

Abstract
Recombination activating gene 2 (RAG2) deficiency results in severe combined immunodeficiency (SCID) with complete lack of T and B lymphocytes. Initial gammaretroviral gene therapy trials for other types of SCID proved effective, but also revealed the necessity of safe vector design. We report the development of lentiviral vectors with the spleen focus forming virus (SF) promoter driving codon-optimized human RAG2 (RAG2co), which improved phenotype amelioration compared to native RAG2 in Rag2−/− mice. With the RAG2co therapeutic transgene, T-cell receptor (TCR) and immunoglobulin repertoire, T-cell mitogen responses, plasma immunoglobulin levels and T-cell dependent and independent specific antibody responses were restored. However, the thymus double positive T-cell population remained subnormal, possibly due to the SF virus derived element being sensitive to methylation/silencing in the thymus, which was prevented by replacing the SF promoter by the previously reported silencing resistant element (ubiquitous chromatin opening element (UCOE)), and also improved B-cell reconstitution to eventually near normal levels. Weak cellular promoters were effective in T-cell reconstitution, but deficient in B-cell reconstitution. We conclude that immune functions are corrected in Rag2−/− mice by genetic modification of stem cells using the UCOE driven codon-optimized RAG2, providing a valid optional vector for clinical implementation.
Introduction
Recombination activating gene 2 (RAG2) deficiency is an autosomal recessive disorder causing a complete lack of mature T and B lymphocytes leading to severe combined immunodeficiency (SCID). HLA-identical bone marrow (BM) transplantation is the treatment of choice, but due to limited availability of HLA-matched donors alternative donor stem cell sources are a necessity for most SCID patients, however limited in efficacy and with variable outcomes.1
Recently, gammaretroviral gene therapy has demonstrated long-term clinical efficacy for X-linked SCID (SCID-X1)2,3,4 and adenosine deaminase-SCID5 by complementation of a correct copy of the defected gene. However, the SCID-X1 trials also highlighted oncogenic risks requiring improved vector design for clinical safety.6,7
RAG2 protein acts in a coordinated fashion with RAG1 (collectively referred to as RAG) to achieve V(D)J recombination of T-cell receptors (TCRs) and immunoglobulins (Igs) to recognize a broad diversity of antigens.8 Gammaretroviral vector RAG2 gene therapy to treat Rag2−/− mice resulted in sustained correction,9 but the use of an LTR mutated Moloney murine leukemia virus enhancer promoter10 still carries the inherent oncogenic risk of modifying proto-oncogene expression.
Recoding the transgene to optimize transcription and translation may improve lentiviral vector titers as well as protein production and has been shown to significantly improve efficacy, e.g. for RAG1 SCID,11 X-linked SCID12 and gp91phox 13 in chronic granulomatous disease. Promoter choice may also be critical in successful and safe restoration of genetic defects, since viral promoters are relatively unsafe and prone to methylation and silencing.14 Therefore, the use of cellular-specific promoters15,16,17 or methylation resistant elements, such as the ubiquitous chromatin opening element (UCOE),11,14,18 should improve long-term and safe intervention.
HIV-based lentiviral (LV) vectors are considered further to have an improved safety profile by a more favorable integration pattern with no preference for proto-oncogenes and transcription start sites,19,20,21 while (pre-)stimulation with growth factors is not a strict prerequisite for efficient transduction, thereby preserving stem cell numbers during the transduction procedure. We investigated whether recoding RAG2 for improved expression benefited phenotype correction of Rag2−/− mice by transplantation of lentiviral vector gene-modified stem cells.
Results
Amelioration of peripheral blood T and B cells
Six- to twelve-week-old female Rag2−/− recipients of male Lin- BM cells transduced with the gene therapy vectors after a sublethal dose of 6–7 Gy total body irradiation showed significant long-term populations of peripheral blood (PB) T-cell numbers for all groups (Table 1, Figure 1a,b). At one month after transplantation, CD3+ numbers were 63-fold increased (P < 0.01) in SF-RAG2co mice compared to SF-RAG2 mice, similar to the other gene therapy treated groups, but tenfold lower (P < 0.001) than those resulting from transplanted wild-type (WT) cells.
Table 1. Absolute peripheral T and B-cell counts in time.
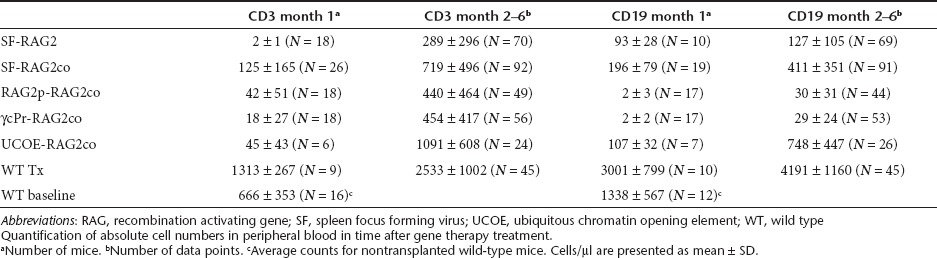
Figure 1.

Reconstitution of T and B cells in peripheral blood (PB). A 6 months follow-up of the absolute number (a) CD3+CD4+, and (b) CD3+CD8+ T-lymphocytes, and (c) CD19+, (d) CD11b−B220+IgM+, (e) CD11b−B220+IgD+ B-lymphocytes. The gray area in the graphs depicts the range of absolute PB cells in untreated wild-type mice.
PB T-cell numbers stabilized two months after transplantation (Table 1, Figure 1a,b), at which time interval PB CD3+ T-cell numbers were on average 2.5-fold higher (P < 0.001) in the SF-RAG2co group than in the SF-RAG2 group, as were the RAG2p-RAG2co and γcPr-RAG2co mice. The UCOE-RAG2co group had cell numbers equivalent to normal WT levels and overall higher than the other groups (P < 0.005), with the exception of the WT group that displayed sustained supranormal levels for both T and B cells.
PB B-cell reconstitution showed differential kinetics depending on the promoter cassette (Table 1, Figure 1c–e). One month after transplantation CD19+ B-cell numbers in SF-RAG2co mice were similar to UCOE-RAG2co and SF-RAG2 treated mice, and ~100-fold higher (P < 0.05) than RAG2p-RAG2co or γcPr-RAG2co mice, which remained barely detectable over time. Of note, B cells in all groups were significantly lower than those in recipients of WT cells (P < 0.001).
B-cell levels continued to increase 2 months after transplantation with the average CD19+ values of SF-RAG2co mice threefold higher than the SF-RAG2 group (P < 0.001), but on average twofold lower than the UCOE-RAG2co group (P < 0.005). The CD19+ B cells in the SF-RAG2co group were significantly lower than WT and untreated WT mice (P < 0.001), whereas the UCOE-RAG2co group eventually reached near normal WT levels.
Thymic development and T-cell responses
Rag2−/− mice have an early arrest at the double negative (DN) 3 stage (CD44−CD25+) in the thymus (Figure 2a). Six months after transplantation, all gene therapy groups displayed a reduction of DN3 percentages. Progression into double positive (DP) T cells was improved in SF and UCOE mice, but percentages were lower in the γcPr and RAG2p treated mice. Variable smaller population sizes of single CD8+ and CD4+ cells were also detected in all groups, except Rag2−/− mice. However, absolute thymocyte cellularity was significantly lower in all gene therapy treated groups relative to recipients of WT cells (Table 2), and a considerable number of mice in the SF treated groups displayed low DP percentages (Table 3).
Figure 2.

Thymic development and T-cell responses to mitogens. (a) After 6 months after transplantation, T-cell differentiation stages in the thymus are gated on the double negative (DN, CD4−CD8−) T-cell population by detection of CD44 and CD25. The differentiation stages CD44+CD25− (DN1), CD44+CD25+ (DN2), CD44−CD25+ (DN3) and CD44−CD25− (DN4) show a block at DN3 in Rag2−/− mice, which was markedly reduced by all promoters (n = 3–9). (b) Further development into double positive CD4+CD8+ cells occurs mainly in the SF-RAG2, RAG2co and UCOE-RAG2co treated mice with progression into single CD4+ and CD8+ cells (n = 3–12). (c) Thymic stainings for hematoxylin and eosin (H&E), CK5 and CK8 assess the cortico-medullary demarcation (CMD; depicted with a broken line in the H&E left panel): AIRE and binding to fucose-specific lectins (UEA) highlights mature mTECs; TdT and CD3 staining show distribution of maturing T lymphocytes, Foxp3 staining highlights medullary mature Treg and caspase 3 (Casp3) have been used as a marker for T-lymphocyte selection. Top panel represents WT thymi, compared to Rag2−/− thymi (middle upper panel) and gene therapy treated mice, SF-RAG2co (middle lower panel) and UCOE-RAG2co (bottom panel). Original magnification ×10 and ×40 (AIRE, UEA and Foxp3), and Bar = 200 µm and Bar = 50 µm. Assessment of T-cell percentages in (d,e) spleen and (f,g) lymph nodes, shows detectable populations for all treatment groups, but the SF-RAG2co and UCOE-RAG2co in general resulted in a larger T-cell subset. (h) Mitogen mediated proliferation response of splenocytes to CD3 stimulation, ConA and IL-2 were similar to wild-type in the SF-RAG2co, γcPr and ubiquitous chromatin opening element (UCOE) treated mice (n = 3–9) (No stim = no addition of mitogens) cpm, counts per minute.
Table 2. Absolute cell counts in hematopoietic tissues.
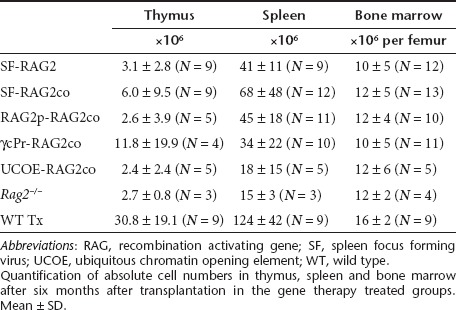
Table 3. Double positive population in thymus.

Thymic architecture in the SF-RAG2co and UCOE-RAG2co mice was assessed by histological and immunohistochemical staining (Figure 2c). In WT mice, hematoxylin and eosin and cytokeratin 5 and 8 (CK5 and CK8) staining highlighted a normal cortico-medullary differentiation with fully mature medullary thymic epithelial cells (mTECs) expressing both Ulex europaeus agglutinin (UEA) and AutoImmune REgulator (AIRE) protein. Terminal deoxynucleotidyl Transferase (TdT) and CD3 staining showed normal distribution of maturing thymocytes. Mature T-regs have been highlighted in the medulla by Foxp3 immunostain and caspase 3 (Casp 3) positive cells were detected into both the cortical and medullary regions, indicating normal lymphocyte selection. Rag2−/− mice showed a virtual lack of cortico-medullary demarcation with only small foci containing rare mature mTECs; CK5, AIRE, UEA, TdT and CD3 staining revealed widespread distribution of immature thymocytes, highlighting the focal mature medullary-like regions. Foxp3+ cells were consistently absent, showing lack of mature regulatory T cells. Moreover, activated caspase 3 were only rarely positive, indicating severe deficiency of thymocyte selection (Figure 2c, higher panels).
At 5 months after transplantation, thymi of both SF-RAG2co and UCOE-RAG2co treated Rag2−/− mice displayed a clear cortico-medullary demarcation along with focal aggregation of fully mature mTEC expressing CK5+, AIRE and UEA binding as well as normal distribution of developing thymocytes expressing TdT and CD3. Foxp3 was present within the medullary regions, and Casp 3 expression was improved compared to the Rag2−/− mice (Figure 2c, lower panels), demonstrating that for both SF-RAG2co and UCOE-RAG2co adequate rescue was achieved.
Six months after transplantation, populations of T cells in the spleen (Figure 2d,e) and lymph nodes (Figure 2f,g) also showed improved rescue in SF-RAG2co and UCOE-RAG2co mice relative to the other treatment groups, despite absolute spleen cell numbers being low compared to recipients of WT cells (Table 2). CD8+ cells were particularly low in spleens of SF-RAG2 mice. Additionally, UCOE-RAG2co mice displayed a normal distribution of naive cells (CD62LhighCD44low), effector cells (CD62LlowCD44high) and central memory cells (CD62LhighCD44high) in the lymph nodes (Supplementary Figure S1).
Spleen cell proliferation responses to mitogens were assessed by in vitro stimulation with CD3 antibody, ConA or interleukin-2 (Figure 2h). The SF-RAG2co, γcPr-RAG2co, and UCOE-RAG2co mice (RAG2p-RAG2co not done) responded similar to WT transplanted mice, showing that T cells were functional, except for a poor response in SF-RAG2 mice.
TCR and Ig repertoire in gene therapy treated mice
In PB, CD4 and CD8 positive TCR Vβ isotypes 4 6, 7, 8.1/8.2, 9, 10b, 13 and 14 were measured by flow cytometry to assess functional RAG recombination to obtain a variety of TCR receptors (Figure 3a,b). All gene therapy treated mice resembled the normal TCR Vβ isotype distribution as observed in WT Tx mice in both CD4+ and CD8+ cells, except for the SF-RAG2 mice. These mice had dominant TCR Vβ isotypes (Supplementary Table S1), which might explain the poor T-cell responses to mitogens.
Figure 3.

T-cell receptor (TCR) and Ig diversity in gene therapy treated mice. Flow cytometric analysis of peripheral blood (a) CD4+ and (b) CD8+ TCR Vβ isotypes at 4 months after transplantation (n = 4–10). The large variation in the SF-RAG2 treated mice was caused by presence of dominant TCR Vβ clones. (c) TCR and Ig repertoire analysis of spleen cDNA derived from mice transplanted with transduced Lin− cells, 4 months after transplantation. Depicted are the polyclonal rearrangement patterns of seven Vβ genes and two immunoglobulin heavy chains Cµ (IgM) and Cγ (IgGs). The other fourteen Vβ genes are presented in Supplementary Figure S2. The gene therapy treated SF-RAG2co and UCOE-RAG2co mice have detectable rearrangements indistinguishable from the WT mice.
Furthermore, TCR rearrangements were determined for 20 Vβ gene and Ig segments (Cµ and Cγ) on spleen cDNA of WT, SF-RAG2co and UCOE-RAG2co mice, as described before.11,22 At 4 months after transplantation, variable peak patterns were observed for both TCR and Ig that were indistinguishable from WT mice, indicating the presence of polyclonal diversity of rearrangements (Figure 3c and Supplementary Figure S2). Consistent with our expectations, analysis of Rag2−/− spleen cDNA resulted in no detectable peaks (data not shown), indicating that the antigen receptor rearrangements were absent.
Additionally, TCRVβ rearrangements were quantified on genomic DNA of a WT, Rag2−/−, SF-RAG2co and UCOE-RAG2co mouse, confirming the potency of the therapeutic approach to acquire full diversity (Supplementary Table S2 and Supplementary Figure S3).
B-cell development and reconstitution
In BM of Rag2−/− mice, B cells arrest at the pro-B cell stage (B220+CD43+IgM−IgD−) as shown (Figure 4a). Following transplantation of WT cells, the pro-B cell number was strongly reduced and B lymphocytes developed into pre-B, immature transitional IgM+ and mature IgD+ expressing cells.
Figure 4.

Bone marrow B lymphocyte maturation populations in spleen and lymph nodes. (a) Bone marrow B-cell differentiation was assessed within B220+ gated cells for pro-B (B220+CD43+IgM−IgD−), pre-B (B220+CD43−IgM−IgD−), immature (Imm; B220+CD43−IgM+IgD−) and mature (B220+CD43−IgM+IgD+) cells (n = 4–9). (b,c)Spleen B-cell percentages of B220+CD19+IgM+ and B220+CD19+IgD+, and (d) specification into CD21+CD23+ follicular B cells and (e) CD21+CD23− marginal zone B cells and (f,g) B-lymphocytes in the lymph nodes. Normalization of the immunoglobulin plasma levels determined for (h) IgM and (i) IgG1 as well as isotypes IgG2a, IgG2b, IgG3, IgA, which are presented in Supplementary Figure S4 (n = 4–10) occurs in the gene therapy treated mice, 4 months after transplantation of transduced Lin- cells. Rag2−/− mice do not have detectable Ig levels (data not shown). Four months after transplantation, T cell dependent specific (j) IgM and (k) IgG responses to antigen were determined by tetanus toxoid administration. Tetanus toxoid was three times injected intraperitoneally at two week intervals, and blood was collected before every injection and two weeks after the last injection. IgM was determined at day 14 after the first injection, IgG at 14 days after the last injection. Shown are the fold changes in optical density relative to pre-immunization values. Furthermore, T-cell independent immune response was induced by Pneumo23, and fold changes are presented for (l) IgM and (m) IgG at day 10 after immunization.
In the gene therapy treatment groups, reductions in the proportion of pro-B cells was smaller, and appreciable populations of immature and mature B cells were observed only in SF-RAG2, SF-RAG2co, and UCOE-RAG2co mice. In the same groups, peripheral B lymphocytes in spleen (Figure 4b–e) and lymph nodes (Figure 4f,g) were increased substantially to near normal levels. SF-RAG2co treated mice presented with large quantities of cells expressing complement receptor CD21 and the low-affinity receptor IgE (CD23, Figure 4d,e), but in the UCOE-RAG2co and the SF-RAG2 the numbers were slightly reduced (Figure 4d,e). Plasma Ig isotypes were quantified at 5 months after gene therapy treatment (Figure 4h,i and Supplementary Figure S4). All mice were able to produce significant levels of Ig isotypes, despite low B cells numbers in peripheral organs.
Regaining specific antibody responses requires diversity in TCR receptor repertoire and Ig, and was analyzed by subjecting mice to T-cell dependent tetanus immunization as well as a T-cell independent Pneumo23 immunization. Significant IgM responses were observed for both tetanus (Figure 4j) and Pneumo23 (Figure 4l) for WT transplanted mice, SF-RAG2co and UCOE-RAG2co mice. As expected, a much more profound IgG response was observed for tetanus toxoid as well as Pneumo23 (Figure 4k,m). Although SF-RAG2 mice had significant B-cell numbers, tetanus toxoid IgG responses were not observed, in line with the few functional T cells present in these mice.
Hematopoietic chimerism, vector copy number per cell and promoter methylation
The male contribution to the overall blood cell production (chimerism) as well as copy number per cell is shown (Figure 5). The proportion of Y-chromosome positive BM cells in the gene therapy treated mice is consistent with the sublethal irradiation dose applied to the mice before transplantation (Figure 5a). SF-RAG2co mice had a lower average copy number per male BM cell (2.3 ± 2.0, n = 17; Figure 5b) than SF-RAG2 treated mice (3.0 ± 0.9, n = 7). Lower copy numbers were calculated for RAG2p (0.5 ± 0.4, n = 9) and γcPr groups (0.5 ± 0.5, n = 11), consistent with the lower T and B cells numbers in the peripheral organs. It is of note that the initial transduction efficiencies in Lin− cells in these treatment groups were on average similar and ranged from 1 to 5 (data not shown), but RAG2 protein expression could have been too low for positive selection towards T and B cells. Finally, the UCOE-RAG2co treated mice had on average near 1 copy per cell, which confirmed the relatively low initial Lin− transduction efficiency of ~25% of the cells determined by qPCR (data not shown).
Figure 5.

Bone marrow chimerism and copy number. (a) Bone marrow chimerism determined by qPCR for male Y-chromosome. (b) Lentiviral vector copy number per cell determined on genomic bone marrow DNA, and corrected for chimerism (n = 6–17). (c) Quantification of methylation of the spleen focus forming virus (SF) promoter determined by pyrosequencing of the 5′ end of integrated vector 6 months after transplantation. Lin− cells have low methylation levels, with a significant increase 6 months after reconstitution in bone marrow (BM) and spleen gDNA, and a further significant increase in thymus (THY). Average methylation levels of the ubiquitous chromatin opening element (UCOE) were below 5–7%, and could be considered non-methylated.
Strong RAG2co expression clearly increased reconstitution, but some SF-RAG2co mice barely had detectable DP T-cell populations in the thymus. We therefore investigated whether methylation and potentially subsequent silencing would explain the small DP populations, and eventually would jeopardize long-term efficacy of treatment. To this end, we performed high-throughput quantification covering 50% of the CpG sites of the SF element, which were averaged and compared for BM, spleen and thymus gDNA. As expected, transduced and cultured Lin− cells (Figure 5c) displayed low methylation percentages, but significant increases in methylation were observed in gDNA of BM and spleen of LV vector treated mice at 6 months after transplantation. Remarkably, the methylation percentage was elevated even more in the thymus (Figure 5c). Additionally, the individual CpG sites did not considerably differ in the level or distribution of methylation within this SF region (Supplementary Table S3). On average, the methylation of the UCOE element was below values of 5–7%, to be considered as not methylated in view of the sensitivity of the method used (Figure 5c and Supplementary Table S4).
Adverse effects
Vector related adverse effects have not been observed during follow-up monitoring of the cohorts presented here, which consisted of 56 mice treated with SF-RAG2co, 18 with SF-RAG2, 18 with RAG2p-RAG2co, 18 with ycPr-RAG2co and 28 with UCOE-RAG2co. In the SF-RAG2co mice, five mice died inadvertently. Two mice died at days 197 and 129, respectively, with an enlarged spleen and thymus, attributable to expansion of CD4/CD8 positive cells, identified as recipient cells harboring no vector copy as determined by qPCR on the HIV 5′LTR. A third mouse died at day 145 with an enlarged thymus, spleen and liver, and neither had detectable vector copies in spleen and thymus cells, while the fourth mouse died at day 251 with an enlarged thymus and liver and a reduced peripheral blood white cell count without detectable vector copies in BM. A fifth mouse died at day 144, had a slight expansion of CD11b/GR-1 positive cells with an enlarged spleen and vector copy numbers ranging between 0.3 and 1.9 per cells in BM, spleen, and thymus, death being attributed to an infectious complication. We conclude that the majority, if not all of these undue deaths are attributable to the underlying phenotype and/or the irradiation used as conditioning rather than to the genetic modification of the transplanted cells.
Discussion
RAG2 immunodeficiency is a disorder that results in an arrest in T- and B-cell differentiation, thereby compromising immunity. Gene therapy may be a valid option if complementation of RAG2 is efficient and safe. In this paper, we report a recoded human RAG2 sequence that resulted in sufficient expression in lentivirus vector transduced Lin- cells transplanted in sublethally irradiated Rag2−/− mice to improve T and B cells in peripheral blood, BM, thymus, and spleen, if expression was driven by the SF and UCOE elements.14,18 Codon-optimization of RAG2 improved protein production and provided a robust reconstitution of lymphocyte subsets with superior restoration of B-cell function, as well as recovery of T-cell dependent, but less profoundly T-cell independent immune responses. The use of more cellular restricted promoters, such as the γcPr, that provided significant therapeutic correction for SCID-X1,12 or the native RAG2 promoter did result in T-cell reconstitution, but often contained low CD8+ T cells in spleen or limited diversity of T-cell isotypes, as well as lacked proper B-cell reconstitution, and consequently, B-cell function was insufficient to respond to antigen presentation.
Recoding transgenes has resulted in significant increases in titers as well as protein expression, depending on the parameters included in the optimization algorithm, as was previously shown for RAG1,11 gp91phox,13 and interleukin-2 receptor γ gene (IL2RG).12 Inclusion of RAG2co in the lentiviral vector constructs did not increase titer, did modestly increase protein expression in HeLa cells, but was sufficient to robustly improve reconstitution in vivo. The detection of RAG2 in HeLa cells may have been an underestimation, since this protein is periodically degraded during cell-cycling by a conserved signal.23
In the gene therapy treated mice studied, overcoming the early arrest at the DN3 stage restored T-cell development completely or partially. Further differentiation into DP thymic cells was restored in the UCOE treated mice, in most mice with SF-containing vectors, was only partial in the γcPr mice, and rare with the RAG2 promoter. Since the γcPr and RAG2p are weak promoters, this observation underlines that relatively strong expression of RAG2 is required for optimal reconstitution. However, a significant number of SF promoter treated mice did not have DP T-cell populations at 6 months after transplantation. As previously described, SF promoter methylation may increase significantly over time in vitro as well as in vivo.14 We confirmed by high-throughput analyses that SF sequences are strongly methylated in BM, spleen and even more profoundly in thymus gDNA, which might be related to physiologic methylation intrinsic to T-cell differentiation.24 Since the cell fate of developing lymphocytes requires changes in overall programs of gene expression, in part regulated by DNA methylation, developing T cells may be prone to DNA methylation. Specific entrapment of developing precursor T cells with methylation induced reduced RAG2 expression may result in the significantly higher methylation levels in the thymus. The presence of the existing peripheral T cells in older mice should then have been derived from developing progenitors in the initial months after transplantation. All mice treated with the UCOE element did have normal thymic DP T-cell populations, even at the lower average copy number per cell, which may be due to its natural resistance to methylation and silencing as confirmed in this paper.14 Since RAG2 expression by the UCOE element is lower than the SF promoter, transgene toxicity to hematopoietic stem cells may also have played a role in the presence of DP populations at older age.
Additionally, absolute thymic cell numbers were low in all gene therapy treated groups compared to WT Tx mice, similar to observations made by Yates et al.9 Nonetheless, substantial T-cell populations were found in peripheral blood, spleen, and lymph nodes with a high diversity in TCRs, and normal distribution of naive, effector and memory T cells similar to previously reported.9 Additional pretreatments may be required to improve thymic reconstitution. Despite the low numbers, restoration of compartmentalization of the thymus was achieved in SF-RAG2co and UCOE-RAG2co treated mice as shown by immunohistological staining of phenotypic markers, and the functionality of the T cells was demonstrated by responses to mitogens, to T-cell dependent vaccination and by the distributions of TCR Vβ isotypes in PB, which were similar to those in WT mice, except for the SF-RAG2 treated mice. A more thorough analysis into the diversity of the TCR Vβ repertoire showed that the SF-RAG2co and UCOE-RAG2co treated mice acquired highly diverse polyclonal TCR repertoires.
BM B-cell development in Rag2−/− mice is arrested at the pro-B cell stage, and consequently, there are no peripheral B cells. As opposed to the T-cell numbers, B-cell development was more difficult to recover. In the SF groups and the UCOE group, small but detectable B-cell percentages were observed in the BM, and larger populations of mature B cells were observed in spleen and lymph nodes. In contrast, the weaker γcPr and RAG2p containing vector treated mice had barely detectable B-cell numbers, pointing out that there is a threshold of RAG2 expression required for substantial B-cell development. In the SF-RAG2co and UCOE-RAG2co mice, the T-cell independent immune responses were less strongly than in the WT transplanted mice as expected, but were significantly increased compared to SF-RAG2, γcPr or RAG2p treated mice, consistent with the notion that the latter groups had reduced B-cell numbers.
All groups had on average normal plasma Ig isotype levels similar to WT transplanted mice. Low B-cell counts in the periphery apparently did not correlate with the Ig levels in vivo, i.e., low circulating B cells can provide normal Ig levels, as has been previously shown by us and others.9,11,12,25 Additionally, the gene therapy mice did not present any obvious features resembling Omenn syndrome, which is caused by mutations that impair but not abolish RAG1 and RAG2 functions. Omenn patients often have normal T-lymphocyte levels26 with a large proportion of activated T cells and skewing of TCR repertoires,27 not observed in the UCOE-RAG2co treated mice. In a previously published Omenn mouse model, B cells were substantially reduced similar to Omenn patients, but certain isotype Ig levels were within normal range.27,28 Finally, the diversity in Ig repertoire in the SF-RAG2co and UCOE-RAG2co mice was similar to that in WT or WT transplanted mice.
It was previously shown that retroviral vector copy number in gene-modified transplanted stem cells and nonlymphoid cells was significantly lower than in lymphoid cells9 pointing out that lymphoid cells containing more than one vector copy conferring high transgene expression have a selective advantage in vivo, consolidating our rationale to codon-optimize the RAG2 transgene. Similar observations that both T- and B-lymphoid cells have a proliferative advantage with high copy number or expression levels were made for RAG1.11,29 In the current study, SF-RAG2 and SF-RAG2co mice had several vector copies per cell on average. Methylation of the SF promoter may have contributed to the proliferative advantage of cells containing high copy number. In contrast, the UCOE-RAG2co mice had near one vector copy per cell. This minimal number was sufficient to correct the murine phenotype. Although RAG2 shares 88% identity with the murine Rag2 gene,9 interaction with the human RAG1 may even improve phenotype correction at the desirable one therapeutic transgene copy per cell.
During TCR and Ig rearrangements executed by the RAG complex, RAG1 represents the catalytic subunit while RAG2 acts as a regulatory subunit that is essential for all activities.30 Both RAG1 and RAG2 are strictly regulated during T- and B-cell development. In follow-up studies, it will be important to determine if constitutive RAG2 expression may lead to pathological abnormalities or malignancies, whether caused by RAG2 or LV integration. However, RAG2 is regulated post-translational during cell-cycle,31 which may itself provide an additional safety feature for cells during rapid cell division. Additionally, RAG is also indirectly controlled by restricting its access to recombination signal sequences substrates within the appropriate antigen receptor locus and developmental stages.32
Although retroviral gene therapy has been shown to be effective to treat immune deficiencies such as X-linked SCID,3,4 Wiskott–Aldrich syndrome33 and adenosine deaminase SCID,5 5 out of 20 patients developed T-cell leukemia in the SCID-X1 trials, four of which were treated successfully with maintenance of immune functions.3,7 In a more recently initiated Wiskott–Aldrich gene therapy trial, 9 out of 10 children were corrected, but one patient developed acute T-cell leukemia related to the treatment (Press Release Hannover Medical School, 2010), and in these trials integrations near LMO2 were observed. Additionally, the SF promoter was involved in insertional activation of the oncogene EVI1 and the resulting clonal expansion in chronic granulomatous disease gene therapy.34 In the SF promoter treated groups of the present study, undue deaths were observed in 5 out of 56 mice, of which three developed leukemia, but these were all recipient derived and did not contain vector as determined by qPCR on the HIV 5′LTR. Potentially oncogenic events were not observed in a total of 28 UCOE-RAG2co treated mice. Safety analysis is under thorough investigation, as will be reported separately in conjunction with the ongoing integration analysis. However, the use of SIN lentiviral vectors with an internal promoter cassette reduces the genotoxic potential significantly as determined by cell based assays in vitro20,35 and in vivo in mice,19,21 showing that physiologic promoters reduce the risk of insertional oncogenesis significantly,36 in large part by a reduction in the ability to deregulate gene expression near integration sites.37 Since the UCOE element provides bidirectional expression, the use in its current form may not be preferred, and further modifications may be necessary to improve safety of this element. In the present study, the copy number of the UCOE-RAG2co treated mice was kept at a minimum, while still conveying full therapeutic efficacy. In follow-up studies it will be important to determine the minimum corrected hematopoietic stem cell numbers required, further improve stem cell transduction, preferably restricting transduction to “true” hematopoietic stem cells, e.g., by using single-chain antibodies targeting the viral vector to hematopoietic stem cell-specific antigens,38 and expand the stem cells by ex vivo stimulation with cytokines to reduce the number of transduced cells required.
From the present study we conclude that the UCOE element in combination with RAG2co ameliorated the disease phenotype sufficiently to warrant its further development for clinical implementation.
Material and Methods
Construction of lentiviral vector plasmids. The human RAG2 consensus cDNA was recoded (RAG2co) with OptimumGene technology (Genscript, Piscataway, NJ), with the addition of a Kozak sequence and two TGA stop codons to improve transcription and translation. EGFP was replaced by RAG2co in the LV pRRL.PPT.SF.EGFP.bPRE4*.SIN described previously,39 to obtain SF-RAG2co, which contains the spleen focus forming virus promoter (SF). The SF-RAG2co vector was compared to the SF-RAG2 vector containing native RAG2 sequence. For the weak cellular promoters SF was replaced by a 1505 bp human RAG2 promoter sequence (RAG2p;15,16 primer sequences see Supplementary Table S4) or a ~1.1kb γ chain promoter element12 (γcPr), that provided significant expression to correct SCID-X1 mice.12 Finally, a UCOE14,18 was combined with RAG2co. All constructs were verified by restriction digests and sequencing. LV production was done according to standard procedures.12,39,40,41 and titered as previously described.42
Animals. Congenic Rag2−/− mice were derived from 10th generation backcross of BALB/c Rag2−/−/γc−/− mice43 with syngenic WT BALB/c mice (WT) and bred in the Erasmus MC Experimental Animal Center (Rotterdam, The Netherlands). The animals completely lack mature T and B-lymphocytes, which mirrors the human disease.44 All experiments were approved by an ethical committee of Erasmus MC, Rotterdam in accordance with legislation in the Netherlands.
Lentiviral hematopoietic stem cell transduction. Donor BM hematopoietic progenitors from four- to twelve-week-old male Rag2−/− mice were enriched by lineage depletion (Lin− ; BD Biosciences, Santa Clara, CA). These Lin− cells were subjected to overnight LV vector transduction at a cell density of 106 cells/ml and a multiplicity of infection of 2–10, in serum free medium with standard cocktail of growth factors.39 The following day, 5 × 105–1 × 106 transduced Lin− cells were injected in the tail vein of 6–7 Gy sublethally irradiated six- to ten-week-old female Rag2−/− recipients.
Immunological phenotyping. PB cells were counted monthly using VET ABC counter (Scil animal care company GmbH, Viernheim, Germany), and leukocytes were prepared as described12 for staining T- and B-cell markers. At the end of the experiments, spleen, thymus, BM and lymph nodes were prepared for flow cytometry with anti-mouse antibodies to: CD3, CD4, CD8, CD25, CD44, CD62L, CD21/CD35, CD23, B220, CD19, CD43, IgM, IgD, and CD11b (all antibodies BD Biosciences), and measured on a FACSCalibur or FACS LSRII (Becton Dickinson, Erembodegem, Belgium).
Histopathology. Two micrometers of formalin fixed paraffin embedded thymic sections were subjected to routine hematoxylin and eosin staining for assessment of basic histopathological changes and immunohistochemistry. Briefly, sections were dewaxed, rehydrated, endogenous peroxidase activity blocked by 0.3% H2O2/methanol for 20 minutes and heat induced antigen-retrieval was performed by either microwave treatment or incubation in a thermostatic bath using EDTA buffer pH 8.0 or citrate buffer pH 6.0. The following primary antibodies were used: rabbit anti-cytokeratin 5 (CK5, clone AF 138, 1:100; Covance, Princeton, NJ); rat anti-cytokeratin 8 (CK8, clone TROMA-I, 1:200); rabbit anti-caspase 3 active (Casp 3 act., 1:600; R&D systems, Minneapolis, MN); rabbit anti-CD3 (1:100; Dako Cytomation, Glostrup, Denmark); rabbit anti-Terminal Deoxynucleotidyl Transferase (TdT, 1:100; Dako), rabbit anti-AIRE (AIRE, 1:2000; rat anti-Foxp3 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA), or Ulex europaeus agglutinin I (UEA, 1:600; Vector Laboratories, Burlingame, CA). Immunostains were revealed using Real EnVision Rabbit HRP Labeled Polymer system (Dako) or biotinylated rabbit anti-rat mouse pre-absorbed (1:200; Vector Laboratories) followed by streptavidin conjugated HRP and chromogen diaminobenzidine (Dako). Nuclei were counterstained with Hematoxylin. Digital imagines were acquired by Olympus DP70 camera mounted on Olympus Bx60 microscope using CellF Imaging software (Soft Imaging System GmbH, Muenster, Germany).
Detection of RAG2 protein, immunoglobulin levels and humoral immune response. SF-RAG2 or SF-RAG2co transduced HeLa cells were kept in culture for 7 days, and enzyme-linked immunosorbent assays were performed as previously described.39 Briefly, enzyme-linked immunosorbent assay plates were coated with serially diluted cell lysates at room temperature. After washing and blocking, wells were incubated with rabbit polyclonal anti-human RAG2 (GeneTex, Irvine, CA) for 1 hour and washed again. Signal was detected after incubation with goat anti-rabbit HRP by staining with TMB substrate (Kirkegaard and Perry Laboratories, Gaithersburg, MD) and the reaction was stopped by adding 1 mol/l phosphoric acid. The absorbance was read at 405 nm using a FLUOstar Optima plate reader (BMG LABTECH GmbH, Ortenberg, Germany). The relative fold increase in RAG2 expression was determined, normalized for protein levels, and adjusted for copy number per cell.
Baseline levels of IgM, IgG1, IgG2a, IgG2b, IgG3, IgA were measured in mouse plasma by multiplex assay kit (Beadlyte Mouse Immunoglobulin Isotyping kit, Millipore, Billerica, MA) and run using a Bio-Plex reader (BioRad Laboratories, Hercules, CA).
Mice were immunized with Streptococcus pneumoniae (PNEUMO 23, Sanofi Pasteur MSD) and tetanus toxoid. Enzyme-linked immunosorbent assay was performed as described previously.12
In vitro spleen cell proliferation assay. Spleen cells were cultured at 1 × 106 cells/ml in DMEM supplemented with 10% heat-inactivated fetal calf serum, antibiotics penicillin/streptomycin and concanavalin A (Con A, 2.5 µg/ml) or human interleukin-2 (1000 units/ml) or in precoated wells with mouse anti-CD3ε antibody (250 ng/well, clone 145-2C11, BioLegend, San Diego, CA). On day 3, 0.5µCi 3H-thymidine was added to each well. On day 4 cells were washed and 3H-thymidine incorporation was measured on a TopCount microplate scintillation counter (GMI, Ramsey, MN).
TCR and Ig rearrangement analysis. Repertoire analysis of murine TCR β (TCRβ) and Ig segments was performed on cDNA synthesized with the QuantiTect Reverse Transcription kit (Qiagen, Venlo, The Netherlands) derived from 2 µg total RNA fresh purified spleen cells using the Allprep DNA/RNA kit (Qiagen). PCR products were obtained by amplifying the cDNA using a constant β gene specific primer (5′-FAM-CTTGGGTGGAGTCACATTTCTC-3′) in combination with 20 Vβ gene segment-specific primers or FAM-labeled constant Cµ (IgM) or Cγ (IgG) gene segment-specific primer (Cµ: 5′-FAM-TCTCTGCGACAGCTGGAATGG-3′ and Cγ: 5′-FAM-GGACAGGGATCCAGAGTTCCA-3′) with a consensus VH primer (5′-AGGTCAAACTGCAGCAGTCTGG-3′) as described previously.11,12,22 All PCR products were analyzed on an ABI 3130XL sequencer, and graphs generated with PeakScanner (Applied Biosystems, Foster City, CA).
T-lymphocyte repertoire diversity on DNA template was measured using ImmunTraCkeR test (ImmunIDTechnologies, Grenoble, France). Diagnostic of Divpenia and NDL score were analyzed using the Constel'ID software (ImmunID). Multiplex PCR was performed as described45,46 using an upstream primer specific of all functional members of a given V family and a downstream primer specific of a given J segment. This assay allows the simultaneous detection and resolution of several V–J rearrangements in the same reaction. By using this technique, it is possible to detect near 100% of the possible TRβVJ combinatorial rearrangements. In order to perform semiquantitative analysis, PCR reactions were stopped at the exponential step of the PCR.
Quantitative PCR of lentiviral integrations. The LV copy number per cell was determined on 100 ng genomic BM DNA (gDNA) by qPCR with SYBR Green PCR Master Mix (Applied Biosystems) and measured in a ABI PRISM 7900 HT sequence detection system (Applied Biosystems) as previously described.39,42 Integrated HIV provirus copy number was corrected for chimerism determined by qPCR on the Sry locus of mouse Y-chromosome, normalized for mouse Gapdh (primer sequences provided in Supplementary Table S4). Samples were analyzed with SDS2.3 software.
Promoter methylation assay. Pyrosequencing of eleven CpG sites of the SF enhancer/promoter of SF-RAG2 and SF-RAG2co vectors and 43 CpG sites of the UCOE element was performed by Varionostic GmbH (Ulm, Germany) based on techniques previously described47 (Supplementary Figures S5 and S6 and Supplementary Tables S5 and S6). Briefly, genomic DNA was extracted from 7-day cultured Lin- cells, and from BM, spleen and thymus 6 months after transplantation. The sequences before and after bisulfite conversion are depicted (Supplementary Figures S5 and S6) with the amplification and sequencing oligonucleotides (Supplementary Table S5). For the PCR, HotStar master mix (Qiagen), forward primer, 5′-biotinylated reverse primer and bisulfite converted DNA template were mixed, and amplified accordingly: 95 °C for 15 minutes, followed by 50 cycles of 94 °C for 35 seconds, 54 °C for 35 seconds, and 72 °C for 1 minute, and finally 72 °C for 10 minutes. Sample preparation was carried out using a Vacuum Prep Tool; 40–50 µl PCR product was immobilized to 3µl Streptavidin Sepharose HP beads (GE Healthcare, formerly Amersham Biosciences, Hoevelaken, The Netherlands) followed by annealing to 2 µl sequencing primer (5 µmol/l) for 2 minutes at 80 °C. The PSQ ID System was used with the dispensation orders assigned for the assays. CpG analyses were done with Pyro Q-CpG software.
Statistical analysis. Statistical analysis was performed with SPSS 11 (IBM SPSS Statistics, Armonk, NY). Significance of differences was determined by one-way analysis of variance or Mann–Whitney U test for comparing two groups. A significant difference was assumed if P < 0.05.
SUPPLEMENTARY MATERIAL Figure S1. Peripheral distribution of naive, effector and central memory T cells. Figure S2. Spleen TCR repertoire analysis on cDNA. Figure S3. Spleen TCR repertoire analysis on genomic DNA. Figure S4. Immunoglobulin plasma values in gene therapy treated mice. Figure S5. Original and converted DNA sequence used for SF methylation assay. Figure S6. Original and converted DNA sequence used for UCOE methylation assay. Table S1. Dominant TCR Vβ clones in peripheral blood. Table S2. Mouse β VJ rearrangements determined on genomic DNA of gene therapy treated mice. Table S3. Methylation assay of the SF promoter. Table S4. Methylation assay of the UCOE promoter. Table S5. Primer sequences for the promoters and quantitative PCR. Table S6. Oligo sequences for methylation assay.
Acknowledgments
The authors would like to thank Luigi Naldini (HSR-TIGET) for kindly providing the third generation LV vector plasmids, Axel Schambach and Christopher Baum (Department of Experimental Hematology, Hannover Medical School) for the mutated WPRE element, Frank Staal and Karin Pike-Overzet (Leiden University Medical Center) for the SF lentiviral vector containing the native RAG2 sequence, Neil Berinstein (Department of Immunology, University of Toronto) for the RAG2 promoter plasmid, U.H. von Andrian (Harvard Medical School) for the anti-CK8 antibody, Pärt Peterson (University of Tartu) for the anti-AIRE antibody, Roya Sarwari, Adri van Oudenaren and Hans Hoogerbrugge for technical assistance, and Egied Simons for designing the graphics (Department of Hematology, Erasmus Medical Center). This work was supported by the European Commission's 5th, 6th and 7th Framework Programs, Contracts QLK3-CT-2001-00427-INHERINET, LSHB-CT-2004-005242-CONSERT, 222878-PERSIST and 261387-CELL-PID. Additionally, this work was also funded by The Netherlands Organization for Health Research ZonMW, program grant 434-00-010, Fondazione CARIPLO to P.L.P. and A.V., and Ministry of Health RF-2009-1485896 to A.V. The authors declare no competing financial interests.
Supplementary Material
Peripheral distribution of naive, effector and central memory T cells.
Spleen TCR repertoire analysis on cDNA.
Spleen TCR repertoire analysis on genomic DNA.
Immunoglobulin plasma values in gene therapy treated mice.
Original and converted DNA sequence used for SF methylation assay.
Original and converted DNA sequence used for UCOE methylation assay.
Dominant TCR Vβ clones in peripheral blood.
Mouse β VJ rearrangements determined on genomic DNA of gene therapy treated mice.
Methylation assay of the SF promoter.
Methylation assay of the UCOE promoter.
Primer sequences for the promoters and quantitative PCR.
Oligo sequences for methylation assay.
REFERENCES
- Smith AR, Gross TG., and, Baker KS. Transplant outcomes for primary immunodeficiency disease. Semin Hematol. 2010;47:79–85. doi: 10.1053/j.seminhematol.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P.et al. (2000Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease Science 288669–672. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Le Deist F, Carlier F, Bouneaud C, Hue C, De Villartay JP.et al. (2002Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy N Engl J Med 3461185–1193. [DOI] [PubMed] [Google Scholar]
- Gaspar HB, Parsley KL, Howe S, King D, Gilmour KC, Sinclair J.et al. (2004Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector Lancet 3642181–2187. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L.et al. (2009Gene therapy for immunodeficiency due to adenosine deaminase deficiency N Engl J Med 360447–458. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E.et al. (2008Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1 J Clin Invest 1183132–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H.et al. (2008Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients J Clin Invest 1183143–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- Yates F, Malassis-Séris M, Stockholm D, Bouneaud C, Larousserie F, Noguiez-Hellin P.et al. (2002Gene therapy of RAG-2−/− mice: sustained correction of the immunodeficiency Blood 1003942–3949. [DOI] [PubMed] [Google Scholar]
- Robbins PB, Skelton DC, Yu XJ, Halene S, Leonard EH., and, Kohn DB. Consistent, persistent expression from modified retroviral vectors in murine hematopoietic stem cells. Proc Natl Acad Sci USA. 1998;95:10182–10187. doi: 10.1073/pnas.95.17.10182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike-Overzet K, Rodijk M, Ng YY, Baert MR, Lagresle-Peyrou C, Schambach A.et al. (2011Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer Leukemia 251471–1483. [DOI] [PubMed] [Google Scholar]
- Huston MW, van Til NP, Visser TP, Arshad S, Brugman MH, Cattoglio C.et al. (2011Correction of murine SCID-X1 by lentiviral gene therapy using a codon-optimized IL2RG gene and minimal pretransplant conditioning Mol Ther 191867–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Carranza B, Gentsch M, Stein S, Schambach A, Santilli G, Rudolf E.et al. (2009Transgene optimization significantly improves SIN vector titers, gp91phox expression and reconstitution of superoxide production in X-CGD cells Gene Ther 16111–118. [DOI] [PubMed] [Google Scholar]
- Zhang F, Frost AR, Blundell MP, Bales O, Antoniou MN., and, Thrasher AJ. A ubiquitous chromatin opening element (UCOE) confers resistance to DNA methylation-mediated silencing of lentiviral vectors. Mol Ther. 2010;18:1640–1649. doi: 10.1038/mt.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrin AA, Fong I, Malkin L, Marsden PA., and, Berinstein NL. Cloning and characterization of the human recombination activating gene 1 (RAG1) and RAG2 promoter regions. J Immunol. 1997;159:4382–4394. [PubMed] [Google Scholar]
- Fong IC, Zarrin AA, Wu GE., and, Berinstein NL. Functional analysis of the human RAG 2 promoter. Mol Immunol. 2000;37:391–402. doi: 10.1016/s0161-5890(00)00056-0. [DOI] [PubMed] [Google Scholar]
- Markiewicz S, Bosselut R, Le Deist F, de Villartay JP, Hivroz C, Ghysdael J.et al. (1996Tissue-specific activity of the gammac chain gene promoter depends upon an Ets binding site and is regulated by GA-binding protein J Biol Chem 27114849–14855. [DOI] [PubMed] [Google Scholar]
- Zhang F, Thornhill SI, Howe SJ, Ulaganathan M, Schambach A, Sinclair J.et al. (2007Lentiviral vectors containing an enhancer-less ubiquitously acting chromatin opening element (UCOE) provide highly reproducible and stable transgene expression in hematopoietic cells Blood 1101448–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C.et al. (2006Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration Nat Biotechnol 24687–696. [DOI] [PubMed] [Google Scholar]
- Modlich U, Navarro S, Zychlinski D, Maetzig T, Knoess S, Brugman MH.et al. (2009Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors Mol Ther 171919–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini E, Cesana D, Schmidt M, Sanvito F, Bartholomae CC, Ranzani M.et al. (2009The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy J Clin Invest 119964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannetier C, Cochet M, Darche S, Casrouge A, Zöller M., and, Kourilsky P. The sizes of the CDR3 hypervariable regions of the murine T-cell receptor beta chains vary as a function of the recombined germ-line segments. Proc Natl Acad Sci USA. 1993;90:4319–4323. doi: 10.1073/pnas.90.9.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Dordai DI, Lee J., and, Desiderio S. A conserved degradation signal regulates RAG-2 accumulation during cell division and links V(D)J recombination to the cell cycle. Immunity. 1996;5:575–589. doi: 10.1016/s1074-7613(00)80272-1. [DOI] [PubMed] [Google Scholar]
- Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW.et al. (2001A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival Immunity 15763–774. [DOI] [PubMed] [Google Scholar]
- Zhou S, Mody D, DeRavin SS, Hauer J, Lu T, Ma Z.et al. (2010A self-inactivating lentiviral vector for SCID-X1 gene therapy that does not activate LMO2 expression in human T cells Blood 116900–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa A, Sobacchi C, Notarangelo LD, Bozzi F, Abinun M, Abrahamsen TG.et al. (2001V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations Blood 9781–88. [DOI] [PubMed] [Google Scholar]
- Marrella V, Poliani PL, Casati A, Rucci F, Frascoli L, Gougeon ML.et al. (2007A hypomorphic R229Q Rag2 mouse mutant recapitulates human Omenn syndrome J Clin Invest 1171260–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassani B, Poliani PL, Marrella V, Schena F, Sauer AV, Ravanini M.et al. (2010Homeostatic expansion of autoreactive immunoglobulin-secreting cells in the Rag2 mouse model of Omenn syndrome J Exp Med 2071525–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagresle-Peyrou C, Yates F, Malassis-Séris M, Hue C, Morillon E, Garrigue A.et al. (2006Long-term immune reconstitution in RAG-1-deficient mice treated by retroviral gene therapy: a balance between efficiency and toxicity Blood 10763–72. [DOI] [PubMed] [Google Scholar]
- Fugmann SD. The origins of the Rag genes–from transposition to V(D)J recombination. Semin Immunol. 2010;22:10–16. doi: 10.1016/j.smim.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., and, Desiderio S. Cyclin A/CDK2 regulates V(D)J recombination by coordinating RAG-2 accumulation and DNA repair. Immunity. 1999;11:771–781. doi: 10.1016/s1074-7613(00)80151-x. [DOI] [PubMed] [Google Scholar]
- Sleckman BP, Bassing CH, Bardon CG, Okada A, Khor B, Bories JC.et al. (1998Accessibility control of variable region gene assembly during T-cell development Immunol Rev 165121–130. [DOI] [PubMed] [Google Scholar]
- Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Díez IA, Dewey RA.et al. (2010Stem-cell gene therapy for the Wiskott-Aldrich syndrome N Engl J Med 3631918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A.et al. (2010Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease Nat Med 16198–204. [DOI] [PubMed] [Google Scholar]
- Modlich U, Bohne J, Schmidt M, von Kalle C, Knöss S, Schambach A.et al. (2006Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity Blood 1082545–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zychlinski D, Schambach A, Modlich U, Maetzig T, Meyer J, Grassman E.et al. (2008Physiological promoters reduce the genotoxic risk of integrating gene vectors Mol Ther 16718–725. [DOI] [PubMed] [Google Scholar]
- Maruggi G, Porcellini S, Facchini G, Perna SK, Cattoglio C, Sartori D.et al. (2009Transcriptional enhancers induce insertional gene deregulation independently from the vector type and design Mol Ther 17851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anliker B, Abel T, Kneissl S, Hlavaty J, Caputi A, Brynza J.et al. (2010Specific gene transfer to neurons, endothelial cells and hematopoietic progenitors with lentiviral vectors Nat Methods 7929–935. [DOI] [PubMed] [Google Scholar]
- van Til NP, Stok M, Aerts Kaya FS, de Waard MC, Farahbakhshian E, Visser TP.et al. (2010Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype Blood 1155329–5337. [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D.et al. (1998A third-generation lentivirus vector with a conditional packaging system J Virol 728463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L.et al. (1998Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery J Virol 729873–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Til NP, Markusic DM, van der Rijt R, Kunne C, Hiralall JK, Vreeling H.et al. (2005Kupffer cells and not liver sinusoidal endothelial cells prevent lentiviral transduction of hepatocytes Mol Ther 1126–34. [DOI] [PubMed] [Google Scholar]
- Gimeno R, Weijer K, Voordouw A, Uittenbogaart CH, Legrand N, Alves NL.et al. (2004Monitoring the effect of gene silencing by RNA interference in human CD34+ cells injected into newborn RAG2−/− gammac−/− mice: functional inactivation of p53 in developing T cells Blood 1043886–3893. [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M.et al. (1992RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement Cell 68855–867. [DOI] [PubMed] [Google Scholar]
- Marodon G, Desjardins D, Mercey L, Baillou C, Parent P, Manuel M.et al. (2009High diversity of the immune repertoire in humanized NOD.SCID.gamma c−/− mice Eur J Immunol 392136–2145. [DOI] [PubMed] [Google Scholar]
- Pasqual N, Gallagher M, Aude-Garcia C, Loiodice M, Thuderoz F, Demongeot J.et al. (2002Quantitative and qualitative changes in V-J alpha rearrangements during mouse thymocytes differentiation: implication for a limited T cell receptor alpha chain repertoire J Exp Med 1961163–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Liloglou T, Rogers SN, Brown JS, Vaughan ED, Lowe D.et al. (2006Promoter methylation of P16, RARbeta, E-cadherin, cyclin A1 and cytoglobin in oral cancer: quantitative evaluation using pyrosequencing Br J Cancer 94561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Peripheral distribution of naive, effector and central memory T cells.
Spleen TCR repertoire analysis on cDNA.
Spleen TCR repertoire analysis on genomic DNA.
Immunoglobulin plasma values in gene therapy treated mice.
Original and converted DNA sequence used for SF methylation assay.
Original and converted DNA sequence used for UCOE methylation assay.
Dominant TCR Vβ clones in peripheral blood.
Mouse β VJ rearrangements determined on genomic DNA of gene therapy treated mice.
Methylation assay of the SF promoter.
Methylation assay of the UCOE promoter.
Primer sequences for the promoters and quantitative PCR.
Oligo sequences for methylation assay.