

Background: The urokinase receptor (uPAR) is a modular receptor containing three LU domains.
Results: Ligand-free uPAR is inherently flexible with a detached N-terminal domain (DI).
Conclusion: Allosteric regulation of uPAR is driven by uPA-induced compaction of the intact receptor and a concomitant stabilization of DI.
Significance: This flexibility and ligand-induced allostery are expected to impact future studies on uPAR function and targeted intervention.
Keywords: Allosteric Regulation, Extracellular Matrix, Protein Dynamics, Protein-Protein Interactions, Tumor Microenvironment, Urokinase Receptor, SAXS, Hydrogen-Deuterium Exchange, Surface Plasmon Resonance, Vitronectin, CD87, Cancer Invasion and Metastasis, Cell Adhesion and Migration
Abstract
The urokinase-type plasminogen activator receptor (uPAR) provides a rendezvous between proteolytic degradation of the extracellular matrix and integrin-mediated adhesion to vitronectin. These processes are, however, tightly linked because the high affinity binding of urokinase regulates the binding of uPAR to matrix-embedded vitronectin. Although crystal structures exist to define the corresponding static bi- and trimolecular receptor complexes, it is evident that the dynamic property of uPAR plays a decisive role in its function. In the present study, we combine small angle x-ray scattering, hydrogen-deuterium exchange, and surface plasmon resonance to develop a structural model describing the allosteric regulation of uPAR. We show that the flexibility of its N-terminal domain provides the key for understanding this allosteric mechanism. Importantly, our model has direct implications for understanding uPAR-assisted cell adhesion and migration as well as for translational research, including targeted intervention therapy and non-invasive tumor imaging in vivo.
Introduction
The urokinase-type plasminogen activator receptor (uPAR)3 focuses uPA-mediated plasminogen activation to cell surfaces in vivo, which plays an important physiological role in, for example, hepatic fibrin clearance and liver homeostasis by attenuating fibrin-induced chronic inflammation (1). Several studies have also implicated uPAR in pathological conditions, such as focal segmental glomerulosclerosis (2, 3), paroxysmal nocturnal hemoglobinuria (4), rheumatoid arthritis (5), and cancer invasion and metastasis (6–8). In the latter group of diseases, uPAR is predominantly expressed by cells residing in the tumor-stromal interface of the invasive front, and high levels of uPAR in either resected tumor tissue or circulating in blood are concordantly a general biomarker for poor prognosis in many types of human cancer (9). Following this line of evidence, several intervention strategies targeting uPAR have been developed, and some have shown promising therapeutic effects in preclinical mouse model systems (10–13). To assist their further clinical translation, a non-invasive imaging modality for visualizing uPAR expression in vivo using positron emission tomography has been developed recently (13–15).
One complicating factor in targeting uPAR is the dual function this receptor exerts on both degradation of and adhesion to the extracellular matrix (16). These distinct functional properties are, however, interrelated because the low affinity binding between uPAR and the somatomedin B domain (SMB) of vitronectin (17) is regulated by uPAR occupancy with its high affinity protease ligand uPA (18, 19). This raised the unexpected conundrum that small molecules targeting the uPA binding site may unleash undesirable agonist effects by stimulating the uPAR·vitronectin interaction (13, 19). This should be considered in future drug discovery programs aiming at controlling uPAR function by a given intervention therapy. The molecular mechanisms underpinning this phenomenon therefore define one of the most important challenges in our present understanding of the structure-function relationships of uPAR in cell biology and in pathophysiology.
The ligand-binding part of the glycolipid-anchored uPAR (20) comprises three homologous, three-fingered modules designated DI, DII, and DIII, which belong to the Ly6/uPAR/α-neurotoxin (LU) protein domain family as defined by four conserved disulfide bonds (21, 22). The structures of several uPAR complexes have been solved by x-ray crystallography, including those with a synthetic antagonist peptide (23), the amino-terminal fragment of uPA (ATF) (24–26), and the ternary complex with ATF and the SMB domain from vitronectin (27). In these structures, the β-hairpin of the growth factor-like domain (GFD) of uPA is buried deeply within a large hydrophobic cavity assembled by all three LU domains in uPAR (25, 26). In contrast, the SMB domain binds a small hydrophobic patch located at the interface between uPAR DI and DII (17, 27). We consider that the overall architectures of these two ligand binding sites on uPAR open the possibility that prior uPA binding may prime a subsequent SMB binding event by altering its composite interdomain binding site. Circumstantial cell biological (18, 19, 28) and biochemical (17) evidence exists to favor this scenario, including our design of a constitutively active receptor mutant (uPARH47C/N259C), where DI is tethered to DIII via an interdomain disulfide located distant from to the SMB binding site (29). This constrained uPAR mutant promotes lamellipodia formation on vitronectin-rich matrices in the absence of uPA (29), which suggests that a regulatory structural transition occurs at the SMB binding site when uPA binds.
The unoccupied receptor has so far evaded structural determination despite prolonged efforts aimed at crystallizing it. Only recently has the structure of a constitutively active receptor mutant (uPARH47C/N259C) (29) been solved by x-ray crystallography in the absence of bound ligands (30). As alluded to above, this structure is, nevertheless, unlikely to represent the prevalent conformation populated by unoccupied uPARwt because the constraints we engineered into uPARH47C/N259C render it a structural and functional surrogate for the receptor conformation selected in the uPA·uPAR complex (29, 30).
To better understand the structural transition(s) regulating the interaction between uPAR and vitronectin, we therefore in this study embark on a more dynamic approach by probing the conformation(s) of the ligand-free state of uPAR with small angle x-ray scattering (SAXS) and hydrogen-deuterium exchange (HDX). These data are then integrated with functional data obtained by surface plasmon resonance. We find that ligand-free uPARwt is highly extended in solution, where uPAR DI is markedly flexible compared with the rest of the protein. This open structure presents a suboptimal vitronectin binding site and explains how uPA regulates the affinity for vitronectin without sharing a direct contact surface. Trapping uPAR in the open state therefore provides a new avenue for therapeutic inhibition of the proteolytic activity of the plasminogen activation system without stimulating cellular migration.
EXPERIMENTAL PROCEDURES
Chemicals and Protein Preparations
Recombinant human uPARwt and uPARH47C/N259C were expressed as secreted proteins (residues 1–283) by Drosophila melanogaster S2 cells and affinity-purified as described (29). Recombinant uPAR DI (residues 1–92) and DIIDIII (residues 88–283) were likewise expressed in S2 cells and affinity-purified using anti-uPAR mAbs R21 and R2, respectively (19). Receptor-binding fragments of uPA (GFD(1–48) and ATF(1–143)) and the synthetic peptide antagonists AE105 (DXFsrYLWS) and AE234 (cNKYFSNICW) were prepared as described (19, 31, 32), where lowercase letters correspond to d-enantiomers, X is cyclo-hexyl-l-alanine, and the two cysteine residues in AE234 are oxidized to form a cyclic peptide. D2O (99.9% atom % D) was purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA), and tris(2-carboxyethyl)phosphine was obtained from Sigma-Aldrich.
SAXS Data Collection
Synchrotron radiation x-ray scattering data were collected on the X33 beamline of the EMBL (DESY, Hamburg) (33) using a 1M PILATUS pixel detector (DECTRIS, Baden, Switzerland) and four frames of 30-s exposure time. To ensure maximal monodispersity, all receptor preparations were purified by size exclusion chromatography in 20 mm sodium phosphate buffer, pH 7.2, 50 mm NaSO4, 5% (w/v) glycerol and kept on ice. SAXS data were collected within 48 h in the same buffer at 25 °C using protein concentrations of 1.5–4.4 mg/ml. The sample-to-detector distance was 2.7 meters, covering a range of momentum transfer 0.08 ≤ s ≥ 6.0 nm−1 (s = 4π sinθ/λ, where 2θ is the scattering angle, and λ = 0.15 nm is the x-ray wavelength). Based on comparison of successive 30-s frames, no detectable radiation damage was observed. Data from the detectors were normalized to the transmitted beam intensity and averaged, and the scattering of buffer solutions was subtracted. The difference curves were scaled for solute concentration. All data manipulations were performed using the PRIMUS software package (34). The forward scattering I(0) and radius of gyration, Rg were determined from Guinier analysis (35), assuming that at very small angles (s ≤ 1.3/Rg), the intensity is represented as I(s) = I(0)exp((sRg)⅔). These parameters were also estimated from the full scattering curves using the indirect Fourier transform method implemented in the program GNOM (36), along with the distance distribution function p(r) and the maximum particle dimensions, Dmax. Molecular masses of solutes were estimated from SAXS data by comparing the extrapolated forward scattering with that of a reference solution of bovine serum albumin. Due to the uncertainty in molecular mass estimation from SAXS data that results from uncertainty in the measured protein concentrations, an excluded volume of the solutes was also determined from the ab initio modeling program DAMMIF (37). This estimation is independent of protein concentration and can be obtained in an automated fashion with minimal user bias. For globular proteins, this hydrated particle volume in nm3 is ∼2 times the molecular mass in kDa (38).
Fit of the uPAR Crystal Structures to the SAXS Data
The fits of the available high resolution crystal structures of uPAR, 2FD6 (25), 1YWH (23), and 3U74 (30), to the recorded uPARwt and uPARH47C/N259C SAXS data were conducted using the program CRYSOL (39). CRYSOL calculates the partial scattering amplitudes of proteins from their atomic coordinates, taking into account the hydration layer and excluded solvent volume. The number of harmonics was set to 50, and 256 points were computed across the entire data range (0 < s < 5 nm−1).
Analysis of Flexibility
Analysis of the interdomain flexibility and size distribution of possible conformers consistent with the measured scattering data for uPAR was conducted using the ensemble optimization method (EOM) (40). This method selects an ensemble of possible conformations from a pool of randomly generated models (10,000), using CRYSOL to calculate the theoretical scattering profiles and a genetic algorithm, GAJOE, to select the representative set. The input structures for analysis of the scattering data employing ensemble optimization used the DI, DII, and DIII domains from the 2FD6 uPAR crystal structure as rigid bodies. Linkers between the modules were represented as a flexible chain of dummy residues.
Ab Initio Shape Determination and Molecular Modeling
Low resolution shape envelopes for all constructs were determined using the ab initio bead-modeling program DAMMIF (35). DAMMIF represents the particle as a collection of M (≫1) densely packed beads inside an adaptable and loosely constrained search volume compatible with the experimentally determined Rg. Each bead was randomly assigned to solvent (index = 0) or solute (index = 1), and the particle structure in solution was described by a binary string of length M. Disconnected strings of beads were rejected, and the scattering amplitudes were calculated. Simulated annealing was then used to search for a compact model that minimizes the discrepancy,
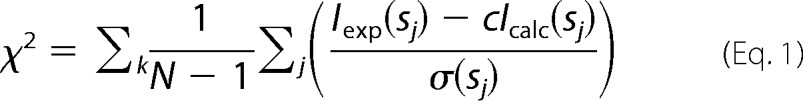 |
where N is the number of experimental points, Iexp(sj) and Icalc(sj) are the experimental and calculated intensities, c is a scaling factor, and σ(sj) is the experimental error at the momentum transfer sj. The results of 10 independent DAMMIF reconstructions were compared using the alignment program SUPCOMB13 (41) to determine the most representative (typical) model. Averaged DAMMIF models were also determined using the program DAMAVER (42), and these models were adjusted such that they agree with the experimentally determined excluded volume using the program DAMFILT (42).
Molecular modeling was conducted using, as rigid bodies and where appropriate, the same structures as were used for EOM. Rigid body models were generated for all of the constructs using the program CORAL, an advanced version of the rigid body modeling program BUNCH (43), where linkers/loops between the individual subunits are represented as random polypeptide chains. The results of 10 multiple CORAL runs were analyzed using the programs SUPCOMB13 (41) and DAMAVER (42) to identify the most representative/typical models.
Hydrogen Deuterium Measurements
Purified uPAR (33 μm) was dissolved in PBS in the presence or absence of a 2.5-fold molar excess of ligand and incubated for 10 min at room temperature. Isotopic exchange was initiated by 20-fold dilution into deuterated PBS at 25 °C. Aliquots of 100 pmol of protein were withdrawn after 10, 100, and 1000 s, and further exchange was quickly quenched on ice by acidification (pH 2.5) using formic acid. Aliquots were subsequently snap frozen in liquid nitrogen and stored at −80 °C until analysis. Duplicate analyses were performed throughout the study. Prior to mass spectrometric analysis, samples were thawed, and disulfide bonds were reduced at pH 2.5 by adding 1 volume of 800 mm tris(2-carboxyethyl)phosphine (adjusted to pH 2.5 using NaOH) and incubation for 2 min on ice. The quenched and reduced samples were injected into a nanoACQUITY UPLC reversed-phased chromatographic system equipped with HDX technology (Waters Corp., Milford, MA) coupled to a Synapt G1 electrospray ionization mass spectrometer (Waters Corp.). For further details on instrumentation and processing of HDX profiles, see the supplemental Materials and Methods.
Surface Plasmon Resonance
Real-time interaction kinetics were determined by surface plasmon resonance using a Biacore T200TM system (GE Healthcare) operated at 20 °C with a flow rate of 50 μl/min running buffer (10 mm HEPES, 150 mm NaCl, 3 mm EDTA, and 0.05% (v/v) surfactant P-20 at pH 7.4). Purified uPAR DIIDIII(88–283) was immobilized covalently onto a carboxymethylated dextran matrix (CM5 sensor chip) using N-hydroxysuccinimide/N-ethyl-N′-(3-(diethylamino)propyl)-carbodiimide coupling chemistry, yielding a surface density of 218 resonance units (RU), ∼9 fmol/mm2. Binding of either DI(1–92) or ATF(1–143) were subsequently measured in real time by injecting serial 2-fold dilutions in running buffer (4 nm to 2 μm) for 140 s, followed by a dissociation phase of 900 s. Binding of preformed DI(1–92)·ATF(1–143) complexes was measured after incubating a serial 2-fold dilution series of DI(1–92) (8 nm to 2 μm) containing 1 μm ATF(1–143) in running buffer for at least 2 h before injection over the sensor surface. To certify reproducibility of the analyses, at least one concentration was retested at the end of each experiment. After each run, the sensor chip was regenerated by two consecutive injections of 0.1 m acetic acid in 0.5 m NaCl.
Determination of the kinetic rate constants, ka and kd, were subsequently derived from these real-time interaction analyses by non-linear regression fitting of the association and dissociation phases to a bimolecular interaction model assuming pseudo-first order reaction conditions using the BIAevaluation version 4.1 software (Biacore, Uppsala, Sweden). For low affinity interactions, the equilibrium binding constants Kd and Rmax were calculated from the corresponding equilibrium binding isotherms by nonlinear curve fitting, assuming saturation of a single binding site, Req = (Rmax[DI])/(Kd + [DI]), where Req is the binding level at equilibrium, and Rmax is the binding capacity of the chip.
RESULTS
Structural Studies Using SAXS
To study the structure of uPAR in solution, small angle x-ray scattering data were collected for soluble uPARwt in complex with two different antagonist peptides, AE105 (31) and AE234 (32), a receptor-binding fragment of the high affinity protease ligand uPA (ATF), and for the unoccupied receptor alone. Importantly, we also collected SAXS data for the disulfide-constrained receptor mutant (uPARH47C/N259C), where domains I and III are linked by a disulfide bridge, which maintains uPAR in a conformation mimicking the uPA-bound state both functionally (29) and structurally (30). The overall SAXS parameters for all samples were in agreement with a monomeric protein of the expected size, with the exception of uPARwt, where the apparent mass is slightly larger than expected (Table 1). This discrepancy between the predicted molecular mass and that estimated from the scattering data could theoretically indicate the presence of a small proportion of oligomers; however, the shape parameters Rg and Dmax remain constant with increasing concentration, arguing against the presence of such reversible self-association (see supplemental Fig. S1 and Table S1). The apparent overestimation of the molecular weight may be explained by the uPARwt monomer being predominantly extended and intrinsically flexible. Accordingly, the expected particle volumes calculated from a compact glycosylated uPAR structure and a highly extended theoretical model are ∼50 and ∼69 nm3, respectively, where the latter is consistent with the experimental data suggesting that the SAXS data for uPARwt is consistent with a monomeric protein.
TABLE 1.
SAXS parameters for uPAR
RgautoRg and RgGNOM are the radii of gyration estimated from the SAXS data using the automated Guinier analysis routine and GNOM, respectively. Dmax, Vp, and MMSAXS are the maximum particle dimensions, hydrated particle volumes, and molecular masses estimated from I(0), respectively. Extracted parameters are taken from the merged SAXS curve from all data sets and extrapolated to infinite dilution, and the molecular masses calculated from the sequence (including four standard glycosylations) are shown (MMseq). The experimentally determined masses for the glycosylated uPARwt and uPARH47C/N259C preparations used in this study are both within 35.1–35.4 kDa, where the differences from the calculated masses most likely relate to incomplete fucosylations of the innermost chitobiose moieties of the glycans.
| Protein | RgautoRg | RgGNOM | Dmax | Vp | MMSAXS | MMseq |
|---|---|---|---|---|---|---|
| nm | nm | nm | nm3 | kDa | kDa | |
| uPARwt | 2.8 ± 0.1 | 2.9 ± 0.1 | 9.7 ± 0.5 | 69 ± 10 | 47 ± 5 | 35.6 |
| uPARH47C/N259C | 2.2 ± 0.1 | 2.2 ± 0.1 | 7.0 ± 0.5 | 57 ± 10 | 34 ± 5 | 35.6 |
| uPARwt·AE105 | 2.5 ± 0.1 | 2.5 ± 0.1 | 8.5 ± 0.5 | 61 ± 5 | 37 ± 5 | 36.8 |
| uPARwt·AE234 | 2.4 ± 0.1 | 2.4 ± 0.1 | 8.3 ± 0.5 | 57 ± 5 | 42 ± 5 | 36.9 |
| uPARwt·ATF | 2.8 ± 0.1 | 2.8 ± 0.1 | 9.4 ± 0.5 | 102 ± 10 | 58 ± 5 | 51.8 |
The SAXS data can be represented in real space as an intraparticle distance distribution function, p(r), which enables a model-independent comparison of structures of uPAR in different states. For the ligand-free uPARwt, the p(r) is positively skewed with a tail at large distances consistent with that of an extended particle (Fig. 1a). In contrast, both the different ligand-bound states and the constrained uPARH47C/N259C mutant have roughly symmetrical p(r) functions indicative of a more compact and globular structure. Notably, the dimensions of the uPAR·ATF complex are even smaller than uPARwt alone despite the addition of two structural domains, causing a mass increment of 16.2 kDa. This demonstrates that large scale structural changes occur in uPARwt, resulting in compaction of the receptor upon binding of ligands.
FIGURE 1.

Comparison between SAXS data and crystal structures. The pairwise interatomic distance distributions were determined from the scattering profiles for uPAR in six different states and are displayed as a p(r) function in a. The p(r) functions clearly show that uPARwt is the most extended species and that the disulfide-constrained uPARH47C/N259C represents the most compact species. The complexes with two different 9–10-mer peptide antagonists (AE105 and AE234) display an intermediate compactness. The best fits of the known crystal structures to the SAXS data are shown for uPARwt (b), for uPARH47C/N259C (c), and for uPARwt·AE105 complexes (d). Both uPARH47C/N259C and uPARwt·AE105 complexes fit the corresponding crystal structures solved for these states, whereas the unoccupied uPARwt is not adequately described by any of the known structures.
Comparison with Known Crystal Structures of uPAR
Theoretical scattering profiles can be calculated from structural models, and the experimental SAXS curves can therefore readily be compared with those derived from published structures. Crystal structures have been solved for several uPARwt complexes (23–25, 27) as well as uPARH47C/N259C in the absence of bound ligand (30). These structures were fit to our experimental scattering data using the software CRYSOL (39). The scattering curves from uPARH47C/N259C (Fig. 1c), uPAR·ATF (see supplemental Fig. S2f), and uPAR·AE105 (Fig. 1d) agree with those calculated from the corresponding known crystal structures, demonstrating that the protein structure in solution is indeed similar to that in the crystals. Furthermore, the scattering profile for the uPAR·AE234 complex deviates only slightly from that of uPAR·AE105 (supplemental Fig. S2, compare c and d), suggesting that the latter complex is marginally less compact. We next attempted to fit the appropriate uPAR components of the available crystal structures to the data collected for unoccupied uPARwt. None of these structures yielded a good fit to the SAXS data (Fig. 1b), all being significantly too compact. Thus, under the conditions used in this study, the free uPAR receptor is observed to adopt an extended conformation distinct from the one of ligand-occupied uPAR in the published crystal structures.
Modeling of uPAR from SAXS Data
To understand the structural transitions of uPAR upon ligand binding, we calculated various models from the SAXS data recorded for uPARwt. To test the modeling methodology on a similar protein of known structure, models of uPARH47C/N259C were generated in parallel. Initially, dummy atom models were reconstructed ab initio from the SAXS data using the bead modeling program DAMMIF (37). The molecular shapes reconstructed from the uPARH47C/N259C SAXS scattering data are compact and globular with an intriguingly pronounced surface depression on one face, which is consistently present in independent reconstructions (Fig. 2, b and c). This cavity most likely corresponds to the hydrophobic uPA binding site observed in the high resolution structures of uPAR in complex with uPA fragments (25, 26) and peptide antagonists (23). Indeed, when a representative model is superimposed upon the uPARH47C/N259C crystal structure (30) using the program SUPCOMB13 (41), the uPA binding site overlays well with this cavity (Fig. 2d). Thus, the ab initio SAXS modeling approach is able to reproduce quite detailed structural features of the protein. Models were subsequently reconstructed from the SAXS data of uPARwt, and, in contrast to the compact disulfide-restrained mutant, they consistently adopt a more extended conformation (Fig. 2, a and c).
FIGURE 2.

Ab initio modeling of the ligand-free uPAR variants. Bead models representing the ab initio surfaces were constructed from the experimental scattering curves for uPARwt (a) and uPARH47C/N259C (b). The resulting models are depicted in c, which highlights the elongated shape of uPARwt (red) compared with the more compact nature of uPARH47C/N259C (blue). In the latter reconstruction, the corresponding ab initio surface even reveals a small but distinct surface depression in its shape at a position corresponding to the unoccupied ligand binding cavity. The crystal structure of uPARH47C/N259C fits the ab initio shape envelope (d) for the closed mutant.
As alluded to previously, uPAR is a modular protein consisting of three homologous LU domains. Structural plasticity in such multidomain proteins is mainly due to differences in the relative orientation of their individual domains. Accordingly, we sought to explore the potential flexibility of unoccupied uPAR using an ensemble based modeling procedure, assuming the structures of each of its three LU domains were similar to those observed in the published crystal structures. Initially, these LU domains were simulated as randomly orientated rigid bodies connected by flexible linkers of appropriate lengths. The p(r) function for the simulated ensemble of random domain orientations is more extended than that derived from the experimental curve, demonstrating that uPARwt has some degree of interdomain structure (Fig. 3a). In contrast, the p(r) function has an approximately correct degree of compactness when two domains are kept in the relative orientation seen in the crystal structure and the third domain is in a random orientation. Subsequently, sets of conformations that best fit the experimental SAXS data were selected from the initial random pool of flexible models using the EOM (40). The procedure was repeated with only one of the LU domains unconstrained (DI or DIII), yielding excellent fits of selected ensembles to the SAXS data in each case (Fig. 3b). The resulting Rg size distribution is narrow and homogeneous for uPARH47C/N259C, whereas it is wide and heterogeneous for uPARwt (Fig. 3c). This suggests a flexible domain structure for uPARwt, which is furthermore consistent with the relatively smooth p(r) function observed for this multidomain receptor; such smoothness is often displayed by modular systems with significant interdomain dynamics (44).
FIGURE 3.

Rigid body modeling of the ligand-free uPAR variants. a, p(r) functions calculated for ensembles of rigid body models of uPAR where the interdomain orientation of either one or all three domains are randomized. The experimental p(r) function for uPARwt (solid red line) is approximately as compact as the structures, where only one of the domains is randomly orientated. b, fits of individual uPARwt models and ensembles to the SAXS data from CORAL rigid body refinement and EOM ensemble selection are shown, respectively, keeping two domains fixed in their relative orientation from the crystal structure and allowing one domain to be flexible. c, Rg distribution of the selected EOM ensembles. The closed mutant is best represented by a compact and relatively homogeneous ensemble, whereas the wild-type receptor is best fit by a heterogeneous ensemble. This suggests the presence of interdomain dynamics in the wild-type protein. Representative models resulting from rigid body modeling to the SAXS data are shown in d. The SAXS data fit well to models where either domain I or domain III is detached from the rest of the protein and projects away from the protein.
Given the extended and flexible nature of uPARwt suggested above, a spatial configuration of the three LU domains providing the best fit to the SAXS data were sought in a rigid body modeling procedure (43). When this procedure is applied to the constrained uPARH47C/N259C, a closed form resembling that observed in the crystal structure is reproducibly obtained. When the rigid body modeling procedure is applied to the wild-type protein, however, several configurations that fit the SAXS data equally well can be generated. These solutions consistently have a stretched configuration in which two of the LU domains are in close contact and the third extends away from this core structure. Next, we fitted the data while maintaining two domains fixed in the configuration observed in the crystal structure and the third domain flexible. This procedure resulted in an equally good fit regardless of whether DI-DII or DII-DIII was treated as the rigid body (Fig. 3b). We can thus conclude that one of the domains appears to extend away from the two others on average, but we cannot discriminate between models with a flexible DI or DIII based on SAXS data alone. It is furthermore unlikely that this state corresponds to a single unique structure because the flexible domains in these models maintain no discernible interdomain contacts with the remaining two rigid domains. The structures in Fig. 3d therefore most likely represent the average of a conformational ensemble alluded to in the EOM analysis.
Hydrogen-Deuterium Exchange of uPAR
To explore the structure and dynamics of uPAR in more detail, the HDX patterns of the various uPAR states were compared by mass spectrometry. Through this strategy, we are probing the solvent accessibility and hydrogen bonding of the backbone amides as a measure of the presence and stability of secondary structure elements. Decoding the local hydrogen exchange patterns was accomplished after reduction of disulfide bonds followed by pepsin digestion under quench conditions, where the labeling information on the backbone amides is well preserved. The deuterium contents of the HPLC-separated peptic peptides were subsequently determined by mass spectrometry analysis and were calculated relative to a fully deuterated uPAR control, which was analyzed in parallel (see supplemental Fig. S3). In the case of uPAR, we analyzed 17 peptic peptides providing a sequence coverage of 44%, which importantly yields information representing all three LU domains (Fig. 4d). A careful inspection of the compiled exchange data set represented by the mirror plots in Fig. 4 demonstrates that particularly one peptide (residues 62–68) exhibits markedly reduced exchange kinetics in the ligand-complexed forms of uPAR. Therefore, this peptide is particularly sensitive to the conformational changes induced upon uPA binding. Altogether, these exchange profiles reveal that ligand-induced transition into a more compact receptor mainly affects structural elements residing in DI (Fig. 4a). Importantly, this ligand-induced stabilization of DI is closely recapitulated by the constitutively active uPARH47C/N259C in the absence of bound ligand (Fig. 4b). Subsequent GFD binding confers only a marginal additional attenuation on the exchange kinetics of residues 62–68 in this constrained uPAR mutant, demonstrating that the predominant shift in the HDX profile for uPARwt upon ligation is caused by structural changes in uPAR per se and not by solvent exclusion by the bound ligand. The reporter peptide covers the last β-strand (β1F) in DI before the connecting linker to DII, and together with β1E, it forms the last β-hairpin (“third finger”) of the LU domain in DI (Fig. 4d). This particular peptide sequence therefore occupies a diagnostic position in the uPAR structure because the corresponding β-turn connecting β1E and β1F points toward the SMB binding site, and the third finger in DI participates in the assembly of the hydrophobic uPA binding cavity as well as the DI-DII interface. Importantly, these exchange data allow us to distinguish between the two possible uPAR models that are consistent with the SAXS data. Because β1E is the only region recovered in our HDX-MS spectra of peptic peptides that significantly changes solvent exposure upon ligand binding, it is implicit that DI is the flexible domain and not DIII. Nevertheless, this diagnostic peptide does not form a direct interdomain contact with DII but rather resides in the central β-sheet of DI (Fig. 4d). As a consequence, our HDX data indicate that the proposed domain flexibility of DI also comprise a certain element of local unfolding and disruption of the hydrogen bonding in the three-finger fold.
FIGURE 4.

Flexibility of local structural elements in uPAR as assessed by HDX-MS. Purified uPARwt or uPARH47C/N259C (33 μm) was incubated alone or in the presence of a 2.5-fold molar excess of one of the two high affinity ligands, AE105 or GFD(1–48) (>1,000-fold above their Kd for uPAR). The solvent exposure of the backbone amides in these protein preparations was probed after exposure to deuterated PBS for 10 s (▾, blue), 100 s (■, red), and 1000 s (●, green), and the local exchange profiles for the corresponding peptic peptides were delineated by HDX-MS. a, exchange profiles for unoccupied uPARwt compared with uPARwt·AE105 and uPARwt·GFD(1–48), respectively. The deuterium uptake is shown relative to a fully deuterated control protein preparation. b compares the exchange profiles for unoccupied uPARwt and uPARH47C/N259C either alone or in complex with GFD(1–48). c, exchange kinetics of DI in intact uPAR, recombinant DI(1–92), and DI(1–87) in the presence of DIIDIII(88–283) generated by limited chymotrypsin cleavage. The domain locations of the peptic peptides analyzed in this study are highlighted on the y axis in a as yellow (DI), blue (DII), and red (DIII), and their actual positions in the uPAR structure are highlighted by green/cyan in the model for uPAR·ATF·SMB in d. For clarity, the ATF and SMB moieties have been omitted. The reporter peptide 62–68 in βIF is colored cyan in the uPAR model and is indicated by cyan arrows in a and b.
A comparison of the exchange kinetics recorded for intact uPARwt, recombinant DI(1–92), and a mixture of DI(1–87) and DIIDIII(88–283) generated by limited chymotrypsin cleavage of uPARwt reveals comparable exchange profiles for all peptic peptides derived from DI (Fig. 4c). As judged by these criteria, the conformation of uPAR DI is thus equally solvent-exposed whether it is present in the context of intact uPARwt or it is released as a solitary domain. This suggests that DI does not form a stable interface with the remaining receptor in the absence of ligand.
Functional Measurements by Surface Plasmon Resonance
One important functional ramification of this structural model for unoccupied uPAR is the possibility that this inherently “detached DI” conformer actually forms the initial encounter complex with uPA. A subsequent tightening of this transient complex is predicted to form the high affinity complex leading to the exposure of an optimal vitronectin binding site. To challenge this proposition, we tested the interaction between a receptor-binding uPA fragment (ATF(1–143)) and an artificial uPAR model simulating the ultimate detachment of DI (i.e. being composed of immobilized recombinant uPAR DIIDIII(88–283) and recombinant uPAR DI(1–92) in solution). As shown in Fig. 5, a and b, we were unable to detect any measurable interaction between either uPAR DI(1–92) or ATF(1–143) and immobilized uPAR DIIDIII(88–283) when they were analyzed in concentrations up to 2 μm by surface plasmon resonance. Despite this lack of interaction, mixtures of DI and ATF nevertheless exhibit a tight and concentration-dependent binding to the immobilized DIIDIII (Fig. 5d), and the stability of the formed trimolecular DI·ATF·DIIDIII complex equals that of the corresponding genuine bimolecular ATF·uPAR complex with dissociation rate constants (kd) of 1.9 × 10−4 and 1.8 × 10−4 s−1, respectively (Fig. 5, compare d and e). Separate surface plasmon resonance experiments revealed that a transient bimolecular DI·ATF complex can be formed with a Kd of 1.7 μm (Fig. 5c). Assuming that an equivalent bimolecular complex initially forms in solution with similar kinetics before it subsequently binds the immobilized DIIDIII (Fig. 5d) allows us to calculate the second order association rate constant (ka ∼0.7 × 105 m−1 s−1) for the formation of the (DI·ATF)·DIIDIII complex. Notably, this rate constant is only 3-fold slower than that measured for the corresponding ATF·uPAR complex (i.e. ka ∼2.0 × 105 m−1 s−1) (Fig. 5f). Comparable effects are observed when ATF(1–143) is replaced by the smaller GFD(1–48) or pro-uPA (data not shown), illustrating that the assembly of this trimolecular complex is indeed driven by the occupancy of the uPA binding cavity per se. HDX studies reveal that the solvent exposure of DI(1–92) in such trimolecular DI·ATF·DIIDIII complexes resembles that of DI in the genuine bimolecular GFD·uPAR complexes (Fig. 5g) as well as that in the constitutively active uPARH47C/N259C mutant (Fig. 4b). In combination, these kinetic and HDX experiments thus illustrate that it is possible for a completely detached DI to form an initial encounter complex with uPA, and in the presence of uPAR DIIDIII this will induce the formation of a highly stable complex.
FIGURE 5.

Ligand binding kinetics for intact uPAR and its fragments DI and DIIDIII using surface plasmon resonance. To probe the uPA binding potential of unoccupied uPAR with a detached DI, we assessed the binding kinetics of DI(1–92) and ATF(1–143) to immobilized DIIDIII(88–283) either alone or in combination. a and b show that neither DI nor ATF binds as a single component to immobilized DIIDIII (218 resonance units (RU), ∼9 fmol/mm2) when tested up to 2 μm. c shows that DI(1–92) binds weakly to immobilized ATF with an equilibrium dissociation constant (Kd) of 1.7 μm, which is similar to the one we measure for ATF(1–143) binding to immobilized DI (Kd of 1.6 μm; data not shown). In contrast, the sensorgrams in d reveal a tight binding to immobilized DIIDIII, when a serial 2-fold dilution of DI(1–92) (8 nm to 2 μm) is tested in the presence of 1 μm ATF(1–143). The second order rate constants for the interaction of ATF·DI complexes with immobilized DIIDIII were calculated assuming a Kd of 1.7 μm for the ATF·DI interaction in solution (c). For comparison, the interaction between a serial 2-fold dilution of intact uPAR (0.6–200 nm) and immobilized ATF is shown in e. Repeat analyses are shown as blue curves, and buffer runs are shown as green curves in c–e. Fits to dissociation and association phases are shown as thin red lines, and the derived kinetic rate constants are shown in f. g, the solvent accessibility of DI(1–92) was measured by HDX-MS either as an isolated component or in a trimolecular complex, DI·ATF·DIIDIII, which was preassembled by incubating a mixture of the individual component (30, 60, and 60 μm, respectively) for 60 min before exchange was initiated for 10, 100, and 1000 s using same color coding as in Fig. 4.
DISCUSSION
The role of uPAR in cell surface-associated proteolysis and adhesion is increasingly well described in molecular terms due to extensive biochemical studies (19, 29, 45, 46) and several published crystal structures of ligand-bound states of the receptor (23–27, 30). Important challenges, nevertheless, still remain to be solved. These concern the structure of the ligand-free state of uPAR, which is essentially undefined, and the molecular basis underlying the allosteric regulation of the protein. Our current study reveals these two questions to be closely interrelated. Because traditional methods for high resolution structure determination for various reasons have failed for unoccupied uPARwt, we have in the present study adopted an untraditional approach to define the conformation(s) of uPARwt in solution by combining SAXS with hydrogen-deuterium exchange monitored by mass spectrometry. These two technologies are essentially complementary because SAXS data provide information on the overall molecular shapes and domain arrangements, whereas the HDX data allow us to assess solvent accessibility of the backbone amides. Comparison of HDX patterns of several states of uPAR allows us to discriminate between different models consistent with the SAXS data, thus buttressing the conclusions from molecular modeling. The present combination of SAXS and HDX may therefore represent a generally applicable strategy for defining the structures of multidomain proteins that fail to crystallize due to dynamics.
It is becoming increasingly evident that non-proteolytic roles of uPAR in cell migration and adhesion (28, 47–49) to a large extent are regulated by receptor occupancy with uPA (16, 18, 19). Recently, we showed that constraining uPAR to a structure resembling the one selected by complex formation with uPA (30) has the same effect on adhesion and lamellipodia formation on vitronectin as uPA-binding (29). This demonstrates that it is the structural transition induced in uPAR per se and not direct contributions from the bound uPA that drives this regulatory potential. In this study, we show that DI of uPAR is flexible relative to the rest of the receptor. Upon uPA binding, our data suggest that DI docks onto DII, and this consequently enables an allosteric regulation of the affinity for secondary ligands engaging the interface between these domains, as illustrated by vitronectin binding (17, 27). Beyond a mere docking event, our HDX data also imply that uPA binding is accompanied by a stabilization of the three-finger fold, suggesting that DI in unoccupied uPAR exists in a partially unfolded state. Such local unfolding is not likely to be detectable by SAXS because this technique is most sensitive to the overall shape properties of the molecule. Ligand-induced folding of DI is furthermore concordant with our previous observation demonstrating that GFD binding protects the N-linked glycan on Asn52 against N-glycanase cleavage (50). This glycosylation is confined to the outer convex surface of the central β-sheet in DI (βIE) and is thus not facing the central uPA binding cavity, as illustrated in Fig. 2d. In contrast, there seems to be little ligand-induced structural change in the orientation/stability of DII and DIII as disclosed by their almost invariant HDX patterns. Although a plethora of ligands have been proposed for uPAR, uPA binding will most likely only influence those binding sites that localize to DI and the interface between DI and DII in particular. uPAR is encoded by a small gene cluster containing at least six homologous proteins containing 2–4 LU-domains (13, 21). Interestingly, these multidomain homologs all possess an atypical DI, which is lacking one particular consensus disulfide that is essential for the stability of the corresponding single domain proteins. From an evolutionary perspective, this suggests that the multidomain structure precedes gene duplication within this gene locus and that the allosteric switch between DI and DII could potentially be shared among the family members.
Protein binding reactions that are coupled to a large structural transition generally follow one of two mechanisms, conformational selection or induced fit, depending on whether the structural changes take place before or after protein binding (51). Site-directed mutagenesis studies show that a large proportion of the thermodynamic driving force for the uPA·uPAR complex formation is provided by the defined interaction between GFD and uPAR DI (26, 45). Accordingly, the uPA derivatives GFD and ATF do bind, albeit weakly, to isolated uPAR DI as opposed to DIIDIII, where no specific binding is detected. Given that DI is detached from the rest of the protein in the unoccupied receptor, it is likely that DI acts as bait, forming a transient intermediate complex with GFD. After the association reaction, the receptor closes and forms the final complex in a mechanism resembling induced fit. The constrained uPARH47C/N259C mutant nonetheless binds uPA with kinetics similar to those of uPARwt, although it is trapped in the closed conformation and hence is forced to bind by a “conformational selection”-dominated mechanism (29). This demonstrates that the binding mechanism between uPA and uPAR is plastic and can proceed through both routes. This is in keeping with recent suggestions that the two schematic mechanisms are only extreme cases and that most binding reactions proceed as a composite of the two (52).
The involvement of uPAR in cancer invasion and metastasis has prompted several academic institutions and pharmaceutical companies to embark on drug development programs aimed at targeting and controlling uPAR function (31, 32, 53, 54). The majority of the small molecules developed so far target the central uPAR cavity to antagonize uPA binding. The Achilles heel of this approach is, nevertheless, that these inhibitors may induce and stabilize the closed form of the receptor and thus stimulate non-proteolytic functions of uPAR (19, 29). Alternative strategies to circumvent this agonistic effect would be to either target both sites simultaneously or to stabilize the open unoccupied state of the receptor. Recently, a number of monoclonal anti-uPAR antibodies sharing epitopes located in the proximity of the DI-DIII interface were found to prevent both uPA and vitronectin binding, presumably because the bulkiness of the bound antibodies impedes closure of the receptor by steric hindrance (19, 29). This mode of action is particularly evident for the inhibitory anti-uPAR mAb H2 (55), which has Asn-259 in uPAR DIII as its primary hot spot (29). In the uPA·uPAR complexes, this residue stabilizes the DI-DIII interface by hydrogen bonding to His-47 in DI (25), which is the rational basis for our design of the closed and constitutively active uPARH47C/N259C. Our present demonstration of a flexible and temporarily detached DI opens yet another intervention strategy where the domain interface between DI and DII serves as the targeting site for inhibitors preventing uPA and vitronectin binding by trapping the open conformation of uPAR. This proposition is excellently aligned with the circumstantial observation that a monoclonal anti-uPAR antibody, denoted IIIF10, recognizing a linear epitope in unfolded uPAR (residues 52–60, which is equivalent to βIE), also binds the native glycolipid-anchored uPAR on the cell surface and that this inhibits subsequent uPA binding (56). Previously, this reactivity of IIIF10 was not easily reconciled with the limited surface exposure of βIE in the x-ray structures available for intact uPAR (25, 27, 30), where this β-strand is buried in the domain interface with βIID. This observation thus lends further credit to our proposition to explore this particular domain interface as a new druggable target site in uPAR.
Acknowledgments
We thank Gitte Juhl-Funch, Haldis Egholm Mønsted, and John Post for excellent technical assistance. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by National Institutes of Health Grant P41 RR001081). All plots were created using the software package GRACE.
This work was supported by a fellowship from the EMBL Interdisciplinary Postdocs program (EIPOD) (to H. D. T. M.) and a grant from the Danish National Research Foundation (Danish-Chinese Centre for Proteases and Cancer) (to M. P. and H. G.).

This article contains supplemental Materials and Methods, Table S1, and Figs. S1–S3.
- uPAR
- uPA receptor
- uPA
- urokinase-type plasminogen activator
- ATF
- amino-terminal fragment of uPA
- GFD
- growth factor-like domain
- HDX
- hydrogen-deuterium exchange
- LU domains
- Ly6/uPAR-like domains
- SAXS
- small angle x-ray scattering
- SMB
- somatomedin B domain of vitronectin
- EOM
- ensemble optimization method.
REFERENCES
- 1. Connolly B. M., Choi E. Y., Gårdsvoll H., Bey A. L., Currie B. M., Chavakis T., Liu S., Molinolo A., Ploug M., Leppla S. H., Bugge T. H. (2010) Selective abrogation of the uPA-uPAR interaction in vivo reveals a novel role in suppression of fibrin-associated inflammation. Blood 116, 1593–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wei C., Möller C. C., Altintas M. M., Li J., Schwarz K., Zacchigna S., Xie L., Henger A., Schmid H., Rastaldi M. P., Cowan P., Kretzler M., Parrilla R., Bendayan M., Gupta V., Nikolic B., Kalluri R., Carmeliet P., Mundel P., Reiser J. (2008) Modification of kidney barrier function by the urokinase receptor. Nat. Med. 14, 55–63 [DOI] [PubMed] [Google Scholar]
- 3. Wei C., El Hindi S., Li J., Fornoni A., Goes N., Sageshima J., Maiguel D., Karumanchi S. A., Yap H. K., Saleem M., Zhang Q., Nikolic B., Chaudhuri A., Daftarian P., Salido E., Torres A., Salifu M., Sarwal M. M., Schaefer F., Morath C., Schwenger V., Zeier M., Gupta V., Roth D., Rastaldi M. P., Burke G., Ruiz P., Reiser J. (2011) Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat. Med. 17, 952–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ploug M., Plesner T., Rønne E., Ellis V., Høyer-Hansen G., Hansen N. E., Danø K. (1992) The receptor for urokinase-type plasminogen activator is deficient on peripheral blood leukocytes in patients with paroxysmal nocturnal hemoglobinuria. Blood 79, 1447–1455 [PubMed] [Google Scholar]
- 5. Serratì S., Margheri F., Chillà A., Neumann E., Müller-Ladner U., Benucci M., Fibbi G., Del Rosso M. (2011) Reduction of in vitro invasion and in vivo cartilage degradation in a SCID mouse model by loss of function of the fibrinolytic system of rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 63, 2584–2594 [DOI] [PubMed] [Google Scholar]
- 6. Stephens R. W., Nielsen H. J., Christensen I. J., Thorlacius-Ussing O., Sørensen S., Danø K., Brünner N. (1999) Plasma urokinase receptor levels in patients with colorectal cancer. Relationship to prognosis. J. Natl. Cancer Inst. 91, 869–874 [DOI] [PubMed] [Google Scholar]
- 7. Heiss M. M., Allgayer H., Gruetzner K. U., Funke I., Babic R., Jauch K. W., Schildberg F. W. (1995) Individual development and uPA receptor expression of disseminated tumor cells. Nat. Med. 1, 1035–1039 [DOI] [PubMed] [Google Scholar]
- 8. Hu J., Jo M., Cavenee W. K., Furnari F., VandenBerg S. R., Gonias S. L. (2011) Cross-talk between the urokinase-type plasminogen activator receptor and EGF receptor variant III supports survival and growth of glioblastoma cells. Proc. Natl. Acad. Sci. U.S.A. 108, 15984–15989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lund I. K., Illemann M., Thurison T., Christensen I. J., Høyer-Hansen G. (2011) uPAR as anti-cancer target. Evaluation of biomarker potential, histological localization, and antibody-based therapy. Curr. Drug Targets 12, 1744–1760 [DOI] [PubMed] [Google Scholar]
- 10. Bauer T. W., Liu W., Fan F., Camp E. R., Yang A., Somcio R. J., Bucana C. D., Callahan J., Parry G. C., Evans D. B., Boyd D. D., Mazar A. P., Ellis L. M. (2005) Targeting of urokinase plasminogen activator receptor in human pancreatic cancer. Cancer Res. 65, 7775–7781 [DOI] [PubMed] [Google Scholar]
- 11. Kenny H. A., Leonhardt P., Ladanyi A., Yamada S. D., Montag A., Im H. K., Jagadeeswaran S., Shaw D. E., Mazar A. P., Lengyel E. (2011) Targeting the urokinase plasminogen activator receptor inhibits ovarian cancer, Clin. Cancer Res. 17, 459–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gondi C. S., Lakka S. S., Dinh D. H., Olivero W. C., Gujrati M., Rao J. S. (2004) RNAi-mediated inhibition of cathepsin B and uPAR leads to decreased cell invasion, angiogenesis and tumor growth in gliomas. Oncogene 23, 8486–8496 [DOI] [PubMed] [Google Scholar]
- 13. Kriegbaum M. C., Persson M., Haldager L., Alpizar-Alpizar W., Jacobsen B., Gårdsvoll H., Kjaer A., Ploug M. (2011) Rational targeting of the urokinase receptor (uPAR). Development of antagonists and non-invasive imaging probes. Curr. Drug Targets 27, 27. [DOI] [PubMed] [Google Scholar]
- 14. Persson M., Madsen J., Østergaard S., Jensen M. M., Jørgensen J. T., Juhl K., Lehmann C., Ploug M., Kjaer A. (2012) Quantitative PET of human urokinase-type plasminogen activator receptor with 64Cu-DOTA-AE105. Implications for visualizing cancer invasion. J. Nucl. Med. 53, 138–145 [DOI] [PubMed] [Google Scholar]
- 15. Li Z. B., Niu G., Wang H., He L., Yang L., Ploug M., Chen X. (2008) Imaging of urokinase-type plasminogen activator receptor expression using a 64Cu-labeled linear peptide antagonist using microPET. Clin. Cancer Res. 14, 4758–4766 [DOI] [PubMed] [Google Scholar]
- 16. Smith H. W., Marshall C. J. (2010) Regulation of cell signaling by uPAR. Nat. Rev. Mol. Cell Biol. 11, 23–36 [DOI] [PubMed] [Google Scholar]
- 17. Gårdsvoll H., Ploug M. (2007) Mapping of the vitronectin-binding site on the urokinase receptor. Involvement of a coherent receptor interface consisting of residues from both domain I and the flanking interdomain linker region. J. Biol. Chem. 282, 13561–13572 [DOI] [PubMed] [Google Scholar]
- 18. Waltz D. A., Chapman H. A. (1994) Reversible cellular adhesion to vitronectin linked to urokinase receptor occupancy. J. Biol. Chem. 269, 14746–14750 [PubMed] [Google Scholar]
- 19. Gårdsvoll H., Jacobsen B., Kriegbaum M. C., Behrendt N., Engelholm L., Østergaard S., Ploug M. (2011) Conformational regulation of urokinase receptor function. Impact of receptor occupancy and epitope-mapped monoclonal antibodies on lamellipodia induction. J. Biol. Chem. 286, 33544–33556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ploug M., Rønne E., Behrendt N., Jensen A. L., Blasi F., Danø K. (1991) Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosylphosphatidylinositol. J. Biol. Chem. 266, 1926–1933 [PubMed] [Google Scholar]
- 21. Galat A. (2008) The three-fingered protein domain of the human genome. Cell Mol. Life Sci. 65, 3481–3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ploug M. (2003) Structure-function relationships in the interaction between the urokinase-type plasminogen activator and its receptor. Curr. Pharm. Des. 9, 1499–1528 [DOI] [PubMed] [Google Scholar]
- 23. Llinas P., Le Du M. H., Gårdsvoll H., Danø K., Ploug M., Gilquin B., Stura E. A., Ménez A. (2005) Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J. 24, 1655–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barinka C., Parry G., Callahan J., Shaw D. E., Kuo A., Bdeir K., Cines D. B., Mazar A., Lubkowski J. (2006) Structural basis of interaction between urokinase-type plasminogen activator and its receptor. J. Mol. Biol. 363, 482–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huai Q., Mazar A. P., Kuo A., Parry G. C., Shaw D. E., Callahan J., Li Y., Yuan C., Bian C., Chen L., Furie B., Furie B. C., Cines D. B., Huang M. (2006) Structure of human urokinase plasminogen activator in complex with its receptor. Science 311, 656–659 [DOI] [PubMed] [Google Scholar]
- 26. Lin L., Gårdsvoll H., Huai Q., Huang M., Ploug M. (2010) Structure-based engineering of species selectivity in the interaction between urokinase and its receptor. Implication for preclinical cancer therapy. J. Biol. Chem. 285, 10982–10992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huai Q., Zhou A., Lin L., Mazar A. P., Parry G. C., Callahan J., Shaw D. E., Furie B., Furie B. C., Huang M. (2008) Crystal structures of two human vitronectin, urokinase and urokinase receptor complexes. Nat. Struct. Mol. Biol. 15, 422–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Madsen C. D., Ferraris G. M., Andolfo A., Cunningham O., Sidenius N. (2007) uPAR-induced cell adhesion and migration. Vitronectin provides the key. J. Cell Biol. 177, 927–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gårdsvoll H., Kjaergaard M., Jacobsen B., Kriegbaum M. C., Huang M., Ploug M. (2011) Mimicry of the regulatory role of urokinase in lamellipodia formation by introduction of a non-native interdomain disulfide bond in its receptor, J. Biol. Chem. 286, 43515–43526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu X., Gårdsvoll H., Yuan C., Lin L., Ploug M., Huang M. (2012) Crystal structure of the urokinase receptor in a ligand-free form. J. Mol. Biol. 416, 629–641 [DOI] [PubMed] [Google Scholar]
- 31. Ploug M., Østergaard S., Gårdsvoll H., Kovalski K., Holst-Hansen C., Holm A., Ossowski L., Danø K. (2001) Peptide-derived antagonists of the urokinase receptor. Affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry 40, 12157–12168 [DOI] [PubMed] [Google Scholar]
- 32. Schmiedeberg N., Schmitt M., Rölz C., Truffault V., Sukopp M., Bürgle M., Wilhelm O. G., Schmalix W., Magdolen V., Kessler H. (2002) Synthesis, solution structure, and biological evaluation of urokinase type plasminogen activator (uPA)-derived receptor binding domain mimetics. J. Med. Chem. 45, 4984–4994 [DOI] [PubMed] [Google Scholar]
- 33. Roessle M. W., Klaering R., Ristau U., Robrahn B., Jahn D., Gehrmann T., Konarev P. V., Round A., Fiedler S., Hermes S., Svergun D. I. (2007) Upgrade of the small angle x-ray scattering beamline X33 at the European Molecular Biology Laboratory. Hamburg. J. Appl. Crystallogr. 40, 190–194 [Google Scholar]
- 34. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., Svergun D. I. (2003) PRIMUS. A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 35. Guinier A. (1939) La diffraction des rayons X aux tres petits angles. Application a l'etude de phenomenes ultramicroscopiques. Ann. Phys. 12, 161–237 [Google Scholar]
- 36. Semenyuk A. V., Svergun D. I. (1991) GNOM. A program package for small-angle scattering data processing. J. Appl. Crystallogr. 24, 537–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D. T., Konarev P. V., Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Svergun D. I., Barberato C., Koch M. H. (1995) CRYSOL. A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates, J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 40. Bernadó P., Mylonas E., Petoukhov M. V., Blackledge M., Svergun D. I. (2007) Structural characterization of flexible proteins using small-angle x-ray scattering, J. Am. Chem. Soc. 129, 5656–5664 [DOI] [PubMed] [Google Scholar]
- 41. Kozin M. B., Svergun D. I. (2001) Automated matching of high and low resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 42. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Petoukhov M. V., Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bernadó P. (2010) Effect of interdomain dynamics on the structure determination of modular proteins by small-angle scattering. Eur. Biophys. J. 39, 769–780 [DOI] [PubMed] [Google Scholar]
- 45. Gårdsvoll H., Gilquin B., Le Du M. H., Ménèz A., Jørgensen T. J., Ploug M. (2006) Characterization of the functional epitope on the urokinase receptor. Complete alanine scanning mutagenesis supplemented by chemical cross-linking. J. Biol. Chem. 281, 19260–19272 [DOI] [PubMed] [Google Scholar]
- 46. Ellis V. (1996) Functional analysis of the cellular receptor for urokinase in plasminogen activation. Receptor binding has no influence on the zymogenic nature of pro-urokinase. J. Biol. Chem. 271, 14779–14784 [DOI] [PubMed] [Google Scholar]
- 47. Fazioli F., Resnati M., Sidenius N., Higashimoto Y., Appella E., Blasi F. (1997) A urokinase-sensitive region of the human urokinase receptor is responsible for its chemotatic activity. EMBO J. 16, 7279–7286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Resnati M., Pallavicini I., Wang J. M., Oppenheim J., Serhan C. N., Romano M., Blasi F. (2002) The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc. Natl. Acad. Sci. U.S.A. 99, 1359–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wei Y., Eble J. A., Wang Z., Kreidberg J. A., Chapman H. A. (2001) Urokinase receptors promote β1 integrin function through interactions with integrin α3β1. Mol. Biol. Cell 12, 2975–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ploug M., Rahbek-Nielsen H., Nielsen P. F., Roepstorff P., Dano K. (1998) Glycosylation profile of a recombinant urokinase-type plasminogen activator receptor expressed in Chinese hamster ovary cells. J. Biol. Chem. 273, 13933–13943 [DOI] [PubMed] [Google Scholar]
- 51. Boehr D. D., Nussinov R., Wright P. E. (2009) The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hammes G. G., Chang Y. C., Oas T. G. (2009) Conformational selection or induced fit. A flux description of reaction mechanism. Proc. Natl. Acad. Sci. U.S.A. 106, 13737–13741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mazar A. P., Ahn R. W., O'Halloran T. V. (2011) Development of novel therapeutics targeting the urokinase plasminogen activator. Curr. Pharm. Des. 17, 1970–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Khanna M., Wang F., Jo I., Knabe W. E., Wilson S. M., Li L., Bum-Erdene K., Li J., W Sledge G., Khanna R., Meroueh S. O. (2011) Targeting multiple conformations leads to small molecule inhibitors of the uPAR·uPA protein-protein interaction that block cancer cell invasion. ACS Chem. Biol. 6, 1232–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kroon M. E., Koolwijk P., van Goor H., Weidle U. H., Collen A., van der Pluijm G., van Hinsbergh V. W. (1999) Role and localization of urokinase receptor in the formation of new microvascular structures in fibrin matrices. Am. J. Pathol. 154, 1731–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Luther T., Magdolen V., Albrecht S., Kasper M., Riemer C., Kessler H., Graeff H., Müller M., Schmitt M. (1997) Epitope-mapped monoclonal antibodies as tools for functional and morphological analyses of the human urokinase receptor in tumor tissue. Am. J. Pathol. 150, 1231–1244 [PMC free article] [PubMed] [Google Scholar]