

Background: Wnts control development and differentiation; their secretion may be regulated.
Results: All Wnt activity absolutely requires post-translational modification by the O-acyltransferase PORCN, whose abundance or activity modulates signaling tightly and over a large dynamic range.
Conclusion: Subtle changes in PORCN activity have major consequences and cause the human disease focal dermal hypoplasia.
Significance: PORCN is a key regulator of all Wnt signaling.
Keywords: beta-Catenin, Development, Protein Secretion, Wnt Pathway, Wnt Signaling, FDH, MBOAT, PORCN, Zinc Finger Nuclease
Abstract
Gradients of diverse Wnt proteins regulate development, renewal, and differentiation. Porcupine (PORCN) is a membrane-bound O-acyltransferase that is required for post-translational modification of all Wnts to enable their transport, secretion, and activity. Mutations in PORCN are associated with focal dermal hypoplasia (FDH), whereas gene deletion causes embryonic lethality in mice. To study the protein in more detail, zinc finger nucleases were used to edit the PORCN genomic locus, establishing two HT1080 fibrosarcoma clones null for PORCN activity that facilitate the study of PORCN structure and function. We establish that PORCN is a key non-redundant node for the regulation of global Wnt signaling because PORCN null cells are completely incapable of autocrine Wnt signaling. The strength of Wnt signaling is exquisitely sensitive to PORCN expression, with a dynamic range of at least 3 orders of magnitude, suggesting that PORCN activity is a key modulator of all Wnt ligand activity. Consistent with this, we find that multiple FDH-associated mutants have only subtle alterations in enzyme activity yet are associated with a severe FDH phenotype. These studies support an essential regulatory role of PORCN in shaping Wnt signaling gradients.
Introduction
Wnts are secreted protein ligands that direct the activation of diverse signaling pathways, of which β-catenin-mediated transcription is the best known and understood. Multiple Wnt-triggered pathways coordinately regulate key developmental processes as well as adult tissue homeostasis and repair and are dysregulated in diverse diseases, including cancer, malformation syndromes, osteoporosis, diabetic retinopathy, and pulmonary fibrosis (1–5). Wnts act locally and regionally, with both quantitative and qualitative regulation of downstream signaling dependent on local Wnt concentration (6, 7). Different Wnts can have opposing actions in the same tissue. The local Wnt concentration, and the balance of varying Wnt activities at any given target cell are dependent on diverse factors, including transcription and translation rates, post-translational modification, secretion, and extracellular transport, and thus these are topics of active investigation (reviewed in Refs. 8 and 9). Careful control of global Wnt activity may be critical for development and homeostasis because changes in the core pathway can affect diverse Wnt-dependent signaling pathways. The rate of Wnt secretion is one potential control point for Wnt signaling. Whereas there are 19 distinct Wnt genes and multiple Wnt receptors, there appears to be a single conserved and essential Wnt biogenesis pathway. This core pathway includes PORCN (porcupine), ER2 to Golgi transit-promoting p24 proteins (10, 11), the Wnt chaperone WLS (Wntless; also known as Evi and GPR177) (12–14), and the retromer complex and sorting nexins, which are necessary for recycling WLS (15). The first step in Wnt protein production is translation in the endoplasmic reticulum, followed by post-translational modification by the ER-resident enzyme PORCN (16). PORCN is a membrane-bound O-acyltransferase (MBOAT), and it catalyzes the palmitoylation of the serine corresponding to Ser-209 of WNT3A (17, 18). This modification is absolutely required for the next step in Wnt secretion, binding to the carrier protein WLS. In addition, palmitoylation is essential for the ability of Wnts to interact with Frizzled receptors at the cell surface (19–21). Whereas Wnts were reported to be palmitoylated at a conserved cysteine corresponding to Cys-77 of WNT3A (22, 23), in the crystal structure of Xenopus Wnt8 in complex with Fz8-CRD, the Cys-77-equivalent residue was instead engaged in a disulfide bond (21). This disulfide formation probably explains why mutations of Cys-77 caused decreased activity of Wnt. It is notable that all Wnts examined from diverse organisms, with the sole exception of Drosophila WntD, require PORCN or at least maintain the serine corresponding to Ser-209 of WNT3A. WntD does not contain this serine (replaced by glutamine) and is secreted independently of either Porcn or Wls, although it retains dependence on the p24 protein Opm (11).
Little is known about the regulation of PORCN expression and palmitoyltransferase activity. PORCN is a member of the MBOAT family (24), and in most species, it appears to be a single copy X-linked gene most closely related (but with <35% identity) to a family of lysophospholipid acyltransferases (52), and no evidence for functional redundancy in Drosophila, zebrafish, mice, or humans (16, 25–28). The enzyme is expressed in most tissues of the body, with four tissue-specific splice variants of unknown significance (29, 30). Because of the importance of Wnt signaling in human disease, small molecule inhibitors of PORCN have been developed that inhibit Wnt signaling in cultured cells and in intestinal organoids (31, 32).
Consistent with its critical role in Wnt signaling, germ line PORCN mutations are associated with the X-linked dominant genetic disease focal dermal hypoplasia (FDH; OMIM 305600) (33, 34). The disease occurs primarily in heterozygous females due to the location of PORCN on the X chromosome, and its phenotypic manifestations are variable due to patterns of X-inactivation. Complete deletion of PORCN is embryonic lethal in mice and also presumably in humans, although postzygotic mutations do occur in males and cause FDH but allow survival. Heterozygous deletion of PORCN in mice also recapitulates the phenotypes seen in FDH (26–28). FDH is associated with defects in ecto-mesodermally derived tissues, leading to superficial skin striation and atrophy, fat herniation, and papillomas of the mucous membranes as well as more severe defects, such as limb shortening and syndactyly. Combined with the knowledge that the known function of PORCN is in Wnt modification, it is presumed, but not yet proven, that FDH is ultimately a disease of Wnt malfunction. Whether there is a discernible genotype-phenotype correlation is not clear (35). Over 100 different PORCN mutations have been reported in FDH (see the Human Genome Variation Society Locus Specific Mutation Databases Web site). Some PORCN mutations in FDH patients are clearly loss of function because they are small or large deletions and/or cause premature protein termination. However, a number of PORCN alterations are missense mutations that fall in residues of unknown function. Because PORCN may have an essential role beyond the Wnt pathway (36), it is also possible that some of the FDH mutations in PORCN do not alter the Wnt signaling pathway.
In this report, we have used zinc finger nucleases to functionally delete PORCN from the human (male) fibrosarcoma cell line HT1080. There appears to be no functional redundancy for Wnt palmitoylation because these cells absolutely require PORCN for any Wnt secretion or signaling. Further, we use these cells to demonstrate that physiologically relevant expression levels of PORCN can fine tune Wnt signaling over a large dynamic range. Although all FDH mutants tested alter Wnt signaling, importantly, a subset of mutations demonstrates nearly normal activity until titrated to very low but physiologically relevant levels. Splice variants of PORCN display small changes in activity similar to those seen in some FDH mutants. These findings suggest that in humans, small changes in Wnt gradients due to subtle mutations in PORCN can have major phenotypic effects.
EXPERIMENTAL PROCEDURES
Reagents
HT1080 cells were acquired from the American Type Culture Collection (Manassas, VA) and grown in DMEM (Nacali Tesque, Japan) containing 4.5 g/liter glucose, penicillin/streptomycin, 10% FBS, and 1 mm sodium pyruvate in a humidified 37 °C atmosphere. The zinc finger nuclease was purchased from Sigma-Aldrich and is composed of an array of six zinc fingers upstream and five zinc fingers downstream of a 5-bp spacer cut site at the beginning of exon 9 (Fig. 1A). Antibody to WNT3A was a kind gift from Shinji Takada. Antibody against HA tag was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) (sc-7392). Dvl2 antibodies (diluted 1:500) were from Santa Cruz Biotechnology, Inc. (catalog no. 13974). The Surveyor mutation detection kit was from Transgenomic (Omaha, NE). Super8xTopflash reporter was a gift from Randy Moon (37). pGK-WNT3A plasmid was provided by Karl Willert. Other Wnt expression constructs were prepared as part of the Open Source Wnt project (38). pMKIT-3xHA-mPORC-A as well as isoforms B, C, and D were gifts from Tatsuhiko Kadowaki. Xenopus laevis PORCN (isoform A) expression vector (clone ID 6638385) was purchased from Open Biosystems/Thermo Scientific (Lafayette, CO). Point mutants and small deletions in PORCN plasmids were created with the Stratagene QuikChange site-directed mutagenesis kit (La Jolla, CA). DNA sequencing was performed by AITBiotech (Singapore). The human tissue array mRNA set was from Clontech, lot number 1001243A.
FIGURE 1.
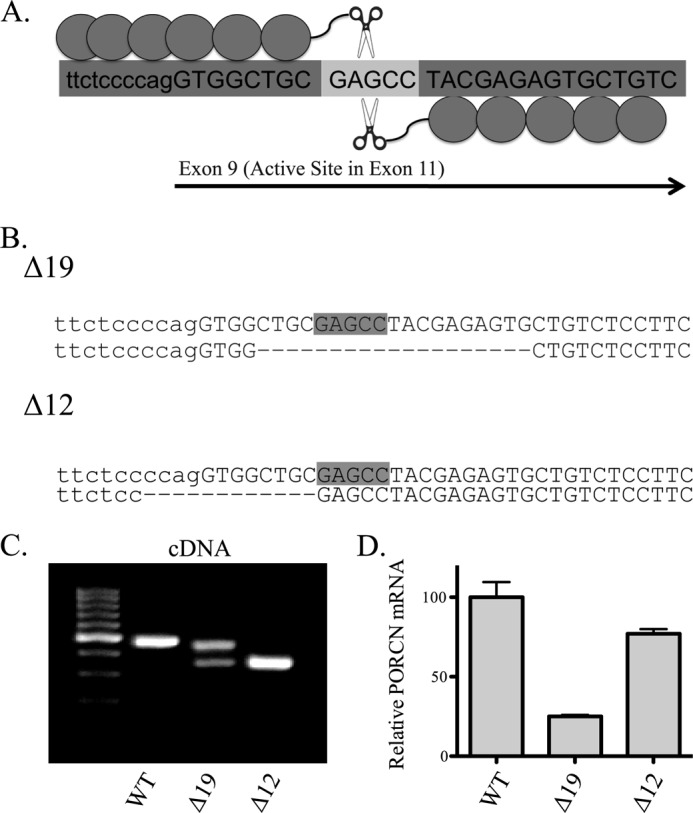
Zinc finger nuclease-mediated deletion of PORCN from HT1080 cells. A, graphic representation of ZFN targeting strategy. Capital letters indicate exonic coding sequence, and the lighter gray 5-base pair spacer is the site of initial cleavage. B, sequence of genomic DNA at the PORCN locus for two edited clones. Not shown is a third, unedited clone. C, PORCN cDNA. The region around exon 9 was amplified, and bands were excised and sequenced. The Δ19 clone upper band matches the deletion seen in B, whereas the lower band sequence lacks exon 9. The Δ12 band also lacks exon 9. D, quantitative PCR for PORCN. Results have been normalized to actin levels, and error bars represent S.D.
Zinc Finger Nuclease-mediated PORCN Deletion
In order to delete PORCN, HT1080 cells were transfected in 6-well dishes with 2 μg each of ZFN1 and ZFN2, creating the heterodimeric endonuclease. 48 h after transfection, genomic DNA was isolated, and a 382-bp fragment of PORCN surrounding exon 9 was amplified using the forward primer TTT CTT CTC TGC TGC CTT CC and reverse primer ACA CTT CTG TCT TGG CCG TT. This product was subjected to the Transgenomic cel1-based endonuclease assay to detect gene editing. Following confirmation of significant editing within the pooled population, single cells were cloned by limiting dilution into 96-well plates. After ∼2 weeks of growth, gDNA stabilizing lysate was prepared using Lyse-and-go reagent (Promega, Madison, WI). This lysate was used for amplification of the same 382-bp region surrounding exon 9. A 13% polyacrylamide TBE gel was used to resolve products, and mutant clones were identified by faster migration. Two clones with significant deletion were isolated, in addition to an isogenic clone with no modification in the PORCN gene.
Wnt Secretion and SuperTOPFlash Assays
To monitor Wnt secretion into culture medium, cells were grown in 6-well plates and transfected with 1 μg of WNT3A, along with mCherry and 100 ng of 3xHAmPORCN-D encoding plasmids as indicated. Six hours after transfection, medium was changed to low serum 2% FBS at half volume (1 ml). Lipofectamine 2000 was used for transfections (Invitrogen). For SuperTOPFlash (β-catenin-activated TCF/LEF transcriptional reporter) (STF) assays, experiments were performed in 24-well plates, transfecting 800 ng of total plasmid/well, composed of 50 ng of Wnt, 100 ng of mCherry, 550 ng of SuperTOPFlash, and up to 100 ng of PORCN plasmid (pMSCV-puro empty vector used as filler). Lysates were prepared in PBS with 0.6% IGEPAL-CA630, and firefly luciferase was measured with a luciferase assay kit according to the manufacturer's recommendations (Promega), using a Tecan Infinite M200 plate reader (San Jose, CA).
Dvl2 Mobility Shift Assay
For measurement of Dvl2 mobility shift, transfection in 24-well dishes was with 200 ng of each Wnt, 600 ng of mCherry, and 10 ng of mPORCN-D expression plasmids. Lysates were prepared in 100 mm sodium phosphate, pH 7.5, 150 mm NaCl, 1% IGEPAL-CA630, complete protease inhibitor mixture (Roche Applied Science) and analyzed by SDS-PAGE on 10% gels followed by Western blotting to detect Dvl2.
Quantitative PCR
RNA was isolated using the RNEasy kit (Qiagen), reverse transcribed with the i-script RT kit (Bio-Rad), and quantified on a Bio-Rad CFX96 real-time cycling machine using the SsoFast EvaGreen PCR assay (Bio-Rad). Primers used for AXIN2 were CTC CCC ACC TTG AAT GAA GA (forward) and TGG CTG GTG CAA AGA CAT AG (reverse). Primers for PORCN for quantitative PCR were GGA GGC TCG GGT TGC CAT CCT (forward) and GCC CCT CGC ATC TTG TGC CA (reverse). Primers for ACTB were AAG GAT TCC TAT GTG GGC GAC G (forward) and GCC TGG ATA GCA ACG TAC ATG G (reverse). Primers for GUSB were TTG CTC ACA AAG GTC ACA GG (forward) and CGT CCC ACC TAG AAT CTG CT (reverse). Primers for PLA2G4A were TTC TTT GGC ACT CTA GCG TCT (forward) and GAA TTC TCC GGA GCT GAA AA (reverse). Primers for PORCN reverse transcription in exons 6, 7, and 10 were CTC CTT CGC AAC AAG AAA CG (forward) and AGT GGC TTG GAC ACC GTC AGG (reverse).
PORCN Null MEF Isolation and Assays
Mice with loxP sites surrounding exon 3 of PORCN were acquired from Janet Rossant and Steffen Biechele. MEFs were isolated at day 14.5 from a homozygous and hemizygous cross. Briefly, fetal heads and internal organs were removed, and remaining tissue was minced and then trypsinized to yield a single-cell suspension. These cells were grown in DMEM and spontaneously immortalized by serial passaging and growth to high density with several changes of medium. After immortalization, PORCN was deleted by adenoviral Cre-GFP (Vector Biolabs, Philadelphia, PA) excision was confirmed by genomic analysis and RT-PCR. Adenoviral GFP expression was used as control. To perform SuperTOPFlash assays in PORCN null MEFs, 2 × 106 cells/data point were transfected by nucleofection using the VPD-1004 MEF 1 nucleofector kit with an Amaxa Nucleofector I (Lonza).
RESULTS
Creation of PORCN Null Cells
In order to perform rigorous characterization of PORCN function and test the hypothesis that all FDH mutants affect the Wnt signaling pathway, we chose to create PORCN null HT1080 fibrosarcoma cancer cells using zinc finger nuclease technology. HT1080 cells were selected because they are derived from mesoderm, the germ layer that is most affected in FDH, male and hence hemizygous for PORCN, readily transfected, and highly responsive to Wnt ligands. Although they normally express PORCN, their growth rate is unaffected by inhibition of PORCN by RNAi and small molecule inhibitors of PORCN (36). Despite some reports indicating that HT1080 cells have an autocrine Wnt signaling loop (39, 40), we were unable to detect any endogenous STF activity without the co-expression of Wnt.
A zinc finger nuclease targeting exon 9 of PORCN was generated to create double-stranded breaks in and thereby inactivate the PORCN gene (Fig. 1A). Exon 9 is predicted to encode a membrane-spanning region. Its importance in PORCN function is indicated by the fact that two missense mutations, a single amino acid insertion, and three frameshift mutations as well as mutations in splice donor and acceptor sites occur in exon 9 in FDH patients (35). Additionally, because the catalytic active site of PORCN exists within exon 11, a frameshift in the coding sequence anywhere upstream is expected to disrupt enzymatic activity.
Single-cell dilution cloning allowed for the recovery of two separate ZFN-modified HT1080 sublines with defined 12- and 19-base pair deletions (hereafter referred to as Δ12 and Δ19), along with an isogenic unedited wild-type clone (Fig. 1B). The 12-base pair deletion of Δ12 extends into the intron upstream of exon 9, thus removing the AG splice acceptor, whereas the Δ19 deletion is fully exonic, causing a predicted frameshift. RT-PCR analysis and sequencing of the mRNA in this region reveal that the Δ12 clone transcript lacks exon 9 (and hence one predicted membrane-spanning domain), whereas the Δ19 clone produces two transcripts, one with an internal deletion (and hence a frameshift) and one that skips exon 9 (Fig. 1C). Quantitative RT-PCR of PORCN mRNA in edited cells showed that the Δ12 clone has a slightly reduced transcript level, whereas Δ19 has only ∼25% remaining, probably due to nonsense-mediated RNA decay (Fig. 1D). To ensure that the observed phenotypes were due to mutation of the PORCN gene and not a second site mutation, the two clones were tested side-by-side and found to behave identically in all subsequent assays. As anticipated, inactivation of PORCN in these cells had no effect on cell growth or viability (data not shown).
PORCN Is Essential and Non-redundant for Wnt Ligand Activity
Given that PORCN is a member of the MBOAT multigene family, one important question is whether there is redundancy of PORCN function. To test this, PORCN null or wild-type cells were transfected with plasmid encoding WNT3A, and culture media and lysates were subsequently isolated to probe for Wnt secretion. As seen in Fig. 2A, PORCN null cells were able to express WNT3A as well as their wild-type counterparts, but could not effectively secrete WNT3A into the medium. Secretion of WNT3A was rescued by expression of mouse 3xHA epitope-tagged PORCN-D, indicating that the secretion defect was due to the mutation of the PORCN gene (Fig. 2D). Even more dramatically, PORCN null cells were entirely unable to increase signaling from the STF reporter in the presence of transfected WNT3A expression plasmid, whereas wild-type cells responded with a >200-fold increase in luciferase activity (Fig. 2B). Although PORCN null cells could not produce active WNT3A, they remained fully competent to respond to exogenous Wnt from conditioned medium produced from PORCN-positive cells (Fig. 2C).
FIGURE 2.
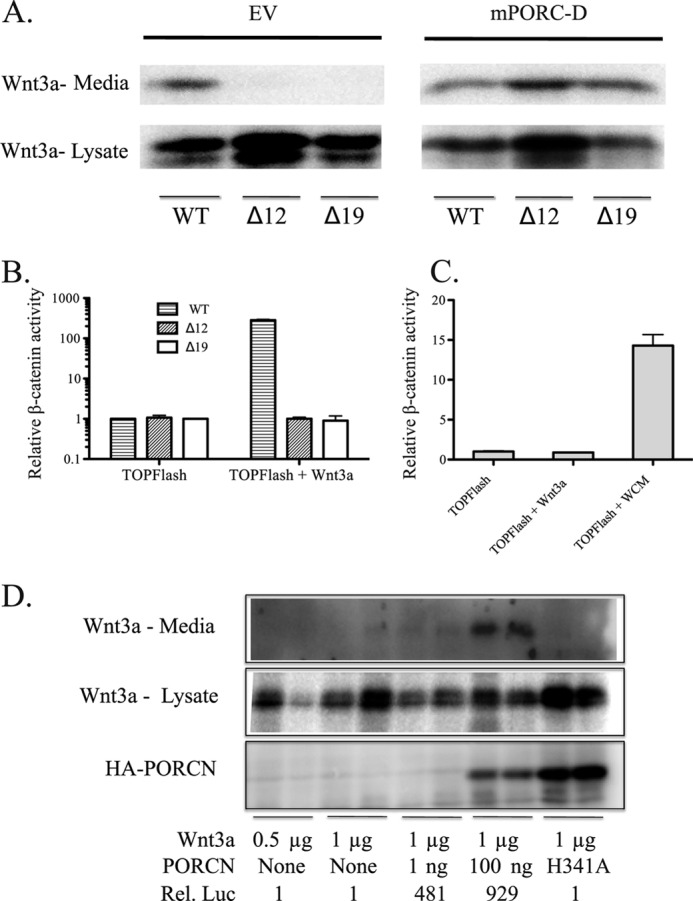
PORCN null cells cannot activate WNT3A. A, PORCN null cells do not secrete WNT3A. The cloned cells indicated were transfected with WNT3A, and culture media and lysates were prepared 24 h later. Western blot was performed to detect WNT3A. Left, no PORCN. Right, 3xHA-tagged mPORCN-D was co-transfected with Wnt and rescued secretion. B, cells were transfected with SuperTOPFlash reporter in the absence or presence of WNT3A. mCherry transfection was used to ensure equivalent transfection. 24 h after transfection, lysates were prepared and probed for firefly luciferase. C, luciferase assay as in B. Δ12 cells are shown. WNT3A-conditioned medium (WCM) was added in the right bar and is able to activate signaling. D, WT PORCN can rescue Wnt secretion and STF activity (number shown at bottom indicates -fold signal above background), but there is a non-linear relationship between detectable secreted Wnt and STF activity. The catalytic active site mutant PORCN H341A (100 ng transfected) cannot rescue secretion or STF activity. Duplicates are shown. Error bars, S.D.
MBOAT proteins have a putative catalytic histidine (His-341) residue that in PORCN and other MBOAT proteins (ghrelin O-acyltransferase) is required for activity. Neither Wnt secretion nor β-catenin signaling was rescued by PORCN with an active site (H341A) mutation, both confirming the importance of His-341 to MBOAT enzyme activity and showing that the secretion and activity defect was due to a loss of enzyme activity. As has been reported previously (e.g. see Ref. 6), there was not a direct correlation between detectable Wnt secretion and signaling activity because even 1 ng of PORCN-encoding plasmid (per well in a 24-well culture dish, typical of assays and results to follow) could robustly rescue STF activity but was not sufficient to produce immunologically detectable WNT3A in the conditioned medium (Fig. 2D). This reinforces the conclusion that the detection of free Wnt in culture medium is an insensitive measure of active Wnt. We conclude that mutation or skipping of PORCN exon 9 creates cells null for PORCN catalytic activity and that active PORCN is absolutely essential for production of active Wnts in HT1080 cells.
Stringent Control of Wnt Signaling by PORCN
PORCN expression and splicing patterns vary widely between tissues, during development (26, 29, 30), and in some disease states (36, 41). This suggests that PORCN expression regulates the strength of Wnt signaling independent of Wnt gene expression. To test this experimentally, a saturating amount of WNT3A plasmid was co-transfected with increasing amounts of PORCN plasmid into PORCN null cells. Impressively, as little as 1 pg of PORCN plasmid (mixed with carrier plasmid) was sufficient to restore detectable expression from the STF reporter. This single pg of PORCN plasmid, transfected into a 24-well dish containing ∼150,000 cells, is calculated to be about one expression plasmid per cell. The Wnt/β-catenin signal output varied linearly with PORCN expression over 3 orders of magnitude (Fig. 3A). This broad dynamic response to PORCN indicates that tissues could finely modulate Wnt signaling by varying PORCN activity. Analysis of the dynamic range of signal mediated by Wnt at a saturating concentration of PORCN revealed, as expected, that levels of the Wnt also produce a strong control over resulting signal. To test this further, we examined the relative contributions of PORCN and Wnt expression to overall signal output. Between 400 pg and 50 ng of WNT3A expression plasmid was transfected in different combinations with between 100 pg and 3 ng of PORCN plasmid. Fig. 3B shows that each of these variables has a similar amount of control over signal output. This exquisite control of Wnt signaling is not simply a function of the synthetic β-catenin-responsive STF reporter because the natural target gene AXIN2 demonstrates a similar dose-response curve, albeit we are comparing mRNA in one case and a reporter enzyme in another (Fig. 3C). Thus, fine tuning of both high and moderate response Wnt/β-catenin target genes can be similarly modulated by the abundance of both Wnt ligand and its activator, PORCN. This strongly suggests that the strength of Wnt signaling in development and disease will be critically dependent on both PORCN and Wnt protein abundance.
FIGURE 3.
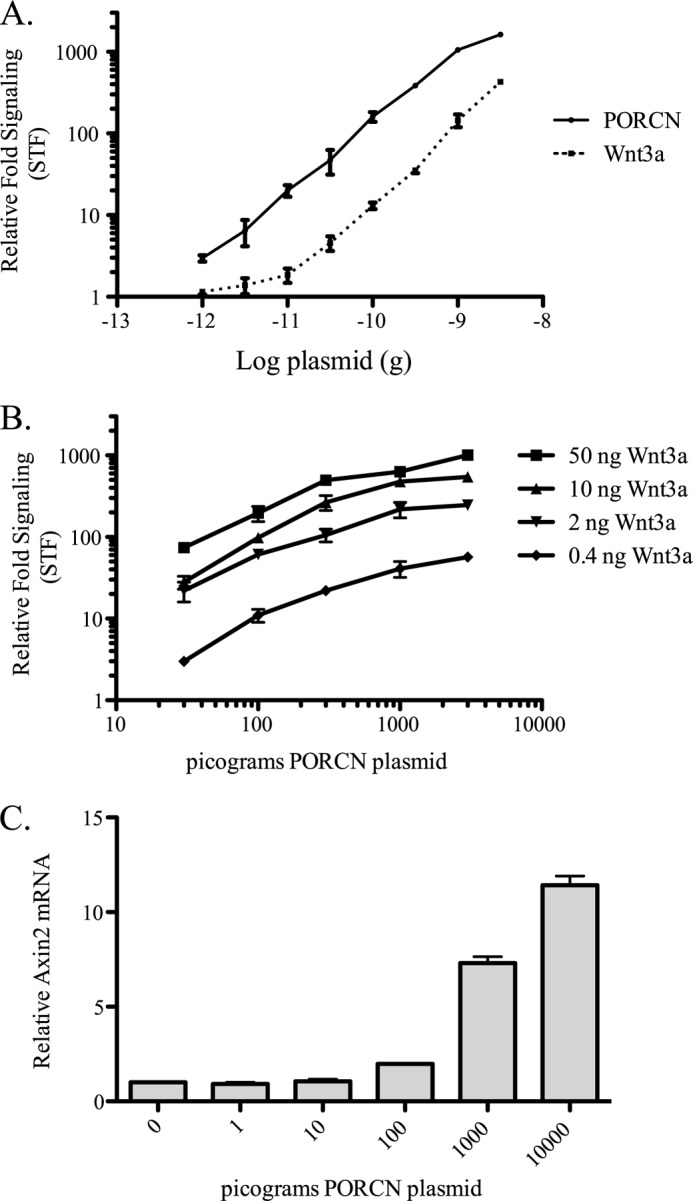
PORCN governs a broad dynamic range of Wnt signaling. A, in a 24-well dish, Δ19 cells were transfected with WNT3A, SuperTOPFlash, mCherry transfection control, and amounts of 3xHA mPORCN-D plasmid as indicated. When titrating PORCN, 50 ng of WNT3A was used; when titrating WNT3A, 3 ng of PORCN was used. The luciferase assay was performed 24 h after transfection. B, same as A, except with variable Wnt expression. C, Δ19 cells were treated as in A, without transfection of STF. Instead, the natural reporter gene AXIN2 was measured by quantitative RT-PCR. 24 h after transfection, RNA was isolated and probed for AXIN2, using actin (ACTB) as control. Error bars, S.D.
Analysis of Conserved Amino Acids in PORCN and Influence on Activity
Sequence conservation in PORCN suggests several amino acid residues that are likely to be crucial to its acyltransferase activity (24). The MBOAT family contains highly conserved residues, including Asn-306, Ser-337, and His-341 of PORCN-D. In the MBOAT protein ghrelin O-acyltransferase, which transfers octanoate onto the appetite-stimulating hormone ghrelin, the asparagine and histidine equivalent to PORCN Asn-306 and His-341 are absolutely required for enzyme activity (42). PORCN is predicted to palmitoylate Wnt either co-translationally or post-translationally, so that in either case, the catalytic site is likely to be in the membrane or in the lumen of the ER. However, in most models of PORCN topology, Asn-306 and Ser-337 are predicted to be in the cytoplasm, whereas most FDH point missense mutants are predicted to be in the lumen of the ER (Fig. 4). Thus, it was important to test if Asn-306 and Ser-337 are indeed critical to PORCN function in the same manner in which His-341 demonstrably is. In our assay, unlike their role in ghrelin O-acyltransferase, the N306A and S337A PORCN mutants were virtually identical to wild-type PORCN in their ability to restore Wnt/β-catenin activity to PORCN null HT1080 cells (Fig. 5A). These mutants were fully active with several Wnts regardless of whether they were β-catenin-activating (Fig. 5, B and D). Confirming that their activity was not an artifact of the HT1080 cell line, these mutants also fully restored Wnt/β-catenin signaling activity in MEFs deleted for PORCN (Fig. 5C). This was not an artifact of mammalian PORCN because independent mutation of the Asn-306 and Ser-337 sites in Xenopus PORCN had no discernible effect on activity (Fig. 5E). Thus, these highly conserved, putative cytoplasmic loop residues may play an evolutionarily important role distinct from direct involvement in Wnt acyltransferase activity.
FIGURE 4.

Schematic representation of PORCN structure and topology model. FDH mutations studied in this report are highlighted in yellow, red, and green, whereas the highly conserved residues Asn-306 and Ser-337 are highlighted in white on black. Splice variants resulting from selective inclusion of exons 7 and 8 are indicated by lines between amino acids, and resulting different protein sequences are shown below. Exon 9 is highlighted in light blue, with the ZFN binding site indicated. Colored amino acid residues indicate mutations associated with FDH. Colors indicate classification: low protein expression (yellow), loss of activity and near normal expression (red), and normal activity and expression at higher transfection levels (green). The N terminus is at the left, and the predicted ER lumenal side is below. Only missense mutations are indicated here. We have borrowed heavily from a previous publication in constructing this model (44).
FIGURE 5.

Highly conserved residues Asn-306 and Ser-337 of PORCN are not required for Wnt activation, and PORCN splice variants exert subtle control of activation. A, Wnt/β-catenin reporter (STF) assay assessing PORCN mutants N306A and S337A relative to WT. Δ19 cells were transfected with the following plasmids: 50 ng of WNT3A expression, 100 ng of mCherry expression, 550 ng of SuperTOPFlash reporter, and wild-type or mutant 3xHA-mPORCN-D expression plasmids as indicated. B, STF assay of N306A and WT PORCN plasmids (300 pg transfected) comparing multiple Wnts as indicated. C, PORCN null mouse embryonic fibroblasts were used to confirm results for N306A mutant PORCN. MEFs were nucleofected with plasmids as indicated under “Experimental Procedures.” 24 h after transfection, luciferase activity was measured. This assay was performed with multiple replicates twice with similar results. D, conserved PORCN residues are not required for non-canonical Wnt signaling to Dvl2. The indicated PORCN expression constructs were co-expressed in PORCN null HT1080 cells with WNT4, and Dvl2 mobility shift was subsequently assessed (see “Experimental Procedures”). E, conserved residues in Xenopus PORCN equivalent to Asn-306 and Ser-337 are not required for Wnt/β-catenin signaling. The STF assay was performed in Δ19 cells transfected with 50 ng of WNT3A, 100 ng of mCherry, 650 ng of STF reporter, and the indicated 500 pg of Xenopus PORCN-A expression constructs. F, the STF assay was performed after transfection of 50 ng of the indicated Wnt, in combination with 300 pg each of 3xHA-mPORCN splice variants A–D as indicated. Data are presented as percentage of maximal signal obtained for each Wnt (the PORCN isoform producing maximal signal varies). Fold activity is dependent on Wnt transfected. Maximal signals were as follows: WNT1, 122×; WNT2, 24×; WNT3, 302×; WNT3A, 147×; WNT6, 21×; WNT7A, 3×; WNT7B, 4×; WNT9A, 5×; WNT9B, 10×; WNT10B, 52×. Expression of isoform mutants from a separate experiment with 100 ng of transfection/well is shown below (duplicates). Error bars, S.D.
PORCN Splice Variants Differ Globally in Signaling Activity
PORCN exists as four different isoforms due to the alternative splicing of exons 7 and 8. This results in splice variants B, C, and D with the addition of 7, 5, or 12 amino acids with net positive charge not present in variant A (six of the 12 added residues are Arg or Lys). This charged patch is predicted to reside in the ER lumen (Fig. 4). Tissue-specific expression patterns of these isoforms have been reported (30). One possibility is that the isoforms allow selectivity for specific Wnts; alternatively, they could globally modulate activity, or they could confer sensitivity to different regulatory pathways. To differentiate between these options, we tested the ability of each splice variant to restore Wnt/β-catenin signaling activity in the PORCN null HT1080 cells. There is no detectable signaling activity for any of these Wnts in the absence of PORCN (38). As Fig. 5F shows, the different splice variants globally affect Wnt ligand activity and show more subtle differences between Wnts. For example, isoform B has a reduced capacity to signal relative to isoforms C and D for WNT1, -2, and -3. Most notably, isoform A (lacking both exons 7 and 8) is approximately half as active as the other splice variants. This suggests that the basic region missing in isoform A globally up-regulates PORCN enzymatic activity. Because isoform A is the predominant variant expressed in kidney, spleen, uterus, testis, and corpus callosum (30), these tissues may down-regulate global Wnt signaling by alternative splicing of PORCN mRNA.
Analysis of PORCN Mutations Associated with FDH
Inherited and postzygotic mutations in the PORCN gene cause the disorder focal dermal hypoplasia (33, 34). Since the initial association was discovered, over 100 mutations have been identified (34, 35). It is presumed but not yet demonstrated that all of these mutations cause disease through a loss of PORCN enzymatic function and hence loss of Wnt signaling. Consistent with this, several cases of FDH are associated with either large deletions in the PORCN gene or early nonsense mutations predicted to entirely eliminate PORCN expression. However, in just over 20 phenotypically similar patients, the disease is associated with missense or small in-frame deletions in PORCN coding sequence. Whether and how these mutations affect PORCN function is less clear. In order to address this experimentally, we recreated 18 of the disease-associated point mutations and small deletions within the coding sequence of a 3xHA-mPORC-D plasmid, highlighted in Fig. 4. The point mutations associated with PORCN are largely predicted to exist in intramembrane space or aligned on the same predicted side of the ER membrane. As such, the FDH mutation-prevalent side of PORCN is predicted to be in the ER lumen ((44) (Fig. 4).
Knowing that the PORCN expression level would have a significant effect on overall signal, we tested the ability of WNT3A to activate STF signaling in the absence or presence of 300 pg of PORCN plasmid, with the goal of measuring activity in the middle of dynamic range. Due to the limits of antibody sensitivity, we measured protein abundance of the mutants by transfecting 100 ng of plasmid encoding the PORCN mutants (Fig. 6A). We find that FDH mutants fall into three categories (Table 1). Class 1 mutants show low expression and low activity, indicating that these mutations affect protein stability. Five of 18 mutants tested fall into this category. Class 2 mutants (5 of 18 tested) are well expressed but have low specific activity (<50% of wild type). We presume that these mutants affect enzyme function. Consistent with this, the FDH mutant H341Y, affecting the putative active site residue, is in this class. A third, unexpectedly common class of mutants (8 of 18) have both significant Wnt/β-catenin signaling activity (>50% of wild type) and detectable PORCN protein expression. Consistent with our work, the S136F mutant was recently reported to also have significant activity, using a different assay method (28). Remarkably, patients with these high activity disease-associated mutations appear to be phenotypically indistinguishable from those with null mutations (44–47).
FIGURE 6.

Focal dermal hypoplasia-associated PORCN mutations disrupt activity. A, SuperTOPFlash assay of the ability of different PORCN mutants to rescue signaling mediated by WNT3A. Top, all wells receive 50 ng of WNT3A, 100 ng of mCherry, 300 pg of the 3xHA-mPORCN-D variant indicated, and 650 ng of SuperTOPFlash reporter. Bottom, for analysis of expression levels, all PORCN variants were transfected in a separate experiment at 100 ng. B, for select FDH mutant PORCN variants, a TOPFlash assay was performed while titrating down the total amount of PORCN from 100 ng to 100 pg. Results are displayed as a percentage of WT signaling activity at the corresponding equivalent transfection quantity. Δ12 cells were used in this experiment. C, STF assay was performed in Δ19 cells transfected with 50 ng of Wnt indicated, along with 100 ng of mCherry, 650 ng of STF reporter, and 300 pg of the 3xHA-mPORCN-D variant indicated. Results are displayed as a percentage of the signal obtained from indicated Wnt when co-transfected with WT PORCN. D, Dvl2 mobility shift induced by WNT4. The experiment was done at the same time as that in Fig. 5D, and hence the WT and EV controls are shown again here for clarity. Error bars, S.D.
TABLE 1.
Categories of FDH mutants
| Class 1: Low expression (presumed protein stability mutant) | Class 2: Expression but low activity (mutations primarily affect MBOAT activity) | Class 3: Nearly normal specific activity (reference) |
|---|---|---|
| G60R | V258E | S136F (44) |
| G168R | S297L | L331R (46) |
| Δ349–355 | H341Y | L347P (45) |
| Δ355–360 | W439X | E361V (47) |
| R365Q | W448X | A369V (45) |
| A374P (45) | ||
| C385R (44) | ||
| W439R (44) |
We tested if the class 3 mutants might be less active at very low but still physiologically relevant PORCN expression levels. As shown in Fig. 6B, this is what we observed; reduced amounts of class 3 FDH mutant PORCN had disproportionately reduced activity compared with the equivalent amount of wild-type PORCN plasmid. For instance, the mutant V258E had ∼50% WT activity when 100 ng of plasmid was transfected, but the activity quickly fell to <10% of WT when just 300 pg was transfected. PORCN with L331R, L347P, or A369V mutations surprisingly had near wild-type activity at high expression levels, but the relative activity was reduced when the more physiologically relevant 100 pg PORCN plasmid was used. Interestingly, for several of these disease mutations, the reductions in activity are similar to those seen when splice variant A is compared with splice variants B–D. These results were generalizable to other Wnts because we found that an active (L331R) and inactive (V258E) FDH mutant behaved consistently with five additional Wnts (Fig. 6C). Finally, we found that the S136F and L331R mutants are also capable of activating β-catenin-independent signaling, whereas the H341Y mutant is not (Fig. 6D).
We wished to ascertain if our picogram-level transfections produced physiologically relevant levels of PORCN mRNA. We recognize that this is an imperfect measure because the PORCN expression construct does not have native 5′- and 3′-UTRs and will vary in translation efficiency. By performing quantitative RT-PCR to compare absolute PORCN mRNA abundance in a panel of human tissue samples, in HT1080 cells, and exogenously expressed in HT1080 cells by transient transfections, we calculate that 2.5 ng of transfected PORCN expression plasmid results in approximately endogenous mRNA levels in HT1080 cells. (Fig. 7 and supplemental Fig. 1). HT1080 cells have relatively high expression of PORCN mRNA compared with many of the tissues analyzed. Thus, transfection of lower quantities of PORCN (i.e. in the 100 pg range) is likely to reflect PORCN abundance in many physiological settings. Because several FDH mutants (S136F, L331R, and L347P) have nearly 50% of wild-type activity when 100 pg of expression vector is transfected, our data strongly suggest that the complete FDH phenotype can be caused by as little as a 50% decrease in PORCN activity.
FIGURE 7.

PORCN expression levels vary across human tissues, and HT1080 cells have high expression. A human tissue mRNA panel was used to assess relative levels of PORCN message in HT1080 cells related to these various tissues. PORCN mRNA expression is plotted normalized to input RNA quantity (open bars) or normalized to a combination of GUSB and PLA2G4A abundance (gray bars). Error bars, S.D.
DISCUSSION
In this study, we confirm that PORCN has the non-redundant function of regulating human Wnt activation and secretion. We show using both synthetic and endogenous reporters that Wnt signaling can be robustly and dynamically regulated by PORCN gene expression. We confirm that all known missense and in-frame deletions in the PORCN gene found in the disease FDH alter PORCN function in Wnt/β-catenin signaling. Strikingly, one common class of mutations causes only 2-fold reductions in Wnt/β-catenin signaling and then only at low but physiologically relevant levels of gene expression. Notably, these reductions are similar to those seen comparing normal PORCN splice variants. These findings indicate that subtle alterations in PORCN activity and hence Wnt activation are sufficient to dysregulate development in sensitive tissues, specifically in the mesoderm-derived tissues that are most affected in FDH.
Here we have used zinc finger nuclease technology to create PORCN null cells. By targeting a double-stranded break to the PORCN gene in this manner, we were able to rapidly create two independent clones of HT1080 cells with unique small deletions ultimately disrupting PORCN activity. This validated zinc finger nuclease is now available to create similar deletions in other human cell types. Our results, combined with those in a recent paper (38) show definitively and for all Wnts tested (both β-catenin-stabilizing and “non-canonical”) that PORCN is absolutely required for their signaling activity. Previous studies have come to the same conclusions in a more limited fashion, using siRNA (48), small molecule inhibitors (31), gene trap and assay in embryonic stem cells (26), or loxP targeting and assay with ES cells or MEFs (27). Some reports have indicated that Wnts can be secreted without PORCN activity. One recent study defines a novel β-catenin-independent lengthening of cells that occurs even in the presence of PORCN inhibition or when using a Ser-224 mutant of WNT1 that cannot be palmitoylated, conditions under which Wnt secretion into culture medium is observed (49). Yet another report indicated that WNT3A could be easily detected in medium, whereas free extracellular WNT1 was undetectable, despite having similar or better signaling capacity (50). Instead, the authors observed WNT1 associated with an extracellular matrix. We find that there is a non-linear relationship between Wnt that is easily detected in culture medium compared with an amount sufficient to activate STF signaling; even when Wnt is not detectable in medium, there can be enough active Wnt to produce very robust β-catenin signaling activity. Clearly, there are experimental differences that affect Wnt secretion. One difference between our results, where no Wnt is observed in culture medium in the absence of PORCN, and those results indicating PORCN-independent Wnt secretion may be the quantity of plasmids transfected. In our hands, 1–10 ng of WNT3A plasmid saturates signaling to the STF reporter, whereas other studies often use 1 μg or more of Wnt expression plasmid, possibly allowing Wnt secretion through other lower affinity pathways. In addition, other cell types (e.g. neuron-derived HEK293 cells) may release non-palmitoylated Wnts more readily. This relationship between Wnt palmitoylation, general secretion, and ultimate signaling activity is complex and nuanced, and as such, we would stress caution in overreliance on Wnt secretion as an assay method. Most importantly, the activity that we measure in the form of STF activity, AXIN2 mRNA level, and Dvl2 phosphorylation absolutely depends on PORCN expression. This is entirely consistent with both our observation that palymitoylation is required for all Wnts to bind to WLS (19, 38) and the insight from the co-crystal structure that the palmitoyl moiety is a major contact between Wnt and Frizzled (21).
We made the unexpected observation that the MBOAT highly conserved residues Asn-306 and Ser-337 are not required for PORCN activity on Wnts and validated this result using both MEF cells with deletion of PORCN exon 3, and Xenopus PORCN. The Asn-306 site in particular is completely conserved in PORCN, going back to the primitive eumetazoan Trichoplax and its PORCN-like protein (51). That mutation of this residue does not affect Wnt/β-catenin signaling indicates additional complexities to PORCN structure and function that are not yet understood. A separate function of PORCN has been described that promotes cancer cell proliferation independently of acyltransferase activity because a catalytic mutant PORCN (H341A) was able to rescue siRNA-mediated cell growth inhibition in breast cancer cells (36). Additionally, PORCN is most closely related to acyltransferases that act on lysophospholipid species (52) and was originally suggested to not modify Wnt directly but rather an associated sugar due to this homology (24). PORCN may retain acyltransferase activity toward lipids or other proteins not detected in our current assays. Thus, identifying additional PORCN functions is an area of active investigation.
Our finding that PORCN can rescue Wnt signaling capacity at very low concentrations and then provide a linear relationship between expression level and STF signal capacity over at least 3 orders of magnitude provides several insights. First, PORCN represents a critical and non-redundant node for the control of Wnt signaling; second, due to this apparent importance, it is likely that there are control mechanisms in place to regulate PORCN activity, such as post-translational modification, subcellular localization, and degradation pathways. This finding also provides support to the notion that small molecule targeting of PORCN would be an attractive means to disrupt all Wnt signaling in various Wnt-associated disease conditions. It will be interesting to learn, however, whether treatment of Wnt-high disorders with PORCN inhibitors could maintain a therapeutic window, considering the role that Wnt/β-catenin signaling plays in adult stem cell homeostasis (43).
Our data show that FDH with a typical phenotype can be due to mutations that produce only a 50% decrease in PORCN activity. In the setting of development, where all 19 Wnts may be expressed and multiple Wnts can be expressed from a single cell, this modest decrease in signal evidently leads to dramatic effects on a subset of tissues responding to complex morphogen gradients. The defects associated with FDH originate in this crucial developmental period, and our results indicate that mutations such as L331R, with a 50% decrease in activity, have phenotypic effects equivalent to those of more clearly deleterious mutations like H341Y (45, 46). This is consistent with the concept that 50% changes in morphogen gradients are biologically critical in the developing organism. However, the degree of severity associated with FDH is also a function of X chromosome inactivation, making such direct comparisons more complicated. We recently reported that PORCN has additional functions that require PORCN protein but not PORCN enzyme activity (36). Hence, some manifestations of FDH could result from Wnt-independent pathways. Although the many early termination mutations associated with FDH may be compounded by this non-Wnt function for PORCN, it is less likely that the point mutants discussed here suffer these additional disruptions because catalytically inactive PORCN maintained this moonlighting function. Still, it serves as a reminder that many proteins have more than one function. We are also aware that we have relied on WNT3A-initiated β-catenin signaling as our primary measure of FDH mutant PORCN activity; it remains possible that these mutations could have more significant effects when considering all of the potential Wnts and pathways, both canonical and non-canonical.
In summary, we have used PORCN null cells in order to study the effects of PORCN mutation on Wnt signaling capacity. In so doing, we have uncovered exquisite control of Wnt signaling through this acyltransferase. Further, we have used these null cells to confirm that mutations associated with FDH have reduced activity compared with normal, consistent with the conclusion that FDH is a disease of Wnt malfunction. These null cells will be useful for further study of mutational effects on activity and will help increase understanding of PORCN function.
Acknowledgments
We thank Dr. Janet Rossant and Steffen Biechele for providing PORCNflox/flox mice, Dr. Zahra Kabiri for mouse maintenance and breeding, and Dr. Jitkong Cheong for assistance in acquiring and culturing MEFs. We thank Simran Kaur, Velani Utomo, Hayin Lee, and Chun Wei Chan for technical assistance. We are also grateful to Dr. Gary Coombs and Dr. Tracy Covey, who set the stage for this work and provided invaluable advice.
This work was supported by the Duke-NUS Signature Research Program funded by the Agency for Science, Technology, and Research, Singapore, and the Ministry of Health, Singapore, and a Singapore Translational Research Investigator Award (to D. M. V.) funded by the National Research Foundation and the National Medical Research Council.

This article contains supplemental Fig. 1.
- ER
- endoplasmic reticulum
- FDH
- focal dermal hypoplasia
- ZFN
- zinc finger nuclease
- MBOAT
- membrane-bound O-acyltransferase
- MEF
- mouse embryo fibroblast
- STF
- SuperTOPFlash.
REFERENCES
- 1. MacDonald B. T., Tamai K., He X. (2009) Wnt/beta-catenin signaling. Components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Williams B. O., Insogna K. L. (2009) Where Wnts went. The exploding field of Lrp5 and Lrp6 signaling in bone. J. Bone Miner. Res. 24, 171–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Polakis P. (2007) The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 17, 45–51 [DOI] [PubMed] [Google Scholar]
- 4. Chen Y., Hu Y., Zhou T., Zhou K. K., Mott R., Wu M., Boulton M., Lyons T. J., Gao G., Ma J. X. (2009) Activation of the Wnt pathway plays a pathogenic role in diabetic retinopathy in humans and animal models. Am. J. Pathol. 175, 2676–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lam A. P., Gottardi C. J. (2011) β-Catenin signaling. A novel mediator of fibrosis and potential therapeutic target. Curr. Opin. Rheumatol. 23, 562–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zecca M., Basler K., Struhl G. (1996) Direct and long range action of a wingless morphogen gradient. Cell 87, 833–844 [DOI] [PubMed] [Google Scholar]
- 7. Gao B., Song H., Bishop K., Elliot G., Garrett L., English M. A., Andre P., Robinson J., Sood R., Minami Y., Economides A. N., Yang Y. (2011) Wnt signaling gradients establish planar cell polarity by inducing Vangl2 phosphorylation through Ror2. Dev. Cell 20, 163–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bartscherer K., Boutros M. (2008) Regulation of Wnt protein secretion and its role in gradient formation. EMBO Rep. 9, 977–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harterink M., Korswagen H. C. (2012) Dissecting the Wnt secretion pathway. Key questions on the modification and intracellular trafficking of Wnt proteins. Acta. Physiol. 204, 8–16 [DOI] [PubMed] [Google Scholar]
- 10. Port F., Hausmann G., Basler K. (2011) A genome-wide RNA interference screen uncovers two p24 proteins as regulators of Wingless secretion. EMBO Rep. 12, 1144–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buechling T., Chaudhary V., Spirohn K., Weiss M., Boutros M. (2011) p24 proteins are required for secretion of Wnt ligands. EMBO Rep. 12, 1265–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bänziger C., Soldini D., Schütt C., Zipperlen P., Hausmann G., Basler K. (2006) Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell 125, 509–522 [DOI] [PubMed] [Google Scholar]
- 13. Bartscherer K., Pelte N., Ingelfinger D., Boutros M. (2006) Secretion of Wnt ligands requires Evi, a conserved transmembrane protein. Cell 125, 523–533 [DOI] [PubMed] [Google Scholar]
- 14. Goodman R. M., Thombre S., Firtina Z., Gray D., Betts D., Roebuck J., Spana E. P., Selva E. M. (2006) Sprinter. A novel transmembrane protein required for Wg secretion and signaling. Development 133, 4901–4911 [DOI] [PubMed] [Google Scholar]
- 15. Harterink M., Port F., Lorenowicz M. J., McGough I. J., Silhankova M., Betist M. C., van Weering J. R., van Heesbeen R. G., Middelkoop T. C., Basler K., Cullen P. J., Korswagen H. C. (2011) A SNX3-dependent retromer pathway mediates retrograde transport of the Wnt sorting receptor Wntless and is required for Wnt secretion. Nat. Cell Biol. 13, 914–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kadowaki T., Wilder E., Klingensmith J., Zachary K., Perrimon N. (1996) The segment polarity gene porcupine encodes a putative multitransmembrane protein involved in Wingless processing. Genes Dev. 10, 3116–3128 [DOI] [PubMed] [Google Scholar]
- 17. Zhai L., Chaturvedi D., Cumberledge S. (2004) Drosophila wnt-1 undergoes a hydrophobic modification and is targeted to lipid rafts, a process that requires porcupine. J. Biol. Chem. 279, 33220–33227 [DOI] [PubMed] [Google Scholar]
- 18. Takada R., Satomi Y., Kurata T., Ueno N., Norioka S., Kondoh H., Takao T., Takada S. (2006) Monounsaturated fatty acid modification of Wnt protein. Its role in Wnt secretion. Dev Cell 11, 791–801 [DOI] [PubMed] [Google Scholar]
- 19. Coombs G. S., Yu J., Canning C. A., Veltri C. A., Covey T. M., Cheong J. K., Utomo V., Banerjee N., Zhang Z. H., Jadulco R. C., Concepcion G. P., Bugni T. S., Harper M. K., Mihalek I., Jones C. M., Ireland C. M., Virshup D. M. (2010) WLS-dependent secretion of WNT3A requires Ser-209 acylation and vacuolar acidification. J. Cell Sci. 123, 3357–3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herr P., Basler K. (2012) Porcupine-mediated lipidation is required for Wnt recognition by Wls. Dev. Biol. 361, 392–402 [DOI] [PubMed] [Google Scholar]
- 21. Janda C. Y., Waghray D., Levin A. M., Thomas C., Garcia K. C. (2012) Structural basis of Wnt recognition by Frizzled. Science 337, 59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Willert K., Brown J. D., Danenberg E., Duncan A. W., Weissman I. L., Reya T., Yates J. R., 3rd, Nusse R. (2003) Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 423, 448–452 [DOI] [PubMed] [Google Scholar]
- 23. Kurayoshi M., Yamamoto H., Izumi S., Kikuchi A. (2007) Post-translational palmitoylation and glycosylation of Wnt-5a are necessary for its signaling. Biochem. J. 402, 515–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hofmann K. (2000) A superfamily of membrane-bound O-acyltransferases with implications for wnt signaling. Trends Biochem. Sci. 25, 111–112 [DOI] [PubMed] [Google Scholar]
- 25. Chen Q., Takada R., Takada S. (2012) Loss of Porcupine impairs convergent extension during gastrulation in zebrafish. J. Cell Sci. 125, 2224–2234 [DOI] [PubMed] [Google Scholar]
- 26. Biechele S., Cox B. J., Rossant J. (2011) Porcupine homolog is required for canonical Wnt signaling and gastrulation in mouse embryos. Dev. Biol. 355, 275–285 [DOI] [PubMed] [Google Scholar]
- 27. Barrott J. J., Cash G. M., Smith A. P., Barrow J. R., Murtaugh L. C. (2011) Deletion of mouse Porcn blocks Wnt ligand secretion and reveals an ectodermal etiology of human focal dermal hypoplasia/Goltz syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 12752–12757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu W., Shaver T. M., Balasa A., Ljungberg M. C., Wang X., Wen S., Nguyen H., Van den Veyver I. B. (2012) Deletion of Porcn in mice leads to multiple developmental defects and models human focal dermal hypoplasia (Goltz syndrome). PLoS ONE 7, e32331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tanaka K., Okabayashi K., Asashima M., Perrimon N., Kadowaki T. (2000) The evolutionarily conserved porcupine gene family is involved in the processing of the Wnt family. Eur. J. Biochem. 267, 4300–4311 [DOI] [PubMed] [Google Scholar]
- 30. Caricasole A., Ferraro T., Rimland J. M., Terstappen G. C. (2002) Molecular cloning and initial characterization of the MG61/PORC gene, the human homologue of the Drosophila segment polarity gene Porcupine. Gene 288, 147–157 [DOI] [PubMed] [Google Scholar]
- 31. Chen B., Dodge M. E., Tang W., Lu J., Ma Z., Fan C. W., Wei S., Hao W., Kilgore J., Williams N. S., Roth M. G., Amatruda J. F., Chen C., Lum L. (2009) Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 5, 100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sato T., Vries R. G., Snippert H. J., van de Wetering M., Barker N., Stange D. E., van Es J. H., Abo A., Kujala P., Peters P. J., Clevers H. (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 [DOI] [PubMed] [Google Scholar]
- 33. Grzeschik K. H., Bornholdt D., Oeffner F., König A., del Carmen Boente M., Enders H., Fritz B., Hertl M., Grasshoff U., Höfling K., Oji V., Paradisi M., Schuchardt C., Szalai Z., Tadini G., Traupe H., Happle R. (2007) Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat. Genet. 39, 833–835 [DOI] [PubMed] [Google Scholar]
- 34. Wang X., Reid Sutton V., Omar Peraza-Llanes J., Yu Z., Rosetta R., Kou Y. C., Eble T. N., Patel A., Thaller C., Fang P., Van den Veyver I. B. (2007) Mutations in X-linked PORCN, a putative regulator of Wnt signaling, cause focal dermal hypoplasia. Nat. Genet. 39, 836–838 [DOI] [PubMed] [Google Scholar]
- 35. Lombardi M. P., Bulk S., Celli J., Lampe A., Gabbett M. T., Ousager L. B., van der Smagt J. J., Soller M., Stattin E. L., Mannens M. A., Smigiel R., Hennekam R. C. (2011) Mutation update for the PORCN gene. Hum. Mutat. 32, 723–728 [DOI] [PubMed] [Google Scholar]
- 36. Covey T. M., Kaur S., Tan Ong T., Proffitt K. D., Wu Y., Tan P., Virshup D. M. (2012) PORCN moonlights in a Wnt-independent pathway that regulates cancer cell proliferation. PLoS ONE 7, e34532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Veeman M. T., Slusarski D. C., Kaykas A., Louie S. H., Moon R. T. (2003) Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr. Biol. 13, 680–685 [DOI] [PubMed] [Google Scholar]
- 38. Najdi R., Proffitt K., Sprowl S., Kaur S., Yu J., Covey T. M., Virshup D. M., Waterman M. L. (2012) A uniform human Wnt expression library reveals a shared secretory pathway and unique signaling activities. Differentiation 84, 203–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guo Y., Xie J., Rubin E., Tang Y. X., Lin F., Zi X., Hoang B. H. (2008) Frzb, a secreted Wnt antagonist, decreases growth and invasiveness of fibrosarcoma cells associated with inhibition of Met signaling. Cancer Res. 68, 3350–3360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vijayakumar S., Liu G., Rus I. A., Yao S., Chen Y., Akiri G., Grumolato L., Aaronson S. A. (2011) High frequency canonical Wnt activation in multiple sarcoma subtypes drives proliferation through a TCF/β-catenin target gene, CDC25A. Cancer Cell 19, 601–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen Z., Li J., Li Q. S., Fan J. Q., Dong X. M., Xu J. P., Wang X. M., Yang G. W., Yan P., Wen G. Z., Zhang Y. T., Niu R. G., Nan P. H., He J., Zhou H. M. (2008) Suppression of PPN/MG61 attenuates Wnt/β-catenin signaling pathway and induces apoptosis in human lung cancer. Oncogene 27, 3483–3488 [DOI] [PubMed] [Google Scholar]
- 42. Yang J., Brown M. S., Liang G., Grishin N. V., Goldstein J. L. (2008) Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 132, 387–396 [DOI] [PubMed] [Google Scholar]
- 43. Barker N., Bartfeld S., Clevers H. (2010) Tissue-resident adult stem cell populations of rapidly self-renewing organs. Cell Stem Cell 7, 656–670 [DOI] [PubMed] [Google Scholar]
- 44. Bornholdt D., Oeffner F., König A., Happle R., Alanay Y., Ascherman J., Benke P. J., Boente Mdel C., van der Burgt I., Chassaing N., Ellis I., Francisco C. R., Della Giovanna P., Hamel B., Has C., Heinelt K., Janecke A., Kastrup W., Loeys B., Lohrisch I., Marcelis C., Mehraein Y., Nicolas M. E., Pagliarini D., Paradisi M., Patrizi A., Piccione M., Piza-Katzer H., Prager B., Prescott K., Strien J., Utine G. E., Zeller M. S., Grzeschik K. H. (2009) PORCN mutations in focal dermal hypoplasia. Coping with lethality. Hum. Mutat. 30, E618–E628 [DOI] [PubMed] [Google Scholar]
- 45. Fernandes P. H., Wen S., Sutton V. R., Ward P. A., Van den Veyver I. B., Fang P. (2010) PORCN mutations and variants identified in patients with focal dermal hypoplasia through diagnostic gene sequencing. Genet. Test Mol. Biomarkers 14, 709–713 [DOI] [PubMed] [Google Scholar]
- 46. Froyen G., Govaerts K., Van Esch H., Verbeeck J., Tuomi M. L., Heikkilä H., Torniainen S., Devriendt K., Fryns J. P., Marynen P., Järvelä I., Ala-Mello S. (2009) Novel PORCN mutations in focal dermal hypoplasia. Clin. Genet. 76, 535–543 [DOI] [PubMed] [Google Scholar]
- 47. Harmsen M. B., Azzarello-Burri S., García González M. M., Gillessen-Kaesbach G., Meinecke P., Müller D., Rauch A., Rossier E., Seemanova E., Spaich C., Steiner B., Wieczorek D., Zenker M., Kutsche K. (2009) Goltz-Gorlin (focal dermal hypoplasia) and the microphthalmia with linear skin defects (MLS) syndrome. No evidence of genetic overlap. Eur. J. Hum. Genet. 17, 1207–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Galli L. M., Barnes T. L., Secrest S. S., Kadowaki T., Burrus L. W. (2007) Porcupine-mediated lipid modification regulates the activity and distribution of Wnt proteins in the chick neural tube. Development 134, 3339–3348 [DOI] [PubMed] [Google Scholar]
- 49. Galli L. M., Burrus L. W. (2011) Differential palmit(e)oylation of Wnt1 on Cys-93 and Ser-224 residues has overlapping and distinct consequences. PLoS ONE 6, e26636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Doubravska L., Krausova M., Gradl D., Vojtechova M., Tumova L., Lukas J., Valenta T., Pospichalova V., Fafilek B., Plachy J., Sebesta O., Korinek V. (2011) Fatty acid modification of Wnt1 and Wnt3a at serine is prerequisite for lipidation at cysteine and is essential for Wnt signaling. Cell. Signal. 23, 837–848 [DOI] [PubMed] [Google Scholar]
- 51. Srivastava M., Begovic E., Chapman J., Putnam N. H., Hellsten U., Kawashima T., Kuo A., Mitros T., Salamov A., Carpenter M. L., Signorovitch A. Y., Moreno M. A., Kamm K., Grimwood J., Schmutz J., Shapiro H., Grigoriev I. V., Buss L. W., Schierwater B., Dellaporta S. L., Rokhsar D. S. (2008) The Trichoplax genome and the nature of placozoans. Nature 454, 955–960 [DOI] [PubMed] [Google Scholar]
- 52. Hishikawa D., Shindou H., Kobayashi S., Nakanishi H., Taguchi R., Shimizu T. (2008) Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc. Natl. Acad. Sci. U.S.A. 105, 2830–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]