

Background: Heme bound to the heme degrading enzyme IsdI is distorted in the form of ruffling.
Results: IsdI Trp66 variants are less catalytically active, and heme bound to the W66Y variant is less distorted.
Conclusion: Extensive heme ruffling is required for optimal catalytic activity of IsdI.
Significance: The contribution of heme ruffling to catalysis of heme oxidation is a feature that distinguishes IsdI-like enzymes from heme oxygenase.
Keywords: Electrochemistry, Enzyme Catalysis, Heme, Heme Oxygenase, Site-directed Mutagenesis, X-ray Crystallography
Abstract
IsdG and IsdI are paralogous heme degrading enzymes from the bacterium Staphylococcus aureus. Heme bound by these enzymes is extensively ruffled such that the meso-carbons at the sites of oxidation are distorted toward bound oxygen. In contrast, the canonical heme oxygenase family degrades heme that is bound with minimal distortion. Trp-66 is a conserved heme pocket residue in IsdI implicated in heme ruffling. IsdI variants with Trp-66 replaced with residues having less bulky aromatic and alkyl side chains were characterized with respect to catalytic activity, heme ruffling, and electrochemical properties. The heme degradation activity of the W66Y and W66F variants was approximately half that of the wild-type enzyme, whereas the W66L and W66A variants were inactive. A crystal structure and NMR spectroscopic analysis of the W66Y variant reveals that heme binds to this enzyme with less heme ruffling than observed for wild-type IsdI. The reduction potential of this variant (−96 ± 7 mV versus standard hydrogen electrode) is similar to that of wild-type IsdI (−89 ± 7 mV), so we attribute the diminished activity of this variant to the diminished heme ruffling observed for heme bound to this enzyme and conclude that Trp-66 is required for optimal catalytic activity.
Introduction
IsdI4 is associated with the iron-regulated surface determinant (Isd) system for heme transport and iron release in Staphylococcus aureus. IsdI and its paralog IsdG catalyze the oxidative degradation of heme to the linear tetrapyrroles, 5-oxo-δ-bilirubin and 15-oxo-β-bilirubin (1), with the concomitant release of the central iron atom (2). The cellular fate or function of the two oxobilirubins, also termed staphylobilins, remains unknown. IsdG and IsdI share 64% amino acid sequence identity yet are differentially regulated by a heme-dependent protein degradation mechanism (3). Both enzymes contribute to the virulence of Staphylococcus aureus in a mouse model of infection (3).
IsdI is a homodimer with two heme binding pockets on either side of a β-barrel formed at the dimeric interface (1, 4). The porphyrin ring is bound in a hydrophobic pocket to a histidine as the sole protein-derived iron ligand, whereas the propionate groups form salt bridges to two Arg residues. On the distal side of the heme, the side-chain amide of Asn-6 forms an H-bond to external bound ligands. Within the hydrophobic heme pocket, no other residue is present to be a possible H-bond donor for bound ligands (1, 4).
The prominent structural feature of IsdI-metalloprotoporphyrin IX complexes is the extreme ruffling of the bound heme (normal-coordinate displacements >2.0 Å from planarity) (1, 4, 5). The electronic configuration of the ruffled heme in IsdI has been investigated by 1H NMR spectroscopy and proposed to have a large component of the uncommon (dxz,dyz)4(dxy)1 rather than the (dxy)2(dxz,dyz)3 configuration (5), as ruffling is considered to stabilize the dxz and dyz orbitals (6–10). In addition, both 1H NMR and x-ray crystallography provide evidence for a single orientation of heme binding about the α,γ-axis (5). Heme ruffling has been proposed to direct the β- and δ-meso carbons toward the distal heme pocket for oxidative cleavage at either site, consistent with the formation of two staphylobilins that differ by ring cleavage at the β- and δ-meso carbons (1). Notably, this structural characteristic is not observed among the family of classical heme oxygenases (HO) that degrades heme to biliverdin.
Hydrophobic residues that line the heme binding pocket of IsdI have been proposed to participate in porphyrin ring ruffling (4). Among these residues, Trp-66 is in direct steric contact with the β-meso carbon and is conserved across the IsdG-like enzyme family (11). In IsdG, substitution of the equivalent residue, Trp-67, with alanine abolished heme degrading activity (11). Although this result is consistent with a role for this residue in structural distortion of the bound heme, the structure of this IsdG variant has not been determined. In this work we have prepared the W66Y, W66F, W66L, and W66A variants of IsdI in an effort to manipulate heme ruffling while maintaining the hydrophobicity of the active site. Kinetic, spectroscopic, electrochemical, and crystallographic characterization of these variants provide evidence that Trp-66 plays a key role in ruffling of heme bound to IsdI and in the catalytic activity of the enzyme.
EXPERIMENTAL PROCEDURES
Protein Preparation
Active site variants of IsdI were produced by a megaprimer method involving two PCRs (12, 13). In both steps the plasmid pET15bisdI was used as the template after linearization by BamHI digest. First, a region of the isdi coding sequence was amplified between the mutagenic forward primer and the T7 terminator primer. The amplified DNA was used as a megaprimer for the second step of whole plasmid PCR and transformed into Escherichia coli DH5α. The site-directed mutations were confirmed by DNA sequencing. Recombinant wild-type and variant IsdI were expressed, purified, and reconstituted with heme as reported (4, 11). The final protein preparations are in Buffer A (Tris-HCl (20 mm) containing NaCl (200 mm), pH 7.5).
Activity Assay
The heme degrading activity of IsdI and the IsdI-Trp-66 variants was determined by monitoring the change in absorbance in the Soret region of the electronic spectrum of the enzyme-heme complex. The reaction mixture contained heme-reconstituted protein (10 μm) in buffer A. Protein concentration was determined by the Bradford method using bovine serum albumin as a standard. The reaction was initiated by the addition of sodium ascorbate (1 mm). Electronic spectra were recorded (300–700 nm; 1-cm path length) with a Varian Cary-50 UV-visible spectrophotometer at 10-min intervals for a total of 90 min. First-order rate constants were obtained by fitting the change in absorbance at the Soret maxima to an exponential function (A = Aoe−kt (A, absorbance; Ao, initial absorbance; k, rate constant; t, time)) with GraphPad Prism software (GraphPad Software, La Jolla, CA).
Crystallization and X-ray Diffraction Data Collection
The W66Y variant of IsdI was concentrated (∼17 mg/ml) in Buffer A and mixed (1:1) with reservoir solution (PEG 3350 (25% w/v), ammonium acetate (0.2 m), and Bis-Tris (0.1 m, pH 5.0)). The drops were set up in sitting drop vapor diffusion plates (4 °C), and crystals were formed in 4–7 days. Before data collection, the crystals were looped and dipped in cryoprotectant solution (reservoir solution containing ethylene glycol (20%)). Diffraction data were collected under a cryostream at 100 K on beamline 08ID-1 at the Canadian Light Source (Saskatoon, SK). Data were processed and scaled with MOSFLM (14) and SCALA (15), respectively.
The cyanide complex of the W66Y variant was prepared by adding sodium cyanide (final concentration 10 mm) to heme-reconstituted protein in Buffer A. The protein solution was mixed (1:1) with reservoir solution containing PEG 3350 (25% w/v), magnesium chloride (0.2 m), and Bis-Tris (0.1 m, pH 5.5). The drops were set up at room temperature in hanging drop vapor diffusion plates, and crystals formed in 3 days. Immediately before data collection, the crystals were looped and dipped in cryoprotectant solution (reservoir solution containing ethylene glycol (20%) and sodium cyanide solution (20 mm)). Diffraction data were collected under a cryostream at 100 K with a Rigaku Micromax 007 HF x-ray generator, Varimax-VHF optics, and a Saturn 944+ CCD detector with HKL3000 software for processing and scaling.
Model Building and Crystallographic Refinement
The programs in the CCP4 suite (16) were used to solve and refine the structures. For each structure, initial phases were obtained by molecular replacement with the program MolRep (17) using data to 2.0 Å resolution. The search model was the protein moiety from the IsdI-heme structure (PDB ID 3LGN), which gave unique solutions. For the W66Y structure the R-factors were 51.9 and 54.9% for the top and next unrelated solution, respectively. For W66Y(CN) the corresponding R-factors were 50.4 and 55.5%. Both the W66Y and W66Y(CN) crystals have the IsdI dimer in the asymmetric unit. Model building was carried out with COOT visualization software (18), and the structure was refined with the program Refmac5 (19). Two heme groups were seen in the electron density map and were included during subsequent refinement. Water molecules were added automatically using COOT:findwaters. For W66Y(CN), each of the heme irons was observed to bind a cyanide ion in a Fo − Fc map. The Fe-C-N bond links were defined in the Refmac library without restraints on the Fe-C bond length or Fe-C-N bond angle.
The final structure was validated with the program PROCHECK (20), and figures were prepared with the program PyMOL (21). The extent of heme ruffling in the final structures was analyzed by the normal-coordinate structural decomposition method (22). The final structures were superimposed, and the root mean square deviations were calculated with the program SuperPose (23).
1H NMR Spectroscopy
1H NMR spectra were collected with a Varian INOVA 500 MHz spectrometer. IsdI samples (∼2 mm) were prepared in sodium phosphate buffer (20 mm, 100% D2O, pH 7.0), and a small crystal of KCN was added to prepare the cyanide bound form. Chemical shifts are reported in ppm downfield from sodium 2,2-dimethyl-2-silapentane-5-sulfonate. For the one-dimensional spectra, presaturation was used to suppress the solvent signal. 13C,1H heteronuclear multiple quantum coherence spectra were recorded with a refocusing time of 2.5 ms, 2048 data points in the t2 dimension, and 256 blocks in the t1 dimension with 512 scans/block (30 °C).
Cyclic Voltammetry
Cyclic voltammetry was performed with an Autolab PGSTAT12 potentiostat-galvanostat (Eco Chemie) with an edge-plane pyrrolytic carbon electrode polished with alumina slurry and then sonicated in deionized water for 1 min. Protein solution (3 μl, ∼300 μm) was spread evenly with a micro-syringe onto the surface of the pyrrolytic carbon electrode. The pyrrolytic carbon electrode surface was then covered with a semipermeable membrane and used as the working electrode. An SCE electrode and a platinum wire were employed as the reference and counter electrodes, respectively. All experiments were performed (20 °C) in sodium phosphate buffer (20 mm). Solution pH was adjusted with small volumes of NaOH or HCl (0.5 m). The buffer was purged (≥10 m) with N2 before data collection.
RESULTS
Preparation and Activities of the Trp-66 Variants
Each of the four IsdI variants was expressed and purified as the recombinant apoprotein (≥150 mg/liter of cell culture) and reconstituted with heme, and unbound heme was removed by gel filtration chromatography. After the addition of ascorbate, the electronic spectra of the reconstituted W66Y and W66F variants exhibit Soret bands that are highly similar to that of the wild-type enzyme, whereas the corresponding spectra of the W66L and W66A variants exhibit a significant shoulder at higher energy that is reminiscent of unbound heme (Fig. 1). The change in spectrum of all four variant proteins after initiation of the reaction by aerobic addition of ascorbate (Fig. 1) established that all variants exhibited a diminished rate of substrate oxidation relative to the wild-type enzyme and that the activities of the W66Y and W66F variants were greater than those of the W66L and W66A variants. The pseudo-first order rate constants (min−1) for heme degradation derived from these spectra (Fig. 1F) decreased in the order wild-type (0.066 ± 0.003) > W66Y (0.020 ± 0.002) > W66F (0.015 ± 0.002) > W66L (0.0022 ± 0.0007)) > W66A (0.002 ± 0.002)). Similar rates were obtained from an analysis of the spectra at a single wavelength (396 nm).
FIGURE 1.

Heme degradation by IsdI and the Trp-66 variants. Enzymatic degradation of heme was monitored spectrophotometrically for wild type (A), W66Y (B), W66F (C), W66L (D), and W66A (E) forms of IsdI. Assay mixtures contained enzyme (10 μm) in Tris buffer (0.02 m, pH 7.5) containing NaCl (0.2 m) and catalase (0.1 μm) with ascorbate added (to 1 mm) to initiate the reaction. F, the change in absorbance at the Soret maxima was recorded at 10-min intervals for 90 min, normalized to the initial absorbance for the wild-type, and fit to a single exponential decay function. Abs, absorbance.
Electronic Absorption Spectra
At acidic pH, the spectra of the W66Y and W66F variants were similar to those of classical HOs except that the Soret transition for the W66Y variant was slightly blue-shifted and broader (Fig. 2). At alkaline pH, these variants also exhibited Soret maxima similar to those observed for classical HOs as well as a broad transition in the visible (∼560 nm) that may correspond to the two visible bands observed in spectra of classical heme oxygenases. The pKa values of the acid-alkaline transition of the water molecule coordinated to the heme iron of the W66Y and W66F variants are 0.8 and 0.9 greater than that of the wild-type enzyme (5) and are similar to that of classical HOs (pKa = 7.6 ∼ 9.3 (24–27)) as shown in Table 1. With cyanide bound, the IsdI variants exhibited Soret and Q band maxima at higher energy relative to the spectrum of the wild-type enzyme, and the positions of these maxima are also more similar to those observed for classical HOs. Similar studies of the W66L and W66A variants were prevented by their limited stabilities over this range of pH.
FIGURE 2.

pH dependence of absorption spectra of IsdI variants. The W66Y variant (A) and (B) the W66F variant (sodium phosphate buffer (20 mm) 20 °C). Inset, the change in absorbance (Abs) of the W66Y (397 nm) and W66F (404 nm) variants is plotted against pH. The curve represents the least-squares fit of the data to a titration curve assuming n = 1. C, shown are electronic spectra of the cyanide derivatives of wild-type IsdI (solid line) and the W66Y (dashed line) and W66F (dotted line) variants at pH 7.0.
TABLE 1.
Electronic absorption parameters for wild-type and variant forms of IsdI and for some classical heme oxygenases
pa, Pseudomonas aeruginosa; nm, Neisseria meningococcus.
| Protein | λmax |
pKa | λmax (CN− bound) | |
|---|---|---|---|---|
| Low pH | High pH | |||
| nm | nm | |||
| Wild-type IsdI | 404, 640 | 412, 485, 520, 578 | 7.1 | 421, 558 |
| IsdI W66Y | 397, 506, 635 | 411, ∼567 | 7.9 | 417, 553 |
| IsdI W66F | 404, 504, 630 | 411, ∼560 | 8.0 | 416, 553 |
| Rat HO-1 | 404, 500, 631a | 413, 540, 575a | 7.6a | 418, 536b |
| Human HO-2 | 404, 500, 631c | 413, 540, 575c | 8.5c | |
| pa-HO | 405, 503, 537, 638d | 415, 540, 574e | 8.1e | 419, ∼540d |
| nm-HO | 405, 504, 638d | 9.3d | 419, ∼540d | |
1H NMR Spectra
The 1H NMR spectra (30 °C) of the IsdI W66Y and W66F variants with cyanide bound are compared with that of the wild-type enzyme in Fig. 3. The paramagnetically shifted resonances of the IsdI variants were shifted farther downfield than the corresponding resonances in the spectrum of the wild-type protein. Notably, the heme meso signals observed near −15 ppm in the spectrum of wild-type IsdI are not observed in the high-field region in the spectra of the variant proteins. In the 13C,1H heteronuclear multiple quantum coherence spectrum of the W66Y variant (Fig. 4), eight strong signals appeared in the low-field region that can be attributed to the heme methyl groups, suggesting the occurrence of two heme insertion isomers that are rotated 180° about the α/γ axis in the protein. The average chemical shift of heme methyl signals of the W66Y variant is 11.6 ppm, which is comparable with that of classical HOs (Table 2). The W66F variant exhibits much broader signals for both the paramagnetically shifted resonances and those in the protein region, indicating that the structure of this protein is more dynamic. As a result, we could not obtain two-dimensional NMR spectra for this variant that were of a quality sufficient for analysis.
FIGURE 3.

1H NMR spectra of IsdI cyanide derivatives. Bottom, wild-type IsdI. Center, W66Y variant. Top, W66F variant (sodium phosphate buffer (20 mm) in 10% D2O, pH 7.0, 30 °C).
FIGURE 4.

13C,1H heteronuclear multiple quantum coherence spectrum of cyanide derivative of the W66Y variant. The 8 heme methyl signals are indicated with numbers (sodium phosphate buffer (20 mm) in 100% D2O, pH 7.0, 30 °C).
TABLE 2.
Chemical shifts of heme methyl resonances of the cyanide derivatives of wild-type and variant forms of IsdI and for some classical heme oxygenases
pa, Pseudomonas aeruginosa; nm, Neisseria meningococcus; cd, Corynebacterium diphtheriae.
| Protein | Temperature | Methyl shifts | Average | Reference |
|---|---|---|---|---|
| ° C | ppm | ppm | ||
| Wild-type IsdI | 30 | 12.0, 10.6, 8.0, 4.0 | 8.7 | 5 |
| IsdI W66Y | 30 | 17.7, 17.6, 17.0, 11.4, 9.6, 7.9, 7.7, 4.0 | 11.6 | This work |
| Human HO-1a | 25 | 19.6, 10.5, 9.0, 5.0 | 11.0 | 41 |
| pa-HO | 10 | 27.7, 22.7, 19.0, 4.4 | 18.5 | 42 |
| nm-HO | 25 | 21.4, 10.3, 9.6, 7.9 | 12.3 | 43 |
| cd-HO | 35 | 19.2, 10.6, 8.5, 5.4 | 10.9 | 42 |
| HmuO | 25 | 19.7, 10.4, 8.1, 4.9 | 10.8 | 44 |
| Nitrophorin2 | 35 | 18.1, 13.4, 9.1, 7.6 | 12.1 | 45 |
| SW Mb | 25 | 27.0, 18.6, 12.9, 4.8 | 15.8 | 46 |
a The protein reconstituted with protohemin.
Electrochemistry
Cyclic voltammetry of the IsdI W66Y variant (sodium phosphate buffer (20 mm), pH 7.0) yields a midpoint reduction potential (Em) of −96 ± 7 mV versus standard hydrogen electrode (Fig. 5A), which is slightly more negative but comparable to those of wild-type protein (−88 ± 7 mV) and HOs from rat (−87 mV (28)) and human (−65 mV (29)). No electrochemical response could be elicited for the W66F variant under identical conditions. For wild-type IsdI and the W66Y variant, stable and well-defined peaks were obtained for the pH range of 5.0- 8.0. A negative shift of Em was observed with increasing pH with the slope of about −50 mV/pH for both wild-type and W66Y proteins (Fig. 5B). This slope is similar to the theoretical value (−59 mV/pH) for a simultaneous 1-electron 1-proton transfer reaction.
FIGURE 5.
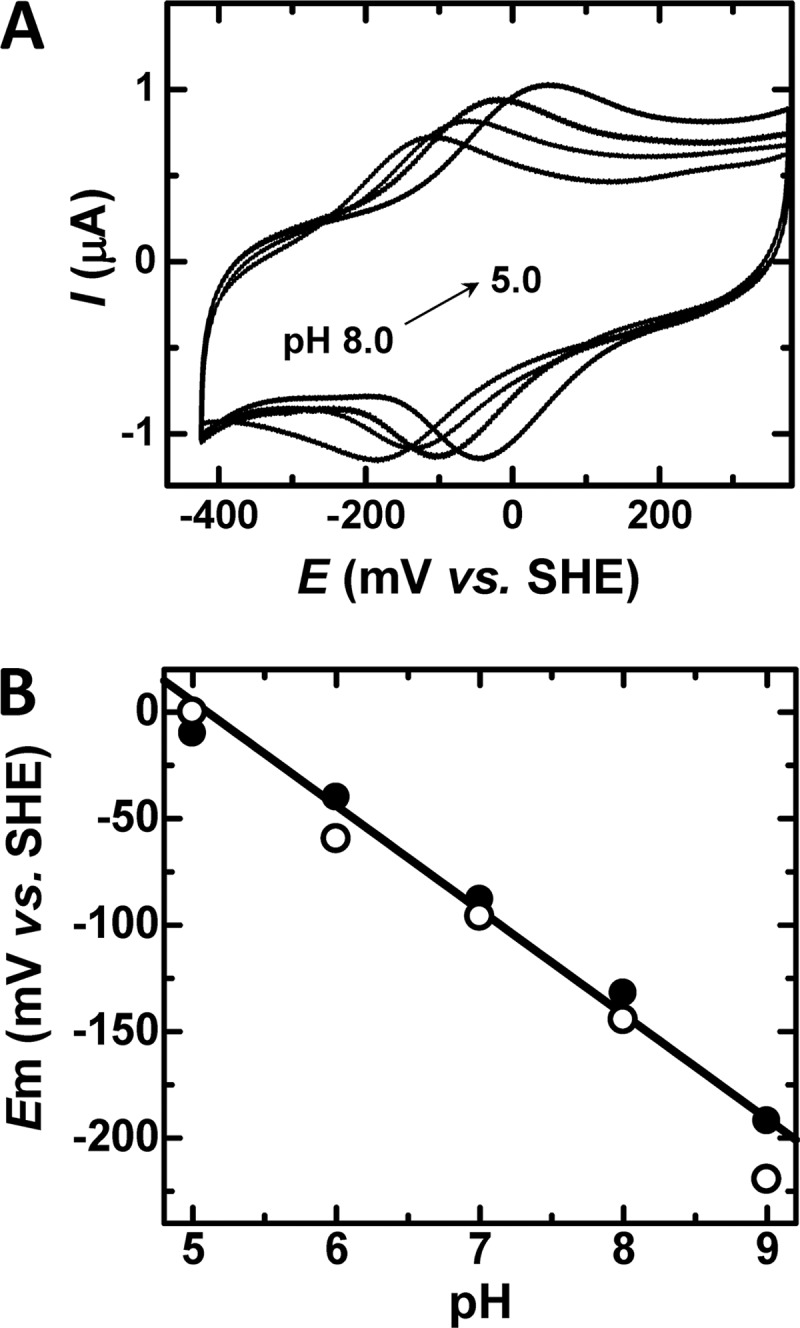
Cyclic voltammetry of the W66Y variant as a function of pH. A, cyclic voltammograms were collected at a scan rate of 50 mV/s (sodium phosphate buffer (20 mm)) at pH 5.0, 6.0, 7.0, and 8.0). B, pH dependence of the Em of wild-type IsdI (filled circle) and the W66Y variant (open circle). The solid line represents the least-squares fit to the data for wild-type IsdI (slope, −49 mV/pH). The least-squares fit for the variant is identical within experimental error. SHE, standard hydrogen electrode.
Crystal Structures of the W66Y and W66Y(CN) Variants
The structures of the W66Y variant in complex with heme (IsdI-W66Y) and its cyanide-bound form (IsdI-W66Y(CN)) were solved to at least 1.9 Å resolution (see Table 3 for a summary of the crystallographic statistics). The asymmetric unit of both crystals contained a homodimer that consists of an α+β-barrel fold similar to wild-type IsdI. The Cα atoms of the IsdI-W66Y and IsdI-W66Y(CN) structures superimposed with those of wild-type IsdI (PDB 3LGN) with root mean square deviations of 0.68 and 0.40 Å, respectively (Fig. 6A).
TABLE 3.
Crystallographic data collection and refinement statistics for the W66Y variant of IsdI and its cyanide derivative
| IsdI-W66Y | IsdI-W66Y(CN) | |
|---|---|---|
| Data collectiona | ||
| Resolution range (Å) | 50.12–1.90 (1.95–1.90) | 48.26–1.80 (1.85–1.80) |
| Space group | P212121 | P212121 |
| Unit cell dimension (Å) | a = 59.49, b = 68.80, c = 73.17 | a = 59.31, b = 67.01, c = 69.57 |
| Unique reflections | 24,334 (3481) | 25,378 (3877) |
| Completeness (%) | 100.0 (100.0) | 99.9 (99.5) |
| Average I/σI | 10.4 (4.2) | 22.5 (2.7) |
| Redundancy | 6.6 (6.7) | 9.9 (5.1) |
| Rmerge | 0.135 (0.430) | 0.095 (0.454) |
| Wilson B-factor (Å2) | 13.2 | 18.7 |
| Refinement | ||
| Rwork (Rfree) | 0.193 (0.230) | 0.197 (0.238) |
| % reflections for Rfree | 5 | 5 |
| Coordinate error estimateb | 0.09 | 0.09 |
| No. atoms (B-factor (Å2)) | 2,208 (15.1) | 2,321 (18.9) |
| Protein | 1,863 (14.2) | 1,892 (17.0) |
| Solvent | 259 (22.3) | 339 (30.0) |
| Heme | 86 (10.8) | 86 (17.0) |
| Cyanide | 4 (16.7) | |
| Root mean square deviations bond length (Å) | 0.010 | 0.012 |
| Ramachandran plot, no. residues | ||
| In most-favorable regions | 202 | 202 |
| In disallowed regions | 1 | 1 |
a Values in parentheses for the data collection statistics are for the highest resolution shell indicated.
b Coordinate error is the standard estimated uncertainty from maximum likelihood refinement.
FIGURE 6.

Structural comparison of wild-type IsdI, the W66Y variant, and its cyanide derivative. A, the Cα trace of the structures of wild-type IsdI (PDBID 3LGN: green, chain A; dark green, chain B), IsdI-W66Y (gray, chain A; dark gray, chain B), and IsdI-W66Y(CN) (cyan, chain A; blue, chain B). The variant forms superimpose to wild type with root mean square deviations of 0.68 Å for IsdI-W66Y and 0.40 Å for IsdI-W66Y(CN). The N and C termini are shown. Shown are the 2 Fo − Fc map (gray) contoured at 1σ for the heme binding residues of chains A for the W66Y variant (gray) (B) and the cyanide derivative of the W66Y variant (cyan) (C). The omit map (green) for cyanide is contoured at 3σ.
Heme bound to the active site in each IsdI monomer exhibits clear electron density for coordination of the heme iron by H76 for both IsdI-W66Y and IsdI-W66Y(CN) (Fig. 6, B and C, respectively). On the other hand, the electron density for the heme vinyl groups is noticeably weak, consistent with heme binding in two orientations that differ by rotation about the α,γ axis. As the occupancy of the two orientations could not be resolved in either structure, the heme groups are modeled in one orientation. Ruffling distortion of heme bound to the W66Y variant and to wild-type IsdI decreases in the following order: cyanide bound wild-type (2.3 Å; PDB ID 3QGP) > wild-type (2.1 Å; PDB ID 3LGN) > IsdI-W66Y(CN) (1.5, 1.9 Å) > IsdI-W66Y (1.3, 1.4 Å) (out-of-plane normal coordinate displacement for each molecule in the asymmetric unit is shown in parentheses).
Several changes occur in the heme pocket of the IsdI-W66Y structure as a result of replacing Trp-66 with Tyr as can be seen by superimposing the two structures (Fig. 7A). The side chain of Tyr-66 in the variant occupies the space of Trp-66 on the proximal side of the heme, and the smaller ring structure of the Tyr residue allows the heme to relax into a more planar conformation. The closest heme-Tyr contact involves Tyr-66 Cϵ2 and the β-meso carbon (4.0 Å), in contrast to the closest heme-Trp contact of 3.5 Å involving the β-meso carbon and Trp-66 Cη2 in wild-type IsdI. The side chain of Tyr-66 is angled toward the back of the heme pocket with the phenol group forming a water-bridged H-bond to a heme propionate group (Fig. 7B). In the wild-type structure, Trp-66 Nϵ1 provides the equivalent H-bond to a water molecule interacting with the propionate group.
FIGURE 7.

Comparison of the heme pocket structures of wild-type IsdI, the W66Y variant, and its cyanide derivative. A, superposition of the heme pocket residues is shown. Residues with an altered conformation in the IsdI-W66Y structure (Asn-6, Trp-66, His-76, and Ile-53) are highlighted. B, a water bridged H-bond interaction of Trp-66 with a heme propionate group is mimicked by Tyr-66 in the variant. Amino acid carbon atoms are colored according to the main chain of each structure: gray, IsdI-W66Y; cyan, IsdI-W66Y(CN); green, wild-type IsdI (PDB ID 3LGN).
Three other active site residues are also influenced by replacement of Trp-66 with Tyr. In the distal heme pocket, the side chain of Asn-6 adopts an alternate conformation more distant from the iron center (Fig. 7A) such that the amides of Asn-6 and Asn-95 are within H-bonding distance (3.1 Å) of each other. The Cδ1 atom of Ile-53 is displaced to occupy the position of a distal heme iron ligand. The salt bridge between Arg-25 and the heme propionate group is disrupted, and the side chain of Arg-25 is redirected into the solvent (Fig. 7B). These conformational changes are accompanied by the addition of a water molecule (B-factor 9 Å2) to the heme pocket that can form an H-bond to T55 Oγ1 (∼2.8 Å).
Inspection of the Fo − Fc map of the cyanide derivative of the W66Y variant reveals elongated density on the distal side of the heme-iron that was modeled as a fully occupied cyanide ion 1.9 Å from the iron atom (Fig. 6C). This cyanide ligand is bent in relation to the heme-iron at angles of 159° and 158° in chains A and B, respectively. This orientation is comparable to that reported for the cyanide derivative of the wild-type enzyme in which the cyanide is bent at angles of 158° and 171°. The average B-factors for cyanide (16.7 and 17.0 Å2) are similar to those of the heme (Table 3).
Residues in the heme pocket of the cyanide derivative of the W66Y variant maintain similar positions to the wild-type protein (Fig. 7A). Specifically, Ile-53 adopts the wild-type conformation so that the Cδ1 atom does not interfere with cyanide binding. Also, the conformation of Asn-6 is unaltered, enabling an H-bond with cyanide (3.1 and 3.3 Å in chains A and B, respectively). Although the side chain conformations are less perturbed in the structure of the cyanide derivative of the W66Y variant, the additional water molecule located in the distal heme pocket of the IsdI-W66Y structure remains (Fig. 6C). In addition, the heme pocket of chain A contains electron density for a second water molecule (B-factor of 33 Å2) in the vicinity of T55 (4.6 Å), closer to the bound cyanide (2.5 Å).
DISCUSSION
Heme Degrading Activity of the IsdI Trp-66 Variants
All four IsdI variants involving replacement of Trp-66 bound and retained heme after gel filtration implying that this residue is not required for substrate binding. Trp-66 is needed for full activity of the enzyme, in agreement with the loss in heme-degrading activity of the W66A variant of IsdG (4). In IsdI, the more conservative W66Y and W66F variants retained about half the activity of the wild-type enzyme. The other two variants, W66L and W66A, exhibited an altered heme environment as indicated by the visible electronic absorbance spectrum and suffered a nearly complete loss of activity. Thus, the data suggest that the heme degrading activity of IsdI is dependent on the presence of amino acids with large side chains at position 66. To better understand the loss in activity of the W66Y and W66F variants, these variants were investigated further with respect to heme environment, electronic structure, and electrochemical behavior.
Heme Environment of the IsdI W66Y and W66F Variants
Electronic absorption spectra of IsdI variants obtained as a function of pH and in the absence and presence of KCN were similar to those of the classic HOs except that the spectrum of the W66Y variant at acidic pH exhibited a broader and blue-shifted Soret band, that might be attributable to a five-coordinate heme center or to a mixture of five- and six-coordinate heme centers as reported for some myoglobin variants lacking a distal His residue (Fig. 2) (30, 31). The pKa values obtained for our IsdI variants (7.9 for W66Y and 8.0 for W66F (Table 1)) are also similar to those reported for HOs but contrast with the much lower pKa of the wild-type enzyme (7.1), which exhibits a different spectroscopic pattern. This low pKa value for wild-type IsdI is probably the result of the unusually hydrophobic nature of the heme binding site that stabilizes the electrostatically neutral ferric-hydroxide heme iron center (5). Therefore, the higher pKa values we observe for the current IsdI variants suggest a more hydrophilic heme environment in these proteins.
Of the Trp-66 variants of IsdI, crystals suitable for x-ray analysis could be obtained only for the W66Y variant in the presence and absence of cyanide. The observed decreased ruffling of heme bound to this variant relative to the native enzyme provides an explanation for the similarity of electronic spectra of the variant to that of classic HOs. Furthermore, the introduction of additional bound water molecules in proximity to the heme in the structure of the W66Y variant in the absence of bound cyanide results in a more hydrophilic heme environment. An important finding from this structure is that the hydroxyl group of Tyr-66 is able to mimic the interaction of W66Nϵ1 with the propionate group. This interaction may be essential to stabilizing heme binding and enabling crystallization of this protein. Nonetheless, heme binding by the W66Y variant is more flexible, as evidenced by the observation of eight heme methyl signals in the NMR spectrum and the absence of well defined crystallographic density for the heme vinyl groups.
Soret and Q-band maxima of cyanide-bound forms of the IsdI variants were blue-shifted relative to the corresponding maxima in the spectrum of wild-type IsdI (Fig. 2). The red shift of absorption maxima is commonly observed for distorted model heme compounds (32–34). However, these model compounds typically involve non-ferrous metal centers or porphyrin ligands with peripheral substituents different from those of protoheme IX that are required to achieve heme distortion. Consequently, these models differ from protoheme IX in several respects in addition to the extent of heme distortion, a situation that prevents unambiguous assignment of these spectroscopic differences to differences in heme distortion alone (32–34). For the forms of IsdI studied here, heme distortion is achieved without metal ion substitution or porphyrin ring side-chain substitution, so the shifts in the absorption maxima of the cyanide derivatives of these proteins can be attributed primarily to heme distortion.
Electronic Structure of IsdI W66Y and W66F
The paramagnetic shifts of the heme methyl protons observed in the 1H NMR spectrum of wild-type IsdI are considerably smaller, and the shifts observed for the meso-protons are considerably greater than those observed for other heme proteins, indicating that this protein is in the less common (dxz,dyz)4(dxy)1 electronic configuration (5). However, the heme methyl signals in the corresponding spectrum of the W66Y variant exhibited greater paramagnetic shifts than the wild-type enzyme and an average chemical shift for the heme methyl signals that is comparable with that of classical HOs (Fig. 3 and Table 2). This observation indicates that the W66Y variant has much less (dxz,dyz)4(dxy)1 character than the wild-type protein, consistent with the smaller degree of heme ruffling observed in the crystal structure of this variant. Although we could not obtain a crystal structure or the assignments of heme methyl resonances for the W66F variant, it is reasonable to conclude that the heme in this variant is even less ruffled relative to the heme at the active site of the W66Y variant because for the W66F variant 1) the paramagnetic 1H NMR signals are shifted to a lower field than seen for the corresponding signals in the spectrum of the W66Y variant, 2) replacement of Trp-66 with Phe introduces a residue smaller than Tyr, and 3) broader NMR signals imply a heme binding site with a more disordered structure. Systematic variation in the extent of heme distortion in a heme protein has been achieved only recently for H-NOX variants (35), and the range of distortion is up to ∼1 Å, which is not sufficient to obtain a protein with a predominant (dxz,dyz)4(dxy)1 electronic configuration. With wild-type IsdI and the W66Y variant, on the other hand, we have achieved a variation in heme distortion of 1.3–2.3 Å, a range that reveals the contribution of heme ruffling to heme protein functional and spectroscopic properties.
Electrochemical Properties of IsdI W66Y
The Em of wild-type IsdI and the W66Y variant both decreased with increasing pH with a slope of about −50 mV/pH (Fig. 5B). The value of the slope is similar to the theoretical value (−59 mV/pH) expected for a system that exhibits proton-coupled electron transfer. Both spectroscopic and x-ray crystallographic data indicated that the heme pockets of the IsdI variants possess less constraining and more hydrophilic heme binding environments than that of the wild-type enzyme, so a significant decrease in Em values was expected for the variants. Nevertheless, the W66Y variant exhibited an Em that is only slightly more negative than that of wild-type IsdI. This finding supports our previous proposal that the relatively low Em of wild-type IsdI can be attributed primarily to heme ruffling as observed for model porphyrin compounds (36) and other heme proteins (21, 35, 37). The W66F variant exhibited no direct electrochemistry under identical experimental conditions probably as the result of structural dynamics, but the less ruffled heme at the active site of this variant may also be a contributory factor. Density functional theory calculations show that in the ruffled porphyrinate the iron carries ∼50% of the unpaired electron density, and the remainder resides in the dxy orbital, whereas ∼85% of the spin density resides on the iron in a planar porphyrinate (38). This characteristic has led to the proposal that electron transfer through the porphyrin ring is facilitated by heme ruffling. Although the heme at the active site of the W66Y variant is ruffled less than that of the wild-type enzyme, the degree of distortion (1.3–1.4 Å) is still much greater than that of most other heme proteins so heme ruffling should facilitate electron transfer through the porphyrin ring in a manner similar to the wild-type enzyme.
Significance of Heme Distortion in Heme Oxidation by IsdI
This study demonstrates that the properties of the W66Y and W66F variants more closely approach those of classical HOs than those of wild-type IsdI. The diminished heme ruffling, electronic absorption spectrum, pKa of the distally coordinated water molecule, the paramagnetic shifts of the heme substituents, the presence of a less constraining and more hydrophilic heme binding site, and the Em value of IsdI W66Y are all more similar to the characteristics of the classical HOs. The inactivity of the W66Y variant in heme degradation despite these similarities to the classical HOs, however, strongly supports the conclusion that heme distortion contributes significantly to the ability of wild-type IsdI to oxidize heme.
Acknowledgments
We thank Professor Lawrence McIntosh for use of the NMR spectrometer. Research described in this paper was performed using beamline 08ID-1 at the Canadian Light Source, which is supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan.
This work was supported by a postdoctoral fellowship from the Uehara Memorial Foundation (to S. J. T.), a Canada Research Chair (to A. G. M.), Canadian Institutes of Health Research (CIHR) Operating Grant MOP-49597 (M. E. P. M.), a Canadian Blood Services-CIHR partnership grant (to A. G. M.), and the Canadian Foundation for Innovation (to A. G. M. and M. E. P. M.).
The atomic coordinates and structure factors (codes 4FNH and 4FNI) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- IsdI
- the ferriheme complex of IsdI
- HO
- heme oxygenase
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Reniere M. L., Ukpabi G. N., Harry S. R., Stec D. F., Krull R., Wright D. W., Bachmann B. O., Murphy M. E., Skaar E. P. (2010) The IsdG-family of haem oxygenases degrades haem to a novel chromophore. Mol. Microbiol. 75, 1529–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Skaar E. P., Humayun M., Bae T., DeBord K. L., Schneewind O. (2004) Iron-source preference of Staphylococcus aureus infections. Science 305, 1626–1628 [DOI] [PubMed] [Google Scholar]
- 3. Reniere M. L., Skaar E. P. (2008) Staphylococcus aureus haem oxygenases are differentially regulated by iron and haem. Mol. Microbiol. 69, 1304–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee W. C., Reniere M. L., Skaar E. P., Murphy M. E. (2008) Ruffling of metalloporphyrins bound to IsdG and IsdI, two heme-degrading enzymes in Staphylococcus aureus. J. Biol. Chem. 283, 30957–30963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Takayama S. J., Ukpabi G., Murphy M. E., Mauk A. G. (2011) Electronic properties of the highly ruffled heme bound to the heme degrading enzyme IsdI. Proc. Natl. Acad. Sci. 108, 13071–13076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nakamura M. (2006) Electronic structures of highly deformed iron(III) porphyrin complexes. Coord. Chem. Rev. 250, 2271–2294 [Google Scholar]
- 7. Cai S., Shokhireva T. Kh, Lichtenberger D. L., Walker F. A. (2006) NMR and EPR studies of chloroiron(III) tetraphenyl-chlorin and its complexes with imidazoles and pyridines of widely differing basicities. Inorg. Chem. 45, 3519–3531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yatsunyk L. A., Shokhirev N. V., Walker F. A. (2005) Magnetic resonance spectroscopic investigations of the electronic ground and excited states in strongly nonplanar iron(III) dodecasubstituted porphyrins. Inorg. Chem. 44, 2848–2866 [DOI] [PubMed] [Google Scholar]
- 9. Walker F. A., Nasri H., Turowska-Tyrk I., Mohanrao K., Watson C. T., Shokhirev N. V., Debrunner P. G., Scheidt W. R. (1996) Π acid ligands in iron(III) porphyrinates. Characterization of low-spin bis(tert-butylisocyanide)(porphyrinato)iron(III) complexes having (dxz,dyz)4(dxy)1 ground states. J. Am. Chem. Soc. 118, 12109–12118 [Google Scholar]
- 10. Safo M. K., Gupta G. P., Watson C. T., Simonis U., Walker F. A., Scheidt W. R. (1992) Models of the cytochromes b. Low-spin bis-ligated (porphinato)iron(III) complexes with unusual molecular structures and NMR, EPR, and Mössbauer spectra. J. Am. Chem. Soc. 114, 7066–7075 [Google Scholar]
- 11. Wu R., Skaar E. P., Zhang R., Joachimiak G., Gornicki P., Schneewind O., Joachimiak A. (2005) Staphylococcus aureus IsdG and IsdI, heme-degrading enzymes with structural similarity to monooxygenases. J. Biol. Chem. 280, 2840–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miyazaki K. (2003) Creating random mutagenesis libraries by megaprimer PCR of whole plasmid (MEGAWHOP). Methods Mol. Biol. 231, 23–28 [DOI] [PubMed] [Google Scholar]
- 13. MacPherson I. S., Rosell F. I., Scofield M., Mauk A. G., Murphy M. E. (2010) Directed evolution of copper nitrite reductase to a chromogenic reductant. Protein Eng. Des. Sel. 23, 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leslie A. G. W., Powell H. R. (2007) in Evolving Methods for Macromolecular Crystallography, pp. 41–51, Springer, Dordrecht, The Netherlands [Google Scholar]
- 15. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 16. Potterton E., Briggs P., Turkenburg M., Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 17. Vagin A., Teplyakov A. (1997) MOLREP. An automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 18. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 20. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK. A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 21. Roberts S. A., Weichsel A., Qiu Y., Shelnutt J. A., Walker F. A., Montfort W. R. (2001) Ligand-induced heme ruffling and bent NO geometry in ultra-high resolution structures of nitrophorin 4. Biochemistry 40, 11327–11337 [DOI] [PubMed] [Google Scholar]
- 22. Jentzen W., Song X. Z., Shelnutt J. A. (1997) Structural characterization of synthetic and protein-bound porphyrins in terms of the lowest frequency normal coordinates of the macrocycle. J. Phys. Chem. B 101, 1684–1699 [Google Scholar]
- 23. Maiti R., Van Domselaar G. H., Zhang H., Wishart D. S. (2004) SuperPose. A simple server for sophisticated structural superposition. Nucleic Acids Res. 32, W590–W594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takahashi S., Wang J., Rousseau D. L., Ishikawa K., Yoshida T., Host J. R., Ikeda-Saito M. (1994) Heme-heme oxygenase complex. Structure of the catalytic site and its implication for oxygen activation. J. Biol. Chem. 269, 1010–1014 [PubMed] [Google Scholar]
- 25. Caignan G. A., Deshmukh R., Zeng Y., Wilks A., Bunce R. A., Rivera M. (2003) The hydroxide complex of Pseudomonas aeruginosa heme oxygenase as a model of the low-spin iron(III) hydroperoxide intermediate in heme catabolism. 13C NMR spectroscopic studies suggest the active participation of the heme in macrocycle hydroxylation. J. Am. Chem. Soc. 125, 11842–11852 [DOI] [PubMed] [Google Scholar]
- 26. Chu G. C., Tomita T., Sönnichsen F. D., Yoshida T., Ikeda-Saito M. (1999) The heme complex of HmuO, a bacterial heme degradation enzyme from Corynebacterium diphtheriae. J. Biol. Chem. 274, 24490–24496 [DOI] [PubMed] [Google Scholar]
- 27. Ishikawa K., Takeuchi N., Takahashi S., Matera K. M., Sato M., Shibahara S., Rousseau D. L., Ikeda-Saito M., Yoshida T. (1995) Heme oxygenase-2. J. Biol. Chem. 270, 6345–6350 [DOI] [PubMed] [Google Scholar]
- 28. Sato H., Higashimoto Y., Sakamoto H., Sugishima M., Takahashi K., Palmer G., Noguchi M. (2007) Electrochemical reduction of ferrous α-verdoheme in complex with heme oxygenase-1. J. Inorg. Biochem. 101, 1394–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu Y., Moënne-Loccoz P., Hildebrand D. P., Wilks A., Loehr T. M., Mauk A. G., Ortiz de Montellano P. R. (1999) Replacement of the proximal histidine iron ligand by a cysteine or tyrosine converts heme oxygenase to an oxidase. Biochemistry 38, 3733–3743 [DOI] [PubMed] [Google Scholar]
- 30. Yamamoto Y., Suziki T., Hori H. (1993) Dynamics and thermodynamics of acid-alkaline transitions in metmyoglobins lacking the usual distal histidine residue. Biochim. Biophys. Acta 1203, 267–275 [DOI] [PubMed] [Google Scholar]
- 31. Ikeda-Saito M., Hori H., Andersson L. A., Prince R. C., Pickering I. J., George G. N., Sanders C. R., 2nd, Lutz R. S., McKelvey E. J., Mattera R. (1992) Coordination structure of the ferric heme iron in engineered distal histidine myoglobin mutants. J. Biol. Chem. 267, 22843–22852 [PubMed] [Google Scholar]
- 32. Zhou Z., Cao C., Liu Q., Jiang R. (2010) Hybrid orbital deformation (HOD) effect and spectral red-shift property of nonplanar porphyrin. Organic Letters 12, 1780–1783 [DOI] [PubMed] [Google Scholar]
- 33. Haddad R. E., Gazeau S., Pécaut J., Marchon J. C., Medforth C. J., Shelnutt J. A. (2003) Origin of the red shifts in the optical absorption bands of nonplanar tetraalkylporphyrins. J. Am. Chem. Soc. 125, 1253–1268 [DOI] [PubMed] [Google Scholar]
- 34. Ryeng H., Ghosh A. (2002) Do nonplanar distortions of porphyrins bring about strongly red-shifted electronic spectra? Controversy, consensus, new developments, and relevance to chelatases. J. Am. Chem. Soc. 124, 8099–8103 [DOI] [PubMed] [Google Scholar]
- 35. Olea C., Jr., Kuriyan J., Marletta M. A. (2010) Modulating heme redox potential through protein-induced porphyrin distortion. J. Am. Chem. Soc. 132, 12794–12795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shelnutt J. A., Song X. Z., Ma J. G., Jia S. L., Jentzen W., Medforth C. J. (1998) Nonplanar porphyrins and their significance in proteins. Chem. Soc. Rev. 27, 31–41 [Google Scholar]
- 37. Ma J. G., Zhang J., Franco R., Jia S. L., Moura I., Moura J. J., Kroneck P. M., Shelnutt J. A. (1998) The structural origin of nonplanar heme distortions in tetraheme ferricytochromes c3. Biochemistry 37, 12431–12442 [DOI] [PubMed] [Google Scholar]
- 38. Ghosh A., Gonzalez E., Vangberg T. (1999) Theoretical studies of low spin six-coordinate iron(III) porphyrins relevant to cytochrome b. Variable electronic configurations, ligand noninnocence, and macrocycle ruffling. J. Phys. Chem. B 103, 1363–1367 [Google Scholar]
- 39. Hawkins B. K., Wilks A., Powers L. S., Ortiz de Montellano P. R., Dawson J. H. (1996) Ligation of the iron in the heme-heme oxygenase complex. X-ray absorption, electronic absorption, and magnetic circular dichroism studies. Biochim. Biophys. Acta 1295, 165–173 [DOI] [PubMed] [Google Scholar]
- 40. Zeng Y., Caignan G. A., Bunce R. A., Rodríguez J. C., Wilks A., Rivera M. (2005) Azide-inhibited bacterial heme oxygenases exhibit an S = 3/2(dxz,dyz)3(dxy)1(dz2)1 spin state. Mechanistic implications for heme oxidation. J. Am. Chem. Soc. 127, 9794–9807 [DOI] [PubMed] [Google Scholar]
- 41. Gorst C. M., Wilks A., Yeh D. C., Ortiz de Montellano P. R., La Mar G. N. (1998) Solution 1H NMR investigation of the molecular and electronic structure of the active site of substrate-bound human heme oxygenase. The nature of the distal hydrogen bond donor to bound ligands. J. Am. Chem. Soc. 120, 8875–8884 [Google Scholar]
- 42. Caignan G. A., Deshmukh R., Wilks A., Zeng Y., Huang H. W., Moënne-Loccoz P., Bunce R. A., Eastman M. A., Rivera M. (2002) Oxidation of heme to β- and δ-biliverdin by Pseudomonas aeruginosa heme oxygenase as a consequence of an unusual seating of the heme. J. Am. Chem. Soc. 124, 14879–14892 [DOI] [PubMed] [Google Scholar]
- 43. Ma L. H., Liu Y., Zhang X., Yoshida T., La Mar G. N. (2006) 1H NMR study of the magnetic properties and electronic structure of the hydroxide complex of substrate-bound heme oxygenase from Neisseria meningitidis. Influence of the axial water deprotonation on the distal H-bond network. J. Am. Chem. Soc. 128, 6657–6668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li Y., Syvitski R. T., Chu G. C., Ikeda-Saito M., Mar G. N. (2003) Solution 1H NMR investigation of the active site molecular and electronic structures of substrate-bound, cyanide-inhibited HmuO, a bacterial heme oxygenase from Corynebacterium diphtheriae. J. Biol. Chem. 278, 6651–6663 [DOI] [PubMed] [Google Scholar]
- 45. Yang F., Knipp M., Shokhireva T. K., Berry R. E., Zhang H., Walker F. A. (2009) 1H and 13C NMR spectroscopic studies of the ferriheme resonances of three low-spin complexes of wild-type nitrophorin 2 and nitrophorin 2(V24E) as a function of pH. J. Biol. Inorg. Chem. 14, 1077–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Emerson S. D., La Mar G. (1990) Solution structural characterization of cyanometmyoglobin. Resonance assignment of heme cavity residues by two-dimensional NMR. Biochemistry 29, 1545–1556 [DOI] [PubMed] [Google Scholar]