

Background: C. rodentium and E. coli induce noncanonical Nlrp3 inflammasome activation through caspase-11.
Results: TLR4-TRIF are important for caspase-11 expression, caspase-1 activation, and downstream IL-1β and IL-18 production.
Conclusion: TLR4-TRIF axis plays an important role in the up-regulation of caspase-11 and activation of noncanonical inflammasome.
Significance: Our study identifies novel molecules upstream of caspase-11 that are involved in activation of noncanonical inflammasome.
Keywords: Caspase, Immunology, Innate Immunity, Nod-like receptor (NLR), Toll-like receptor (TLR), Caspase-1, Caspase-11, Noncanonical Inflammasome, TLR4, TRIF
Abstract
Enteric pathogens represent a major cause of morbidity and mortality worldwide. Toll-like receptor (TLR) and inflammasome signaling are critical for host responses against these pathogens, but how these pathways are integrated remains unclear. Here, we show that TLR4 and the TLR adaptor TRIF are required for inflammasome activation in macrophages infected with the enteric pathogens Escherichia coli and Citrobacter rodentium. In contrast, TLR4 and TRIF were dispensable for Salmonella typhimurium-induced caspase-1 activation. TRIF regulated expression of caspase-11, a caspase-1-related protease that is critical for E. coli- and C. rodentium-induced inflammasome activation, but dispensable for inflammasome activation by S. typhimurium. Thus, TLR4- and TRIF-induced caspase-11 synthesis is critical for noncanonical Nlrp3 inflammasome activation in macrophages infected with enteric pathogens.
Introduction
Enteric pathogens such as enterohemorrhagic Escherichia coli and enteropathogenic E. coli are responsible for a large number of cases of diarrhea, which causes significant morbidity and mortality among infants and children in the developing world (1–3). Orchestration of an appropriate immune response against these bacterial pathogens is accomplished in part through their recognition by a limited number of germ line-encoded pattern recognition receptors expressed by immune and epithelial cells (4). Activation of members of the mammalian Toll-like receptor (TLR)3 family at the cell surface and within endosomes triggers NF-κB activation through the adaptor proteins MyD88 and TRIF (5). NF-κB target genes include proinflammatory cytokines such as members of the interleukin (IL) family, the transcriptional up-regulation of which drives induction of host responses contributing to effective eradication of the bacterial pathogen (5). This is illustrated by the observation that mice with mutations in TLR4 are hyporesponsive to lipopolysaccharide (LPS)-induced cytokine production (6, 7) and that Tlr4−/− and Myd88−/− mice are highly resistant to E. coli-induced septic shock (8).
In addition to TLRs, intracellular pattern recognition receptors of the NOD-like receptor (NLR), HIN-200, and RIG-I-like protein families are increasingly recognized as critical sensors that detect conserved microbial components and endogenous danger-associated molecules in intracellular compartments (4, 9). A subset of NLR and HIN-200 proteins assembles inflammasomes, cytosolic multiprotein complexes that drive the proteolytic maturation of caspase-1, a proinflammatory protease whose activity is implicated in a variety of infectious and autoinflammatory diseases (10). Caspase-1 contributes to inflammatory responses by proteolytically maturing the proinflammatory cytokines IL-1β and IL-18, and by initiating pyroptosis, a rapid proinflammatory cell death mode in infected myeloid cells (10). Genetic studies in mice indicate that at least four inflammasomes are assembled depending on the infectious agent or danger-associated molecule that is encountered (9, 10). Notably, the Nlrp3 inflammasome responds to a wide variety of insults, including the detection of bacterial mRNA in intracellular compartments (11, 12). Recently, the Nlrp3 inflammasome was demonstrated to be responsible for inflammasome-mediated IL-1β and IL-18 production in macrophages infected with the enteric pathogens E. coli, Citrobacter rodentium, and Vibrio cholerae (13). Moreover, the Nlrp3 inflammasome plays a critical role in induction of immune and host responses against C. rodentium infection in vivo (14). However, how TLR and inflammasome activation are integrated during infection with enteric bacterial pathogens remains unclear.
Here, we show that Nlrp3 inflammasome activation and caspase-1 processing in macrophages infected with the enteric pathogens E. coli and C. rodentium specifically required the TLR adaptor TRIF downstream of TLR4. In contrast, TLR4 and TRIF were dispensable for Salmonella typhimurium-induced caspase-1 activation, which proceeded through the Nlrc4 inflammasome. TLR4 and TRIF mediated synthesis of caspase-11, a protease that is critical for E. coli- and C. rodentium-induced inflammasome activation. Thus, we identified TRIF-induced caspase-11 production as a new immune pathway that integrates TLR4- and Nlrp3 inflammasome-mediated recognition of enteric pathogens in macrophages.
EXPERIMENTAL PROCEDURES
Mice
All mice were fully backcrossed to C57BL/6 and housed at St. Jude Children's Research Hospital and Ghent University in a specific pathogen-free animal care facility. Tlr2−/− (15), Tlr4−/− (16), Tlr7−/− (17), Myd88−/− (18), Trif−/− (19), and Myd88−/−Trif−/− (19) mice were kindly provided by Dr. Akira (Osaka University) and have been described previously. Caspase-11−/− mice (13) were a kind gift of Dr. Vishva Dixit (Genentech). Nlrp3−/−, Nlrc4−/−, Asc−/−, Nod1−/−, Nod2−/−, Mavs−/−, Nlrp6−/−, and Nlrp12−/− mice used in this study have all been described previously (20–29). Animal studies were conducted under protocols approved by St. Jude Children's Research Hospital and Ghent University Committee on Use and Care of Animals.
Macrophage Culture and in Vitro Infections
Bone marrow-derived macrophages were prepared as described previously (14, 22, 30). Briefly, bone marrow cells were cultured in L cell-conditioned Iscove's modified Dulbecco's medium supplemented with 10% FBS, 1% nonessential amino acid, and 1% penicillin-streptomycin for 5 days to differentiate into macrophages. Bone marrow-derived macrophages were seeded in 6-well cell culture plates, stimulated with or without LPS (500 ng/ml) for 5 h, and infected with E. coli, C. rodentium, or S. typhimurium at the indicated multiplicity of infection (m.o.i.) for 24 h in a CO2 incubator at 37 °C. 1 h after infection, gentamycin (50 μg/ml) was added to the culture medium. As a positive control, macrophages were stimulated with LPS (1 μg/ml) for 4 h, the last half-hour of which in the presence of ATP (5 mm).
C. rodentium in Vivo Infection
C. rodentium (ATCC 51459) was grown in LB broth overnight with shaking at 37 °C. For in vivo experiments, mice were infected with 1 × 1010 c.f.u. by oral gavage. Food and water intake were stopped 8 h prior to infection and allowed to resume 1 h after infection. To determine bacterial counts, serial dilutions of homogenized feces were plated on MacConkey agar plates and incubated at 37 °C for 24 h.
Cytokine Analysis
Concentrations of mouse cytokines and chemokines in cell supernatants were determined using multiplex ELISA (Millipore), IL-1β ELISA (eBioscience), and IL-18 ELISA (MBL International).
Real-time Quantitative PCR
Total RNA was isolated using the TRIzol method (Invitrogen). First strand cDNA was generated from total RNA using High Capacity cDNA Reverse Transcription reagents (Applied Biosystems). cDNA samples were prepared in duplicate and subjected to real-time quantitative RT-PCR on an ABI Prism 7900 instrument using SYBR Green PCR Master Mix (Applied Biosystems) and normalized to the housekeeping gene Gapdh. The following primer pairs were used for quantitative RT-PCR analysis: mouse Gapdh forward, 5′-CGTCCCGTAGACAAAATGGT-3′ and reverse, 5′-TTGATGGCAACAATCTCCAC-3′; mouse caspase-11 forward, 5′-ACGATGTGGTGGTGAAAGAGGAGC-3′ and reverse, 5′-TGTCTCGGTAGGACAAGTGATGTGG-3′; mouse Nlrp3 forward, 5′-TGCAGAAGACTGACGTCTCC-3′ and reverse, 5′-CGTACAGGCAGTAGAACAGTTC-3′. Quantitative RT-PCR data were reported according to the standard curve method.
Western Blotting
For protein analysis of Nlrp3, caspase-1, and caspase-11, cell lysates were denatured with SDS plus 100 mm DTT and boiled for 5 min. SDS-PAGE-separated proteins were transferred to PVDF membranes and immunoblotted with primary antibodies against caspase-1, caspase-11, and Nlrp3 followed by secondary anti-rabbit HRP antibodies as described previously (23).
Statistics
GraphPad Prism 5.0 software was used for data analysis. Data are represented as mean ± S.E. or S.D. Statistical significance was determined by Student's t test; p < 0.05 was considered statistically significant.
RESULTS
Caspase-11 Is Required for C. rodentium- and E. coli-induced Caspase-1 Activation and Secretion of IL-1β and IL-18
C. rodentium is an enteropathogen of the murine gastrointestinal tract and triggers inflammatory responses resembling those of humans infected with enteropathogenic and enterohemorrhagic E. coli (31). Macrophages infected with these pathogens induce activation of the Nlrp3 inflammasome (13), and we recently described a critical role for this inflammasome in regulating host responses against C. rodentium in vivo (14). In agreement with previously published results from Kayagaki et al. (13), we show here that C. rodentium- and E. coli-induced caspase-1 activation was defective in caspase-11-deficient macrophages infected in vitro (Fig. 1, A and B). Interestingly, S. typhimurium-induced caspase-1 activation was intact in caspase-11-deficient macrophages (Fig. 1C), suggesting a specific role for caspase-11 in noncanonical inflammasome activation during C. rodentium and E. coli infection, as described before. In addition to caspase-1 processing, secretion of mature IL-1β and IL-18 in response to E. coli and C. rodentium infection was dependent on caspase-11 (Fig. 1, D–G), whereas S. typhimurium-induced secretion of IL-1β and IL-18 was not (Fig. 1, H and I). The role of caspase-11 in regulating production of IL-1β and IL-18 was specific because production of the inflammasome-independent cytokines TNF-α and IL-6 was similar in WT and caspase-11 macrophages infected with E. coli and C. rodentium (supplemental Fig. 1).
FIGURE 1.

Caspase-11 is required for inflammasome signaling in macrophages infected with C. rodentium and E. coli, but dispensable during S. typhimurium infection. Bone marrow-derived macrophages from WT and caspase-11−/− mice were infected with C. rodentium (m.o.i. 20), E. coli (m.o.i. 20), and S. typhimurium (m.o.i. 5) as described under “Experimental Procedures.” A–C, caspase-1 activation was determined by Western blotting of cell lysates. D–I, secreted IL-1β and IL-18 in supernatants of infected macrophages were quantified by ELISA. Data represent means ± S.E. (error bars) and are representative of three independent experiments.
Role of NLRs and AIM2 in E. coli- and C. rodentium-induced Activation of Noncanonical Inflammasome Signaling
Both the recent report by Kayagaki et al. (13) and our results (Fig. 1) demonstrate that caspase-11 is required for activation of the Nlrp3 inflammasome upon E. coli and C. rodentium infection. However, the adaptor molecules regulating Nlrp3 inflammasome activation upstream of caspase-11 are not known. To determine whether additional NLRs were involved in activation of noncanonical Nlrp3 inflammasome upstream of caspase-11, we examined caspase-1 activation in macrophages deficient for various NLR receptors and the HIN-200 protein AIM2. As expected, both C. rodentium- and E. coli-induced caspase-1 activation and IL-1β production were dependent on NLRP3 and ASC, whereas S. typhimurium-induced caspase-1 activation was dependent on NLRC4 and ASC (Fig. 2). Similar results were observed for LPS+ATP-induced caspase-1 activation, which required NLRP3 and ASC (supplemental Fig. 2). However, none of the other molecules tested (AIM2, NLRC4, NOD1, NOD2, NLRP6, and NLRP12) was required for caspase-1 activation in E. coli- and C. rodentium-infected macrophages, suggesting that these cytoplasmic adaptors do not play an important role in noncanonical inflammasome activation.
FIGURE 2.

Role of NLRs and adaptor molecules in caspase-1 activation and IL-1β secretion from macrophages infected with Gram-negative bacteria. Bone marrow-derived macrophages from WT, Asc−/−, Aim2−/−, Nlrp3−/−, Nlrc4−/−, Nod1−/−, Nod2−/−, Nlrp6−/−, and Nlrp12−/− mice were infected with C. rodentium (m.o.i. 20), E. coli (m.o.i. 20), or S. typhimurium (m.o.i. 5) for 24 h as described under “Experimental Procedures.” Caspase-1 activation was determined by Western blotting of cell lysates (A, C, and E), and secreted IL-1β in supernatants of infected macrophages was quantified by ELISA (B, D, and F). Data represent means ± S.E. (error bars) and are representative of at least three independent experiments.
TLR4 Is Specifically Required for Nlrp3 Inflammasome Activation in Macrophages Infected with C. rodentium and E. coli
Activation of the Nlrp3 inflammasome by extracellular ATP, pore-forming toxins and crystals requires NF-κB-mediated priming (32), but whether TLR-induced NF-κB activation mediates Nlrp3 inflammasome activation in response to enteropathogens is not known. Wild type, Tlr2−/−, and Tlr7−/− macrophages infected with C. rodentium secreted normal levels of IL-1β and IL-18 in the culture medium, but secretion of these inflammasome-dependent cytokines was significantly attenuated in TLR4-deficient macrophages (Fig. 3, A and B). Concurrently, E. coli-induced IL-1β and IL-18 secretion were severely hampered in Tlr4−/− macrophages (Fig. 3, C and D). Notably, Tlr4−/− macrophages infected with S. typhimurium also secreted less IL-1β and IL-18 (Fig. 3, E and F). This suggests that TLR4 is required for production of proIL-1β and proIL-18 in macrophages infected with Gram-negative bacteria irrespective of the inflammasome involved. To examine directly the role of TLR signaling in inflammasome activation by C. rodentium and E. coli, we analyzed caspase-1 maturation in cell lysates of macrophages infected with these pathogens. Caspase-1 processing into the large catalytic subunit (p20) was observed in wild type, Tlr2−/−, and Tlr7−/− macrophages infected with C. rodentium or E. coli, but not in Tlr4−/− macrophages (Fig. 3, G and H). Notably, S. typhimurium-induced caspase-1 activation was not affected in macrophages lacking TLR4 (Fig. 3I). These results suggest that in addition to its general role in inducing proIL-1β synthesis in macrophages infected with bacterial pathogens, TLR4 is specifically required for inflammasome activation by enteric bacterial pathogens.
FIGURE 3.

Role of TLR4 in inflammasome activation and secretion of IL-1β and IL-18 by C. rodentium-, E. coli-, and S. typhimurium-infected macrophages. WT, Tlr2−/−, Tlr4−/−, and Tlr7−/− bone marrow-derived macrophages were infected with C. rodentium (m.o.i. 20), E. coli (m.o.i. 20), and S. typhimurium (m.o.i. 5) for 24 h as described under “Experimental Procedures.” Secreted IL-1β and IL-18 in supernatants of infected macrophages were quantified by ELISA (A–F), and caspase-1 activation was determined by Western blotting of cell lysates (G–I). Data represent means ± S.E. (error bars) and are representative of at least three independent experiments.
TRIF Mediates Nlrp3 Inflammasome Activation by Enteropathogens
To examine further the mechanism by which TLR4 mediates Nlrp3 inflammasome activation in macrophages infected with enteropathogens, cells lacking the TLR adaptors MyD88 (MyD88−/−), TRIF (Trif−/−), or both MyD88 and TRIF (MyD88−/−Trif−/−) were infected with C. rodentium and E. coli as before. Tlr2−/− and Tlr4−/− macrophages were included in these experiments as positive and negative controls, respectively. C. rodentium-induced caspase-1 activation proceeded normally in MyD88−/− macrophages but was abrogated in macrophages lacking TRIF alone or in combination with MyD88 (Fig. 4A). Similarly, Tlr4−/−, Trif−/−, and MyD88−/−Trif−/− macrophages failed to activate caspase-1 when infected with E. coli, whereas wild type, Tlr2−/−, and MyD88−/− macrophages responded with robust caspase-1 processing (Fig. 4B). Unlike these enteropathogens, S. typhimurium-induced caspase-1 activation proceeded unabated in macrophages lacking TRIF (Fig. 4C). Together with our previous results, this suggests TLR4- and TRIF-mediated signaling to be specifically required for enteropathogen-induced inflammasome activation. In agreement with these caspase-1-processing data, induction of pyroptotic cell death was also reduced in Tlr4−/− and Trif−/− macrophages compared with WT macrophages infected with C. rodentium or E. coli, respectively (supplemental Fig. 3). In contrast, pyroptosis in Myd88−/− macrophages was induced to levels comparable with those of WT macrophages infected with C. rodentium or E. coli as determined by lactate dehydrogenase release assay (supplemental Fig. 3).
FIGURE 4.
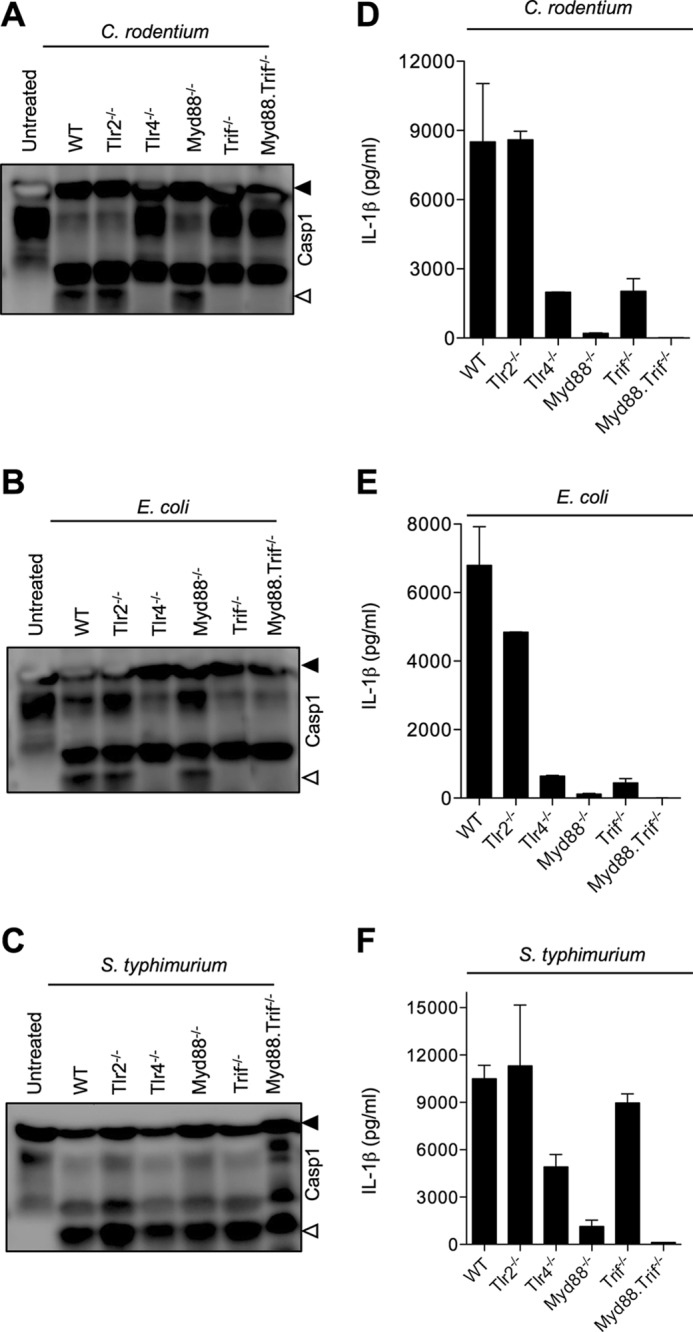
TRIF is critical for C. rodentium- and E. coli-induced inflammasome activation. WT, Tlr2−/−, Tlr4−/−, Myd88−/−, Trif−/−, and Myd88−/−Trif−/− macrophages were infected with C. rodentium (m.o.i. 20), E. coli (m.o.i. 20), or S. typhimurium (m.o.i. 5) as described under “Experimental Procedures.” Caspase-1 activation was determined by Western blotting of cell lysates (A–C), and secreted IL-1β in supernatants of infected macrophages was quantified by ELISA (D–F). Data represent means ± S.E.(error bars) and are representative of at least three independent experiments.
Despite being dispensable for C. rodentium- and E. coli-induced caspase-1 activation, MyD88 was required for IL-1β secretion from macrophages infected with these pathogens (Fig. 4, D and E). Notably, IL-1β secretion was also reduced in Trif−/− macrophages, but its production was completely blunted in cells lacking MyD88 (Fig. 4, D and E). In agreement with a general role in proIL-1β synthesis, MyD88 was also required for S. typhimurium-induced IL-1β secretion (Fig. 4F). The modestly higher level of secreted IL-1β in Tlr4−/− cells relative to MyD88−/− macrophages suggests that MyD88 may contribute to NF-κB-driven production of proIL-1β downstream of multiple TLRs during Gram-negative infections. Unlike MyD88, TRIF was specifically required for secretion of IL-1β from macrophages infected with C. rodentium or E. coli (Fig. 4, D and E), and not in cells infected with S. typhimurium (Fig. 4F). To address further how TRIF regulates IL-1β expression, we determined IL-1β mRNA levels in WT and Trif−/− macrophages infected with C. rodentium or E. coli. IL-1β mRNA synthesis partially depended on TRIF, whereas MyD88 was critical for enteropathogen-induced IL-1β mRNA production (supplemental Fig. 4). Altogether, these results suggest MyD88 to be critical for proIL-1β production in macrophages infected with Gram-negative bacteria, whereas TRIF specifically controls enteropathogen-induced inflammasome activation and partially regulates IL-1β transcription.
TLR4 and TRIF Are Required for Caspase-11 Synthesis and Activation
Activation of the Nlrp3 inflammasome in C. rodentium- and E. coli-infected macrophages was previously shown to rely on caspase-11 (13). In agreement, we found caspase-1 maturation and IL-1β production to be significantly hampered in caspase-11-deficient cells infected with C. rodentium or E. coli (Fig. 1). Having established that caspase-11 is required for inflammasome activation by C. rodentium and E. coli, we explored the hypothesis that TLR4 and TRIF may specifically regulate enteropathogen-induced inflammasome activation by modulating the expression of caspase-11. Relative to levels in C. rodentium- and E. coli-infected wild type and TLR2-deficient macrophages, caspase-11 production in cells lacking MyD88 was largely normal (Fig. 5, A and B). In contrast, caspase-11 mRNA induction was markedly reduced in Tlr4−/−, Trif−/−, and MyD88−/−Trif−/− macrophages infected with C. rodentium or E. coli (Fig. 5, A and B), indicating that TLR4 and TRIF are required for caspase-11 production. In agreement, procaspase-11 protein levels and caspase-11 processing in Tlr4−/− and Trif−/− macrophages that have been infected with C. rodentium or E. coli were significantly reduced relative to infected WT cells (Fig. 5, C and D). Notably, TLR4 and MyD88, but not TRIF, were required for efficient induction of Nlrp3 transcripts in response to these enteropathogens (Fig. 5, C and D) although Nlrp3 protein levels remained relatively stable in infected WT, Tlr4−/−, and Trif−/− macrophages (Fig. 5, G and H). Together, these results suggest that TLR4 and TRIF modulate enteropathogen-induced inflammasome activation by promoting caspase-11 expression and activation.
FIGURE 5.

The TLR4-TRIF axis regulates caspase-11 expression. A, B, E, and F, WT, Tlr2−/−, Tlr4−/−, Myd88−/−, Trif−/−, and Myd88−/−Trif−/− macrophages were infected with C. rodentium and E. coli. RNA was extracted 6 h after infection, and mRNA expression of caspase-11 and Nlrp3 was determined as described under “Experimental Procedures.” RNA expression were normalized to the expression of GAPDH and then depicted as -fold increase. C, D, G, and H, cell lysates of WT, Tlr4−/−, and Trif−/− macrophages infected with C. rodentium or E. coli for 24 h were analyzed for caspase-11 and Nlrp3 expression by Western blotting. Data represent means ± S.E. (error bars) and are representative of at least three independent experiments.
Role of TRIF, MyD88, and Caspase-11 during C. rodentium Infection in Vivo
Previous studies showed that mice deficient in the TLR adaptor MyD88 are highly susceptible to C. rodentium infection (33). We showed here that the TLR4-TRIF signaling axis is important for caspase-11 expression and Nlrp3 inflammasome activation during E. coli and C. rodentium infection of macrophages in vitro. To determine whether TLR4 and TRIF were important for C. rodentium infection in vivo, mice deficient in TLR4, TRIF, MyD88, or both TRIF and MyD88 were infected orally with C. rodentium. Mice lacking only TRIF or MyD88 survived, whereas mice doubly deficient for MyD88 and TRIF were highly susceptible to infection and all succumbed by day 8 (Fig. 6A). These results suggest that TRIF and MyD88 play redundant roles during C. rodentium infection and that deletion of both markedly affected host responses against enteropathogen infection. Although Trif−/− and Myd88−/− mice showed no difference in survival compared with WT mice, bacterial counts in feces were increased, albeit only modestly in Trif−/− mice (Fig. 6B). As expected, bacterial burdens in Myd88−/−Trif−/− mice were more elevated than in animals lacking MyD88 or TRIF alone (Fig. 6B), suggesting that these mice succumbed to infection consequent to uncontrollable bacterial replication. The response of C. rodentium-infected caspase-11−/− mice resembled that of Trif−/− mice in that they also had nearly normal (statistically not significant) bacterial numbers (Fig. 6C), and none of the infected caspase-11−/− mice succumbed to infection (data not shown). These results suggest that unlike in in vitro-infected macrophages, TRIF and caspase-11 play relatively modest roles during C. rodentium infection in vivo.
FIGURE 6.

Roles of TLR4-TRIF and caspase-11 during C. rodentium infection in vivo. A, survival of WT, Tlr4−/−, Myd88−/−, Trif−/−, and Myd88−/−Trif−/− mice infected with 1 × 1010 C. rodentium by oral gavage. B and C, bacterial burden in fecal pellets of infected mice determined at day 7 after infection. D, working model for TLR4-/TRIF-mediated regulation of caspase-11 expression and noncanonical inflammasome activation in C. rodentium- and E. coli-infected macrophages.
DISCUSSION
Kayagaki et al. (13) previously described a critical role for caspase-11 in Nlrp3 inflammasome activation and IL-1β secretion in macrophages infected with the enteropathogens C. rodentium, E. coli, and V. cholerae. We extended these results to an in vivo setting by demonstrating that the Nlrp3 inflammasome contributed importantly to host defense against C. rodentium because mice lacking Nlrp3 or doubly deficient for the inflammasome proteases caspase-1 and -11 were hypersusceptible to C. rodentium infection (14). However, insight into the mechanisms controlling caspase-11-mediated inflammasome activation during infections with enteropathogens in infected macrophages and the relative importance of caspase-11 during C. rodentium infection in vivo was lacking.
Here, we provided genetic and biochemical evidence implicating TLR4 and TRIF in regulating expression of caspase-11 and activation of the Nlrp3 inflammasome in response to infection with enteropathogens such as E. coli and C. rodentium. Notably, our results revealed differential roles for MyD88 and TRIF in modulating enteropathogen-induced Nlrp3 inflammasome activation and IL-1β secretion, suggesting a mechanistic model in which these TLR adaptors integrate TLR and inflammasome responses through nonredundant mechanisms (Fig. 6D). Indeed, the TLR4-TRIF signaling axis was critical for up-regulation of procaspase-11 and its processing during noncanonical activation of the Nlrp3 inflammasome by C. rodentium and E. coli. Caspase-11 activation may result from “spontaneous” autocatalytic processing in cis or trans when a certain concentration threshold has been surpassed (34). More likely, however, caspase-11 activation may require its recruitment in a multiprotein complex that induces proximity-induced oligomerization and activation-promoting conformational changes in caspase-11 zymogens, as described for activation of other initiator caspases (35, 36) In this model, autocatalytic maturation may function to subsequently stabilize activity of assembled caspase homo- and heterodimers.
Our results implicate TRIF signaling in regulating caspase-11 production and activation, but further analysis is required to clarify the molecular mechanism by which the TLR4-TRIF axis regulates caspase-11 expression. TRIF is known to regulate IRF3/7-mediated type I interferon production and has been implicated in delayed NF-κB activation (37, 38). Type I interferon signaling was recently confirmed to contribute to caspase-11 synthesis (39), but the role of delayed NF-κB signaling remains to be examined. Notably, the TLR4-TRIF signaling axis appears to be specifically required during enteropathogen infections, as TRIF and caspase-11 were dispensable for inflammasome activation by S. typhimurium. In contrast, MyD88 mediated transcriptional up-regulation of NLRP3 and proIL-1β, but was dispensable for caspase-11 synthesis during E. coli and C. rodentium infection. Moreover, MyD88 was required for proIL-1β production in response to enteropathogens and other stimuli alike, including LPS+ATP and S. typhimurium infection. Interestingly, macrophages that were stimulated with LPS or infected with S. typhimurium both responded with inducing caspase-11 expression (data not shown), similar to cells infected with C. rodentium or E. coli. However, why caspase-11 is critical for inflammasome activation during C. rodentium and E. coli infections, but not in response to Salmonella or LPS+ATP treatment requires further investigation. Relative to C. rodentium and E. coli, LPS+ATP and S. typhimurium potently induce inflammasome activation in a short time frame (i.e. <3 h). One possibility may therefore be that relatively fast inflammasome activation may bypass the requirement for caspase-11, whereas stimuli that trigger inflammasome activation at later time points (as during E. coli and C. rodentium infection) may proceed through caspase-11. In support of this hypothesis is the observation that a mutant strain of S. typhimurium that lacks flagellin (S. typhimurium flib/c) fails to activate the Nlrc4 inflammasome in the first hours after infection, but instead required caspase-11 to activate the Nlrp3 inflammasome upon prolonged infection (>12 h) (data not shown). An alternative explanation for the differential requirement for caspase-11 is that C. rodentium and E. coli produce yet unknown molecules that trigger caspase-11 activation, which are absent during LPS+ATP stimulation and during the initial phases of S. typhimurium infection. Additional work is needed to dissect these mechanistic models further.
We previously demonstrated a critical role for the Nlrp3 inflammasome in host defense against C. rodentium in vivo (14). Similarly, MyD88-deficient mice are highly susceptible to C. rodentium infection (33). Notably, susceptibility increased further when both TRIF and MyD88 signaling were abrogated simultaneously. In addition to increased bacterial counts in the stool of C. rodentium-infected Myd88−/−Trif−/− mice, this was reflected by a 100% mortality rate. Unlike Myd88−/−Trif−/− mice, however, we found that Trif−/− mice displayed only slightly increased bacterial burdens in the stool without any associated morbidity or mortality. Also, caspase-11−/− mice presented with few signs of increased disease progression, suggesting that noncanonical inflammasome signaling plays a relatively minor role during in vivo infection with C. rodentium. Because Nlrp3 signaling is important for host defense against C. rodentium in vivo (14) and given that caspase-11 is critical for Nlrp3 inflammasome activation in C. rodentium-infected macrophages, this suggests that the role of noncanonical inflammasome signaling in C. rodentium-infected mice might be mitigated by canonical Nlrp3 activation in other cell types. Further analysis is required to examine the mechanism of enteropathogen-induced Nlrp3 inflammasome activation in additional cell types that may contribute to host responses in vivo such as epithelial cells, fibroblasts, and other hematopoietic cell types.
In conclusion, we showed here that TRIF and MyD88 signaling downstream of TLR4 differentially regulate Nlrp3 inflammasome activation and IL-1β secretion in macrophages infected with enteropathogens. The TLR4-TRIF axis regulated noncanonical inflammasome activation by promoting transcription induction of procaspase-11 expression and procaspase-11 processing, whereas the TLR4-MyD88 pathway controlled transcriptional up-regulation of Nlrp3 and proIL-1β. These results highlight the nonredundant roles of MyD88 and TRIF in integrating TLR- and inflammasome activation during enteropathogen infection.
Acknowledgments
We thank Anthony Coyle, Ethan Grant, John Bertin (Millennium Pharmaceuticals), Shizuo Akira (Osaka University), Vishva Dixit (Genentech), Gabriel Nuñez (University of Michigan), and Richard Flavell (Yale University) for a generous supply of mutant mice.
This work was supported, in whole or in part, by National Institutes of Health Grants AR056296, AI101935, and CA163507 (to T.-D. K.). This work was also supported by Marie-Curie Grant 256432 from the European Union, European Research Council Grant 281600, and the Fund for Scientific Research-Flanders Grants G030212N, 1.2.201.10.N.00, and 1.5.122.11.N.00 (to M. L.), and by the American Lebanese Syrian Associated Charities (ALSAC) (to T.-D. K.).

This article contains supplemental Figs. 1–4.
- TLR
- Toll-like receptor
- m.o.i.
- multiplicity of infection
- NLR
- NOD-like receptor
- TRIF
- Toll or interleukin receptor (TIR) domain-containing adaptor-inducing interferon-β.
REFERENCES
- 1. Kaper J. B., Nataro J. P., Mobley H. L. (2004) Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2, 123–140 [DOI] [PubMed] [Google Scholar]
- 2. DuPont H. L. (2009) Clinical practice: bacterial diarrhea. N. Engl. J. Med. 361, 1560–1569 [DOI] [PubMed] [Google Scholar]
- 3. Kappelman M. D., Rifas-Shiman S. L., Porter C. Q., Ollendorf D. A., Sandler R. S., Galanko J. A., Finkelstein J. A. (2008) Direct health care costs of Crohn's disease and ulcerative colitis in US children and adults. Gastroenterology 135, 1907–1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Takeuchi O., Akira S. (2010) Pattern recognition receptors and inflammation. Cell 140, 805–820 [DOI] [PubMed] [Google Scholar]
- 5. Kawai T., Akira S. (2006) TLR signaling. Cell Death Differ. 13, 816–825 [DOI] [PubMed] [Google Scholar]
- 6. Poltorak A., He X., Smirnova I., Liu M. Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 [DOI] [PubMed] [Google Scholar]
- 7. Qureshi S. T., Larivière L., Leveque G., Clermont S., Moore K. J., Gros P., Malo D. (1999) Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4) J. Exp. Med. 189, 615–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roger T., Froidevaux C., Le Roy D., Reymond M. K., Chanson A. L., Mauri D., Burns K., Riederer B. M., Akira S., Calandra T. (2009) Protection from lethal Gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc. Natl. Acad. Sci. U.S.A. 106, 2348–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kanneganti T. D., Lamkanfi M., Núñez G. (2007) Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559 [DOI] [PubMed] [Google Scholar]
- 10. Lamkanfi M., Walle L. V., Kanneganti T. D. (2011) Deregulated inflammasome signaling in disease. Immunol. Rev. 243, 163–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kanneganti T. D., Body-Malapel M., Amer A., Park J. H., Whitfield J., Franchi L., Taraporewala Z. F., Miller D., Patton J. T., Inohara N., Núñez G. (2006) Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 281, 36560–36568 [DOI] [PubMed] [Google Scholar]
- 12. Sander L. E., Davis M. J., Boekschoten M. V., Amsen D., Dascher C. C., Ryffel B., Swanson J. A., Müller M., Blander J. M. (2011) Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature 474, 385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kayagaki N., Warming S., Lamkanfi M., Vande Walle L., Louie S., Dong J., Newton K., Qu Y., Liu J., Heldens S., Zhang J., Lee W. P., Roose-Girma M., Dixit V. M. (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 [DOI] [PubMed] [Google Scholar]
- 14. Liu Z., Zaki M. H., Vogel P., Gurung P., Finlay B. B., Deng W., Lamkanfi M., Kanneganti T. D. (2012) Role of inflammasomes in host defense against Citrobacter rodentium infection. J. Biol. Chem. 287, 16955–16964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takeuchi O., Hoshino K., Akira S. (2000) Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 165, 5392–5396 [DOI] [PubMed] [Google Scholar]
- 16. Hoshino K., Takeuchi O., Kawai T., Sanjo H., Ogawa T., Takeda Y., Takeda K., Akira S. (1999) Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162, 3749–3752 [PubMed] [Google Scholar]
- 17. Hemmi H., Kaisho T., Takeuchi O., Sato S., Sanjo H., Hoshino K., Horiuchi T., Tomizawa H., Takeda K., Akira S. (2002) Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 3, 196–200 [DOI] [PubMed] [Google Scholar]
- 18. Adachi O., Kawai T., Takeda K., Matsumoto M., Tsutsui H., Sakagami M., Nakanishi K., Akira S. (1998) Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9, 143–150 [DOI] [PubMed] [Google Scholar]
- 19. Yamamoto M., Sato S., Hemmi H., Hoshino K., Kaisho T., Sanjo H., Takeuchi O., Sugiyama M., Okabe M., Takeda K., Akira S. (2003) Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643 [DOI] [PubMed] [Google Scholar]
- 20. Franchi L., Amer A., Body-Malapel M., Kanneganti T. D., Ozören N., Jagirdar R., Inohara N., Vandenabeele P., Bertin J., Coyle A., Grant E. P., Núñez G. (2006) Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat. Immunol. 7, 576–582 [DOI] [PubMed] [Google Scholar]
- 21. Jones J. W., Kayagaki N., Broz P., Henry T., Newton K., O'Rourke K., Chan S., Dong J., Qu Y., Roose-Girma M., Dixit V. M., Monack D. M. (2010) Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. U.S.A. 107, 9771–9776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kanneganti T. D., Lamkanfi M., Kim Y. G., Chen G., Park J. H., Franchi L., Vandenabeele P., Núñez G. (2007) Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 26, 433–443 [DOI] [PubMed] [Google Scholar]
- 23. Kanneganti T. D., Ozören N., Body-Malapel M., Amer A., Park J. H., Franchi L., Whitfield J., Barchet W., Colonna M., Vandenabeele P., Bertin J., Coyle A., Grant E. P., Akira S., Núñez G. (2006) Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236 [DOI] [PubMed] [Google Scholar]
- 24. Kobayashi K., Inohara N., Hernandez L. D., Galán J. E., Núñez G., Janeway C. A., Medzhitov R., Flavell R. A. (2002) RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 416, 194–199 [DOI] [PubMed] [Google Scholar]
- 25. Kobayashi K. S., Chamaillard M., Ogura Y., Henegariu O., Inohara N., Nuñez G., Flavell R. A. (2005) Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734 [DOI] [PubMed] [Google Scholar]
- 26. Lamkanfi M., Malireddi R. K., Kanneganti T. D. (2009) Fungal zymosan and mannan activate the cryopyrin inflammasome. J. Biol. Chem. 284, 20574–20581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chamaillard M., Hashimoto M., Horie Y., Masumoto J., Su Q., Saab L., Ogura Y., Kawasaki A., Fukase K., Kusumoto S., Valvano M. A., Foster S. J., Mak T. W., Nuñez G., Inohara N. (2003) An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 4, 702–707 [DOI] [PubMed] [Google Scholar]
- 28. Anand P. K., Malireddi R. K., Lukens J. R., Vogel P., Bertin J., Lamkanfi M., Kanneganti T. D. (2012) NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488, 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zaki M. H., Vogel P., Malireddi R. K., Body-Malapel M., Anand P. K., Bertin J., Green D. R., Lamkanfi M., Kanneganti T. D. (2011) The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20, 649–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Malireddi R. K., Ippagunta S., Lamkanfi M., Kanneganti T. D. (2010) Cutting edge: proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. J. Immunol. 185, 3127–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mundy R., MacDonald T. T., Dougan G., Frankel G., Wiles S. (2005) Citrobacter rodentium of mice and man. Cell. Microbiol. 7, 1697–1706 [DOI] [PubMed] [Google Scholar]
- 32. Bauernfeind F. G., Horvath G., Stutz A., Alnemri E. S., MacDonald K., Speert D., Fernandes-Alnemri T., Wu J., Monks B. G., Fitzgerald K. A., Hornung V., Latz E. (2009) Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gibson D. L., Ma C., Bergstrom K. S., Huang J. T., Man C., Vallance B. A. (2008) MyD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during Citrobacter rodentium-induced colitis. Cell. Microbiol. 10, 618–631 [DOI] [PubMed] [Google Scholar]
- 34. Kang S. J., Wang S., Kuida K., Yuan J. (2002) Distinct downstream pathways of caspase-11 in regulating apoptosis and cytokine maturation during septic shock response. Cell Death Differ. 9, 1115–1125 [DOI] [PubMed] [Google Scholar]
- 35. Boatright K. M., Renatus M., Scott F. L., Sperandio S., Shin H., Pedersen I. M., Ricci J. E., Edris W. A., Sutherlin D. P., Green D. R., Salvesen G. S. (2003) A unified model for apical caspase activation. Mol. Cell 11, 529–541 [DOI] [PubMed] [Google Scholar]
- 36. Salvesen G. S., Dixit V. M. (1999) Caspase activation: the induced-proximity model. Proc. Natl. Acad. Sci. U.S.A. 96, 10964–10967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kawai T., Takeuchi O., Fujita T., Inoue J., Mühlradt P. F., Sato S., Hoshino K., Akira S. (2001) Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 167, 5887–5894 [DOI] [PubMed] [Google Scholar]
- 38. Akira S., Takeda K. (2004) Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511 [DOI] [PubMed] [Google Scholar]
- 39. Rathinam V. A., Vanaja S. K., Waggoner L., Sokolovska A., Becker C., Stuart L. M., Leong J. M., Fitzgerald K. A. (2012) TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by Gram-negative bacteria. Cell 150, 606–619 [DOI] [PMC free article] [PubMed] [Google Scholar]