

Background: Heparin cofactor II (HCII), a serine protease inhibitor, exerts protective actions against cardiovascular remodeling.
Results: HCII-deficient mice manifested insufficient recovery of peripheral circulation after hindlimb ischemia with inactivation of vascular endothelial AMPK and eNOS.
Conclusion: HCII promotes angiogenesis in response to ischemia via the AMPK-eNOS-dependent pathway.
Significance: HCII might be a novel therapeutic target in patients with peripheral arterial disease.
Keywords: AMP-activated Protein Kinase (AMPK), Angiogenesis, Endothelial Dysfunction, Ischemia, Vascular Biology, Heparin Cofactor II
Abstract
We previously clarified that heparin cofactor II (HCII), a serine proteinase inhibitor, exerts various protective actions on cardiovascular diseases in both experimental and clinical studies. In the present study, we aimed to clarify whether HCII participates in the regulation of angiogenesis. Male heterozygous HCII-deficient (HCII+/−) mice and male littermate wild-type (HCII+/+) mice at the age of 12–16 weeks were subjected to unilateral hindlimb ligation surgery. Laser speckle blood flow analysis showed that blood flow recovery in response to hindlimb ischemia was delayed in HCII+/− mice compared with that in HCII+/+ mice. Capillary number, arteriole number, and endothelial nitric-oxide synthase (eNOS), AMP-activated protein kinase (AMPK), and liver kinase B1 (LKB1) phosphorylation in ischemic muscles were decreased in HCII+/− mice. Human purified HCII (h-HCII) administration almost restored blood flow recovery, capillary density, and arteriole number as well as phosphorylation levels of eNOS, AMPK, and LKB1 in ischemic muscles of HCII+/− mice. Although treatment with h-HCII increased phosphorylation levels of eNOS, AMPK, and LKB1 in human aortic endothelial cells (HAECs), the h-HCII-induced eNOS phosphorylation was abolished by compound C, an AMPK inhibitor, and by AMPK siRNA. In a similar fashion, tube formation, proliferation, and migration of HAECs were also promoted by h-HCII treatment and were abrogated by pretreatment with compound C. HCII potentiates the activation of vascular endothelial cells and the promotion of angiogenesis in response to hindlimb ischemia via an AMPK-eNOS signaling pathway. These findings suggest that HCII is a novel therapeutic target for treatment of patients with peripheral circulation insufficiency.
Introduction
Heparin cofactor II (HCII),2 a serine protease inhibitor (serpin), is secreted from hepatocytes and circulates systemically at a concentration of ∼1.0 μm (1). A major role of HCII is exerting its antithrombin action (1, 2). At the injured vascular wall of an atherosclerotic lesion, thrombin action via activation of protease-activated receptors (PARs), especially PAR-1, is enhanced (3), and thrombin action is inhibited by HCII. Antithrombin, a member of the serpin family, inactivates coagulation-related proteases including thrombin, factor Xa, and factor IXa, whereas HCII inhibits only thrombin activation without affecting other proteases involved in cascades of blood coagulation (4). HCII inactivates thrombin by producing a bimolecular complex with dermatan sulfate, and the complex is deposited in the injured vascular wall (5, 6).
We have reported that an elderly female patient with heterozygous HCII deficiency manifested multiple and advanced atherosclerosis (7). To elucidate the clinical significance of HCII in the development of atherosclerosis, we and others have conducted clinical studies and have reported that plasma HCII activity is inversely associated with carotid artery atherosclerosis (8) and with in-stent restenosis of the coronary artery (9) and femoropopliteal artery (10). In experimental animal studies, we and Vicente et al. revealed that HCII-deficient mice showed more prominent intimal hyperplasia after vascular injury than did in wild-type (WT) littermates (11, 12). In addition, plasma HCII activity is inversely associated with acceleration of human and murine cardiac remodeling including atrial enlargement and left ventricular concentric alteration (13, 14). These findings revealed that HCII plays a protective role in the development of vascular remodeling and cardiac remodeling in humans and mice.
Because angiogenesis is an important response for rescue and recovery of ischemic organs (15), promoting angiogenesis is an essential therapeutic strategy in patients with peripheral circulation insufficiency, including peripheral arterial disease and thromboangiitis obliterans. Recently, we have reported that plasma HCII activity is inversely correlated to the prevalence of peripheral arterial disease and that HCII is an independent inhibitory factor against peripheral arterial disease in elderly patients with cardiovascular risk factors (16). From those results, we hypothesized that HCII is involved in the process of angiogenesis in ischemic tissues. To clarify this issue, we examined the effect of HCII action on angiogenesis by using a hindlimb ischemia mice model with or without HCII deficiency and human aortic endothelial cells (HAECs).
EXPERIMENTAL PROCEDURES
Animal Preparation and Ischemic Hindlimb Model
All experimental procedures were performed in accordance with the guidelines of the Animal Research Committee, the University of Tokushima Graduate School. We generated HCII-deficient mice by the method of targeted disruption of the HCII gene (12). HCII+/− mice were backcrossed for 10 generations with the C57BL/6J strain (12). Because our homozygote HCII-deficient mice were embryonically lethal, we used male heterozygote HCII-deficient (HCII+/−) mice and male littermate WT (HCII+/+) mice in all experiments of this study as in our previous studies (12, 14). Our HCII+/− mice showed approximately half of the plasma HCII activity of littermate WT mice, and there is no obviously different phenotype in either HCII+/+ mice or HCII+/− mice at base line (12). The procedure for ischemic hindlimb surgery has been described in detail previously (17).
Antibodies and Reagents
The following commercially available antibodies were purchased for this study: anti-phospho-AMP-activated protein kinase (AMPK) (Thr172), anti-total AMPK, anti-phosphoendothelial nitric-oxide synthase (eNOS) (Ser1177), and anti-phospholiver kinase B1 (LKB1) (Ser428) (Cell Signaling Technology, Beverly, MA); anti-total eNOS and anti-total LKB1 (Santa Cruz Biotechnology, Santa Cruz, CA); anti-α-tubulin as a loading control (Calbiochem); anti-CD31 (PECAM-1) (BD Biosciences); anti-α smooth muscle actin (α-SMA) (Sigma-Aldrich); and anti-hypoxia-inducible factor-1α (HIF-1α) (Cayman Chemical, Ann Arbor, MI). Compound C was purchased from Calbiochem. Matrigel was obtained from BD Biosciences. Human plasma thrombin was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan).
Analysis of Peripheral Blood Flow
Measurement of blood flow in the hindlimb was performed before surgery and on postoperative days 0, 3, 7, 14, and 28 by using a laser speckle blood flow (LSBF) analyzing system (OMEGA ZONE; Omega Wave Co., Tokyo, Japan) (17).
Evaluation of Capillary Density and Number of Arterioles
Capillary density was evaluated by immunohistochemistry with CD31 antibody. In brief, adductor muscles at 14 days after surgery were embedded in OCT compound and frozen in liquid nitrogen-cold isopentane. Samples were sliced into 7-μm-thick sections and stained by CD31 immunohistochemistry with counterstaining of hematoxylin. Capillary density was expressed as the number of CD31-positive cells/high power field (×400) corrected by the number of muscle fibers (17). The density of arterioles was estimated by immunofluorescence staining with α-SMA antibody. Quantification of arteriole number was expressed as the number of α-SMA-positive cells/mm2 of muscle area as described previously (17).
Western Blotting
The methods for protein extraction from HAECs and skeletal muscles at 7 days after surgery were described previously (17, 18). The obtained protein was used for Western blotting.
Blood Pressure Measurement
The tail cuff method was used for measurement of blood pressure as described in detail previously (19).
Endothelial Cell Culture
The method used for culture of HAECs was described previously (17). In some experiments, cells were pretreated with compound C (2 μm) or a vehicle alone for 1 h before stimulation with human purified HCII protein (h-HCII) (20). Cells were also cultured with vehicle or h-HCII for 1 h and then stimulated by thrombin 2 μl/ml with vehicle or h-HCII for 24 h.
Tube Formation Assay
The tube formation assay was performed as described previously (17). In brief, each well in 96-well culture plates was coated with Matrigel (BD Biosciences). HAECs were seeded in coated wells of the plate in the presence of either h-HCII or the vehicle, with or without compound C (2 μm), and incubated in a cell culture incubator for 6 h. Tube formation was captured with a CCD camera and evaluated to measure tube length.
Cell Migration Assay
Cell migration was assessed by using a modified Boyden chamber assay (17). Briefly, cells were pretreated with or without compound C for 1 h, and cell suspensions were added to the Transwell insert and cultured in the presence of h-HCII or the vehicle with or without compound C for 6 h. Cells that migrated on the lower surface of the membrane were fixed, stained, and quantified in five random microscopic fields/well.
Cell Proliferation Assay
Cell proliferation was assessed by an MTS reagent (The CellTiter 96 Aqueous, Promega KK, Tokyo, Japan). HAECs were pretreated with compound C or the vehicle for 1 h and then incubated in the presence of h-HCII or the vehicle with or without compound C for 8 h. Absorbance of each solution was measured at 490 nm on a plate reader at 1 h after addition of the MTS reagent according to the manufacturer's instructions (17).
siRNA Transfection
siRNAs targeting human AMPKα1 and LKB1 and an unrelated-sequence control siRNA were purchased from Sigma-Aldrich, and siRNA transfection was performed as described previously (21). In brief, HAECs were transfected for 24 h with siRNA by using LipofectamineTM RNAiMAX in Opti-MEM (Invitrogen). Then the medium was replaced with EBM-2, after which the cells were incubated for 24 h. For all experiments, transfected HAECs were used 48 h after transfection. The silencing effects of AMPK and LKB1 were more than 50 and 90%, respectively.
Statistical Analysis
All data are expressed as means ± S.E. For comparisons among groups, the statistical significance of each difference was assessed by post hoc testing using Dunnett's method or the Tukey-Kramer method. Statistical significance was considered at p < 0.05.
RESULTS
Insufficient Blood Flow Recovery in HCII+/− Mice after Hindlimb Ischemia
To analyze the effect of HCII on in vivo angiogenesis, we used mice models with unilateral hindlimb ischemia. There were no differences in systolic blood pressure or pulse beat at base line (14) and at 1 week after ischemic surgery between HCII+/− mice and HCII+/+ mice (see supplemental Table 1). Representative LSBF images of blood flow in the hindlimb are shown in Fig. 1A. Serial LSBF analysis showed that blood flow restoration after hindlimb ischemia was delayed in HCII+/− mice compared with that in HCII+/+ mice. Quantitative evaluation of blood flow by LSBF also showed diminished blood flow recovery in the hindlimbs of HCII+/− mice compared with that in HCII+/+ mice from 7 days after ischemic hindlimb surgery. At 28 days after the operation, the ratio of ischemic side to nonischemic side was significantly lower in HCII+/− mice than in HCII+/+ mice (66% versus 84%, p < 0.01) (Fig. 1A).
FIGURE 1.

Evaluation of revascularization in ischemic hindlimbs of HCII+/+ and HCII+/− mice. A. Left, representative LSBF images of ischemic hindlimbs in HCII+/+ and HCII+/− mice. Right, quantitative analysis for LSBF ratio of ischemic hindlimb to control hindlimb in HCII+/+ and HCII+/− mice before surgery and on postoperative days 0, 3, 7, 14, and 28. Values are expressed as means ± S.E. (error bars). *, p < 0.05 and **, p < 0.01 versus HCII+/+ mice, n = 6–8 in each group. B. Upper, representative section of anti-CD31 immunohistochemical staining in ischemic hindlimbs of HCII+/+ and HCII+/− mice. Lower, quantification of capillary density in HCII+/+ and HCII+/− mice at 14 days after surgery. Values are expressed as means ± S.E. *, p < 0.05 and **, p < 0.01, n = 5 in each group. C. Upper, representative immunofluorescence staining with anti-α-SMA in ischemic adductor muscles of HCII+/+ and HCII+/− mice. Lower, quantification of arteriolar numbers in adductor muscles of HCII+/+ and HCII+/− mice. Values are expressed as means ± S.E. **, p < 0.01, n = 8 in each group.
Decreased Capillary Density and Number of Arterioles in Ischemic Muscle of HCII+/− Mice
To evaluate microcirculation and angiogenesis after ischemia in the adductor skeletal muscle, measurement of capillary density was performed. The ratio of CD31-positive cells, indicating the ratio of vascular endothelial cells to muscle fiber number, showed that the capillary density and number of arterioles were significantly reduced in HCII+/− mice compared with those in HCII+/+ mice (Fig. 1, B and C).
Diminished Phosphorylation of eNOS and AMPK in Ischemic Muscles of HCII+/− Mice
We performed Western blotting to evaluate phosphorylation of eNOS and AMPK in adductor muscles at 7 days after the operation. As shown in Fig. 2A, nonischemic adductor muscles showed no differences in eNOS or AMPK phosphorylation between HCII+/+ mice and HCII+/− mice. However, both eNOS phosphorylation and AMPK phosphorylation were significantly decreased in ischemic adductor muscles of HCII+/− mice compared with those in HCII+/+ mice. Taken together, the results indicate that HCII deficiency causes impaired activation of the AMPK-eNOS pathway in response to ischemia. In addition, we examined the expression levels of HIF-1α and VEGF in response to ischemia in HCII+/− mice. As shown in Fig. 2B, HCII+/− mice showed decreased levels of HIF-1α and VEGF expression in ischemic adductor muscles compared with the levels in HCII+/+ mice.
FIGURE 2.

Evaluation of activation and expression of proangiogenic factors in ischemic hindlimbs of HCII+/+ and HCII+/− mice. A, phosphorylated levels of eNOS and AMPK in ischemic hindlimbs of HCII+/+ and HCII+/− mice. Left, representative blots of phospho- and total eNOS, phospho- and total AMPK, and tubulin at 7 days after surgery. Right, densitometric analysis of eNOS and AMPK phosphorylation. Values are expressed as means ± S.E. (error bars). *, p < 0.05 and **, p < 0.01, n = 7 in each group. B, expression levels of HIF-1α and VEGF in hindlimb ischemia of HCII+/+ and HCII+/− mice. Left, representative blots of HIF-1α, VEGF, and tubulin at 7 days after surgery. Right, densitometric analysis of HIF-1α and VEGF expression. Values are expressed as means ± S.E. *, p < 0.05 and **, p < 0.01, n = 7 in each group.
HCII Supplementation Restores Revascularization via AMPK-eNOS Signaling in Ischemic Muscles of HCII+/− Mice
To confirm direct action of HCII on angiogenesis in vivo, we tested whether HCII treatment can improve insufficient angiogenesis after ischemic surgery in HCII+/− mice. HCII+/− mice were administered h-HCII protein (45 mg/kg) 1 day before surgery and three times/week after surgery for 4 weeks as previously described (12, 14). As can be seen in Fig. 3A, HCII+/− mice with h-HCII supplementation showed significant amelioration of blood flow recovery after the ischemic operation compared with HCII+/− mice with vehicle treatment (75% versus 62% at day 28 after the operation, p < 0.01). HCII administration also increased capillary density and the number of arterioles in ischemic muscles (Fig. 3, B and C). In addition, eNOS and AMPK phosphorylation was significantly increased in ischemic adductor muscles of HCII+/− mice with h-HCII treatment compared with those in HCII+/− mice with vehicle treatment (Fig. 4A). Similarly, the expression levels of HIF-1α and VEGF in ischemic muscles were restored in HCII+/− mice with h-HCII supplementation (Fig. 4B). These results suggest that HCII exerts proangiogenic potency via an AMPK-eNOS-dependent pathway in vivo.
FIGURE 3.

Assessment of revascularization in ischemic hindlimbs of HCII+/− mice with h-HCII supplementation or vehicle treatment. A. Left, representative LSBF images of hindlimbs in HCII+/− mice with h-HCII or vehicle treatment after ischemic surgery. Right, quantitative analysis for LSBF ratio of ischemic hindlimb to control hindlimb in HCII+/− mice with h-HCII or vehicle treatment before surgery and on postoperative days 0, 3, 7, 14, and 28. Values are expressed as means ± S.E. (error bars). *, p < 0.05 and **, p < 0.01 versus HCII+/− mice with vehicle, n = 5 in each group. B. Upper, representative section of anti-CD31 immunohistochemical staining in ischemic or control hindlimbs of HCII+/− mice with h-HCII or vehicle treatment. Lower, quantification of capillary density in ischemic or control adductor muscles of HCII+/− mice with h-HCII or vehicle treatment at 14 days after surgery. Values are expressed as means ± S.E. *, p < 0.05 and **, p < 0.01, n = 4 in each group. C. Upper, representative immunofluorescence staining with anti-α-SMA in ischemic or control adductor muscles of HCII+/− mice with vehicle or h-HCII administration. Lower, quantification of arteriolar numbers in adductor muscles of HCII+/− mice with vehicle or h-HCII administration. Values are expressed as means ± S.E. **, p < 0.01, n = 8 in each group.
FIGURE 4.

Evaluation of activation and expression of proangiogenic factors in ischemic hindlimbs of HCII+/+ mice, HCII+/− mice with vehicle administration and HCII+/− mice with h-HCII administration. A, effect of HCII administration on phosphorylated levels of eNOS and AMPK in ischemic adductor muscle. Left, representative blots of phospho- and total eNOS, phospho- and total AMPK, and tubulin at 7 days after surgery in ischemic muscles in HCII+/+ mice, HCII+/− mice with vehicle administration, and HCII+/− mice with h-HCII administration. Right, densitometric analysis of eNOS and AMPK phosphorylation. Values are expressed as means ± S.E. (error bars). *, p < 0.05 and **, p < 0.01, n = 8 in each group. B, expression levels of HIF-1α and VEGF in ischemic adductor muscles of HCII+/− mice with vehicle or h-HCII supplementation. Left, representative blots of HIF-1α, VEGF, and tubulin at 7 days after surgery. Right, densitometric analysis of HIF-1α and VEGF expression. Values are expressed as means ± S.E. *, p < 0.05 and **, p < 0.01, n = 6–7 in each group.
HCII Action on eNOS Phosphorylation in HAECs
Next, we examined HCII-induced eNOS phosphorylation in HAECs. As shown in Fig. 5A, h-HCII stimulated eNOS phosphorylation at 60 and 120 min. AMPK phosphorylation was also induced by h-HCII treatment, although h-HCII did not increase Akt phosphorylation (data not shown). In addition, the h-HCII supplementation augmented eNOS and AMPK phosphorylation in a dose-dependent manner (Fig. 5B). Because AMPK has been shown to be an upstream kinase and regulator in NOS activation (22, 23), we tested whether HCII-induced eNOS phosphorylation was involved in AMPK signaling by using compound C, an AMPK inhibitor. The h-HCII-induced eNOS activation was also completely suppressed by pretreatment with compound C as well as by silencing of AMPK (Fig. 5, C and D). From these results, HCII stimulates eNOS phosphorylation via an AMPK-dependent mechanism.
FIGURE 5.

Analysis of HCII action on eNOS and AMPK phosphorylation in HAECs. A, time-dependent changes in eNOS phosphorylation after h-HCII administration (1 μm). Left, representative blots of phospho-eNOS, total eNOS, phospho-AMPK, total AMPK, and tubulin. Middle, results of densitometric analysis of eNOS phosphorylation Right, results of densitometric analysis of AMPK phosphorylation. Values are expressed as means ± S.E. (error bars). *, p < 0.05 and **, p < 0.01 versus 0 min, n = 4 in each group. B, dose-dependent changes in eNOS and AMPK phosphorylation in HAECs after h-HCII treatment. Left, representative blots of phospho-eNOS, total eNOS, phospho-AMPK, total AMPK, and tubulin. Middle, results of densitometric analysis of eNOS phosphorylation. Right, results of densitometric analysis of AMPK phosphorylation. Values are expressed as means ± S.E. *, p < 0.05 and **, p < 0.01 versus vehicle treatment, n = 8 in each group. C, compound C, an AMPK inhibitor, cancels h-HCII-induced eNOS phosphorylation. HAECs were pretreated with 2 μm compound C 1 h prior to administration of 1 μm h-HCII. Left, representative blots of phospho-eNOS, total eNOS, phospho-AMPK, total AMPK, and tubulin. Right, relative -fold change of phosphorylated levels of eNOS and AMPK determined by densitometric analysis. Values are expressed as means ± S.E. *, p < 0.05 and **, p < 0.01, n = 8 in each group. D, silencing of AMPK gene expression attenuates h-HCII-induced eNOS phosphorylation. HAECs were transfected with 5 nm AMPK siRNA or unrelated control siRNA. After 48 h, 1 μm h-HCII was administered to HAECs. Left, representative blots of phospho-eNOS, total eNOS, phospho-AMPK, total AMPK, and tubulin. Right, relative -fold change of phosphorylated and total levels of eNOS and AMPK determined by densitometric analysis. Values are expressed as means ± S.E. *, p < 0.05 and **, p < 0.01 versus treatment with h-HCII and unrelated siRNA, n = 8 in each group.
HCII Promotes Tube Formation, Proliferation, and Migration of Vascular Endothelial Cells
We performed in vitro assays including tube formation, cell proliferation, and cell migration assays to assess the influence of HCII on vascular endothelial cell activation. As shown in Fig. 6A, representative photographs demonstrated that h-HCII treatment promoted tube formation of HAECs, and quantitative evaluation of tube length showed acceleration of endothelial cell growth by h-HCII treatment (Fig. 6A). Cell proliferation and migration were also augmented by h-HCII treatment (Fig. 6, B and C). Treatment with compound C diminished HCII-stimulated tube formation, cell proliferation, and cell migration (Fig. 6). Taken together, the results indicated that HCII promotes vascular endothelial cell differentiation, proliferation, and migration via an AMPK-mediated pathway.
FIGURE 6.
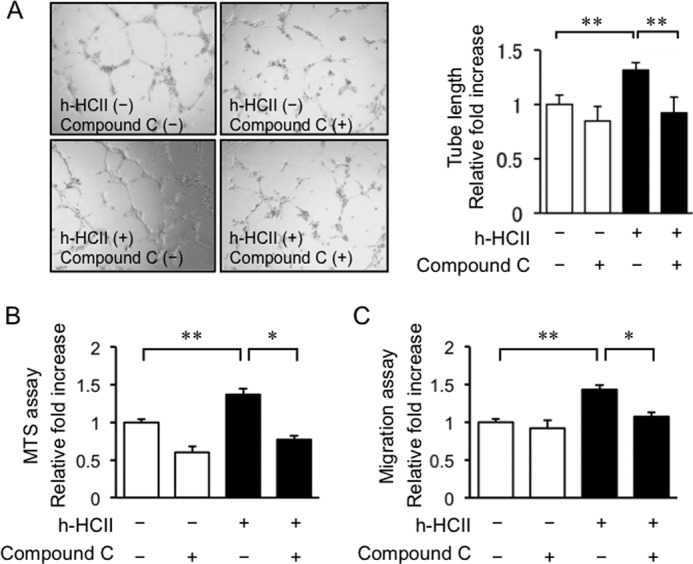
Analysis of functional responses of HAECs to h-HCII stimulation: tube formation (A), cell proliferation (B), and cell migration (C). Human HCII protein augmented tube formation, cell proliferation, and cell migration. These effects of HCII on HAECs were abolished by compound C pretreatment. Values are expressed as means ± S.E. (error bars). *, p < 0.05 and **, p < 0.01, n = 4–6 in each group.
HCII-accelerated AMPK-eNOS Phosphorylation Is Mediated through LKB1
LKB1 is one of the upstream kinases of regulating of AMPKs, and LKB1 is a key regulator of angiogenesis through the AMPK-eNOS pathway (24). We tested whether LKB1 was involved in the HCII-AMPK-eNOS pathway. Heterozygous HCII knock-out mice showed reduced LKB1 phosphorylation in ischemic adductor muscles, which was restored by h-HCII supplementation (Fig. 7, A and B). In an in vitro study, HCII stimulated LKB1 phosphorylation in HAECs (Fig. 7C). These findings suggested that HCII promoted eNOS phosphorylation through a LKB1-AMPK-mediated pathway.
FIGURE 7.

Analysis of HCII action on LKB1 phosphorylation and thrombin-induced interruption of AMPK-eNOS signaling. A, phosphorylated levels of LKB1 in ischemic hindlimbs of HCII+/+ and HCII+/− mice. Upper, representative blots of phospho- and total LKB1 and tubulin at 7 days after surgery. Lower, densitometric analysis of LKB1 phosphorylation. Values are expressed as means ± S.E. (error bars). *, p < 0.05 and **, p < 0.01, n = 8 in each group. B, effect of h-HCII administration on phosphorylated levels of LKB1 in ischemic adductor muscle. Upper, representative blots of phospho- and total LKB1 and tubulin at 7 days after surgery in ischemic muscles in HCII+/+ mice, HCII+/− mice with vehicle administration and HCII+/− mice with h-HCII administration. Lower, densitometric analysis of LKB1 phosphorylation. Values are expressed as means ± S.E. *, p < 0.05 and **, p < 0.01, n = 6–9 in each group. C, time-dependent changes in eNOS, AMPK, and LKB1 phosphorylation after h-HCII administration (1 μm). Left, representative blots of phospho-eNOS, total eNOS, phospho-AMPK, total AMPK, phospho-LKB1, total LKB1, and tubulin. Right, results of densitometric analysis of eNOS, AMPK, and LKB1 phosphorylation. Values are expressed as means ± S.E. *, p < 0.05 and **, p < 0.01 versus 0 min, n = 6 in each group. D, h-HCII protein suppressing thrombin-induced reduction in both eNOS and AMPK phosphorylation and expression. HAECs were pretreated with 1 μm h-HCII protein 1 h prior to stimulation with 2 units/ml human thrombin. Left, representative blots of phospho-eNOS, total eNOS, phospho-AMPK, total AMPK, and tubulin. Right, relative -fold change of phosphorylated and total levels of eNOS and AMPK determined by densitometric analysis. Values are expressed as means ± S.E. *, p < 0.05 and **, p < 0.01, n = 8 in each group.
HCII Restores Thrombin-induced Reduction of eNOS Phosphorylation and Expression in HAECs
Although a short exposure (<15 min) to thrombin increases eNOS phosphorylation through an AMPK-mediated pathway (25, 26), the thrombin-PAR1 pathway has been shown to inhibit eNOS production (27, 28) and eNOS phosphorylation (29). As shown in Fig. 7D, thrombin stimulation for 24 h diminished the phosphorylation and expression of eNOS as well as those of AMPK, which were restored by HCII administration in HAECs. These results confirmed that the preventive action of HCII against thrombin-induced reduction of AMPK-eNOS phosphorylation and expression contributes to the promotion of angiogenesis.
DISCUSSION
Patients and Animals with Peripheral Circulation Insufficiency Have Been Shown to Manifest Reduction of eNOS Activity and/or NO Bioavailability Leading to Endothelial Dysfunction (15, 30, 31)
Several stimuli, such as shear stress, adiponectin, and HMG-CoA reductase inhibitors, increase NO bioavailability through the phosphorylation of eNOS (32–34). These stimuli activate a number of protein kinases, including AMPK, protein kinase A (PKA) Akt, Ca2+/calmodulin-dependent protein kinase II, and PKG, which evoke phosphorylation of eNOS. AMPK has been shown to be crucial not only for regulation of metabolic homeostasis but also for regulation of homeostasis in the cardiovascular system via activating eNOS (35). Indeed, it has been reported that an HMG-CoA reductase inhibitor (36), fenofibrate (37) and adiponectin (38) enhance angiogenesis in response to hindlimb ischemia through activation of the AMPK-eNOS signaling pathway. Previous experiments on genetic or pharmacological inhibition of AMPK activity showed significant reduction in phosphorylation of eNOS, leading to decrease in NO production (39). Therefore, physiological activation of the AMPK-eNOS signaling pathway is required for maintenance of endothelial function and angiogenesis.
We have shown pathophysiological roles of HCII action such as protective effects against cardiovascular remodeling (8, 9, 12–14, 16). In addition, Huang et al. showed that plasma HCII activity is positively correlated with flow-mediated vasodilatation, a marker of endothelial function, suggesting that HCII increases NO bioavailability in humans (40). In the present study, we also revealed that HCII+/− mice showed impairment of revascularization after hindlimb ischemia with reduced eNOS phosphorylation, which was ameliorated by HCII supplementation. Supplementation of HCII enhanced AMPK-eNOS signaling, which promoted activation of endothelial cells including tube formation, migration, and proliferation in in vitro studies. Finally, we found that HCII-induced activation of endothelial cells was abolished by pretreatment with compound C as an AMPK inhibitor and by AMPK gene expression silencing, indicating that HCII action on eNOS activation is mediated by an AMPK-dependent mechanism.
In addition, LKB1 is known as a major kinase that causes phosphorylation of AMPK. Because AMPK activation by agonists, including 5-aminoimidazole-4-carboxamide ribonucleoside, metformin, and phenformin, was abolished in LKB1-deficient cells (41), HCII-induced up-regulation of AMPK-eNOS phosphorylation might be mediated through LKB1.
The present study also showed reduction of VEGF expression in skeletal muscle of HCII+/− mice compared with that in HCII+/+ mice after ischemia. Because Ouchi et al. have demonstrated that the AMPK-p38 mitogen-activated protein kinase signaling cascade increases skeletal muscle VEGF expression, leading to promotion of angiogenesis in response to ischemic injury (42), insufficient AMPK activation may have failed to accelerate VEGF expression in ischemic skeletal muscle of HCII+/− mice. In addition, the impaired angiogenesis in eNOS−/− mice was not restored by vascular endothelial growth factor (VEGF) stimulation, suggesting that eNOS is positioned downstream of VEGF and is essential for angiogenesis after ischemia.
Pathophysiological roles of the endothelial thrombin-PAR-1 axis have been shown to be associated with endothelium-dependent contraction/relaxation, endothelial permeability/barrier function, leukocyte adhesion, angiogenesis, endothelial cell migration, proliferation, and exocytosis leading to vascular inflammation (43). In physiological conditions, thrombin contributes to activation of eNOS phosphorylation leading to promotion of NO production (25, 26). In contrast, excessive thrombin production in pathological conditions such as atherothrombosis results in reduction of NO bioavailability via enhancement of oxidative stress. Indeed, Watts et al. reported that thrombin causes phosphorylation of eNOS-Thr495, which is known as a negative regulatory site of eNOS, leading to inhibition of NO production (28). eNOS phosphorylation and expression in the aorta were reduced by thrombin injection (29). Thus, the thrombin-PAR-1 axis has considerable influence on NO bioavailability in both physiological and pathological conditions, although the relationship between the thrombin-PAR-1 axis and AMPK signaling pathway has remained controversial. In the present study, thrombin stimulation for 24 h diminished the phosphorylation and expression of eNOS as well as those of AMPK, which were restored by HCII administration. HCII exerted a preventive action against thrombin-induced reduction of AMPK-eNOS phosphorylation and expression.
Although further investigations to clarify the detailed molecular mechanism of HCII action on AMPK activation are needed, our results are consistent with the notion that HCII promotes vascular endothelial function via an AMPK-eNOS-mediated pathway, leading to enhancement of angiogenesis after ischemia. Therefore, HCII might be a novel therapeutic target for patients with insufficient peripheral circulation.

This article contains supplemental Table 1.
- HCII
- heparin cofactor II
- AMPK
- AMP-activated protein kinase
- eNOS
- endothelial NOS
- HAEC
- human aortic endothelial cell
- h-HCII
- human purified HCII
- HIF
- hypoxia-inducible factor
- LKB
- liver kinase B
- LSBF
- laser speckle blood flow
- PAR
- protease-activated receptor
- SMA
- smooth muscle actin
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt.
REFERENCES
- 1. Tollefsen D. M., Majerus D. W., Blank M. K. (1982) Heparin cofactor II: purification and properties of a heparin-dependent inhibitor of thrombin in human plasma. J. Biol. Chem. 257, 2162–2169 [PubMed] [Google Scholar]
- 2. Tollefsen D. M., Pestka C. A., Monafo W. J. (1983) Activation of heparin cofactor II by dermatan sulfate. J. Biol. Chem. 258, 6713–6716 [PubMed] [Google Scholar]
- 3. Derian C. K., Damiano B. P., D'Andrea M. R., Andrade-Gordon P. (2002) Thrombin regulation of cell function through protease-activated receptors: implications for therapeutic intervention. Biochemistry 67, 56–64 [DOI] [PubMed] [Google Scholar]
- 4. Parker K. A., Tollefsen D. M. (1985) The protease specificity of heparin cofactor II: inhibition of thrombin generated during coagulation. J. Biol. Chem. 260, 3501–3505 [PubMed] [Google Scholar]
- 5. Tollefsen D. M. (2007) Heparin cofactor II modulates the response to vascular injury. Arterioscler. Thromb. Vasc. Biol. 27, 454–460 [DOI] [PubMed] [Google Scholar]
- 6. Shirk R. A., Parthasarathy N., San Antonio J. D., Church F. C., Wagner W. D. (2000) Altered dermatan sulfate structure and reduced heparin cofactor II-stimulating activity of biglycan and decorin from human atherosclerotic plaque. J. Biol. Chem. 275, 18085–18092 [DOI] [PubMed] [Google Scholar]
- 7. Kanagawa Y., Shigekiyo T., Aihara K., Akaike M., Azuma H., Matsumoto T. (2001) Molecular mechanism of type I congenital heparin cofactor (HC) II deficiency caused by a missense mutation at reactive P2 site: HC II Tokushima. Thromb. Haemost. 85, 101–107 [PubMed] [Google Scholar]
- 8. Aihara K., Azuma H., Takamori N., Kanagawa Y., Akaike M., Fujimura M., Yoshida T., Hashizume S., Kato M., Yamaguchi H., Kato S., Ikeda Y., Arase T., Kondo A., Matsumoto T. (2004) Heparin cofactor II is a novel protective factor against carotid atherosclerosis in elderly individuals. Circulation 109, 2761–2765 [DOI] [PubMed] [Google Scholar]
- 9. Takamori N., Azuma H., Kato M., Hashizume S., Aihara K., Akaike M., Tamura K., Matsumoto T. (2004) High plasma heparin cofactor II activity is associated with reduced incidence of in-stent restenosis after percutaneous coronary intervention. Circulation 109, 481–486 [DOI] [PubMed] [Google Scholar]
- 10. Schillinger M., Exner M., Sabeti S., Mlekusch W., Amighi J., Handler S., Quehenberger P., Kalifeh N., Wagner O., Minar E. (2004) High plasma heparin cofactor II activity protects from restenosis after femoropopliteal stenting. Thromb. Haemost. 92, 1108–1113 [DOI] [PubMed] [Google Scholar]
- 11. Vicente C. P., He L., Tollefsen D. M. (2007) Accelerated atherogenesis and neointima formation in heparin cofactor II-deficient mice. Blood 110, 4261–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aihara K., Azuma H., Akaike M., Ikeda Y., Sata M., Takamori N., Yagi S., Iwase T., Sumitomo Y., Kawano H., Yamada T., Fukuda T., Matsumoto T., Sekine K., Sato T., Nakamichi Y., Yamamoto Y., Yoshimura K., Watanabe T., Nakamura T., Oomizu A., Tsukada M., Hayashi H., Sudo T., Kato S. (2007) Strain-dependent embryonic lethality and exaggerated vascular remodeling in heparin cofactor II-deficient mice. J. Clin. Invest. 117, 1514–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ise T., Aihara K., Sumitomo-Ueda Y., Yoshida S., Ikeda Y., Yagi S., Iwase T., Yamada H., Akaike M., Sata M., Matsumoto T. (2011) Plasma heparin cofactor II activity is inversely associated with left atrial volume and diastolic dysfunction in humans with cardiovascular risk factors. Hypertens. Res. 34, 225–231 [DOI] [PubMed] [Google Scholar]
- 14. Sumitomo-Ueda Y., Aihara K., Ise T., Yoshida S., Ikeda Y., Uemoto R., Yagi S., Iwase T., Ishikawa K., Hirata Y., Akaike M., Sata M., Kato S., Matsumoto T. (2010) Heparin cofactor II protects against angiotensin II-induced cardiac remodeling via attenuation of oxidative stress in mice. Hypertension 56, 430–436 [DOI] [PubMed] [Google Scholar]
- 15. Murohara T., Asahara T., Silver M., Bauters C., Masuda H., Kalka C., Kearney M., Chen D., Symes J. F., Fishman M. C., Huang P. L., Isner J. M. (1998) Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J. Clin. Invest. 101, 2567–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aihara K., Azuma H., Akaike M., Kurobe H., Takamori N., Ikeda Y., Sumitomo Y., Yoshida S., Yagi S., Iwase T., Ishikawa K., Sata M., Kitagawa T., Matsumoto T. (2009) Heparin cofactor II is an independent protective factor against peripheral arterial disease in elderly subjects with cardiovascular risk factors. J. Atheroscler. Thromb. 16, 127–134 [DOI] [PubMed] [Google Scholar]
- 17. Ikeda Y., Tajima S., Yoshida S., Yamano N., Kihira Y., Ishizawa K., Aihara K., Tomita S., Tsuchiya K., Tamaki T. (2011) Deferoxamine promotes angiogenesis via the activation of vascular endothelial cell function. Atherosclerosis 215, 339–347 [DOI] [PubMed] [Google Scholar]
- 18. Ikeda Y., Ohashi K., Shibata R., Pimentel D. R., Kihara S., Ouchi N., Walsh K. (2008) Cyclooxygenase-2 induction by adiponectin is regulated by a sphingosine kinase-1-dependent mechanism in cardiac myocytes. FEBS Lett. 582, 1147–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ikeda Y., Aihara K., Sato T., Akaike M., Yoshizumi M., Suzaki Y., Izawa Y., Fujimura M., Hashizume S., Kato M., Yagi S., Tamaki T., Kawano H., Matsumoto T., Azuma H., Kato S. (2005) Androgen receptor gene knockout male mice exhibit impaired cardiac growth and exacerbation of angiotensin II-induced cardiac fibrosis. J. Biol. Chem. 280, 29661–29666 [DOI] [PubMed] [Google Scholar]
- 20. Yamanaga K., Yuuki T., Tsukada M., Koshiba H., Nakajima T., Takechi K., Nakamura N. (2000) Heparin cofactor II inhibits thrombus formation in a rat thrombosis model. Thromb. Res. 98, 95–101 [DOI] [PubMed] [Google Scholar]
- 21. Ikeda Y., Aihara K., Akaike M., Sato T., Ishikawa K., Ise T., Yagi S., Iwase T., Ueda Y., Yoshida S., Azuma H., Walsh K., Tamaki T., Kato S., Matsumoto T. (2010) Androgen receptor counteracts doxorubicin-induced cardiotoxicity in male mice. Mol. Endocrinol. 24, 1338–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen Z. P., Mitchelhill K. I., Michell B. J., Stapleton D., Rodriguez-Crespo I., Witters L. A., Power D. A., Ortiz de Montellano P. R., Kemp B. E. (1999) AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 443, 285–289 [DOI] [PubMed] [Google Scholar]
- 23. Morrow V. A., Foufelle F., Connell J. M., Petrie J. R., Gould G. W., Salt I. P. (2003) Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J. Biol. Chem. 278, 31629–31639 [DOI] [PubMed] [Google Scholar]
- 24. Ohashi K., Ouchi N., Higuchi A., Shaw R. J., Walsh K. (2010) LKB1 deficiency in Tie2-Cre-expressing cells impairs ischemia-induced angiogenesis. J. Biol. Chem. 285, 22291–22298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thors B., Halldórsson H., Thorgeirsson G. (2004) Thrombin and histamine stimulate endothelial nitric-oxide synthase phosphorylation at Ser1177 via an AMPK-mediated pathway independent of PI3K-Akt. FEBS Lett. 573, 175–180 [DOI] [PubMed] [Google Scholar]
- 26. Stahmann N., Woods A., Carling D., Heller R. (2006) Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase β. Mol. Cell. Biol. 26, 5933–5945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eto M., Barandiér C., Rathgeb L., Kozai T., Joch H., Yang Z., Lüscher T. F. (2001) Thrombin suppresses endothelial nitric-oxide synthase and up-regulates endothelin-converting enzyme-1 expression by distinct pathways: role of Rho/ROCK and mitogen-activated protein kinase. Circ. Res. 89, 583–590 [DOI] [PubMed] [Google Scholar]
- 28. Watts V. L., Motley E. D. (2009) Role of protease-activated receptor-1 in endothelial nitric-oxide synthase-Thr495 phosphorylation. Exp. Biol. Med. 234, 132–139 [DOI] [PubMed] [Google Scholar]
- 29. Ohkawara H., Ishibashi T., Saitoh S., Inoue N., Sugimoto K., Kamioka M., Uekita H., Kaneshiro T., Ando K., Takuwa Y., Maruyama Y., Takeishi Y. (2010) Preventive effects of pravastatin on thrombin-triggered vascular responses via Akt/eNOS and RhoA/Rac1 pathways in vivo. Cardiovasc. Res. 88, 492–501 [DOI] [PubMed] [Google Scholar]
- 30. Brevetti G., Schiano V., Chiariello M. (2008) Endothelial dysfunction: a key to the pathophysiology and natural history of peripheral arterial disease? Atherosclerosis 197, 1–11 [DOI] [PubMed] [Google Scholar]
- 31. Mees B., Wagner S., Ninci E., Tribulova S., Martin S., van Haperen R., Kostin S., Heil M., de Crom R., Schaper W. (2007) Endothelial nitric-oxide synthase activity is essential for vasodilation during blood flow recovery but not for arteriogenesis. Arterioscler. Thromb. Vasc. Biol. 27, 1926–1933 [DOI] [PubMed] [Google Scholar]
- 32. Dimmeler S., Fleming I., Fisslthaler B., Hermann C., Busse R., Zeiher A. M. (1999) Activation of nitric-oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601–605 [DOI] [PubMed] [Google Scholar]
- 33. Chen H., Montagnani M., Funahashi T., Shimomura I., Quon M. J. (2003) Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J. Biol. Chem. 278, 45021–45026 [DOI] [PubMed] [Google Scholar]
- 34. Harris M. B., Blackstone M. A., Sood S. G., Li C., Goolsby J. M., Venema V. J., Kemp B. E., Venema R. C. (2004) Acute activation and phosphorylation of endothelial nitric-oxide synthase by HMG-CoA reductase inhibitors. Am. J. Physiol. Heart Circ. Physiol. 287, H560–566 [DOI] [PubMed] [Google Scholar]
- 35. Levine Y. C., Li G. K., Michel T. (2007) Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells: evidence for an AMPK → Rac1 → Akt → endothelial nitric-oxide synthase pathway. J. Biol. Chem. 282, 20351–20364 [DOI] [PubMed] [Google Scholar]
- 36. Izumi Y., Shiota M., Kusakabe H., Hikita Y., Nakao T., Nakamura Y., Muro T., Miura K., Yoshiyama M., Iwao H. (2009) Pravastatin accelerates ischemia-induced angiogenesis through AMP-activated protein kinase. Hypertens. Res. 32, 675–679 [DOI] [PubMed] [Google Scholar]
- 37. Li P., Shibata R., Maruyama S., Kondo M., Ohashi K., Ouchi N., Murohara T. (2010) Fenofibrate promotes ischemia-induced revascularization through the adiponectin-dependent pathway. Am. J. Physiol. Endocrinol. Metab. 299, E560–566 [DOI] [PubMed] [Google Scholar]
- 38. Kondo M., Shibata R., Miura R., Shimano M., Kondo K., Li P., Ohashi T., Kihara S., Maeda N., Walsh K., Ouchi N., Murohara T. (2009) Caloric restriction stimulates revascularization in response to ischemia via adiponectin-mediated activation of endothelial nitric-oxide synthase. J. Biol. Chem. 284, 1718–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen Z., Peng I. C., Sun W., Su M. I., Hsu P. H., Fu Y., Zhu Y., DeFea K., Pan S., Tsai M. D., Shyy J. Y. (2009) AMP-activated protein kinase functionally phosphorylates endothelial nitric-oxide synthase Ser633. Circ. Res. 104, 496–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang P. H., Leu H. B., Chen J. W., Wu T. C., Lu T. M., Yu-An Ding P., Lin S. J. (2007) Decreased heparin cofactor II activity is associated with impaired endothelial function determined by brachial ultrasonography and predicts cardiovascular events. Int. J. Cardiol. 114, 152–158 [DOI] [PubMed] [Google Scholar]
- 41. Hawley S. A., Boudeau J., Reid J. L., Mustard K. J., Udd L., Mäkelä T. P., Alessi D. R., Hardie D. G. (2003) Complexes between the LKB1 tumor suppressor, STRAD α/β and MO25 α/β are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ouchi N., Shibata R., Walsh K. (2005) AMP-activated protein kinase signaling stimulates VEGF expression and angiogenesis in skeletal muscle. Circ. Res. 96, 838–846 [DOI] [PubMed] [Google Scholar]
- 43. Minami T., Sugiyama A., Wu S. Q., Abid R., Kodama T., Aird W. C. (2004) Thrombin and phenotypic modulation of the endothelium. Arterioscler. Thromb. Vasc. Biol. 24, 41–53 [DOI] [PubMed] [Google Scholar]