroglia-specific GFAP or microglia-specific CD11b (Fig. 3, A and B). Western blot analyses show that gem was capable of increasing the protein expression of PLP, but not GFAP, in human mixed glial cells (Fig. 3C). Time-dependent analyses of gem-treated mixed glial cells demonstrate that gem increased the mRNA expression of MOG, PLP, and MBP, but not GFAP and CD11b, in mixed glial cells (Fig. 3D). This finding is also supported by real time PCR (Fig. 3E). Similarly, gem also up-regulated the protein expression of PLP, MBP, and MOG, but not GFAP, in human mixed glial cells (Fig. 3F). Together, these results suggest that gem is capable of stimulating the expression of myelin genes in oligodendrocytes in the presence of other brain cells and that the effect of gem is specific for oligodendrocytes.
FIGURE 3.

Effect of gem on the expression of myelin genes, CD11b, and GFAP in human fetal mixed glial cultures. A, mixed glial cultures were treated with different concentrations of gem. After 18 h of treatment, total RNA was analyzed for the expression of PLP, MOG, CNPase, CD11b, and GFAP by semiquantitative RT-PCR. B, cells were treated with different concentrations of gem, WY14643, and fenofibrate, and total RNA was analyzed for the expression of PLP, MOG, and CNPase mRNAs by quantitative real time PCR. Results are means ± S.D. of three different experiments. a, p < 0.001 versus control. After 24 h of treatment, the protein level of PLP and GFAP was examined by Western blot (C). Cells were treated with 25 μm gemfibrozil for different time intervals followed by monitoring the mRNA expression of PLP, MOG, MBP, GFAP, and CD11b by semiquantitative RT-PCR (D). The mRNA expression of PLP, GFAP, and CD11b was monitored by quantitative real time PCR (E). Results are means ± S.D. of three different experiments. a, p < 0.001 versus control. At different time intervals, the protein level of PLP, MOG, MBP, and GFAP was examined by Western blot (F). Results represent three independent experiments. Error bars represent S.D.
To test whether other agonists of PPAR-α also induce the expression of myelin genes, mixed glial cultures were treated with different concentrations of fenofibrate and WY14643. As evident from real time data in Fig. 3B, fenofibrate stimulated the expression of only MOG, but not PLP, in mixed glial cells. However, fenofibrate remained less potent than gem in inducing the expression of MOG. On the other hand, WY14643, a synthetic ligand of PPAR-α, did not increase the expression of MOG and PLP mRNAs (Fig. 3B). Because WY14643 is considered as a specific agonist of PPAR-α, these results suggest that PPAR-α signaling pathway may not be required for enhancement of myelin genes in human oligodendrocytes.
Effect of Gem on the Expression of Myelin Genes in Human Fetal Spinal Cord Mixed Cultures
Next we tested whether gem was capable of stimulating the expression of myelin genes in human spinal cord mixed glial cultures. As evident from semiquantitative RT-PCR in Fig. 4A and real time PCR in Fig. 4B, gem markedly increased the mRNA expression of MBP, MOG, PLP, and CNPase in a dose-dependent manner as compared with the control. To determine whether the stimulatory effect of gem was specific for oligodendrocytes, we analyzed the mRNA expression of CD11b and GFAP in gem-treated spinal cord mixed glial cells. Similarly to that found in brain mixed glial cells (Fig. 3), gem had no stimulatory effect on the expression of either GFAP or CD11b in human spinal cord mixed glial cells, suggesting that the stimulatory effect of gem is specific for oligodendroglial genes.
FIGURE 4.
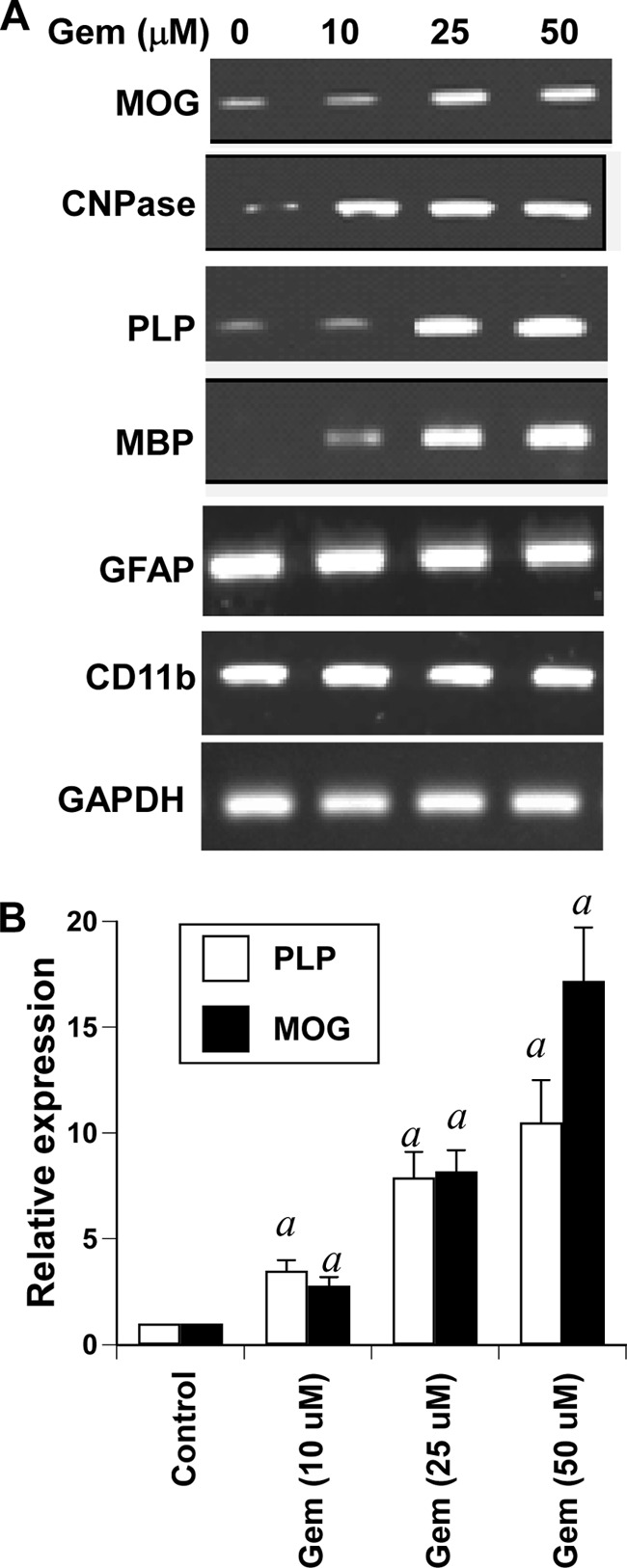
Effect of gem on the expression of myelin genes, CD11b, and GFAP in human fetal spinal cord mixed glial cultures. Mixed spinal cord cultures were treated with different concentrations of gem. After 18 h of treatment, total RNA was analyzed for the expression of MBP, MOG, PLP, CNPase, GFAP, and CD11b by semiquantitative RT-PCR (A). Total RNA was also analyzed for the expression of PLP and MOG mRNAs by quantitative real time PCR (B). Results are means ± S.D. of three different experiments. a, p < 0.001 versus control. Error bars represent S.D.
Effect of Gem on the Expression of Myelin Proteins in Human Fetal Spinal Cord Organotypic Cultures
The above results encouraged us to determine whether gem was capable of increasing the protein level of myelin markers in spinal cord organotypic cultures. For this purpose, thoracic spinal cord slices were treated with 25 μm gem for 96 h followed by immunofluorescence analysis of MOG and PLP. Interestingly, a marked increase in protein expression of MOG and PLP was observed in spinal cord organotypic cultures after gem treatment (Fig. 5). Enhanced myelination of axons is also evident in the magnified image (Fig. 5). These results raise the possibility that gem may be used to remyelinate demyelinated axons in spinal cord and that human fetal spinal cord organotypic slice culture may provide a convenient model for testing conditions or agents for promoting myelination.
FIGURE 5.

Immunofluorescence analysis of MOG and PLP in gem-treated human fetal spinal cord organotypic slice cultures. Spinal cord (thoracic) organotypic tissues (∼1.0 mm thick) were treated with 25 μm gem, and after 4 days of treatment, longitudinal sections were immunostained with antibodies against MOG and PLP as described under “Experimental Procedures.” The selected area was magnified to show myelination of an axon. Figures represent three independent experiments.
Expression of PPARs in Human Fetal Oligodendrocytes, Mixed Glial Cells, and Spinal Cord Organotypic Cultures
Next we investigated mechanisms by which gem increased myelin genes. Gem, a known agonist of PPAR-α, stimulates the β-oxidation of fatty acids in peroxisomes and mitochondria via the activation of PPAR-α (33). At first, we analyzed the level of different PPARs in cultured oligodendrocytes and mixed glial cells. However, despite several attempts, we were unable to detect the expression of PPAR-α mRNA in purified oligodendrocytes by semiquantitative RT-PCR (Fig. 6A). This is also consistent to previous reports that PPAR-α protein or mRNA is not present at a detectable level in rat and mouse primary oligodendrocytes (4, 34, 35). On the other hand, human oligodendrocytes expressed both PPAR-β and PPAR-γ mRNAs (Fig. 6A). However, the expression of PPAR-γ was very low in comparison with PPAR-β (Fig. 6A). We observed a detectable level of PPAR-γ expression in primary oligodendrocytes only at a higher cycle number of PCR (Fig. 6A). Although gem markedly stimulated the expression of PPAR-β, this drug was unable to modulate the expression of PPAR-γ in oligodendrocytes (Fig. 6A). In fetal brain mixed glial cells, the expression of all three PPARs was visible (Fig. 6B). Similarly to that in primary oligodendrocytes, gem stimulated the expression of PPAR-β in mixed glial cells (Fig. 6B). However, surprisingly, gem inhibited the expression of both PPAR-α and PPAR-γ in mixed glial cells (Fig. 6B). Next we examined the protein level of PPAR-β in oligodendrocytes by Western blot (Fig. 6, C and D) and immunofluorescence analysis (Fig. 6E). Consistent with mRNA results, gem dose-dependently increased the level of PPAR-β protein in primary oligodendrocytes (Fig. 6, C–E).
FIGURE 6.

Effect of gem on the expression of different PPARs. Human fetal oligodendrocytes (A) and mixed glial cells (B) were treated with different concentrations of gem for 18 h followed by analysis of PPAR-α, PPAR-β, and PPAR-γ mRNAs by semiquantitative RT-PCR. Oligodendrocytes were treated with different concentrations of gem for 24 h followed by analysis of PPAR-β protein by Western blot (C). Bonds were scanned and represented as relative expression (D). Cells were analyzed for PPAR-β by immunofluorescence (E). Scale bars represent 20 μm. Spinal cord organotypic slices were treated with 25 μm gem for 96 h followed by immunofluorescence analysis with antibodies against PPAR-β and GalC (E). Results represent three independent experiments. Error bars represent S.D.
Similarly, when we tested the level of PPAR-β protein in spinal cord organotypic slice cultures, we also observed a sharp increase of the PPAR-β protein level in gem-treated spinal cord compared with the control (Fig. 6F). This increased PPAR-β co-localized with GalC in gem-treated oligodendrocytes (Fig. 6F). These results clearly demonstrate that gem increases the expression of PPAR-β in human fetal oligodendrocytes, mixed glial cells, and spinal cord organotypic cultures.
Gem Stimulates the Expression of Myelin Genes in Primary Mouse Oligodendrocytes via PPAR-β but Not PPAR-α
The results described above raise the possibility that gem may stimulate the expression of myelin genes in oligodendrocytes without involving PPAR-α. To investigate this possibility, we examined the effect of gem on the expression of myelin genes in oligodendrocytes isolated from wild type, PPAR-α(−/−), and PPAR-β(−/−) mice. We observed that gem was capable of increasing the mRNA expression of MOG and PLP to a similar extent in oligodendrocytes isolated from both WT and PPAR-α(−/−) mice (Fig. 7, A and B). In contrast, gem did not up-regulate the mRNA expression of MOG and PLP in PPAR-β(−/−) oligodendrocytes (Fig. 7, A and B). To further confirm these results, we performed immunofluorescence analysis of PLP and MOG in control and gem-treated oligodendrocytes isolated from WT, PPAR-α(−/−), and PPAR-β(−/−) mice. Consistent with stimulation in mRNA expression, gem increased the expression of PLP and MOG proteins in oligodendrocytes isolated from WT (Fig. 8A) and PPAR-α(−/−) (Fig. 8B), but not PPAR-β(−/−), mice (Fig. 8C). Similarly, gem also increased oligodendroglial processes in oligodendrocytes isolated from WT and PPAR-α(−/−), but not PPAR-β(−/−), mice (Fig. 8D). These findings clearly suggest that gem requires PPAR-β, but not PPAR-α, to stimulate myelin genes in mouse oligodendrocytes.
FIGURE 7.

Gem induces the expression of myelin genes in primary mouse oligodendrocytes via PPAR-β but not PPAR-α. Mouse primary oligodendrocytes isolated from 2–3-day-old wild type, PPAR-α(−/−), and PPAR-β(−/−) pups were treated with 50 μm gem for 18 h followed by analysis of MOG and PLP mRNAs by semiquantitative RT-PCR (A) and real time PCR (B). Results represent the mean ± S.D. of three separate experiments. a, p < 0.001 versus control (Cont)-WT; b, p < 0.001 versus control-PPAR-α(−/−). Error bars represent S.D.
FIGURE 8.

Gem induces the protein expression of PLP and MOG in primary mouse oligodendrocytes via PPAR-β but not PPAR-α. Mouse primary oligodendrocytes isolated from 2–3-day-old wild type (A), PPAR-α(−/−) (B), and PPAR-β(−/−) (C) pups were treated with 50 μm gem for 24 h followed by analysis of PLP and MOG proteins by immunofluorescence. DAPI was used to visualize the nucleus. Phase-contrast images are shown to indicate morphology. Results represent three independent experiments. D, process elongation was monitored as described above. A total of 50 oligodendrocytes per treatment was analyzed for each assay. a, p < 0.001 versus control (Cont)-WT; b, p < 0.001 versus gemfibrozil-WT. Error bars represent S.D.
Gem Stimulates the Expression of Myelin Genes in Primary Human Oligodendrocytes via PPAR-β but Not PPAR-γ
Next we investigated the role of PPAR-β in gem-mediated stimulation of myelin genes in primary human oligodendrocytes. It is difficult to transfect primary human oligodendrocytes with lipid reagents. Phosphorothioate-labeled antisense oligonucleotides provide an important option to knock down specific gene in primary non-dividing cells like oligodendrocytes (48) and neurons (49) without either changing the primary property of cells that is seen with transduction by adenoviral constructs or causing any cell death that is always associated with transfection experiments. Therefore, we used antisense oligonucleotides to knock down PPAR-β and PPAR-γ in primary human oligodendrocytes. As shown in Fig. 9A, PPAR-β was mainly visible in gem-treated oligodendrocytes, and ASO, but not ScO, against PPAR-β inhibited the expression of PPAR-β, but not PPAR-γ, mRNA in gem-treated cells. On the other hand, PPAR-γ was present in equal amount in both control and gem-treated cells (Fig. 9B). As expected, ASO, but not ScO, against PPAR-γ inhibited the expression of PPAR-γ, but not PPAR-β, mRNA (Fig. 9B). Consistent with mouse oligodendrocytes (Figs. 7 and 8), knockdown of PPAR-β markedly suppressed the stimulatory effect of gem on the expression of MOG in human oligodendrocytes (Fig. 9, A and C). Similarly, GSK0660, a specific antagonist of PPAR-β, abrogated the ability of gem to up-regulate the mRNA expression of PLP and MOG in human oligodendrocytes (Fig. 9D). However, PPAR-β knockdown had no effect on the basal level of MOG in control oligodendrocytes (Fig. 9, A and C). In contrast to the knockdown of PPAR-β, ASO against PPAR-γ had no effect on gem-mediated expression of MOG mRNA (Fig. 9, B and C). Taken together, these results suggest that gem stimulates the expression of myelin genes in oligodendrocytes via PPAR-β.
FIGURE 9.

Antisense-mediated knockdown of PPAR-β, but not PPAR-γ, abrogates the stimulatory effect of gem on myelin gene expression in primary human oligodendrocytes. Human fetal oligodendrocytes cultured in complete medium received either ASO or ScO (0.5 μm) against PPAR-β. After 24 h of incubation, cells were treated with 25 μΜ gem under serum-free conditions for 18 h followed by analysis of total RNA for the expression of PPAR-β, PPAR-γ (as control), and MOG by semiquantitative RT-PCR (A). Similar experiments were performed with ASO and ScO against PPAR-γ, and cells were analyzed for the expression of PPAR-γ, PPAR-β (as control), and MOG (B). Total RNA was also analyzed for the expression of MOG mRNA by quantitative real time PCR (C). Results are the mean ± S.D. of three separate experiments. a, p < 0.001 versus gem. D, cells preincubated with GSK0660 (a PPAR-β antagonist) for 30 min were stimulated with gem. After 18 h, the mRNA expression of PLP and MOG was monitored by semiquantitative RT-PCR. Error bars represent S.D.
Gem Stimulates the Recruitment of PPAR-β to the Promoters of MOG and PLP Genes in Primary Human Oligodendrocytes
PPAR-β, a lipid-lowering transcription factor, is known to bind the PPRE present in the promoters of different genes (36, 37). Using MatInspector V2.2 search machinery, we searched the promoter regions of human MBP, PLP, MOG, and CNPase genes for a PPRE. We found that both MOG and PLP promoters harbor consensus PPREs. On the other hand, we did not detect any PPREs in human MBP and CNPase promoters. Therefore, by using ChIP analysis, we examined whether PPAR-β was in fact recruited to the promoters of human MOG and PLP genes. Fig. 10A shows the position of two PPREs in the MOG promoter. First, we examined the recruitment of PPAR-β to the proximal PPRE that is present within −597 to −575 (Fig. 10A). After immunoprecipitation of chromatin fragments by antibodies against PPAR-β, we were able to amplify 144-bp fragments flanking the proximal PPRE in gem-treated, but not control untreated, oligodendrocytes (Fig. 10A). On the other hand, no amplification product was observed in any of the immunoprecipitates obtained with control IgG (Fig. 10A), suggesting the specificity of these interactions. This result was confirmed by real time PCR analysis (Fig. 10B). Next we examined the recruitment of PPAR-β to the distal PPRE that is present within −876 to −854 (Fig. 10C). After immunoprecipitation by antibodies against PPAR-β, we were able to amplify 163-bp fragments flanking the distal PPRE in gem-treated, but not control untreated, oligodendrocytes (Fig. 10C). Again, we did not observe any amplification product in any of the immunoprecipitates obtained with control IgG (Fig. 10C), an effect we confirmed by real time PCR (Fig. 10D). These results suggest that the recruitment of PPAR-β to both proximal and distal PPREs of the MOG promoter is inducible by gem.
FIGURE 10.

ChIP analysis of gem-treated human oligodendrocytes for the recruitment of PPAR-β to the promoters of MOG and PLP genes. A, positions of two PPREs in the human MOG promoter are shown in the upper panel. Cells were treated with 50 μm gem for 6 h under serum-free conditions. Then immunoprecipitated chromatin fragments were amplified by semiquantitative (A, lower panel) and quantitative (B) PCR for the indicated region. Immunoprecipitated chromatin fragments were amplified by semiquantitative (C, lower panel) and quantitative (D) PCR for the indicated region spanning the distal PPRE of the MOG promoter. E, positions of two PPREs in the human PLP promoter are shown in the upper panel. After gem treatment, immunoprecipitated chromatin fragments were amplified by semiquantitative (E, lower panel) and quantitative (F) PCR for the indicated region spanning both PPREs of the PLP promoter. Results are the mean ± S.D. of three separate experiments. a, p < 0.001 versus control-anti-PPAR-β antibody. Error bars represent S.D.
Next we analyzed the PLP promoter. Fig. 10E shows the position of two PPREs in the human PLP promoter. Because these two PPREs are separated by only 84 nucleotides, we analyzed these two PPREs together. After immunoprecipitation of chromatin fragments by antibodies against PPAR-β, we amplified 315-bp fragments flanking both PPREs in gem-treated oligodendrocytes (Fig. 10, E and F). These results suggest that gem stimulates the recruitment of PPAR-β to the human PLP promoter.
Effect of Gem on Demyelination, the Recruitment of PPAR-β to the PLP Promoter in Vivo in the Spinal Cord, and the Disease Process of EAE in PLP-TCR Transgenic Mice
Earlier we have demonstrated that gem enters into the CNS and suppresses the disease process of EAE in female SJL/J mice (29). Here we examined whether gem increased and/or normalized the recruitment of PPAR-β to the myelin gene promoter, restored the level of myelin-associated proteins, and ameliorated the disease process in PLP-TCR transgenic mice. Gem is prescribed in adult humans at a dose of 600–1200 mg/day. Therefore, Tg mice were treated with a dose of 15 mg/kg of body weight/day via gavage. As expected, PLP immunization led to a marked induction of EAE symptoms in female PLP-TCR Tg mice (Fig. 11A). However, gem treatment markedly suppressed clinical symptoms of EAE (Fig. 11A). Next, we examined the recruitment of PPAR-α, PPAR-β, and PPAR-γ to the mouse PLP promoter in vivo in the spinal cord of EAE mice. The mouse PLP promoter harbors one consensus PPRE between −121 and −144 (Fig. 11B). Consistent with cell culture results, only PPAR-β was recruited to the PPRE of the PLP promoter in vivo in the spinal cord of non-Tg control mice (Fig. 11C), suggesting the specificity of the effect. However, this PPAR-β recruitment was inhibited in EAE mice (Fig. 11C), and gem treatment was capable of restoring and/or increasing this recruitment in vivo in the spinal cord of EAE mice (Fig. 11C). Earlier we have demonstrated a decrease in myelin gene expression in CNS tissues of EAE mice (28, 30). Accordingly, here we also saw decreased protein levels of PLP and MOG in the spinal cord of PLP-TCR Tg mice compared with the non-Tg control (Fig. 11D). However, gem treatment restored and/or increased PLP and MOG in vivo in the spinal cord of EAE mice (Fig. 11D). Accordingly, we observed widespread demyelination in the spinal cord of EAE mice; less demyelination was observed after gem treatment (Fig. 11, E and F).
FIGURE 11.

Gemfibrozil treatment restores the recruitment of PPAR-β to the mouse PLP promoter, inhibits demyelination in spinal cord, and suppresses EAE in PLP-TCR transgenic mice. A, PLP-TCR Tg mice were immunized with 10 μg of PLP139–151, and from 8 days postimmunization, mice were treated with either gemfibrozil (15 mg/kg of body weight) or vehicle (0.1% methylcellulose) via gavage. Mice (n = 6 in each group) were scored daily until 30 days postimmunization. B, position of one PPRE in the mouse PLP promoter is shown. C, at 18 days postimmunization, the recruitment of PPAR-α, PPAR-β, and PPAR-γ to the mouse PLP promoter was examined in spinal cord by in situ ChIP. D, at 18 days postimmunization, levels of PLP and MOG were monitored in spinal cord by Western blot. E, at 18 days postimmunization, spinal cord sections of non-Tg control, PLP-TCR Tg, and gemfibrozil-treated PLP-TCR Tg mice were stained with Luxol fast blue. Digital images were collected under bright field setting. F, demyelination in spinal cord is represented quantitatively using a scale as described under “Experimental Procedures.” Data are expressed as the mean ± S.E. of five different mice (n = 5) per group. a, p < 0.001 versus non-Tg control; b, p < 0.001 versus PLP-TCR Tg. Error bars represent S.E. Ab, antibody.
DISCUSSION
In multiple sclerosis, the body's failure to repair myelin is believed to lead to nerve damage, causing disease symptoms and increasing disability. It is expected that by remyelination nerves would be able to send proper signals again, restoring any loss of function as well as preventing further damage. Therefore, delineating mechanisms to encourage remyelination is an important area of multiple sclerosis research. In the CNS, myelin is synthesized by specialized cells called oligodendrocytes. Several lines of evidence presented in this work clearly establish that gemfibrozil, a United States Food and Drug Administration-approved drug for hyperlipidemia in human, stimulates the expression of myelin genes in human primary oligodendrocytes. Our conclusion is based on the following. First, upon serum deprivation, human oligodendrocytes lost processes and underwent cell death. However, gemfibrozil protected oligodendrocytes from serum deprivation-mediated cell death and induced significant elaboration and extension of processes. Second, gemfibrozil stimulated the expression of myelin-specific genes (MBP, MOG, CNPase, and PLP) in human primary oligodendrocytes, mixed glial cells, and spinal cord organotypic cultures. On the other hand, gemfibrozil had no such stimulatory effect on either the astroglial gene GFAP or the microglial gene CD11b in primary mixed glial cells. These studies suggest that the effect of gemfibrozil is oligodendrocyte-specific and that it does not happen in the other two types of glial cells (astroglia and microglia).
Similar to other fibrate drugs, gemfibrozil is a known activator of PPAR-α. However, gemfibrozil does not require PPAR-α to stimulate myelin genes in oligodendrocytes. For example, PPAR-α mRNA was present in primary oligodendrocytes at an undetectable level. This is consistent with a previous report by Granneman et al. (4) that also shows the absence of PPAR-α in rat and mouse oligodendrocytes (4, 35, 38). Gemfibrozil was also unable to stimulate the level of PPAR-α in oligodendrocytes. Although PPAR-α was detected in primary mixed glial cells, it was strongly inhibited by gemfibrozil treatment. Furthermore, gemfibrozil increased the expression of myelin genes in oligodendrocytes isolated from PPAR-α(−/−) mice. On the other hand, human oligodendrocytes expressed both PPAR-β and PPAR-γ. However, gemfibrozil stimulated the expression of PPAR-β, but not PPAR-γ, in oligodendrocytes. Actually, gemfibrozil inhibited the expression of PPAR-γ in mixed glial cells. Recently it has been reported that PPAR-β may function to repress PPAR-α and PPAR-γ target gene expression in certain type of cells (39). Similarly to the stimulation of PPAR-β in pure oligodendrocytes, gemfibrozil was also able to increase the level of PPAR-β in primary mixed glial cells and spinal cord organotypic cultures. Moreover, antisense knockdown of PPAR-β, but not PPAR-γ, abrogated the stimulatory effect of gemfibrozil on the expression of myelin genes in primary human oligodendrocytes. Similarly, genetic knockdown of PPAR-β also eliminated the ability of gem to up-regulate myelin genes in mouse oligodendrocytes. WY14643 is a specific ligand for PPAR-α (40). However, in contrast to gemfibrozil, WY14643 was unable to increase the expression of myelin genes in primary oligodendrocytes. Taken together, these results indicate that gemfibrozil stimulates the expression of myelin genes via PPAR-β but not via either PPAR-α or PPAR-γ. Our results are consistent with published reports that demonstrate reduced myelination of the corpus callosum in PPAR-β-null mice and suggest a role of PPAR-β in oligodendroglial lipid metabolism and myelinogenesis (41, 42).
However, at present, mechanisms by which PPAR-β regulates myelinogenesis are poorly understood. PPAR-β is a transcription factor, and therefore, one possibility could be direct transcriptional regulation of myelin genes by PPAR-β. In fact, while looking at the 5′-upstream regions of MBP, PLP, MOG, and CNPase for regulatory elements, we found that promoter regions of PLP and MOG genes contained consensus PPREs. ChIP analysis also revealed that gem markedly stimulated the recruitment of PPAR-β to the promoters of PLP and MOG in human oligodendrocytes. To understand whether this recruitment of PPAR-β to the myelin gene promoter also occurs in adult oligodendrocytes, we induced EAE in PLP-TCR Tg mice. Similarly to that in cultured oligodendroglia, PPAR-β, but not PPAR-α and PPAR-γ, was also recruited to the mouse PLP promoter in vivo in the spinal cord. The decrease in PPAR-β binding to the PLP promoter during EAE and restoration of this binding after gemfibrozil treatment clearly indicate the importance of PPAR-β recruitment to the myelin gene promoter in physiology and pathophysiology.
Although PLP and MOG genes could be directly regulated by PPAR-β at the transcriptional level, we did not detect any PPREs in the promoters of human MBP and CNPase genes. Accordingly, our real time data also show that gemfibrozil led to a robust increase in mRNA expression of PLP and MOG and a modest increase in mRNA expression of MBP and CNPase in oligodendrocytes. In the absence of any direct transcriptional regulation via PPAR-β, gemfibrozil may increase the myelin gene expression via modulation of other regulators of myelination. Recent studies have delineated that different transcription factors such as cAMP-response element-binding protein, OLIG1, and Sox10 play an important role in the transcription of myelin genes in oligodendrocytes (43–47). Therefore, gemfibrozil may increase the expression of MBP and CNPase via PPAR-β-mediated up-regulation of cAMP-response element-binding protein, OLIG1, and/or Sox10.
In summary, we have demonstrated that gemfibrozil protects oligodendrocytes from serum deprivation-induced cell death and stimulates the expression of myelin genes in oligodendrocytes via PPAR-β. Although the regulation of PPAR-β by gemfibrozil in the CNS microenvironment of patients with demyelinating disorders may differ from that observed in the cultured primary oligodendrocytes and the in vitro situation of human fetal oligodendrocytes in culture and spinal cord organotypic cultures may not truly resemble the in vivo situation of oligodendrocytes in the CNS of patients, our results suggest that gemfibrozil may be used to boost the expression of myelin-specific genes via PPAR-β and promote remyelination in the CNS of patients with demyelinating disorders.
This work was supported, in whole or in part, by National Institutes of Health Grants AT6681, NS64564, and NS71479 (to K. P.). This work was also supported by National Multiple Sclerosis Society Grant RG4170-A-1 (to M. J.).
- MBP
- myelin basic protein
- MOG
- myelin oligodendrocyte glycoprotein
- CNPase
- 2′,3′-cyclic-nucleotide 3′-phosphodiesterase
- PLP
- proteolipid protein
- TCR
- T cell receptor
- PPAR
- peroxisome proliferator-activated receptor
- EAE
- experimental autoimmune encephalomyelitis
- GFAP
- glial fibrillary acidic protein
- ASO
- antisense oligonucleotide
- ScO
- scrambled oligonucleotide
- gem
- gemfibrozil
- GalC
- galactocerebroside
- h
- human
- m
- mouse
- PPRE
- peroxisome proliferator response element
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- Tg
- transgenic.
REFERENCES
- 1. Jana M., Pahan K. (2005) Redox regulation of cytokine-mediated inhibition of myelin gene expression in human primary oligodendrocytes. Free Radic. Biol. Med. 39, 823–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jana M., Jana A., Pal U., Pahan K. (2007) A simplified method for isolating highly purified neurons, oligodendrocytes, astrocytes, and microglia from the same human fetal brain tissue. Neurochem. Res. 32, 2015–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berger T., Reindl M. (2000) Immunopathogenic and clinical relevance of antibodies against myelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis. J. Neural Transm. Suppl. 60, 351–360 [DOI] [PubMed] [Google Scholar]
- 4. Granneman J., Skoff R., Yang X. (1998) Member of the peroxisome proliferator-activated receptor family of transcription factors is differentially expressed by oligodendrocytes. J. Neurosci. Res. 51, 563–573 [DOI] [PubMed] [Google Scholar]
- 5. Saluja I., Granneman J. G., Skoff R. P. (2001) PPARδ agonists stimulate oligodendrocyte differentiation in tissue culture. Glia 33, 191–204 [PubMed] [Google Scholar]
- 6. Woods J. W., Tanen M., Figueroa D. J., Biswas C., Zycband E., Moller D. E., Austin C. P., Berger J. P. (2003) Localization of PPARδ in murine central nervous system: expression in oligodendrocytes and neurons. Brain Res. 975, 10–21 [DOI] [PubMed] [Google Scholar]
- 7. Cullingford T. E., Bhakoo K., Peuchen S., Dolphin C. T., Patel R., Clark J. B. (1998) Distribution of mRNAs encoding the peroxisome proliferator-activated receptor α, β, and γ and the retinoid X receptor α, β, and γ in rat central nervous system. J. Neurochem. 70, 1366–1375 [DOI] [PubMed] [Google Scholar]
- 8. Kastner P., Mark M., Chambon P. (1995) Nonsteroid nuclear receptors: what are genetic studies telling us about their role in real life? Cell 83, 859–869 [DOI] [PubMed] [Google Scholar]
- 9. Kliewer S. A., Xu H. E., Lambert M. H., Willson T. M. (2001) Peroxisome proliferator-activated receptors: from genes to physiology. Recent Prog. Horm. Res. 56, 239–263 [DOI] [PubMed] [Google Scholar]
- 10. Berger J. P., Akiyama T. E., Meinke P. T. (2005) PPARs: therapeutic targets for metabolic disease. Trends Pharmacol. Sci. 26, 244–251 [DOI] [PubMed] [Google Scholar]
- 11. Powell E., Kuhn P., Xu W. (2007) Nuclear receptor cofactors in PPARγ-mediated adipogenesis and adipocyte energy metabolism. PPAR Res. 2007, 53843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arnold R., König W. (2006) Peroxisome-proliferator-activated receptor-γ agonists inhibit the release of proinflammatory cytokines from RSV-infected epithelial cells. Virology 346, 427–439 [DOI] [PubMed] [Google Scholar]
- 13. Alleva D. G., Johnson E. B., Lio F. M., Boehme S. A., Conlon P. J., Crowe P. D. (2002) Regulation of murine macrophage proinflammatory and anti-inflammatory cytokines by ligands for peroxisome proliferator-activated receptor-γ: counter-regulatory activity by IFN-γ. J. Leukoc. Biol. 71, 677–685 [PubMed] [Google Scholar]
- 14. Diab A., Deng C., Smith J. D., Hussain R. Z., Phanavanh B., Lovett-Racke A. E., Drew P. D., Racke M. K. (2002) Peroxisome proliferator-activated receptor-γ agonist 15-deoxy-Δ12,14-prostaglandin J2 ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 168, 2508–2515 [DOI] [PubMed] [Google Scholar]
- 15. Tenenbaum A., Motro M., Fisman E. Z. (2005) Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: the bezafibrate lessons. Cardiovasc. Diabetol. 4, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee C. H., Chawla A., Urbiztondo N., Liao D., Boisvert W. A., Evans R. M., Curtiss L. K. (2003) Transcriptional repression of atherogenic inflammation: modulation by PPARδ. Science 302, 453–457 [DOI] [PubMed] [Google Scholar]
- 17. Polak P. E., Kalinin S., Dello Russo C., Gavrilyuk V., Sharp A., Peters J. M., Richardson J., Willson T. M., Weinberg G., Feinstein D. L. (2005) Protective effects of a peroxisome proliferator-activated receptor-β/δ agonist in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 168, 65–75 [DOI] [PubMed] [Google Scholar]
- 18. Harman F. S., Nicol C. J., Marin H. E., Ward J. M., Gonzalez F. J., Peters J. M. (2004) Peroxisome proliferator-activated receptor-δ attenuates colon carcinogenesis. Nat. Med. 10, 481–483 [DOI] [PubMed] [Google Scholar]
- 19. Peters J. M., Gonzalez F. J. (2009) Sorting out the functional role(s) of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) in cell proliferation and cancer. Biochim. Biophys. Acta 1796, 230–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rubins H. B., Robins S. J., Collins D., Fye C. L., Anderson J. W., Elam M. B., Faas F. H., Linares E., Schaefer E. J., Schectman G., Wilt T. J., Wittes J. (1999) Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N. Engl. J. Med. 341, 410–418 [DOI] [PubMed] [Google Scholar]
- 21. Hsu H. C., Lee Y. T., Yeh H. T., Chen M. F. (2001) Effect of gemfibrozil on the composition and oxidation properties of very-low-density lipoprotein and high-density lipoprotein in patients with hypertriglyceridemia. J. Lab. Clin. Med. 137, 414–421 [DOI] [PubMed] [Google Scholar]
- 22. Pahan K., Jana M., Liu X., Taylor B. S., Wood C., Fischer S. M. (2002) Gemfibrozil, a lipid-lowering drug, inhibits the induction of nitric-oxide synthase in human astrocytes. J. Biol. Chem. 277, 45984–45991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao S. P., Ye H. J., Zhou H. N., Nie S., Li Q. Z. (2003) Gemfibrozil reduces release of tumor necrosis factor-α in peripheral blood mononuclear cells from healthy subjects and patients with coronary heart disease. Clin. Chim. Acta 332, 61–67 [DOI] [PubMed] [Google Scholar]
- 24. Xu J., Storer P. D., Chavis J. A., Racke M. K., Drew P. D. (2005) Agonists for the peroxisome proliferator-activated receptor-α and the retinoid X receptor inhibit inflammatory responses of microglia. J. Neurosci. Res. 81, 403–411 [DOI] [PubMed] [Google Scholar]
- 25. Dasgupta S., Jana M., Zhou Y., Fung Y. K., Ghosh S., Pahan K. (2004) Antineuroinflammatory effect of NF-κB essential modifier-binding domain peptides in the adoptive transfer model of experimental allergic encephalomyelitis. J. Immunol. 173, 1344–1354 [DOI] [PubMed] [Google Scholar]
- 26. Ghosh A., Pahan K. (2012) Gemfibrozil, a lipid-lowering drug, induces suppressor of cytokine signaling 3 in glial cells: implications for neurodegenerative disorders. J. Biol. Chem. 287, 27189–27203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Waldner H., Whitters M. J., Sobel R. A., Collins M., Kuchroo V. K. (2000) Fulminant spontaneous autoimmunity of the central nervous system in mice transgenic for the myelin proteolipid protein-specific T cell receptor. Proc. Natl. Acad. Sci. U.S.A. 97, 3412–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brahmachari S., Pahan K. (2007) Sodium benzoate, a food additive and a metabolite of cinnamon, modifies T cells at multiple steps and inhibits adoptive transfer of experimental allergic encephalomyelitis. J. Immunol. 179, 275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dasgupta S., Roy A., Jana M., Hartley D. M., Pahan K. (2007) Gemfibrozil ameliorates relapsing-remitting experimental autoimmune encephalomyelitis independent of peroxisome proliferator-activated receptor-α. Mol. Pharmacol. 72, 934–946 [DOI] [PubMed] [Google Scholar]
- 30. Mondal S., Roy A., Pahan K. (2009) Functional blocking monoclonal antibodies against IL-12p40 homodimer inhibit adoptive transfer of experimental allergic encephalomyelitis. J. Immunol. 182, 5013–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brahmachari S., Fung Y. K., Pahan K. (2006) Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J. Neurosci. 26, 4930–4939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Roy A., Fung Y. K., Liu X., Pahan K. (2006) Up-regulation of microglial CD11b expression by nitric oxide. J. Biol. Chem. 281, 14971–14980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Asayama K., Sandhir R., Sheikh F. G., Hayashibe H., Nakane T., Singh I. (1999) Increased peroxisomal fatty acid β-oxidation and enhanced expression of peroxisome proliferator-activated receptor-α in diabetic rat liver. Mol. Cell. Biochem. 194, 227–234 [DOI] [PubMed] [Google Scholar]
- 34. Kainu T., Wikström A. C., Gustafsson J. A., Pelto-Huikko M. (1994) Localization of the peroxisome proliferator-activated receptor in the brain. Neuroreport 5, 2481–2485 [DOI] [PubMed] [Google Scholar]
- 35. Moreno S., Farioli-Vecchioli S., Cerù M. P. (2004) Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience 123, 131–145 [DOI] [PubMed] [Google Scholar]
- 36. Basu-Modak S., Braissant O., Escher P., Desvergne B., Honegger P., Wahli W. (1999) Peroxisome proliferator-activated receptor β regulates acyl-CoA synthetase 2 in reaggregated rat brain cell cultures. J. Biol. Chem. 274, 35881–35888 [DOI] [PubMed] [Google Scholar]
- 37. Dressel U., Allen T. L., Pippal J. B., Rohde P. R., Lau P., Muscat G. E. (2003) The peroxisome proliferator-activated receptor β/δ agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol. Endocrinol. 17, 2477–2493 [DOI] [PubMed] [Google Scholar]
- 38. Cimini A., Bernardo A., Cifone M. G., Di Marzio L., Di Loreto S. (2003) TNFα downregulates PPARδ expression in oligodendrocyte progenitor cells: implications for demyelinating diseases. Glia 41, 3–14 [DOI] [PubMed] [Google Scholar]
- 39. Zuo X., Wu Y., Morris J. S., Stimmel J. B., Leesnitzer L. M., Fischer S. M., Lippman S. M., Shureiqi I. (2006) Oxidative metabolism of linoleic acid modulates PPAR-β/δ suppression of PPAR-γ activity. Oncogene 25, 1225–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Forman B. M., Chen J., Evans R. M. (1997) Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc. Natl. Acad. Sci. U.S.A. 94, 4312–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peters J. M., Lee S. S., Li W., Ward J. M., Gavrilova O., Everett C., Reitman M. L., Hudson L. D., Gonzalez F. J. (2000) Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor β(δ). Mol. Cell. Biol. 20, 5119–5128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shi Y., Hon M., Evans R. M. (2002) The peroxisome proliferator-activated receptor δ, an integrator of transcriptional repression and nuclear receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 99, 2613–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Samanta J., Kessler J. A. (2004) Interactions between ID and OLIG proteins mediate the inhibitory effects of BMP4 on oligodendroglial differentiation. Development 131, 4131–4142 [DOI] [PubMed] [Google Scholar]
- 44. Arnett H. A., Fancy S. P., Alberta J. A., Zhao C., Plant S. R., Kaing S., Raine C. S., Rowitch D. H., Franklin R. J., Stiles C. D. (2004) bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science 306, 2111–2115 [DOI] [PubMed] [Google Scholar]
- 45. Liu Z., Hu X., Cai J., Liu B., Peng X., Wegner M., Qiu M. (2007) Induction of oligodendrocyte differentiation by Olig2 and Sox10: evidence for reciprocal interactions and dosage-dependent mechanisms. Dev. Biol. 302, 683–693 [DOI] [PubMed] [Google Scholar]
- 46. Wegner M. (2000) Transcriptional control in myelinating glia: flavors and spices. Glia 31, 1–14 [DOI] [PubMed] [Google Scholar]
- 47. Gao Y., Deng K., Hou J., Bryson J. B., Barco A., Nikulina E., Spencer T., Mellado W., Kandel E. R., Filbin M. T. (2004) Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 44, 609–621 [DOI] [PubMed] [Google Scholar]
- 48. Jana A., Pahan K. (2007) Oxidative stress kills human primary oligodendrocytes via neutral sphingomyelinase: implications for multiple sclerosis. J. Neuroimmune Pharmacol. 2, 184–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jana A., Pahan K. (2004) HIV-1 gp120 induces apoptosis in human primary neurons through redox-regulated activation of neutral sphingomyelinase. J. Neurosci. 24, 9531–9540 [DOI] [PMC free article] [PubMed] [Google Scholar]